
Mineralogy and Geochemistry (HFSE and REE) of the Present-Day Acid-Sulfate Types Alteration from the Active Hydrothermal System of Furnas Volcano, São Miguel Island, The Azores Archipelago

Abstract
:1. Introduction
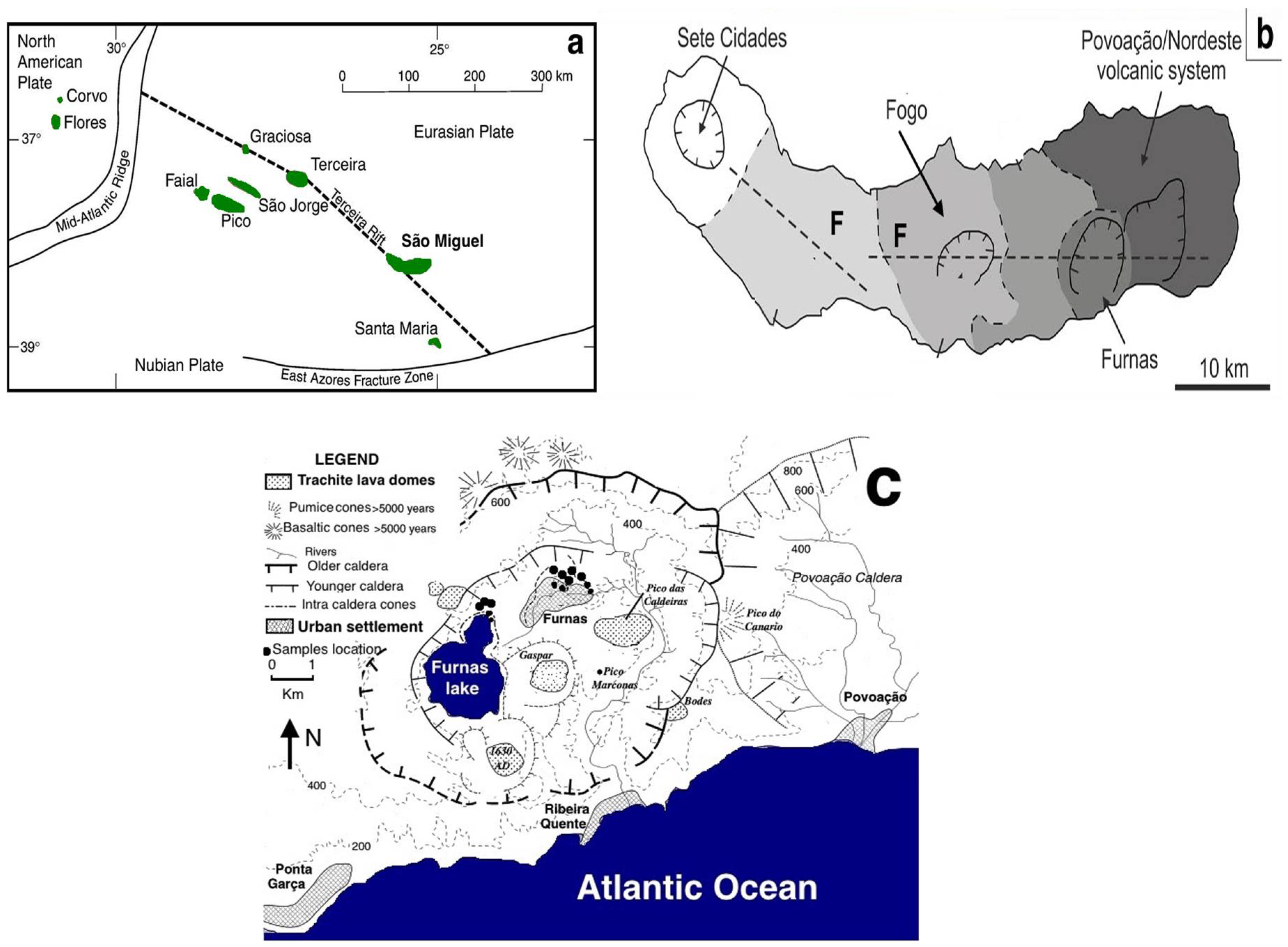
2. Geology
3. Materials and Methods
3.1. Sampling and Field Observation
3.2. Analytical Methods and Samples Preparations
4. Results
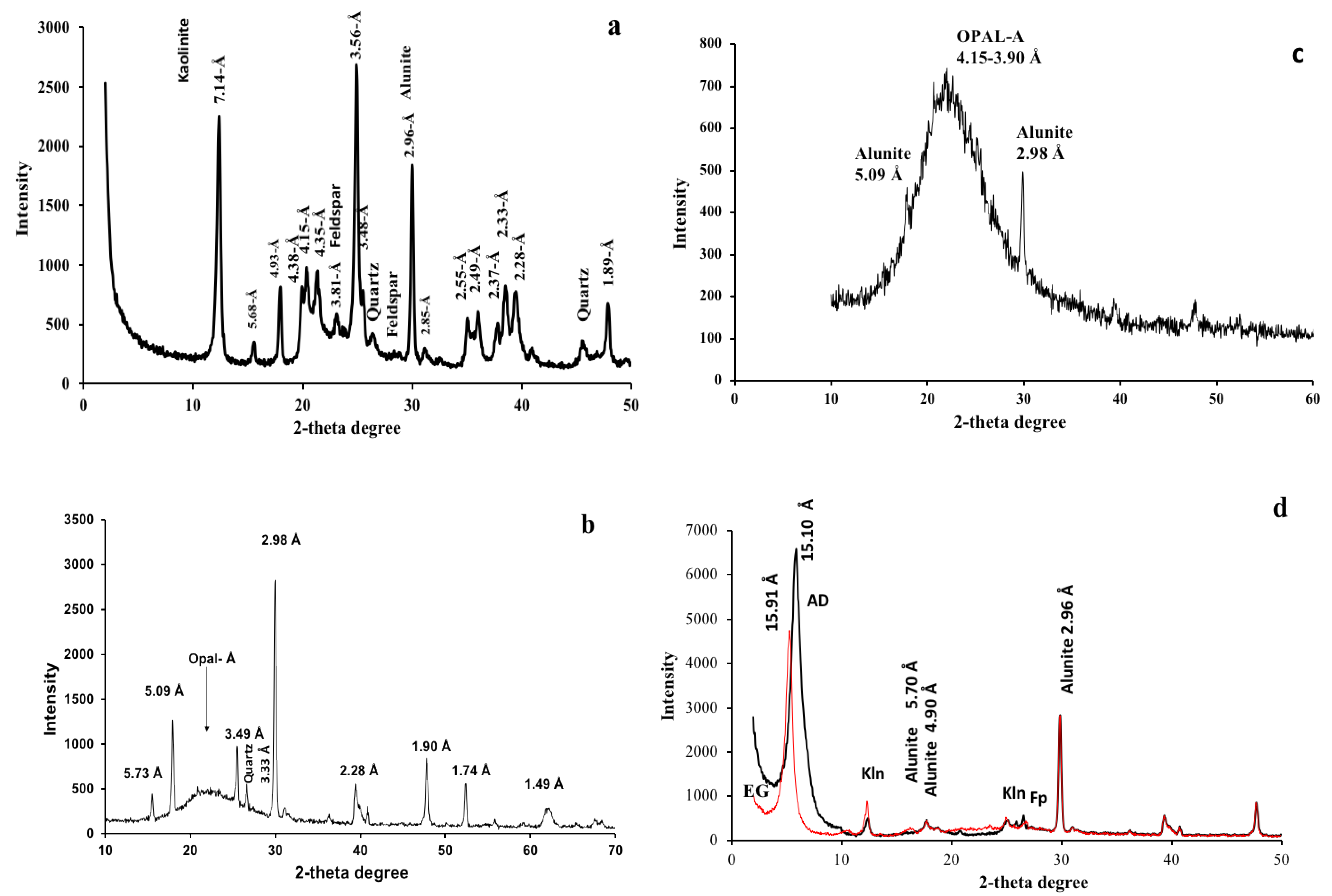
4.1. X-ray Diffraction
4.2. Scanning Electron Microscopy and Electron Microprobe Analysis
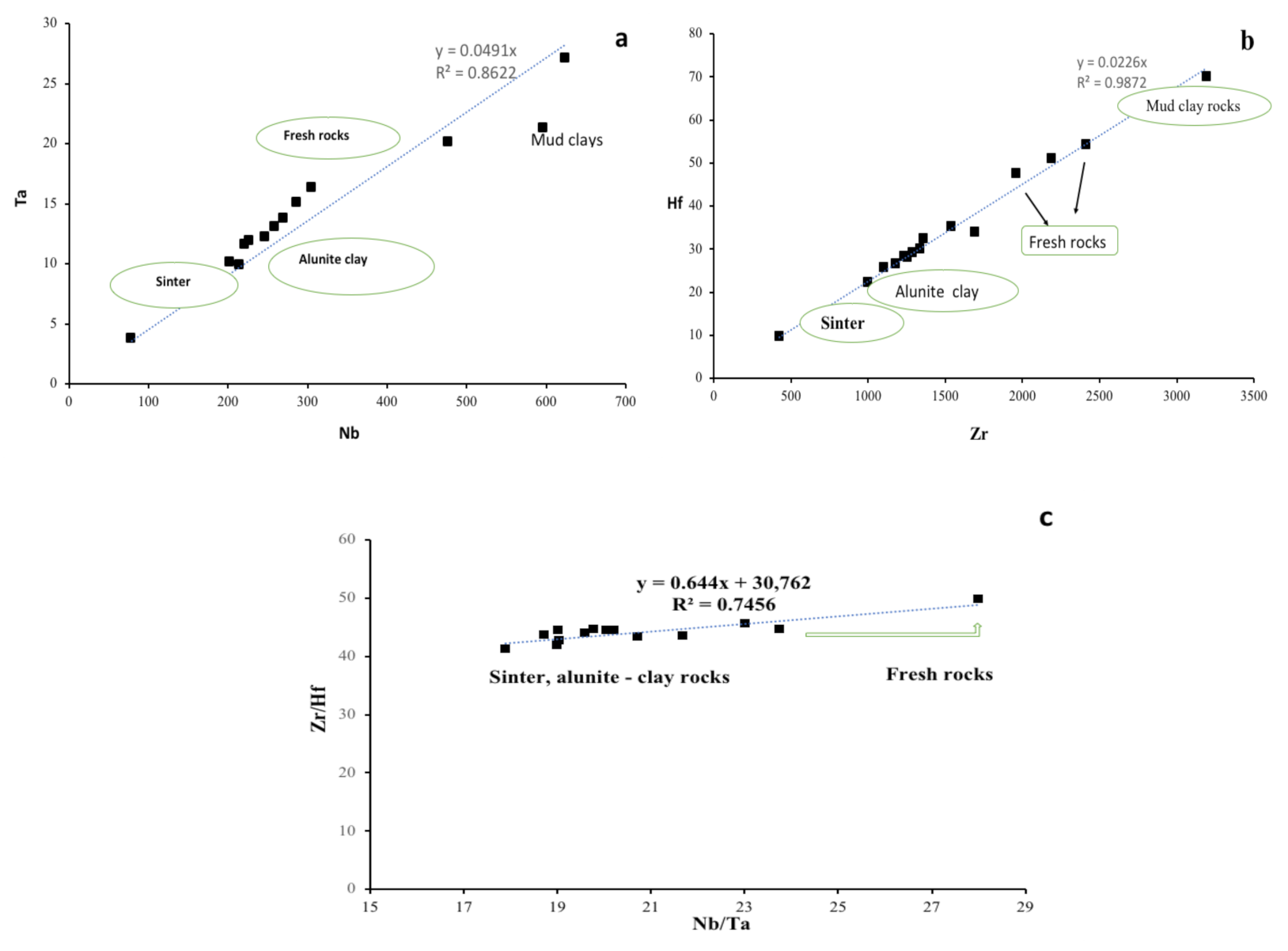
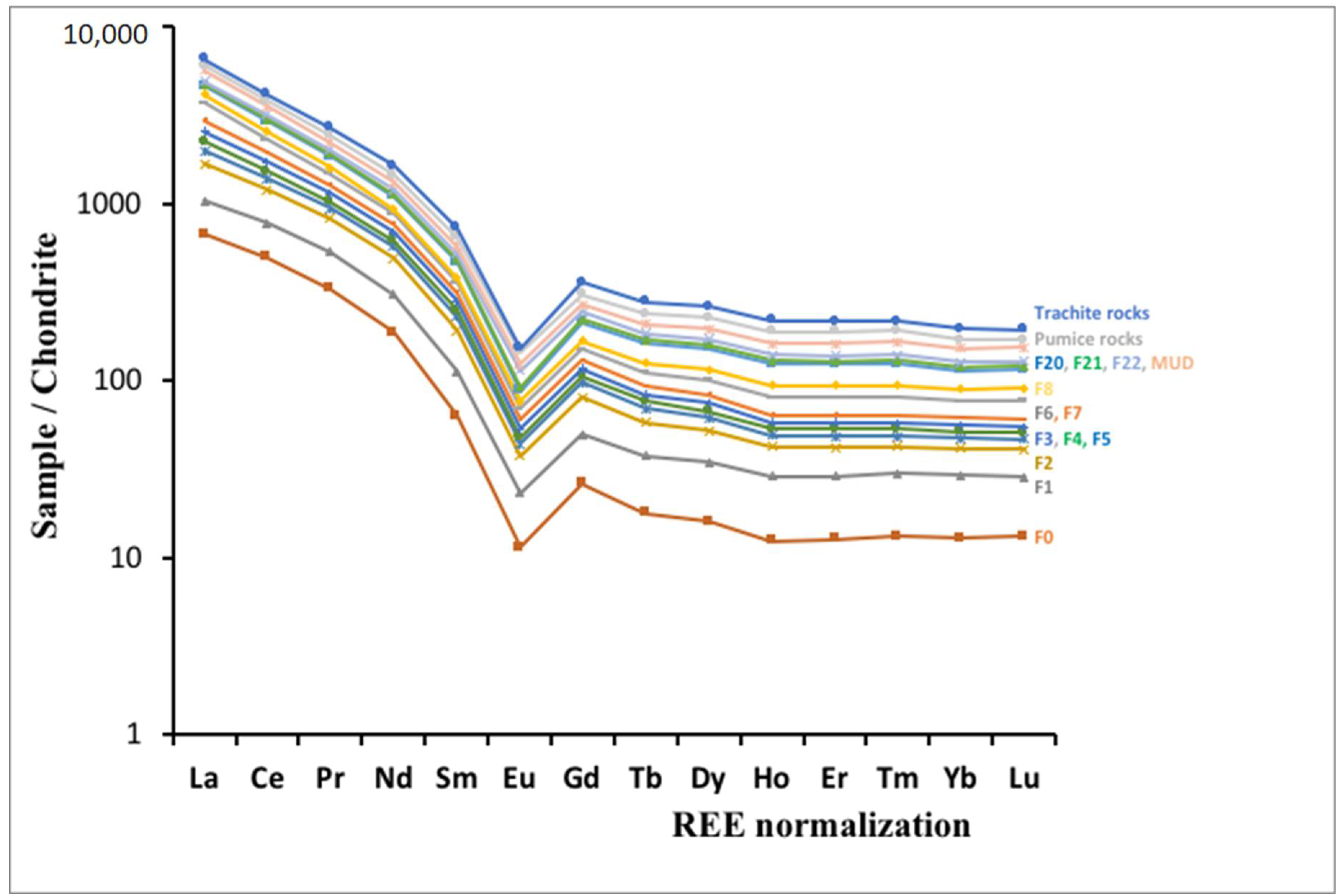
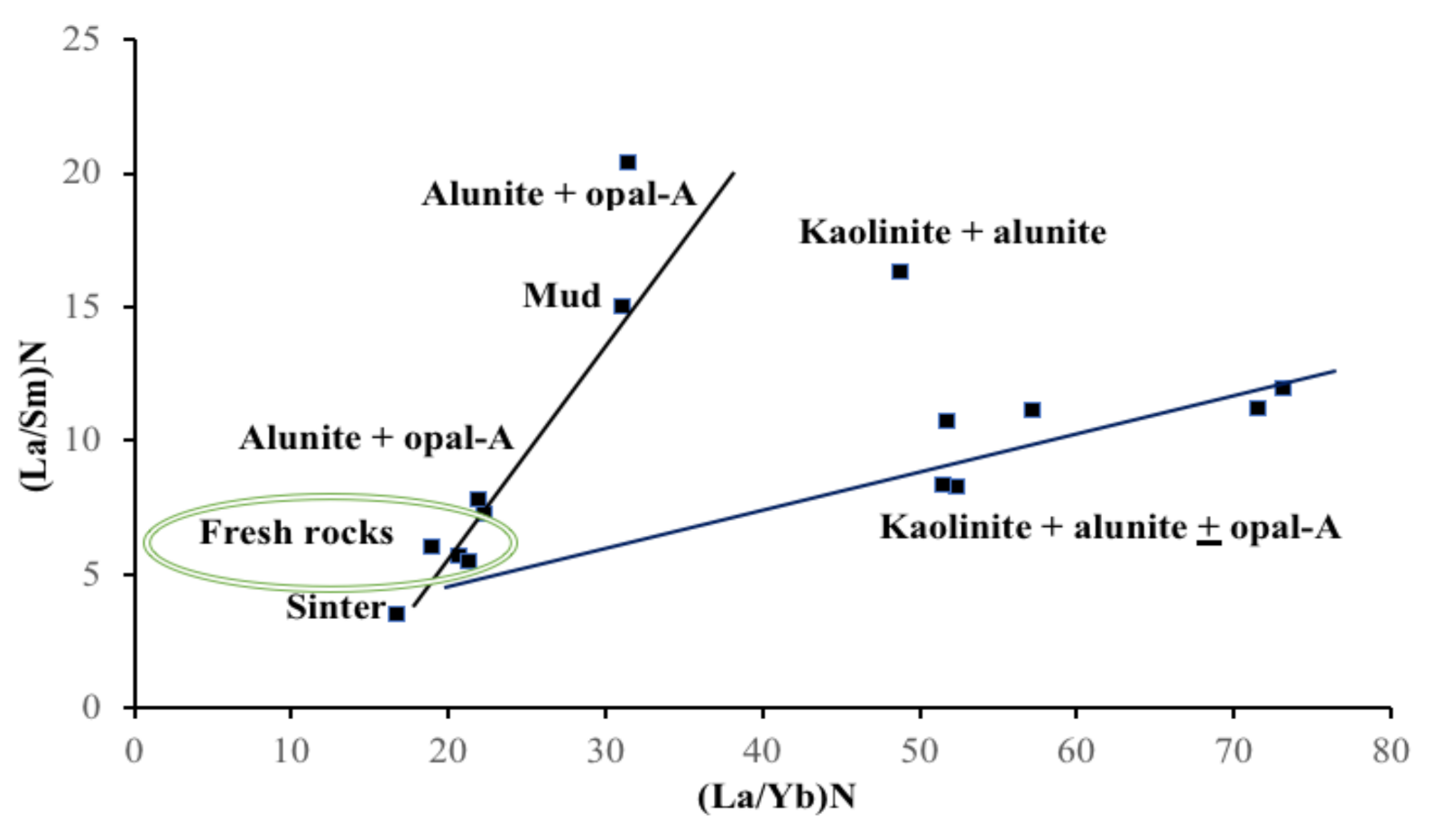
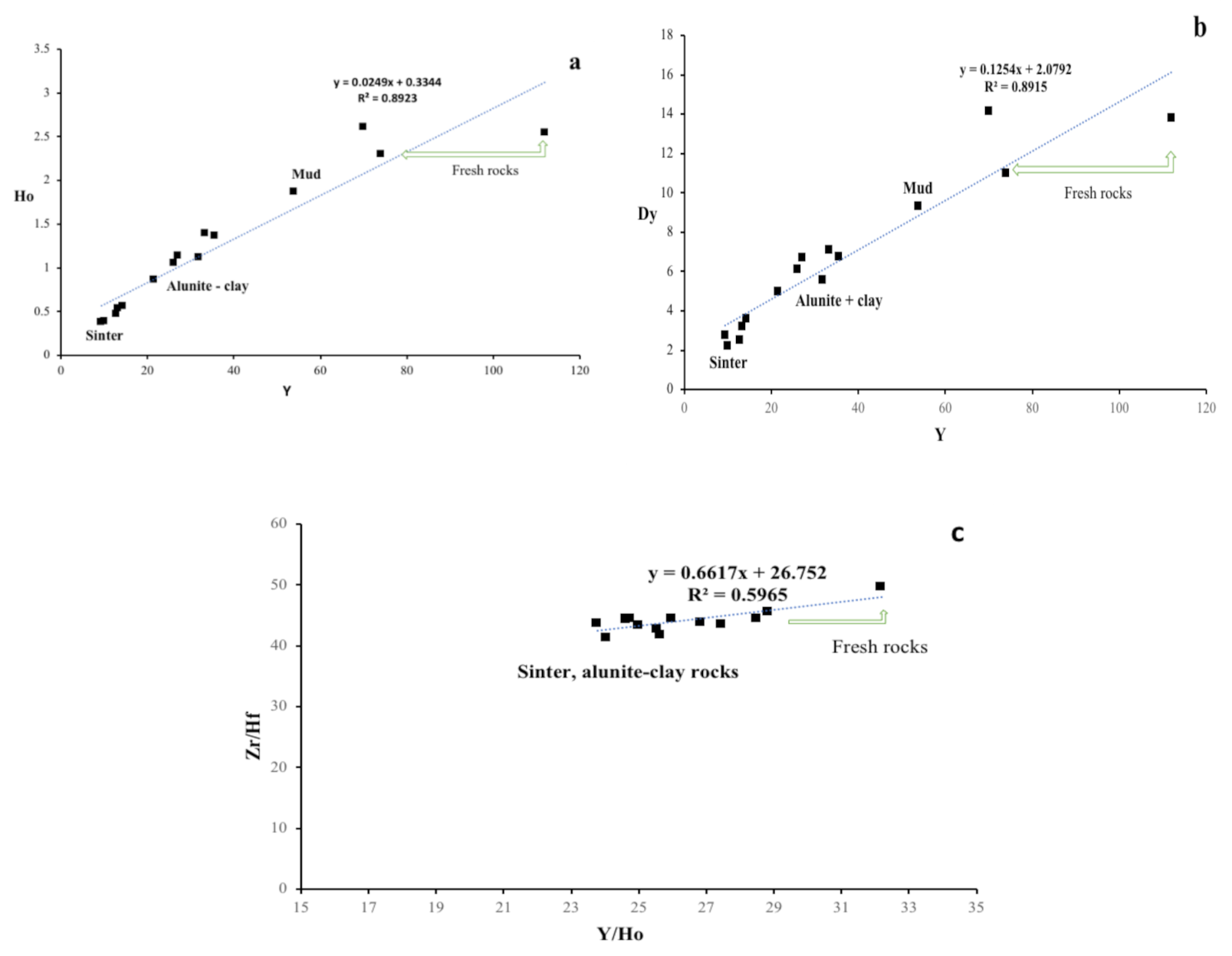
4.3. HFSE and REE Geochemistry
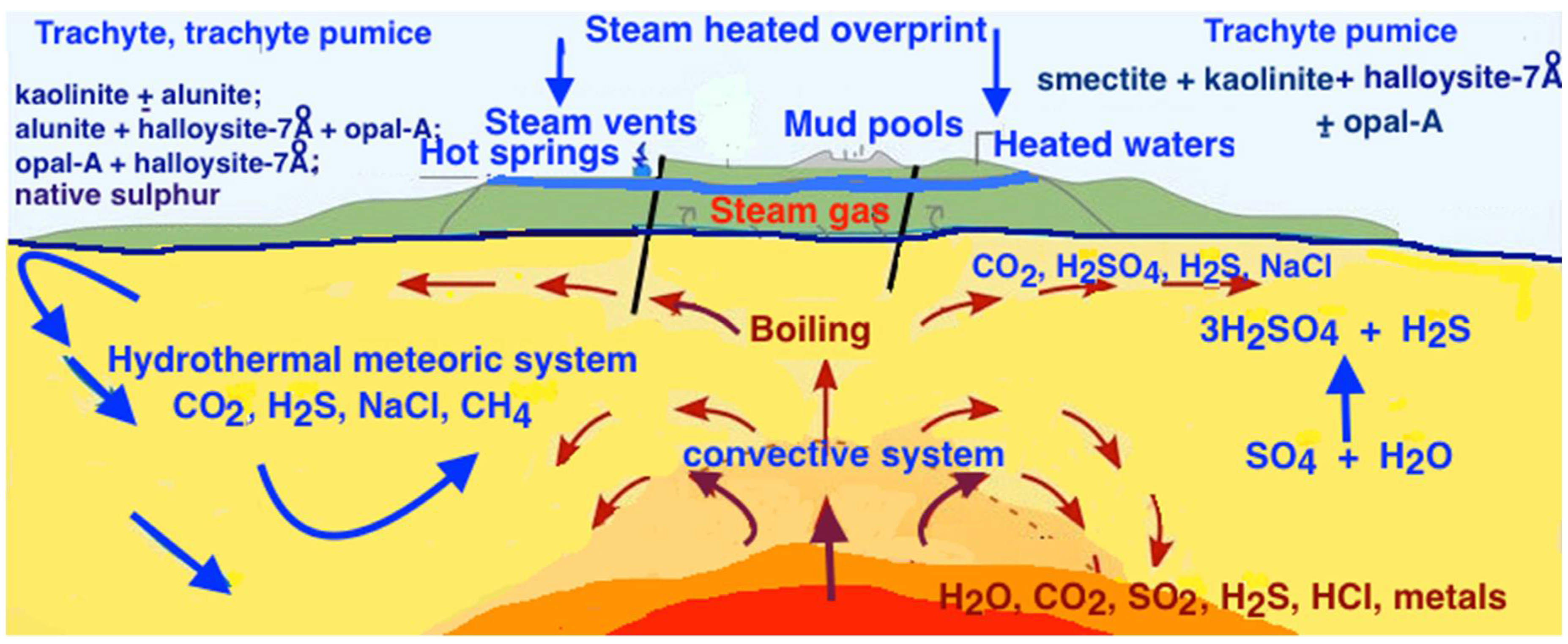
5. Discussion
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Giggenbach, W.F. Magma degassing and mineral deposition in hydrothermal systems along convergent plate boundaries. Econ. Geol. 1992, 97, 1927–1944. [Google Scholar]
- Christenson, B.W.; Wood, C.P. Evolution of a vent-hosted hydrothermal system beneath Ruapehu crater lake, New Zeeland. Bull. Volcanol. 1993, 55, 547–565. [Google Scholar] [CrossRef]
- Delmelle, P.; Bernard, A. Geochemistry, mineralogy, and chemical modelling of the acid crater lake of Kawah Ijen volcano, Indonesia. Geochim. Cosmochim. Acta 1994, 58, 2445–2460. [Google Scholar] [CrossRef]
- Africano, F.; Bernard, A. Acid alteration in the fumarolic environment of Usu volcano, Hokkaido, Japan. J. Volcanol. Geotherm. Res. 2000, 97, 475–495. [Google Scholar] [CrossRef]
- Berger, B.R.; Henley, R.W.; Lowers, H.A.; Pribil, M.J. The Lepanto Cu–Au deposit, Philippines: A fossil hyperacidic volcanic lake complex. J. Volcanol. Geotherm. Res. 2014, 271, 70–82. [Google Scholar] [CrossRef]
