
Transcriptome Analysis of Environmental Pseudomonas Isolates Reveals Mechanisms of Biodegradation of Naphthenic Acid Fraction Compounds (NAFCs) in Oil Sands Tailings

 ,
,
Abstract
:1. Introduction
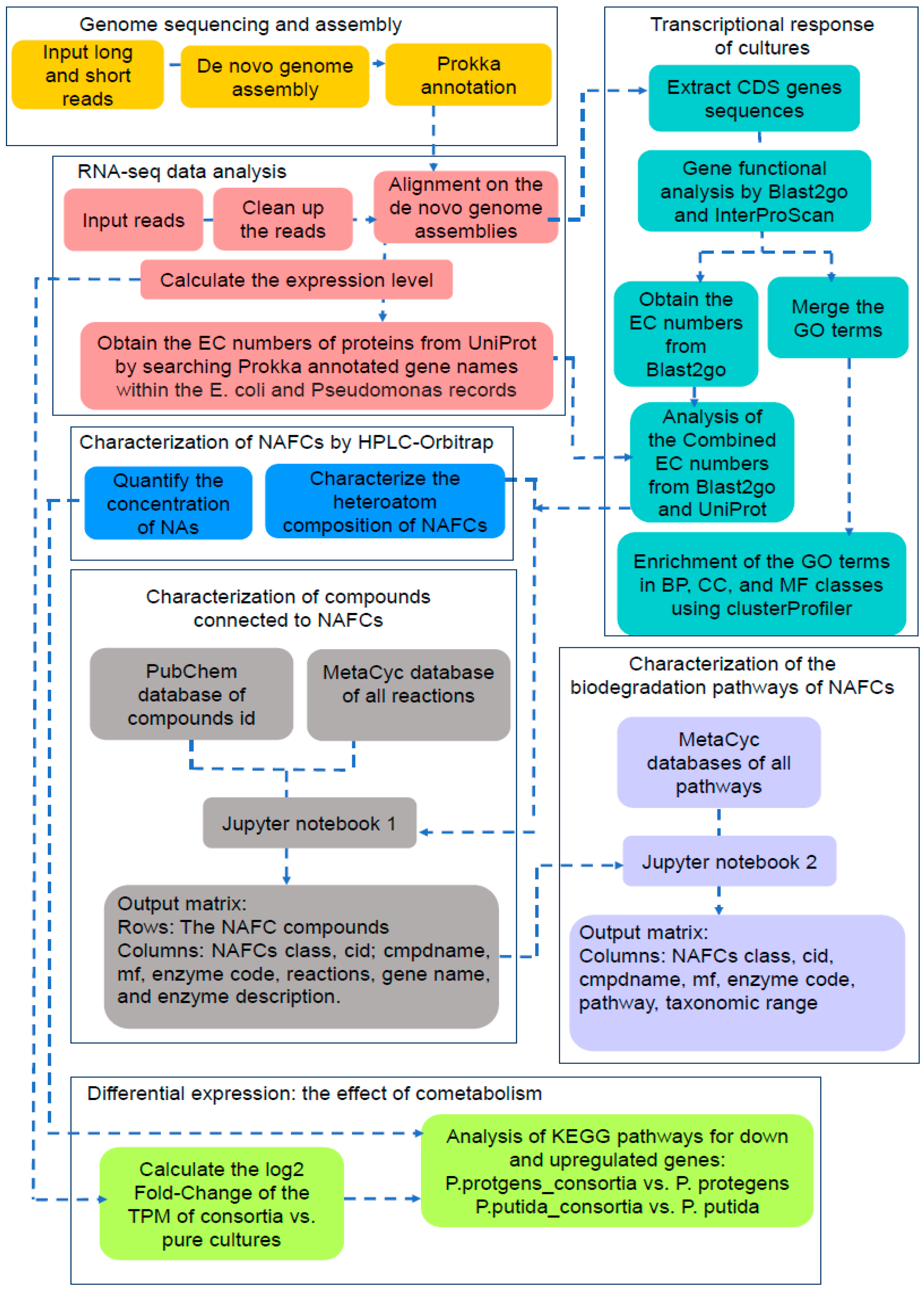
2. Materials and Methods
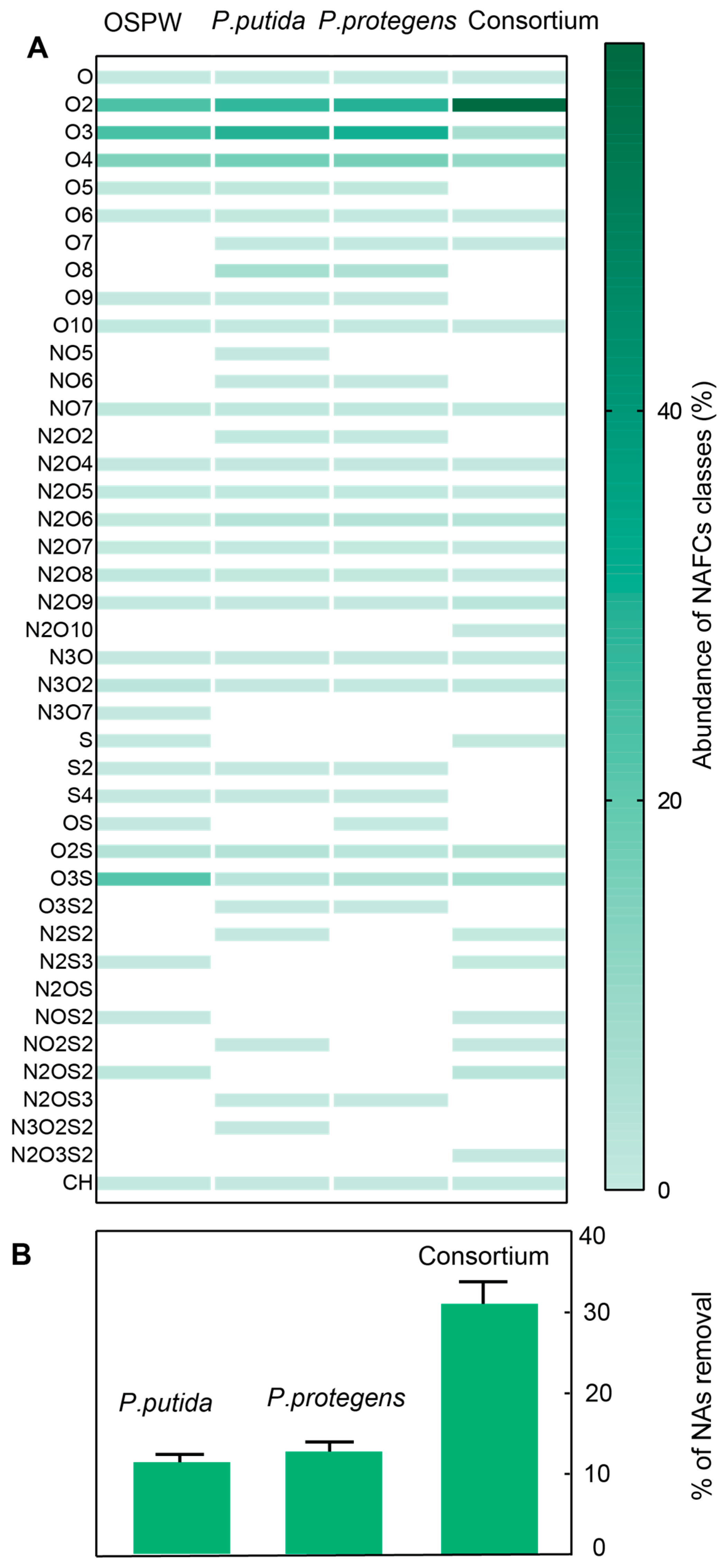
2.1. Extraction and Characterization of NAFCs
2.2. Genome Sequencing and Assembly
2.3. Evaluating Biodegradation of NAFCs by the Microbial Cultures
2.4. RNA Extraction, Library Preparation and Sequencing
2.5. Analysis of the RNA-seq Data
2.6. Characterization of the Genes Involved in Biodegradation of NAFCs
3. Results
3.1. Biodegradation of NAFCs by the Pseudomonas Cultures
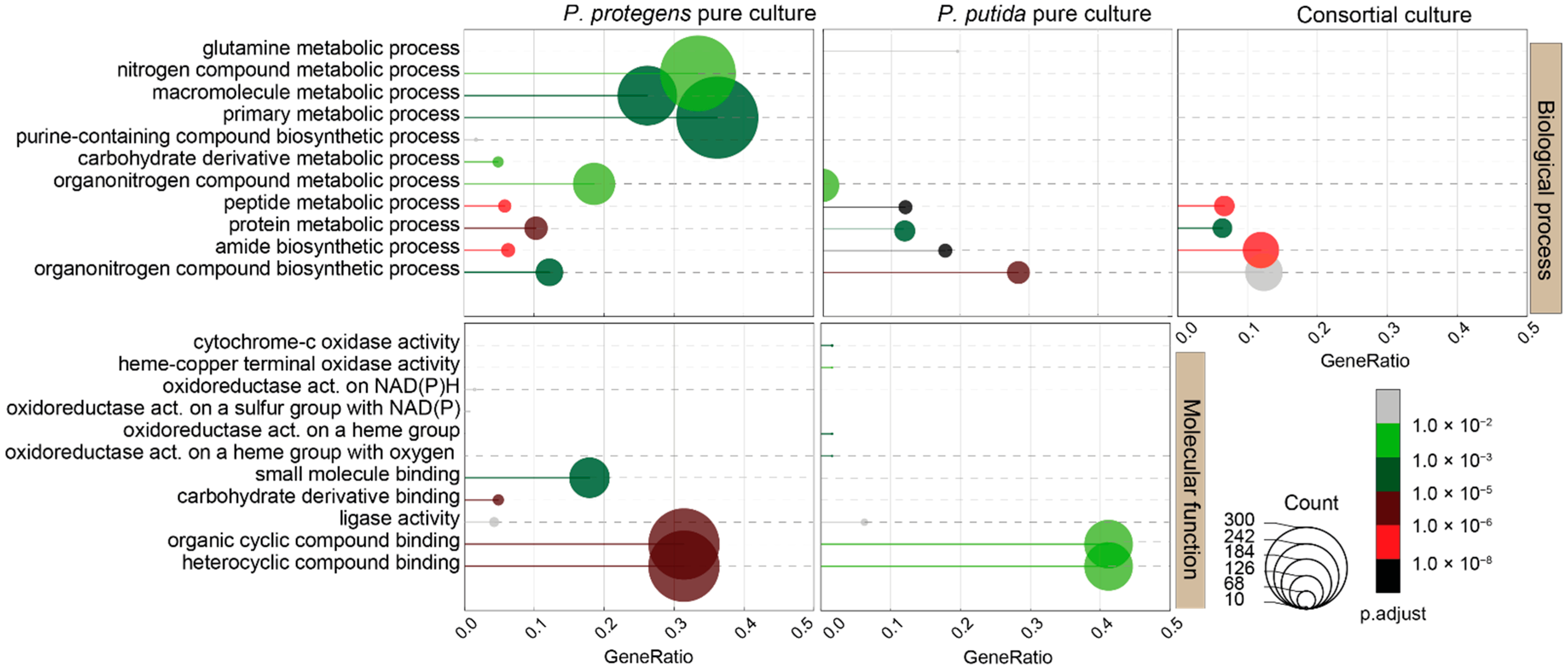
3.2. Transcriptional Responses of Pure Cultures of the Pseudomonas Isolates to NAFCs
3.3. Assessing the Similarity between Substrates of the Expressed Enzymes and NAFCs
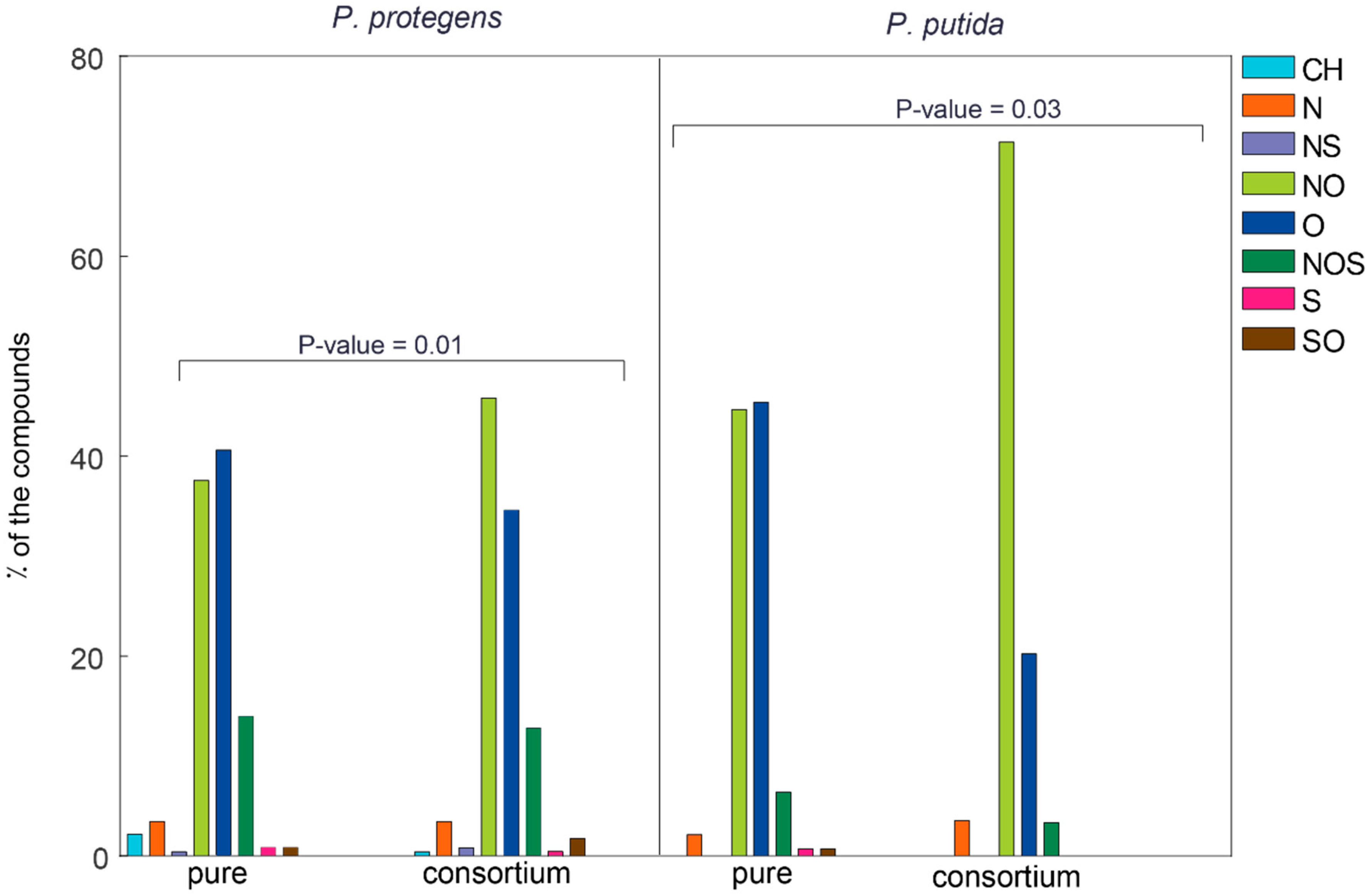
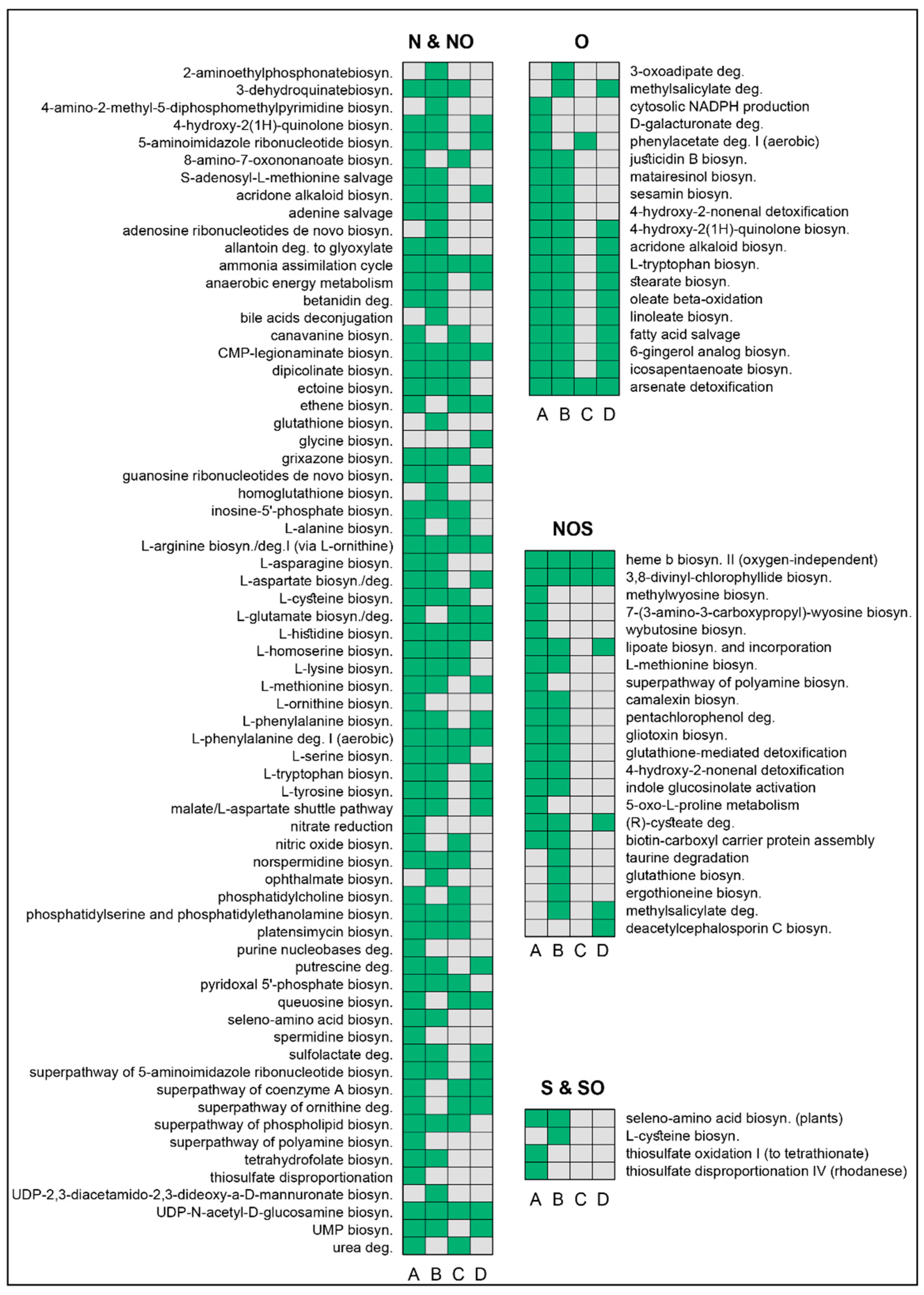
3.4. Characterization of the Pathways That Metabolize NAFCs
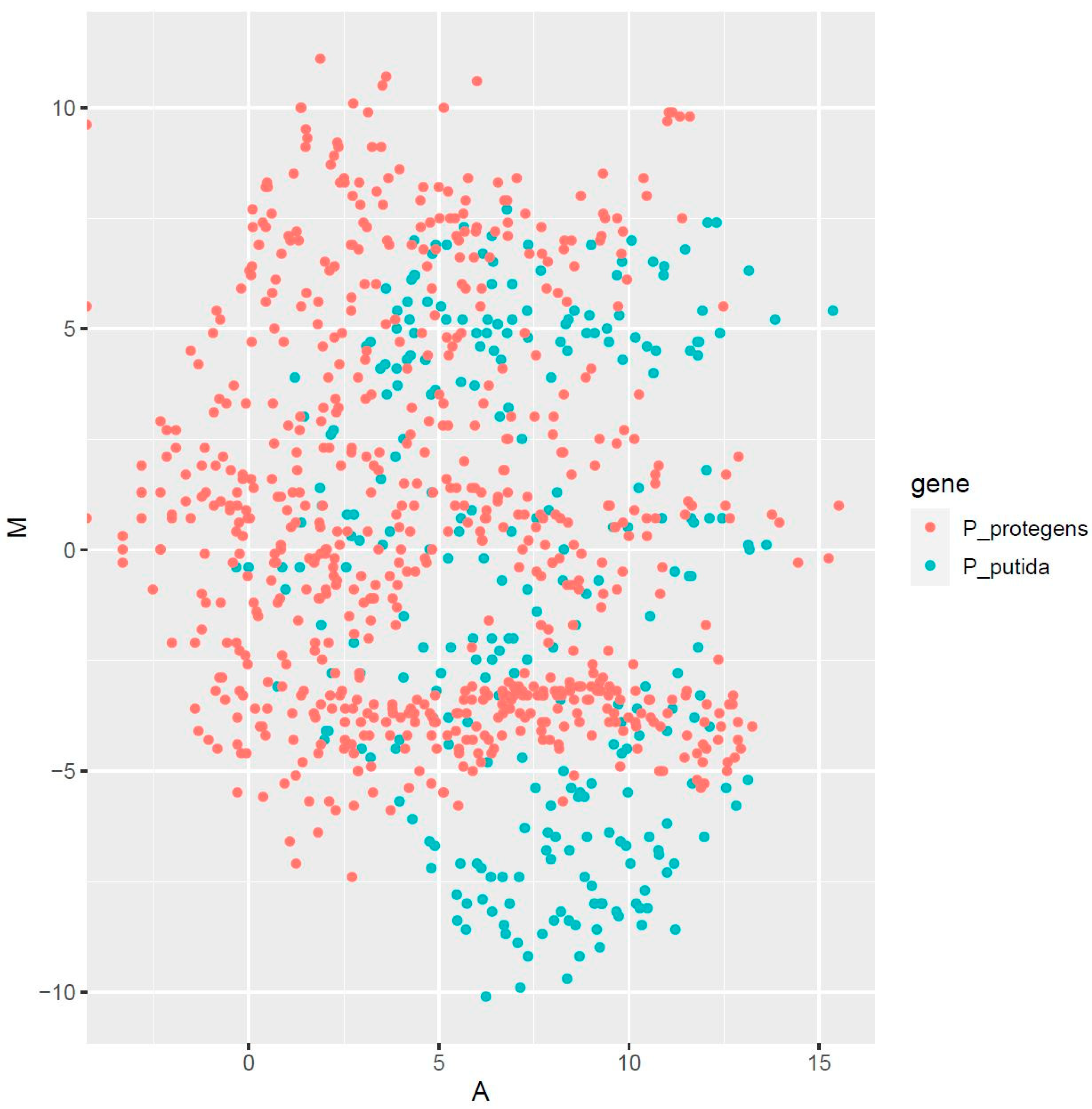
3.5. Analysis of Differential Expression in Pure and Co-Cultures of the Pseudomonads