- Henley, R.W. Hyperacidic Volcanic Lakes, Metal Sinks and Magmatic Gas Expansion in Arc Volcanoes. In Volcanic Lakes, Advances in Volcanology; Springer: Berlin/Heidelberg, Germany, 2015; pp. 155–184. [Google Scholar] [CrossRef]
- Giggenbach, W.F. Isotopic shifts in waters from geothermal and volcanic systems along convergent plate boundaries and their origin. Earth Planet. Sci. Lett. 1992, 113, 495–510. [Google Scholar] [CrossRef]
- Hedenquist, J.W.; Aoki, M.; Shinohara, H. Flux of volatiles and ore-forming metals from the magmatic-hydrothermal system of Satsuma Iwojima volcano. Geology 1994, 22, 585–588. [Google Scholar] [CrossRef]
- Symonds, R.B.; Rose, W.I.; Bluth, G.J.S.; Gerlach, T.M. Volcanic-gas studies: Methods, results and applications. Rev. Mineral. 1994, 30, 1–66. [Google Scholar]
- Shinohara, H. A missing link between volcanic degassing and experimental studies on chloride partitioning. Chem. Geol. 2009, 263, 51–59. [Google Scholar] [CrossRef]
- Delmelle, P.; Henley Richard, W.R.; Opfergelt, S.; Detienne, M. Summit Acid Crater Lakes and Flank Instability in Composite Volcanoes. In Volcanic Lakes; Springer: Berlin/Heidelberg, Germany, 2015; pp. 289–305. [Google Scholar] [CrossRef]
- Muffler, L.J.P. Geothermal reservoir assessment. In Geothermal Systems: Principles and Case Histories; Rybach, L., Muffler, L.J.P., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 1981; p. 181. [Google Scholar]
- Henley, R.W.; McNabb, A. Magmatic vapor plumes and ground water interaction in porphyry copper emplacement. Econ. Geol. 1978, 73, 1–20. [Google Scholar] [CrossRef]
- Hedenquist, J.W.; Arribas, A.; Reynolds, T.J. Evolution of an intrusion-centered hydrothermal system; Far Southeast-Lepanto porphyry and epithermal Cu-Au deposits, Philippines. Econ. Geol. 1998, 93, 373–404. [Google Scholar] [CrossRef] [Green Version]
- Heald, P.; Foley, N.K.; Hayba, D.O. Comparative anatomy of volcanic hosted epithermal deposits: Acid sulfate and adularia-sericite types. Econ. Geol. 1987, 82, 1–26. [Google Scholar] [CrossRef]
- Hemley, J.J.; Jones, J. Chemical aspects of hydrothermal alteration with emphasis on hydrogen metasomatism. Econ. Geol. 1964, 59, 538–569. [Google Scholar] [CrossRef]
- Hedenquist, J.W.; Lowenstern, J.B. The role of magmas in the formation of hydrothermal ore deposits. Nature 1994, 370, 519–527. [Google Scholar] [CrossRef]
- Herdianita, N.R.; Rodgers, K.A.; Browne, P.R.L. Routine procedures for characterizing modern and ancient silica sinter deposits. Geothermics 2000, 29, 367–375. [Google Scholar] [CrossRef]
- Herdianita, N.R.; Browne, P.R.L.; Rodgers, K.A.; Campbell, A. Mineralogical and textural changes accompanying ageing of silica sinter. Mineral. Depos. 2000, 35, 48–62. [Google Scholar] [CrossRef]
- Hedenquist, J.W.; Taran, Y.A. Modeling the formation of advanced argillic lithocaps: Volcanic vapor condensation above porphyry intrusions. Econ. Geol. 2013, 108, 1523–1540. [Google Scholar] [CrossRef]
- Giggenbach, W.F.; Gonfiantini, R.; Jangi, B.L.; Truesdell, A.H. Isotopic and chemical composition of Parbati Valley geothermal discharges, northwest Himalaya, India. Geothermics 1983, 12, 199–222. [Google Scholar] [CrossRef]