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ahad, J.; Pakdel, H.; Gammon, P.; Mayer, B.; Savard, M.; Peru, K.; Headley, J. Distinguishing Natural from Anthropogenic Sources of Acid Extractable Organics in Groundwater near Oil Sands Tailings Ponds. Environ. Sci. Technol. 2020, 54, 2790–2799. [Google Scholar] [CrossRef]
- Lightbown, V. New SAGD Technologies Show Promise in Reducing Environmental Impact of Oil Sand Production. J. Environ. Solut. Oil Gas Min. 2015, 1, 47–58. [Google Scholar] [CrossRef]
- Scarlett, A.G.; Reinardy, H.C.; Henry, T.B.; West, C.E.; Frank, R.A.; Hewitt, L.M.; Rowland, S.J. Acute Toxicity of Aromatic and Non-Aromatic Fractions of Naphthenic Acids Extracted from Oil Sands Process-Affected Water to Larval Zebrafish. Chemosphere 2013, 93, 415–420. [Google Scholar] [CrossRef]
- Morandi, G.D.; Wiseman, S.B.; Guan, M.; Zhang, X.W.; Martin, J.W.; Giesy, J.P. Elucidating Mechanisms of Toxic Action of Dissolved Organic Chemicals in Oil Sands Process-Affected Water (OSPW). Chemosphere 2017, 186, 893–900. [Google Scholar] [CrossRef]
- Morandi, G.D.; Wiseman, S.B.; Pereira, A.; Mankidy, R.; Gault, I.G.M.; Martin, J.W.; Giesy, J.P. Effects-Directed Analysis of Dissolved Organic Compounds in Oil Sands Process-Affected Water. Environ. Sci. Technol. 2015, 49, 12395–12404. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Fu, L.; Stafford, J.; Belosevic, M.; Gamal El-Din, M. The Toxicity of Oil Sands Process-Affected Water (OSPW): A Critical Review. Sci. Total Environ. 2017, 601–602, 1785–1802. [Google Scholar] [CrossRef] [PubMed]
- Loughery, J.R.; Marentette, J.R.; Frank, R.A.; Hewitt, L.M.; Parrott, J.L.; Martyniuk, C.J. Transcriptome Profiling in Larval Fathead Minnow Exposed to Commercial Naphthenic Acids and Extracts from Fresh and Aged Oil Sands Process-Affected Water. Environ. Sci. Technol. 2019, 53, 10435–10444. [Google Scholar] [CrossRef] [PubMed]
- Headley, J.V.; Peru, K.M.; Fahlman, B.; Colodey, A.; McMartin, D.W. Selective Solvent Extraction and Characterization of the Acid Extractable Fraction of Athabasca Oils Sands Process Waters by Orbitrap Mass Spectrometry. Int. J. Mass Spectrom. 2013, 345–347, 104–108. [Google Scholar] [CrossRef]
- Headley, J.V.; Peru, K.M.; Barrow, M.P. Advances in Mass Spectrometric Characterization of Naphthenic Acids Fraction Compounds in Oil Sands Environmental Samples and Crude Oil—A Review. Mass Spectrom. Rev. 2016, 35, 311–328. [Google Scholar] [CrossRef] [PubMed]
- Bowman, D.T.; Jobst, K.J.; Ortiz, X.; Reiner, E.J.; Warren, L.A.; McCarry, B.E.; Slater, G.F. Improved Coverage of Naphthenic Acid Fraction Compounds by Comprehensive Two-Dimensional Gas Chromatography Coupled with High Resolution Mass Spectrometry. J. Chromatogr. A 2018, 1536, 88–95. [Google Scholar] [CrossRef]