- Inskeep, W.; Nordstrom, D.K.; Mogk, D.; Rodman, A.; Macur, E.E.; Fouke, B.; Durães, N.; Guzman, M. Secondary Minerals associated with thermal soils and geothermal features of Yellowstone National Park. In Clays of Yellostone Park; Schroeder, P., Ed.; Clay Minerals Society: Boulder, CO, USA, 2010; pp. 29–47. [Google Scholar]
- Piochi, M.; Kilburn, C.R.J.; Di Vito, M.A.; Mormone, A.; Tramelli, A.; Troise, C.; De Natale, G. The volcanic and geothermally active Campi Flegrei caldera: An integrated multidisciplinary image of its buried structure. Int. J. Earth Sci. 2014, 10, 401–421. [Google Scholar] [CrossRef]
- Piochi, M.; Mormone, A.; Balassone, G.; Strauss, H.; Troise, C.; De Natale, G. Native sulfur, sulfates and sulfides from the active Campi Flegrei volcano (southern Italy): Genetic environments and degassing dynamics revealed by mineralogy and isotope geochemistry. J. Volcanol. Geotherm. Res. 2015, 304, 180–193. [Google Scholar] [CrossRef]
- Piochi, M.; Mormone, A.; Balassone, G. Hydrothermal alteration environments in the recent dynamics of the Ischia volcanic island (Southern Italy): Clues from repeated field, mineralogical and geochemical surveys across the 2017 earthquake of Casamicciola. J. Volcanol. Geotherm. Res. 2019, 376, 104–124. [Google Scholar] [CrossRef]
- Shakeri, A.; Ghoreyshinia, S.; Mehrabi, B.; Delavari, M. Rare earth elements geochemistry in springs from Taftan geothermal area SE Iran. J. Volcanol. Geotherm. Res. 2015, 304, 49–61. [Google Scholar] [CrossRef]
- White, D.E.; Hutchinson, R.A.; Keith, T.E.C. The Geology and Remarkable Thermal Activity of Norris Geyser Basin, Yellowstone National Park, Wyoming; U.S. Geological Survey (USGS): Reston, VA, USA, 1988; p. 1456.
- Moore, R. Volcanic geology and eruption frequency, S. Miguel, Azores. Bull. Volcanol. 1990, 52, 602–614. [Google Scholar] [CrossRef]
- Ferreira, T.; Oskarsson, N. Chemistry and isotopic composition of fumarole discharge of Furnas caldera. J. Volcanol. Geotherm. Res. 1999, 92, 169–179. [Google Scholar] [CrossRef]
- Cruz, J.V.; Coutinho, R.M.; Carvalho, M.R.; Oskarsson, N.; Gislason, S.R. Chemistry of waters from Furnas volcano, São Miguel, Azores: Fluxes of volcanic carbon dioxide and leached material. J. Volcanol. Geotherm. Res. 1999, 92, 151–167. [Google Scholar] [CrossRef]
- Searle, R. Tectonic pattern of the Azores spreading centre and triple junction. Earth Planet Sci. Lett. 1980, 51, 415–434. [Google Scholar] [CrossRef]
- Luis, J.F.; Miranda, J.M.; Galdeano, A.; Patriat, P.; Rossignol, J.C.; Mendes Victor, L.A. The Azores triple junction evolution since 10 Ma from an aerogmagnetic survey of mid-Atlantic Ridge. Earth Planet. Sci. Lett. 1994, 125, 439–459. [Google Scholar] [CrossRef]
- Marques, F.O.; Catalão, J.C.; DeMets, C.; Costa, A.C.G.; Hildenbrand, A. GPS and tectonic evidence for a diffuse plate boundary at the Azores Triple Junction. Earth Planet. Sci. Lett. 2013, 381, 177–187. [Google Scholar] [CrossRef]
- Moore, B.R. Geology of three late Quaternary stratovolcanoes on São Miguel, Azores. U.S. Geol. Surv. Bull. 1991, 1900, 1–46. [Google Scholar]
- Walker, G.P.L.; Croasdale, R. Two plinian-type eruptions in the Azores. J. Geol. Soc. Lond. 1971, 127, 17–55. [Google Scholar] [CrossRef]
- Calvert, A.T.C.; Moore, R.B.; McGeehin, J.P.; Rodrigues da Silva, A.M. Volcanic history and 40Ar/39Ar and 14C geochronology of Terceira Island, Azores, Portugal. J. Volcanol. Geotherm. Res. 2006, 156, 103–115. [Google Scholar] [CrossRef] [Green Version]
- Jeffery, A.J.; Gertisser, R.; O’Driscoll, B.; Pacheco, J.M.; Whitley, S.; Pimentel, S.A. Temporal evolution of a post-caldera, mildly peralkaline magmatic system: Furnas volcano, São Miguel, Azores. Contrib. Mineral. Petrol. 2016, 171, 42–59. [Google Scholar] [CrossRef] [Green Version]
- Cole, P.D.; Queiroz, G.; Wallenstein, N.; Gaspar, J.L.; Duncan, A.M.; Guest, J.E. An historic subplinian to phreatomagmatic eruption: The 1630 eruption of Furnas volcano, São Miguel, Azores. J. Volcanol. Geotherm. Res. 1999, 69, 117–135. [Google Scholar] [CrossRef]
- Guest, J.E.; Gaspar, J.L.; Cole, P.D.; Queiroz, G.; Duncan, M.; Wallenstein, N.; Ferreira, T.; Pacheco, J.M. Volcanic geology of Furnas volcano, São Miguel, Azores. J. Volcanol. Geotherm. Res. 1999, 92, 1–29. [Google Scholar] [CrossRef]
- Booth, B.; Walker, G.P.L.; Croasdale, R. A quantitative study of five thousand years of volcanism on São Miguel, Azores. Philos. Trans. R. Soc. Lond. 1978, 228, 271–319. [Google Scholar]
- Guest, J.E.; Pacheco, J.M.; Cole, P.D.; Duncan, A.M.; Wallenstein, N.; Queiroz, G.; Gaspar, J.L.; Ferreira, T. Volcanic geology of São Miguel Island (Azores Archipelago): Introduction. Geol. Soc. London Mem. 2015, 44, 125–134. [Google Scholar] [CrossRef]
- Cruz, J.V.; França, Z. Hydrogeochemistry of thermal and mineral water springs of the Azores archipelago (Portugal). J. Volcanol. Geotherm. Res. 2006, 151, 382–398. [Google Scholar] [CrossRef]
- Zbyszewski, G.; Almeida, F.M.; Ferreira, O.V.; Assunção, C.T. Carta Geológica de Portugal na escala 1:50,000. Notícia explicativa da folha B, S. Miguel (Açores). Serv. Geol. Port. Lisboa 1958, 37. [Google Scholar]
- Woitischek, J.; Dietzel, M.; Inguaggiato, C.; Böttcher, M.E.; Leis, L.; Cruz, V.J.; Gehre, M. Characterisation and origin of hydrothermal waters at São Miguel (Azores) inferred by chemical and isotopic composition. J. Volcanol. Geotherm. Res. 2017, 346, 104–117. [Google Scholar] [CrossRef]
- Jones, J.B.; Segnit, E.R. The nature of opal I. Nomenclature and constituent phases. J. Geol. Soc. Aust. 1971, 18, 57–68. [Google Scholar] [CrossRef]
- Flörke, O.W.; Graetsch, H.; Martin, B.; Roller, K.; Wirth, R. Nomenclature of micro- and non-crystalline silica minerals, based on structure and microstructure. Neues Jahrbh. Mineral. Abh. 1991, 163, 19–42. [Google Scholar]
- Graetsch, H. Structural characteristics of opaline and microcrystalline silica minerals. In Silica: Physical Behaviour, Geochemistry and Materials Applications; Heaney, P.J., Prewitt, C.T., Gibbs, G.V., Eds.; Mineralogical Society of America: Chantilly, VA, USA, 1994; pp. 209–232. [Google Scholar]
- Taylor, S.R.; McLennan, S.M. The geochemical evolution of the continental crust. Rev. Geophisique 1995, 33, 241–265. [Google Scholar] [CrossRef]
- Hinckley, D.N. Variability in “crystallinity” values among the kaolin deposits of the coastal plain of Georgia and South Carolina. Clays Clay Miner. 1963, 11, 229–235. [Google Scholar] [CrossRef]
- Bigham, J.M.; Schwertmann, U.; Traina, S.J.; Winland, R.L.; Wolf, M. Schwertmannite and the chemical modeling of iron in acid sulfate waters. Geochim. Cosmochim. Acta 1996, 60, 185–195. [Google Scholar] [CrossRef]
- Marschall, H.R.; Dohmen, R.; Ludwig, T. Diffusion-induced fractionation of niobium and tantalum during continental crust formation. Earth Planet. Sci. Lett. 2013, 375, 361–371. [Google Scholar] [CrossRef]
- Jiang, S.-Y.; Wang, R.-C.; Xu, X.-S.; Zhao, K.-D. Mobility of high field strength elements (HFSE) in magmatic-, metamorphic-, and submarine-hydrothermal systems. Phys. Chem. Earth 2005, 30, 1020–1029. [Google Scholar] [CrossRef]
- Münker, C.; Pfänder, J.A.; Weyer, S.; Buchl, A.; Kleine, T.; Mezger, K. Evolution of planetary cores and the Earth–Moon system from Nb/Ta systematics. Science 2003, 301, 84–87. [Google Scholar] [CrossRef]
- Wood, S.A. The geochemistry of rare earth elements and yttrium in geothermal waters. Soc. Econ. Geol. Spec. Pub. 2003, 10, 133–158. [Google Scholar]
- Peiffer, L.; Taran, Y.; Lounejeva, E.; Solís-Pichardo, E.; Rouwet, D.; Bernard-Romero, R.A. Tracing thermal aquifers of El Chichónn volcano-hydrothermal system (Mexico) with 87Sr/87Sr, Ca/Sr and REE. J. Volcanol. Geotherm. Res. 2011, 205, 55–66. [Google Scholar] [CrossRef]
- Wood, S.A. Rare element systematics of acidic geothermal waters from the Taupo Volcanic Zone, New Zealand. J. Geochem. Explor. 2006, 89, 424–427. [Google Scholar] [CrossRef]