- Pereira, A.D.S.; Bhattacharjee, S.; Martin, J.W. Characterization of Oil Sands Process Affected Waters by Liquid Chromatography Orbitrap Mass Spectrometry. Environ. Sci. Technol. 2013, 47, 5504–5513. [Google Scholar] [CrossRef]
- de Oliveira Livera, D.; Leshuk, T.; Peru, K.M.; Headley, J.V.; Gu, F. Structure-Reactivity Relationship of Naphthenic Acids in the Photocatalytic Degradation Process. Chemosphere 2018, 200, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Headley, J.V.; Peru, K.M.; Mohamed, M.H.; Frank, R.A.; Martin, J.W.; Hazewinkel, R.R.O.; Humphries, D.; Gurprasad, N.P.; Hewitt, L.M.; Muir, D.C.G.; et al. Chemical Fingerprinting of Naphthenic Acids and Oil Sands Process Waters-A Review of Analytical Methods for Environmental Samples. J. Environ. Sci. Health-Part A Toxic/Hazard. Subst. Environ. Eng. 2013, 48, 1145–1163. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, Y.; El-Din, M.G. Degradation of Recalcitrant Naphthenic Acids from Raw and Ozonated Oil Sands Process-Affected Waters by a Semi-Passive Biofiltration Process. Water Res. 2018, 133, 310–318. [Google Scholar] [CrossRef]