- Fulignati, A.; Gioncada, A.; Sbrana, A. Rare-earth element (REE) behavior in the alteration facies of the active magmatic hydrothermal system of Vulcano (Aeolian Islands, Italy). J. Volcanol. Geotherm. Res. 1999, 88, 325–342. [Google Scholar] [CrossRef]
- Kulaksız, S.; Bau, M. Contrasting behaviour of anthropogenic gadolinium and natural rare earth elements in estuaries and the gadolinium input into the North Sea. Earth Planet. Sci. Lett. 2007, 260, 361–371. [Google Scholar] [CrossRef]
- Bau, M. Controls on the Fractionation of Isovalent Trace Elements in Magmatic and Aqueous Systems: Evidence from Y/Ho, Zr/Hf and lanthanide tetrad effect. Contrib. Mineral. Petrol. 1996, 123, 323–333. [Google Scholar] [CrossRef]
- Bobos, I.; Gomes, C. Greisen and post-greisen alteration in the São Vicente de Pereira area. Can. Mineral. 1998, 36, 1615–1624. [Google Scholar]
- Bobos, I.; Duplay, J.; Rocha, J.; Gomes, C. Kaolinite to halloysite-7Å transformation in the kaolin deposit of São Vicente de Pereira, Portugal. Clays Clay Miner. 2001, 49, 597–605. [Google Scholar] [CrossRef]
- Gomes, C.S.F.; Massa, M.E. Allophane and sperullitic halloysite, weathering products of trachitic pumice fall-outs in the Caldeira Velha /S.Miguel-Azores). Miner. Petrog. Acta 1992, XXXVA, 283–288. [Google Scholar]
- Hay, R.L.; Iijima, A. Nature and origin of palagonite tuffs of the Honolulu group on Oahu, Hawaii. Mem. Geol. Soc. Am. 1968, 116, 331–376. [Google Scholar]
- Tomita, K.; Yamane, H.; Kawano, M. Synthesis of Smectite from volcanic glass at low temperature. Clays Clay Miner. 1993, 41, 655–661. [Google Scholar] [CrossRef]
- Berger, G.; Meunier, A.; Beaufort, D. Clay mineral formation on Mars: Chemical constraints and possiblecontribution of basalt out-gassing. Planet. Space Sci. 2014, 95, 25–32. [Google Scholar] [CrossRef]
- Berger, G.; Toplis, M.J.; Treguier, E.; d’Uston, C.; Pinet, P. Evidence in favor of small amounts of ephemeral and transient water during alteration at Meridiani Planum, Mars. Am. Mineral. 2009, 94, 1279–1282. [Google Scholar] [CrossRef]
- Mormone, A.; Ghiara, M.R.; Balassone, G.; Poichi, M.; Lonis, R.; Rossi, M. High-silica zeolites in pyroclastic flows from Central Sardinia (Italy): Clues on genetic processes and reserves from a mineralogical study. Miner. Petrol. 2018, 112, 767–788. [Google Scholar] [CrossRef]
- Ikehata, I.; Maruoka, T. Sulfur isotopic characteristics of volcanic products from the September 2014 Mount Ontake eruption, Japan. Earth Planets Space 2016, 68, 116. [Google Scholar] [CrossRef] [Green Version]
- Williams, S.N.; Sturchio, N.C.; Calvache, V.M.L.; Mendez, R.F.; Londonõ, C.A.; Garcia, N.P. Sulfur dioxide from Nevado del Ruiz volcano, Columbia: Total flux and isotopic constraints on its origin. J. Volcanol. Geotherm. Res. 1990, 42, 53–68. [Google Scholar] [CrossRef]
- Raymahashay, B.C. A geochemical study of rock alteration by hot springs in the Paint Pot Hill area, Yellowstone Park. Geochim. Cosmochim. Acta 1968, 32, 499–522. [Google Scholar] [CrossRef]
- Barker, W.W.; Welch, S.A.; Chu, S.; Banfield, J.F. Experimental observations of the effects of bacteria on aluminosilicate weathering. Am. Mineral. 1998, 83, 1551–1563. [Google Scholar] [CrossRef]
- Monterroso, C.; Alvarez, E.; Macías, F. Speciation and solubility control of Al and Fe in mine soil solutions. Sci. Total Environ. 1994, 158, 31–43. [Google Scholar] [CrossRef]
- Durães, N.; Bobos, I.; Ferreira da Silva, E. Speciation and precipitation of heavy metals in the acid mine waters from the Iberian Pyrite Belt (Portuguese sector). Environ. Sci. Pollut. Res. 2017, 24. [Google Scholar] [CrossRef] [PubMed]
- Nordstrom, D.K. Aqueous pyrite oxidation and the consequent formation of secondary iron minerals. Acid Sulfate Weather 1982, 10, 37–56. [Google Scholar]
- Finlow-Bates, T.; Stumpfl, E.F. The behaviour of so-called immobile elements in hydrothermally altered rocks associated with volcanogenic submarine-exhalative ore deposits. Miner. Deposita. 1981, 16, 319–328. [Google Scholar] [CrossRef]
- Rubin, J.N.; Henry, C.D.; Price, J.G. The mobilization of zirconium and other “immobile” elements during hydrothermal alteration. Chem. Geol. 1993, 110, 29–47. [Google Scholar] [CrossRef]
- Aja, S.U.; Wood, S.A.; Williams-Jones, A.E. The aqueous geochemistry of Zr and the solubility of some Zr-bearing minerals. Appl. Geochem. 1995, 10, 603–620. [Google Scholar] [CrossRef]
- Brown, P.; Curti, E.; Grambow, B. Chemical Thermodynamics of Zirconium; Elsevier: Amsterdam, The Netherlands, 2005; Available online: http://www.oecdnea.org/dbtdb/pubs/vol8-zirconium.pdf (accessed on 23 March 2021).
- Michard, A. Rare earth element systematics in hydrothermal fluids. Geochim. Cosmochim. Acta 1989, 53, 745–750. [Google Scholar] [CrossRef]
- Lottermoser, B.G. Rare earth elements and hydrothermal ore formation processes. Ore Geol. Rev. 1992, 7, 25–41. [Google Scholar] [CrossRef]
- Alderton, D.H.M.; Pearce, J.A.; Potts, P.J. Rare earth element mobility during granite alteration: Evidence from southeast England. Earth Planet. Sci. Lett. 1980, 49, 149–165. [Google Scholar] [CrossRef]
- Inguaggiato, C.; Censi, P.; Zuddas, P.; Londoño, J.M.; Chacón, Z.; Alzate, D.; Brusca, L.; D’Alessandro, W. Zirconium–hafnium and rare earth element signatures discriminating the effect of atmospheric fallout from hydrothermal input in volcanic lake water. Chem. Geol. 2016, 433, 1–11. [Google Scholar] [CrossRef]
- Sverjensky, D.A. Europium redox equilibria in aqueous solution. Earth Planet. Sci. Lett. 1984, 67, 70–78. [Google Scholar] [CrossRef]
- Bau, M. Rare-earth element mobility during hydrothermal and metamorphic fluid–rock interaction and the significance of the oxidation state of europium. Chem. Geol. 1991, 93, 219–230. [Google Scholar] [CrossRef]
- Lewis, A.J.; Palmer, M.A.; Sturchio, N.C.; Kemp, A.J. The rare earth element geochemistry of acid-sulfate and acid-sulfate-chloride geothermal systems from Yellowstone National Park, Wyoming, USA. Geochim. Cosmochim. Acta 1997, 61, 695–706. [Google Scholar] [CrossRef]
- Leybourne, M.I.; Goodfellow, W.D.; Boyle, D.R. Hydrogeochemical, isotopic and rare earth element geochemistry of acid–sulphate and acid–sulphate–chloride geothermal systems from Yellowstone. Geochim. Cosmochim. Acta 2000, 61, 695–723. [Google Scholar] [CrossRef]
- Wan, Y.; Liu, C. The effect of humic acid on the adsorption of REE on kaolin. Colloids Surf. A Physicochem. Eng. Asp. 2006, 290, 112–117. [Google Scholar] [CrossRef]
- Coppin, F.; Berger, G.; Bauer, A.; Castet, S.; Loubet, M. Sorption of lanthanides on smectite and kaolinite. Chem. Geol. 2002, 182, 57–68. [Google Scholar] [CrossRef]
- Ellis, A.J.; Mahon, W.A. Geochemistry of Geothermal Systems; Academic Press: New York, NY, USA, 1977; p. 392. [Google Scholar] [CrossRef]
- Merino, E.; Harvey, C.; Murray, H.H. Aqueous-Chemical Control of the Tetrahedral-Aluminum Content of Quartz, Halloysite, and other Low-Temperature Silicates. Clays Clay Miner. 1989, 37, 135–142. [Google Scholar] [CrossRef]
- Inoue, A.; Aoki, M.; Ito, H. Mineralogy of Ohyunuma explosion crater lake, Kokkaido, Japan. Clay Sci. 2000, 11, 169–187. [Google Scholar]
- Sposito, G. Chemical Equilibria and Kinetics in Soils; Oxford University Press: Oxford, UK, 1994; p. 260. [Google Scholar]
- Rimstidt, J.D.; Cole, D.R. Geothermal mineralization I: The mechanism of formation of the Beowawe, Nevada, siliceous sinter deposit. Am. J. Sci. 1983, 283, 861–875. [Google Scholar] [CrossRef]
- Fournier, R.O. The behaviour of silica in hydrothermal solutions. In Geology and Geochemistry of Epithermal Systems; Society of Economic Geologists: Littleton, CO, USA, 1985; pp. 45–60. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Location | Mineral Assemblages |
|---|---|---|
| F0 | Caldeiras-Furnas | Kaolinite, alunite, opal-A |
| F1 | Caldeiras-Furnas | Alunite, halloysite, opal-A |
| F2 | Caldeiras-Furnas | Kaolinite, alunite ± opal-A |
| F3 | Caldeiras-Furnas | Alunite, opal-A |
| F4 | Caldeiras-Furnas | Opal-A, alunite |
| F5 | Caldeiras-Furnas | Alunite, opal-A |
| F6 | Caldeiras-Furnas | Opal-A, alunite |
| F7 | Caldeiras-Furnas | Kaolinite, alunite |
| F8 | Caldeiras-Furnas | Alunite, opal-A |
| F20 | Caldeiras-Furnas | Alunite, (Feldspar) |
| F21 | Caldeiras-Furnas | Sinter (opal-A, alunite) |
| F22 | Caldeiras-Furnas | Sinter (opal-A, alunite) |
| Mud | Lagoa das Furnas | Smectite, kaolinite, alunite, (Feldspar) |
| Trachyte pumice | Caldeiras-Furnas | Volcanic glassy, feldspar |
| Trachyte | Caldeiras-Furnas | Feldspar, clinopyroxene, Fe-Ti oxides, amphibole |
| Oxides | Feldspar n = 5 | Feldspar n = 5 | Feldspar n = 5 | Feldspar n = 5 | Feldspar n = 5 | Feldspar n = 5 | Feldspar n = 5 | Alunite n = 5 | Alunite n = 5 | Alunite n = 5 | Alunite n = 5 | Alunite n = 5 | Kaolinite n = 5 | Kaolinite n = 5 | Kaolinite n =5 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SiO2 | 67.63 | 65.78 | 65.58 | 65.97 | 65.95 | 66.02 | 65.28 | 0.2 | 0.35 | 0.56 | 0.10 | 0.16 | 45.24 | 44.15 | 45.2 |
| TiO2 | 0.28 | 0.12 | 0.14 | 0.19 | 0.15 | 0.14 | 0.11 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.02 | 0.00 |
| Al2O3 | 20.69 | 21.35 | 21.07 | 20.59 | 20.09 | 20.66 | 20.71 | 48.35 | 52.35 | 49.28 | 49.59 | 50.14 | 37.98 | 38.29 | 38.37 |
| FeO | 0.53 | 0.31 | 0.34 | 0.35 | 0.37 | 0.32 | 0.22 | 0.01 | 0.02 | 0.06 | 0.05 | 0.03 | 0.19 | 0.06 | 0.11 |
| MnO | 0.00 | 0.00 | 0.02 | 0.00 | 0.02 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| MgO | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| CaO | 0.40 | 1.17 | 1.04 | 0.59 | 0.28 | 0.75 | 0.99 | 0.03 | 0.01 | 0.03 | 0.01 | 0.01 | 0.00 | 0.00 | 0.00 |
| Na2O | 4.54 | 6.28 | 6.15 | 5.91 | 5.75 | 6.30 | 5.55 | 0.77 | 0.21 | 0.56 | 0.23 | 0.34 | 0.00 | 0.00 | 0.00 |
| K2O | 6.39 | 4.30 | 5.48 | 6.09 | 7.71 | 6.21 | 7.42 | 2.1 | 2 | 2.54 | 2.36 | 2.44 | 0.00 | 0.00 | 0.00 |
| SO2 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 34.89 | 35.32 | 35.32 | 36.51 | 35.82 | 0.00 | 0.00 | 0.00 |
| Total | 100.47 | 99.32 | 99.81 | 99.68 | 100.32 | 100.41 | 100.29 | 86.35 | 90.26 | 88.34 | 88.86 | 88.94 | 83,41 | 83.47 | 83.68 |
| Si | 2.979 | 2.926 | 2.923 | 2.946 | 2.952 | 2.955 | 2.922 | 0.00 | 0.01 | 0.02 | 0.00 | 0.0 | 1.99 | 1.97 | 1.98 |
| Ti | 0.009 | 0.004 | 0.005 | 0.006 | 0.005 | 0.005 | 0.004 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| Al | 1.074 | 1.119 | 1.107 | 1.084 | 1.059 | 1.090 | 1.092 | 1.76 | 1.82 | 1.76 | 1.75 | 1.75 | 1.97 | 2.01 | 2.01 |
| Fe | 0.020 | 0.012 | 0.013 | 0.013 | 0.014 | 0.012 | 0.008 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| Mn | 0.000 | 0.000 | 0.001 | 0.000 | 0.001 | 0.000 | 0.000 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| Mg | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| Ca | 0.019 | 0.056 | 0.050 | 0.028 | 0.013 | 0.036 | 0.048 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| Na | 0.388 | 0.542 | 0.532 | 0.512 | 0.499 | 0.547 | 0.482 | 0.05 | 0.12 | 0.03 | 0.01 | 0.02 | 0.00 | 0.00 | 0.00 |
| K | 0.359 | 0.244 | 0.312 | 0.347 | 0.440 | 0.355 | 0.424 | 0.08 | 0.08 | 0.1 | 0.09 | 0.09 | 0.00 | 0.00 | 0.00 |
| S | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.95 | 0.88 | 0.91 | 0.93 | 0.92 | 0.00 | 0.00 | 0.00 |
| Total | 4.848 | 4.903 | 4.941 | 4.935 | 4.983 | 4.999 | 4.979 | 2.83 | 2.91 | 2.73 | 2.78 | 2.78 | 3.96 | 3.98 | 3.99 |
| An | 2.5 | 6.6 | 5.6 | 3.2 | 1.4 | 3.9 | 5.0 | ||||||||
| Ab | 50.6 | 64.4 | 59.5 | 57.7 | 52.4 | 58.3 | 50.6 | ||||||||
| Or | 46.9 | 29.0 | 34.9 | 39.1 | 46.2 | 37.8 | 44.4 | ||||||||
| Total | 100.0 | 100.0 | 100.0 | 100.0 | 100.0 | 100.0 | 100.0 |
| Samples | Cu | Pb | Zn | As | Ba | Ni | Cr | Co | Cs | Ga | Hf | Nb | Rb | Sn | Sr | Ta | Th | Tl | U | V | W | Zr | Y |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fo | 9 | 11 | 34 | 5 | 247.5 | 11 | 6 | 0.4 | 0.6 | 47.8 | 30 | 259.21 | 44.14 | 8 | 176.9 | 13.1 | 25.6 | 0.2 | 4.2 | 43 | 4.1 | 1336.3 | 26.1 |
| F1 | 11 | 8 | 17 | 5 | 119.9 | 15 | 5 | 0.6 | 0.8 | 39.8 | 47.5 | 361.69 | 23.87 | 12 | 68.2 | 20.2 | 24.1 | 0.5 | 7.6 | 25 | 8.7 | 1958.6 | 33.4 |
| F2 | 3 | 4 | 4 | 5 | 294.4 | 9 | 4 | 0.3 | 0.6 | 50.2 | 35.2 | 305.38 | 25.87 | 10 | 207 | 16.3 | 25.3 | 0.4 | 5.3 | 30 | 5.6 | 1537 | 27.1 |
| F3 | 5 | 8 | 6 | 5 | 211.1 | 13 | 8 | 0.6 | 0.6 | 43.2 | 25.8 | 226.8 | 24.52 | 10 | 140.7 | 11.9 | 12.2 | 0.6 | 4 | 23 | 3.6 | 1101.1 | 14.3 |
| F4 | 4 | 6 | 4 | 5 | 211.8 | 9 | 5 | 0.6 | 0.6 | 39.6 | 32.5 | 287.02 | 25.55 | 8 | 113.3 | 15.1 | 11 | 0.4 | 4.6 | 24 | 4 | 1359.7 | 10 |