- Quesnel, D.M.; Oldenburg, T.B.P.; Larter, S.R.; Gieg, L.M.; Chua, G. Biostimulation of Oil Sands Process-Affected Water with Phosphate Yields Removal of Sulfur-Containing Organics and Detoxification. Environ. Sci. Technol. 2015, 49, 13012–13020. [Google Scholar] [CrossRef]
- Bauer, A.E.; Hewitt, L.M.; Parrott, J.M.; Bartlett, A.J.; Gillis, P.L.; Deeth, L.E.; Rudy, M.D.; Vanderveen, R.; Brown, L.; Campbell, S.D.; et al. The Toxicity of Organic Fractions from Aged Oil Sands Process-Affected Wate to Aquatic Species. Sci. Total Environ. 2019, 669, 702–710. [Google Scholar] [CrossRef]
- Quagraine, E.K.; Peterson, H.G.; Headley, J.V. In Situ Bioremediation of Naphthenic Acids Contaminated Tailing Pond Waters in the Athabasca Oil Sands Region—Demonstrated Field Studies and Plausible Options: A Review. J. Environ. Sci. Health-Part A Toxic/Hazard. Subst. Environ. Eng. 2005, 40, 685–722. [Google Scholar] [CrossRef] [PubMed]
- Allen, E.W. Process Water Treatment in Canada’s Oil Sands Industry: I. Target Pollutants and Treatment Objectives. J. Environ. Eng. Sci. 2008, 7, 123–138. [Google Scholar] [CrossRef] [Green Version]
- COSIA COSIA Challenge: Passive Organics Treatment Technology. Available online: https://bit.ly/3dOJgqE (accessed on 2 April 2020).
- Yu, X.; Lee, K.; Ma, B.; Asiedu, E.; Ulrich, A.C. Indigenous Microorganisms Residing in Oil Sands Tailings Biodegrade Residual Bitumen. Chemosphere 2018, 209, 551–559. [Google Scholar] [CrossRef]
- Siddique, T.; Stasik, S.; Mohamad Shahimin, M.F.; Wendt-Potthoff, K. Microbial Communities in Oil Sands Tailings: Their Implications in Biogeochemical Processes and Tailings Management; McGenity, T.J., Ed.; Springer: Berlin, Germany, 2018. [Google Scholar]
- Foght, J.M.; Gieg, L.M.; Siddique, T. The Microbiology of Oil Sands Tailings: Past, Present, Future. FEMS Microbiol. Ecol. 2017, 93, fix034. [Google Scholar] [CrossRef] [Green Version]
- Xue, J.; Zhang, Y.; Liu, Y.; Gamal El-Din, M. Dynamics of Naphthenic Acids and Microbial Community Structures in a Membrane Bioreactor Treating Oil Sands Process-Affected Water: Impacts of Supplemented Inorganic Nitrogen and Hydraulic Retention Time. RSC Adv. 2017, 7, 17670–17681. [Google Scholar] [CrossRef] [Green Version]
- Xue, J.; Huang, C.; Zhang, Y.; Liu, Y.; Gamal El-Din, M. Bioreactors for Oil Sands Process-Affected Water (OSPW) Treatment: A Critical Review. Sci. Total Environ. 2018, 627, 916–933. [Google Scholar] [CrossRef]
- Martin, J.W. The Challenge: Safe Release and Reintegration of Oil Sands Process-Affected Water. Environ. Toxicol. Chem. 2015, 34, 2682. [Google Scholar] [CrossRef] [Green Version]
- Del Rio, L.F.; Hadwin, A.K.M.; Pinto, L.J.; MacKinnon, M.D.; Moore, M.M. Degradation of Naphthenic Acids by Sediment Micro-Organisms. J. Appl. Microbiol. 2006, 101, 1049–1061. [Google Scholar] [CrossRef] [PubMed]
- Hadwin, A.K.M.; Del Rio, L.F.; Pinto, L.J.; Painter, M.; Routledge, R.; Moore, M.M. Microbial Communities in Wetlands of the Athabasca Oil Sands: Genetic and Metabolic Characterization. FEMS Microbiol. Ecol. 2006, 55, 68–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; McPhedran, K.N.; Gamal El-Din, M. Pseudomonads Biodegradation of Aromatic Compounds in Oil Sands Process-Affected Water. Sci. Total Environ. 2015, 521–522, 59–67. [Google Scholar] [CrossRef]
- Johnson, R.J.; West, C.E.; Swaih, A.M.; Folwell, B.D.; Smith, B.E.; Rowland, S.J.; Whitby, C. Aerobic Biotransformation of Alkyl Branched Aromatic Alkanoic Naphthenic Acids via Two Different Pathways by a New Isolate of Mycobacterium. Environ. Microbiol. 2012, 14, 872–882. [Google Scholar] [CrossRef]
- Alberta Energy Regulator. Directive 085: Fluid Tailings Management for Oil Sands Mining Projects; Alberta Energy Regulator: Calgary, AB, Canada, 2017. [Google Scholar]
- Quinlan, P.J.; Tam, K.C. Water Treatment Technologies for the Remediation of Naphthenic Acids in Oil Sands Process-Affected Water. Chem. Eng. J. 2015, 279, 696–714. [Google Scholar] [CrossRef]
- Chegounian, P.; Zerriffi, H.; Yadav, V.G. Engineering Microbes for Remediation of Oil Sands Tailings. Trends Biotechnol. 2020, 38, 1192–1196. [Google Scholar] [CrossRef]
- Wilde, M.J.; Rowland, S.J. Naphthenic Acids in Oil Sands Process Waters: Identification by Conversion of the Acids or Esters to Hydrocarbons. Org. Geochem. 2018, 115, 188–196. [Google Scholar] [CrossRef]
- Aitken, C.M.; Head, I.M.; Jones, D.M.; Rowland, S.J.; Scarlett, A.G.; West, C.E. Comprehensive Two-Dimensional Gas Chromatography-Mass Spectrometry of Complex Mixtures of Anaerobic Bacterial Metabolites of Petroleum Hydrocarbons. J. Chromatogr. A 2018, 1536, 96–109. [Google Scholar] [CrossRef] [Green Version]
- Rowland, S.J.; Scarlett, A.G.; Jones, D.; West, C.E.; Frank, R.A. Diamonds in the Rough: Identification of Individual Naphthenic Acids in Oil Sands Process Water. Environ. Sci. Technol. 2011, 45, 3154–3159. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Scott, A.C.; Fedorak, P.M.; Bataineh, M.; Martin, J.W. Influence of Molecular Structure on the Biodegradability of Naphthenic Acids. Environ. Sci. Technol. 2008, 42, 1290–1295. [Google Scholar] [CrossRef]
- Misiti, T.M.; Tezel, U.; Pavlostathis, S.G. Effect of Alkyl Side Chain Location and Cyclicity on the Aerobic Biotransformation of Naphthenic Acids. Environ. Sci. Technol. 2014, 48, 7909–7917. [Google Scholar] [CrossRef]