| F5 | 2 | 7 | 5 | 5 | 104 | 16 | 11 | 0.5 | 0.7 | 31.8 | 28.2 | 220.74 | 17.96 | 5 | 64.3 | 11.6 | 13.3 | 0.2 | 2.6 | 17 | 2.6 | 1254.8 | 9.4 |
| F6 | 2 | 4 | 3 | 5 | 182.1 | 12 | 3 | 0.4 | 0.4 | 35 | 26.6 | 246.8 | 22.72 | 8 | 117.4 | 12.2 | 13.2 | 0.4 | 2.9 | 13 | 4.1 | 1179.7 | 13.3 |
| F7 | 9 | 12 | 7 | 5 | 282 | 8 | 12 | 0.5 | 0.2 | 89.6 | 54.2 | 477.45 | 9.71 | 10 | 224.5 | 20.1 | 29.3 | 0.2 | 4.3 | 16 | 7.6 | 2414.4 | 35.6 |
| F8 | 4 | 5 | 2 | 5 | 169.1 | 10 | 7 | 3.5 | 0.8 | 28.5 | 22.4 | 202.58 | 19.41 | 7 | 85.9 | 10.1 | 19.9 | 0.3 | 7.3 | 21 | 3.3 | 996.1 | 31.9 |
| F20 | 5 | 3 | 23 | 5 | 24.3 | 5 | 4 | 0.3 | 1.9 | 37.9 | 29.3 | 270.5 | 218.64 | 8 | 11.1 | 13.8 | 23.6 | 0.1 | 7.3 | 10 | 4 | 1285.2 | 70 |
| F21 | 3 | 4 | 2 | 5 | 310.2 | 11 | 8 | 0.4 | 0.8 | 10.2 | 28.4 | 214.7 | 29.36 | 5 | 50.3 | 9.9 | 9.4 | 0.2 | 2.7 | 23 | 4.3 | 1235.3 | 12.9 |
| F22 | 1 | 3 | 74 | 5 | 279.5 | 9 | 5 | 1.5 | 0.4 | 28.7 | 9.8 | 78.77 | 117.48 | 3 | 85.7 | 3.8 | 7.7 | 0.1 | 2.3 | 20 | 0.5 | 424.6 | 21.5 |
| Mud | 3 | 3 | 8 | 5 | 360.6 | 14 | 6 | 0.4 | 0.4 | 84.2 | 70 | 623.8 | 17.72 | 11 | 184.4 | 27.1 | 32.8 | 0.2 | 5.6 | 34 | 8.5 | 3192.5 | 53.9 |
| Trachyte pumice | 4 | 11 | 66 | 5 | 169.1 | 4 | 3 | 1.8 | 2.1 | 112.8 | 34 | 326.75 | 221.32 | 9 | 126.52 | 21.3 | 34 | 0.6 | 7.8 | 38 | 5.1 | 1690.26 | 74 |
| Trachyte | 6 | 9 | 82 | 5 | 286.3 | 8 | 6 | 1 | 0.9 | 94.7 | 51 | 391.56 | 263.98 | 6 | 148.31 | 27.5 | 41 | 0.9 | 6.3 | 59 | 7.4 | 2189 | 112 |
| Samples | La | Ce | Pr | Nd | Sm | Eu | Gd | Tb | Dy | Ho | Er | Tm | Yb | Lu | Y |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| F0 | 245.2 | 473.2 | 44.87 | 132.2 | 14.5 | 0.99 | 8.01 | 1.03 | 6.11 | 1.06 | 3.16 | 0.47 | 3.2 | 0.5 | 26.1 |
| F1 | 132.3 | 262.8 | 27.71 | 85.3 | 11.6 | 1.02 | 7.19 | 1.14 | 7.06 | 1.39 | 3.98 | 0.59 | 3.99 | 0.58 | 33.4 |
| F2 | 235.4 | 409.6 | 39.66 | 131.1 | 18 | 1.24 | 9.37 | 1.21 | 6.68 | 1.14 | 3.33 | 0.44 | 3.03 | 0.47 | 27.1 |
| F3 | 114.6 | 178.2 | 16.58 | 57.8 | 8.7 | 0.55 | 5.28 | 0.63 | 3.6 | 0.56 | 1.62 | 0.23 | 1.5 | 0.23 | 14.3 |
| F4 | 88.1 | 123.4 | 10.63 | 33.2 | 5 | 0.38 | 2.7 | 0.4 | 2.19 | 0.39 | 1.18 | 0.16 | 1.04 | 0.16 | 10 |
| F5 | 122 | 192.1 | 16.33 | 48.4 | 6.9 | 0.46 | 3.13 | 0.44 | 2.72 | 0.38 | 1.1 | 0.16 | 1.15 | 0.15 | 9.4 |
| F6 | 139.9 | 201.2 | 17.31 | 53.6 | 7.4 | 0.56 | 4.11 | 0.52 | 3.19 | 0.54 | 1.46 | 0.2 | 1.29 | 0.21 | 13.3 |
| F7 | 279.2 | 357.7 | 27.15 | 76.7 | 10.8 | 0.85 | 6.4 | 1.07 | 6.75 | 1.37 | 4.17 | 0.61 | 3.86 | 0.64 | 35.6 |
| F8 | 148.5 | 242.2 | 18.15 | 37.5 | 4.6 | 0.58 | 4.71 | 0.79 | 5.55 | 1.12 | 3.44 | 0.5 | 3.18 | 0.49 | 31.9 |
| F20 | 184.2 | 345.4 | 35.03 | 123.8 | 20.5 | 0.92 | 14.61 | 2.24 | 14.16 | 2.61 | 7.26 | 1.07 | 5.98 | 0.94 | 70 |
| F21 | 45.7 | 79.9 | 7.37 | 23.1 | 3.7 | 0.4 | 2.32 | 0.36 | 2.51 | 0.47 | 1.4 | 0.21 | 1.4 | 0.21 | 12.9 |
| F22 | 48.4 | 148.3 | 11.92 | 47.6 | 8.8 | 1.9 | 6.16 | 0.84 | 4.96 | 0.86 | 2.32 | 0.32 | 1.95 | 0.33 | 21.5 |
| Mud | 276.1 | 376.5 | 29.69 | 82.1 | 11.6 | 0.94 | 7.49 | 1.35 | 9.3 | 1.87 | 5.92 | 0.96 | 5.98 | 0.93 | 53.9 |
| Trachyte | 146 | 232 | 28.3 | 101 | 17 | 1.79 | 12 | 1.8 | 11 | 2.3 | 6.4 | 0.85 | 4.6 | 0.62 | 74 |
| Trachyte pumice | 178.6 | 346.1 | 37.43 | 119.82 | 18.77 | 0.61 | 15.89 | 2.33 | 13.81 | 2.55 | 7.11 | 0.91 | 6.34 | 0.93 | 112 |
| Samples | SUMREE | LREE | HREE | MREE | (LREE/HRRE)N | (La/Yb)N | (La/Ce)N | Ce/Ce* | Eu/Eu* | (La/Sm)N | (Tb/Yb)N | (Gd/Gd)N | Y/Ho | Y/Dy |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fo | 934.50 | 895.47 | 7.33 | 31.7 | 18.36 | 51.78 | 1.35 | 1.06 | 0.28 | 10.64 | 1.38 | 0.80 | 24.62 | 4.27 |
| F1 | 546.65 | 508.11 | 9.14 | 29.4 | 9.46 | 22.41 | 1.31 | 1.02 | 0.34 | 7.18 | 1.22 | 0.79 | 24.03 | 4.73 |
| F2 | 860.67 | 815.76 | 7.27 | 37.64 | 17.06 | 52.50 | 1.49 | 0.99 | 0.29 | 8.23 | 1.71 | 0.77 | 23.77 | 4.057 |
| F3 | 390.08 | 367.18 | 3.58 | 19.32 | 9.80 | 51.63 | 1.67 | 0.96 | 0.25 | 8.29 | 1.79 | 0.87 | 25.53 | 3.97 |
| F4 | 268.93 | 255.33 | 2.54 | 11.06 | 7.48 | 57.24 | 1.86 | 0.94 | 0.32 | 11.09 | 1.64 | 0.75 | 25.64 | 4.56 |
| F5 | 395.42 | 378.83 | 2.56 | 14.03 | 10.91 | 71.68 | 1.65 | 1.01 | 0.30 | 11.13 | 1.64 | 0.68 | 24.74 | 3.46 |
| F6 | 431.49 | 412.01 | 3.16 | 16.32 | 11.47 | 73.28 | 1.81 | 0.96 | 0.31 | 11.90 | 1.72 | 0.81 | 24.63 | 4.17 |
| F7 | 777.27 | 740.75 | 9.28 | 27.24 | 14.13 | 48.88 | 2.03 | 0.96 | 0.31 | 16.27 | 1.18 | 0.75 | 25.98 | 5.27 |
| F8 | 471.31 | 446.35 | 7.61 | 17.35 | 9.05 | 31.56 | 1.59 | 1.09 | 0.38 | 20.32 | 1.06 | 0.98 | 28.48 | 5.75 |
| F20 | 758.72 | 688.43 | 15.25 | 55.04 | 9.52 | 20.81 | 1.39 | 1.01 | 0.16 | 5.65 | 1.60 | 0.86 | 26.82 | 4.94 |
| F21 | 169.05 | 156.07 | 3.22 | 9.76 | 4.22 | 22.06 | 1.49 | 1.02 | 0.42 | 7.77 | 1.09 | 0.80 | 27.45 | 5.14 |
| F22 | 284.66 | 256.22 | 4.92 | 23.52 | 5.60 | 16.77 | 0.85 | 1.45 | 0.79 | 3.46 | 1.84 | 0.90 | 25 | 4.33 |
| Mud | 810.73 | 764.39 | 13.79 | 32.55 | 11.60 | 31.20 | 1.912 | 0.97 | 0.31 | 14.98 | 0.96 | 0.76 | 28.82 | 5.79 |
| Trachyte | 565.66 | 507.3 | 12.47 | 45.89 | 11.72 | 21.45 | 1.64 | 0.84 | 0.38 | 5.40 | 1.67 | 0.87 | 32.17 | 6.73 |
| Trachyte pumice | 751.2 | 681.95 | 15.29 | 53.96 | 12.39 | 19.04 | 1.34 | 0.99 | 0.11 | 5.99 | 1.57 | 0.96 | 43.92 | 8.11 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bobos, I.; Gomes, C. Mineralogy and Geochemistry (HFSE and REE) of the Present-Day Acid-Sulfate Types Alteration from the Active Hydrothermal System of Furnas Volcano, São Miguel Island, The Azores Archipelago. Minerals 2021, 11, 335. https://0-doi-org.brum.beds.ac.uk/10.3390/min11040335
Bobos I, Gomes C. Mineralogy and Geochemistry (HFSE and REE) of the Present-Day Acid-Sulfate Types Alteration from the Active Hydrothermal System of Furnas Volcano, São Miguel Island, The Azores Archipelago. Minerals. 2021; 11(4):335. https://0-doi-org.brum.beds.ac.uk/10.3390/min11040335
Chicago/Turabian StyleBobos, Iuliu, and Celso Gomes. 2021. "Mineralogy and Geochemistry (HFSE and REE) of the Present-Day Acid-Sulfate Types Alteration from the Active Hydrothermal System of Furnas Volcano, São Miguel Island, The Azores Archipelago" Minerals 11, no. 4: 335. https://0-doi-org.brum.beds.ac.uk/10.3390/min11040335