- Blakley, E.R. The Microbial Degradation of Cyclohexanecarboxylic Acid: A Pathway Involving Aromatization to Form p-Hydroxybenzoic Acid. J. Microbiol. 1974, 20, 1297–1306. [Google Scholar] [CrossRef]
- Wang, X.; Chen, M.; Xiao, J.; Hao, L.; Crowley, D.E.; Zhang, Z.; Yu, J.; Huang, N.; Huo, M.; Wu, J. Genome Sequence Analysis of the Naphthenic Acid Degrading and Metal Resistant Bacterium Cupriavidus Gilardii CR3. PLoS ONE 2015, 10, e0132881. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.J.; Smith, B.E.; Sutton, P.A.; McGenity, T.J.; Rowland, S.J.; Whitby, C. Microbial Biodegradation of Aromatic Alkanoic Naphthenic Acids Is Affected by the Degree of Alkyl Side Chain Branching. ISME J. 2011, 5, 486–496. [Google Scholar] [CrossRef] [Green Version]
- Scott, A.C.; MacKinnon, M.D.; Fedorak, P.M. Naphthenic Acids in Athabasca Oil Sands Tailings Waters Are Less Biodegradable than Commercial Naphthenic Acids. Environ. Sci. Technol. 2005, 39, 8388–8394. [Google Scholar] [CrossRef] [PubMed]
- Franzosa, E.A.; Hsu, T.; Sirota-Madi, A.; Shafquat, A.; Abu-Ali, G.; Morgan, X.C.; Huttenhower, C. Sequencing and beyond: Integrating Molecular “omics” for Microbial Community Profiling. Nat. Rev. Microbiol. 2015, 13, 360–372. [Google Scholar] [CrossRef] [Green Version]
- Huang, N.; Mao, J.; Zhao, Y.; Hu, M.; Wang, X. Multiple Transcriptional Mechanisms Collectively Mediate Copper Resistance in Cupriavidus Gilardii CR3. Environ. Sci. Technol. 2019, 53, 4609–4618. [Google Scholar] [CrossRef] [PubMed]
- Chegounian, P.; Yadav, V.G. Biodegradation of Toxic Organic Compounds in Contaminated Environments. U.S. Patent 2020/0318163 A1, 8 October 2020. [Google Scholar]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Thomas, K.V.; Langford, K.; Petersen, K.; Smith, A.J.; Tollefsen, K.E. Effect-Directed Identification of Naphthenic Acids as Important in Vitro Xeno-Estrogens and Anti-Androgens in North Sea Offshore Produced Water Discharges. Environ. Sci. Technol. 2009, 43, 8066–8071. [Google Scholar] [CrossRef] [PubMed]
- Diatchenko, L.; Lau, Y.F.; Campbell, A.P.; Chenchik, A.; Moqadam, F.; Huang, B.; Lukyanov, S.; Lukyanov, K.; Gurskaya, N.; Sverdlov, E.D.; et al. Suppression Subtractive Hybridization: A Method for Generating Differentially Regulated or Tissue-Specific CDNA Probes and Libraries. Proc. Natl. Acad. Sci. USA 1996, 93, 6025–6030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A Flexible Suite of Utilities for Comparing Genomic Features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Hunter, S.; Jones, P.; Mitchell, A.; Apweiler, R.; Attwood, T.K.; Bateman, A.; Bernard, T.; Binns, D.; Bork, P.; Burge, S.; et al. InterPro in 2011: New Developments in the Family and Domain Prediction Database. Nucleic Acids Res. 2012, 40, 306–312. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. ClusterProfiler: An R Package for Comparing Biological Themes among Gene Clusters. OMICS A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Ornston, L.; Stanier, R. The Conversion of Catechol and Protocatechuate to Beta-Ketoadipate by Pseudomonas Putida. J. Biol. Chem. 1966, 241, 3776–3786. [Google Scholar] [CrossRef]
- Cámara, B.; Bielecki, P.; Kaminski, F.; Dos Santos, V.M.; Plumeier, I.; Nikodem, P.; Pieper, D.H. A Gene Cluster Involved in Degradation of Substituted Salicylates via Ortho Cleavage in Pseudomonas Sp. Strain MT1 Encodes Enzymes Specifically Adapted for Transformation of 4-Methylcatechol and 3-Methylmuconate. J. Bacteriol. 2007, 189, 1664–1674. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Zhang, Y.; Patterson, J.; Arslan, M.; Zhang, Y.; Gamal El-Din, M. Biofiltration of Oil Sands Process Water in Fixed-Bed Biofilm Reactors Shapes Microbial Community Structure for Enhanced Degradation of Naphthenic Acids. Sci. Total Environ. 2020, 718, 137028. [Google Scholar] [CrossRef]
- Friedman, J.; Higgins, L.M.; Gore, J. Community Structure Follows Simple Assembly Rules in Microbial Microcosms. Nat. Ecol. Evol. 2017, 1, 0109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzmán, G.I.; Sandberg, T.E.; LaCroix, R.A.; Nyerges, Á.; Papp, H.; de Raad, M.; King, Z.A.; Hefner, Y.; Northen, T.R.; Notebaart, R.A.; et al. Enzyme Promiscuity Shapes Adaptation to Novel Growth Substrates. Mol. Syst. Biol. 2019, 15, e8462. [Google Scholar] [CrossRef] [PubMed]
- Senior, E.; Bull, A.; Slater, J. Enzyme Evolution in a Microbial Community Growing on the Herbicide Dalapon. Nature 1976, 263, 476–479. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme Class | P. putida | P. putida in Co-Culture | P. protegens | P. protegens in Co-Culture |
|---|---|---|---|---|
| EC 1—Oxidoreductases | 41 | 25 | 72 | 62 |
| EC 2—Transferases | 54 | 40 | 122 | 126 |
| EC 3—Hydrolases | 70 | 77 | 276 | 305 |
| EC 4—Lyases | 15 | 6 | 23 | 25 |
| EC 5—Isomerases | 15 | 6 | 18 | 19 |
| EC 6—Ligases | 21 | 10 | 33 | 19 |
| EC 7—Translocases | 16 | 6 | 23 | 17 |
| Top 5 Pathways with the Greatest Increase in TPM | |||
| Name | ID | Enzyme Count | Genes |
| Fatty acid degradation | map00071 | 8 | fadB_2, betB_2, fadD_3, mmgC_1, dmdC_5 |
| Valine, leucine and isoleucine degradation | map00280 | 7 | davT_1, fadB_2, betB_2, dmdC_5, aidB, lpdG_2 |
| Glycolysis/gluconeogenesis | map00010 | 6 | betB_2, aceE, ppsA, cbbA, lpdG_2 |
| Tryptophan metabolism | map00380 | 6 | fadB_2, betB_2, mmgC_1, DJCOHBMJ_04543, lpdG_2 |
| Amino sugar and nucleotide sugar metabolism | map00520 | 6 | wbpA, capD, rkpK_1, gtaB, glmM, gtaB |
| Top 5 Pathways with the Greatest Decrease in TPM | |||
| Name | ID | Enzyme Count | Genes |
| Purine metabolism | map00230 | 10 | recA, uvrD, zapE_3, lepA, uvrA_2, rep_2, lon_1, DJCOHBMJ_04023, apxIB, ettA, gyrB, ffh, DJCOHBMJ_04017, recD, amn, cysC_1guaA, relA_2, apt, DJCOHBMJ_06051, nrdB, purL, hpt |
| Propanoate metabolism | map00640 | 10 | fadJ_2, acnD, menB_1, bauC_1, accD, bkdA2, acs, bauC_1, lpdG_1, sucD |
| Carbon fixation pathways in prokaryotes | map00720 | 7 | fadJ_2, accD, acs, acnD, fumC_1, sucD |
| Valine, leucine and isoleucine degradation | map00280 | 6 | fadJ_2, menB_1, bauC_1, bkdA2, lpdG_1 |
| Drug metabolism—other enzymes | map00983 | 6 | yfcG_2, gstB_1, guaA, rnk_1, ileS, nrdB, hpt |
| Top 5 Pathways with the Greatest Increase in TPM | |||
| Name | ID | Enzyme Count | Genes |
| Pyruvate metabolism | map00620 | 5 | maeB |
| Glutathione metabolism | map00480 | 3 | ldc, ggt_1, ggt_1 |
| Aminoacyl-tRNA biosynthesis | map00970 | 3 | metG, lysS, ndk |
| Arginine and proline metabolism | map00330 | 3 | ldc, map_1, HNFJGDPB_03903 |
| Lysine biosynthesis | map00300 | 2 | dapL |
| Top 5 Pathways with the Greatest Decrease in TPM | |||
| Name | ID | Enzyme Count | Genes |
| Glycolysis/Gluconeogenesis | map00010 | 4 | pckA, pfkB |
| Pyruvate metabolism | map00620 | 3 | pckA, hchA |
| Arginine biosynthesis | map00220 | 3 | gdhB, glnA |
| Amino sugar and nucleotide sugar metabolism | map00520 | 3 | wbpI, pfkB |
| Nitrogen metabolism | map00910 | 3 | gdhB, glnA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chegounian, P.; Flibotte, S.; Peru, K.; Headley, J.; McMartin, D.; Gramlich, B.; Yadav, V.G. Transcriptome Analysis of Environmental Pseudomonas Isolates Reveals Mechanisms of Biodegradation of Naphthenic Acid Fraction Compounds (NAFCs) in Oil Sands Tailings. Microorganisms 2021, 9, 2124. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9102124
Chegounian P, Flibotte S, Peru K, Headley J, McMartin D, Gramlich B, Yadav VG. Transcriptome Analysis of Environmental Pseudomonas Isolates Reveals Mechanisms of Biodegradation of Naphthenic Acid Fraction Compounds (NAFCs) in Oil Sands Tailings. Microorganisms. 2021; 9(10):2124. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9102124
Chicago/Turabian StyleChegounian, Parisa, Stephane Flibotte, Kerry Peru, John Headley, Dena McMartin, Bryne Gramlich, and Vikramaditya G. Yadav. 2021. "Transcriptome Analysis of Environmental Pseudomonas Isolates Reveals Mechanisms of Biodegradation of Naphthenic Acid Fraction Compounds (NAFCs) in Oil Sands Tailings" Microorganisms 9, no. 10: 2124. https://0-doi-org.brum.beds.ac.uk/10.3390/microorganisms9102124