
First Steps towards Understanding the Non-Linear Impact of Mg on Calcite Solubility: A Molecular Dynamics Study


Abstract
:1. Introduction
2. Methods
3. Results
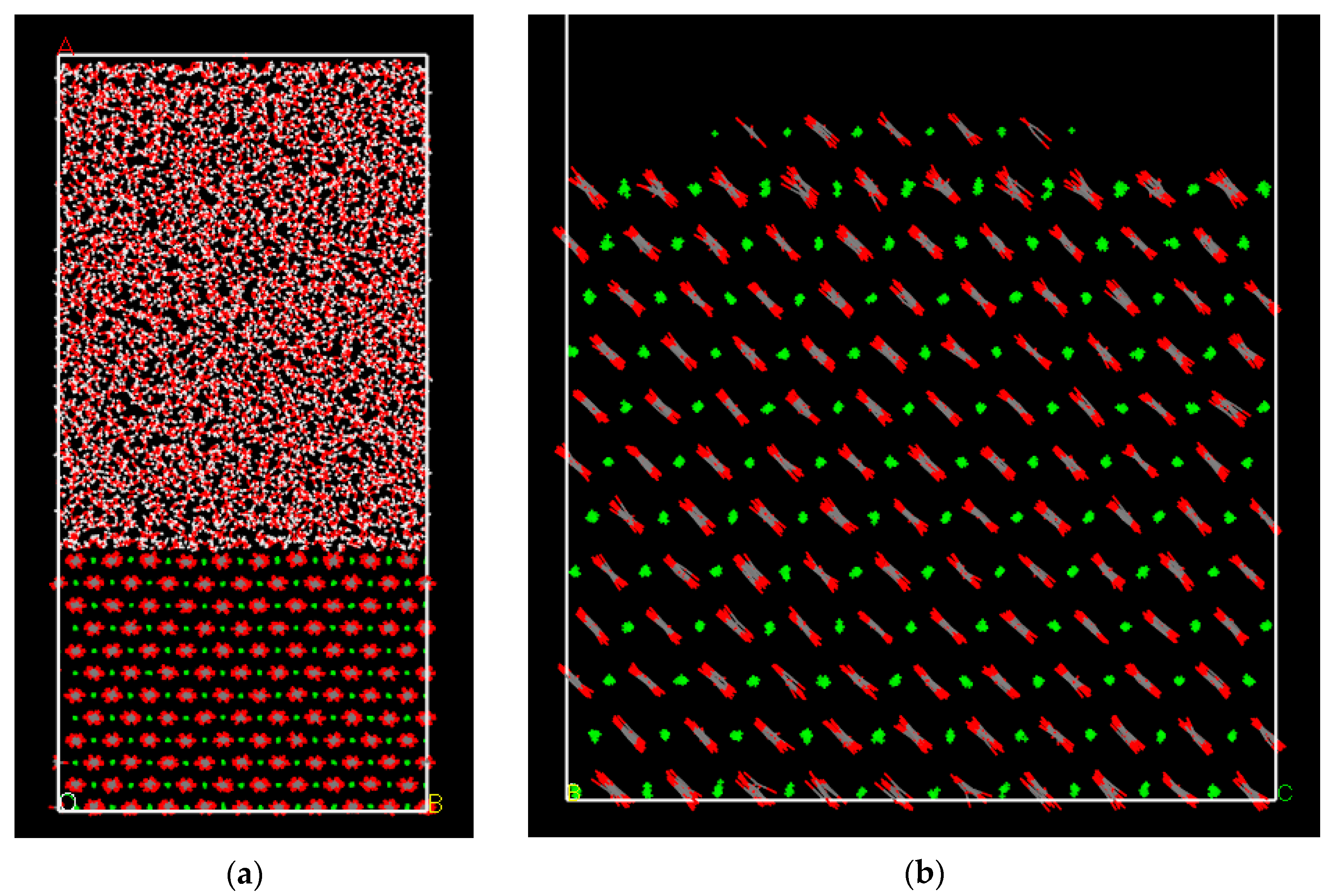
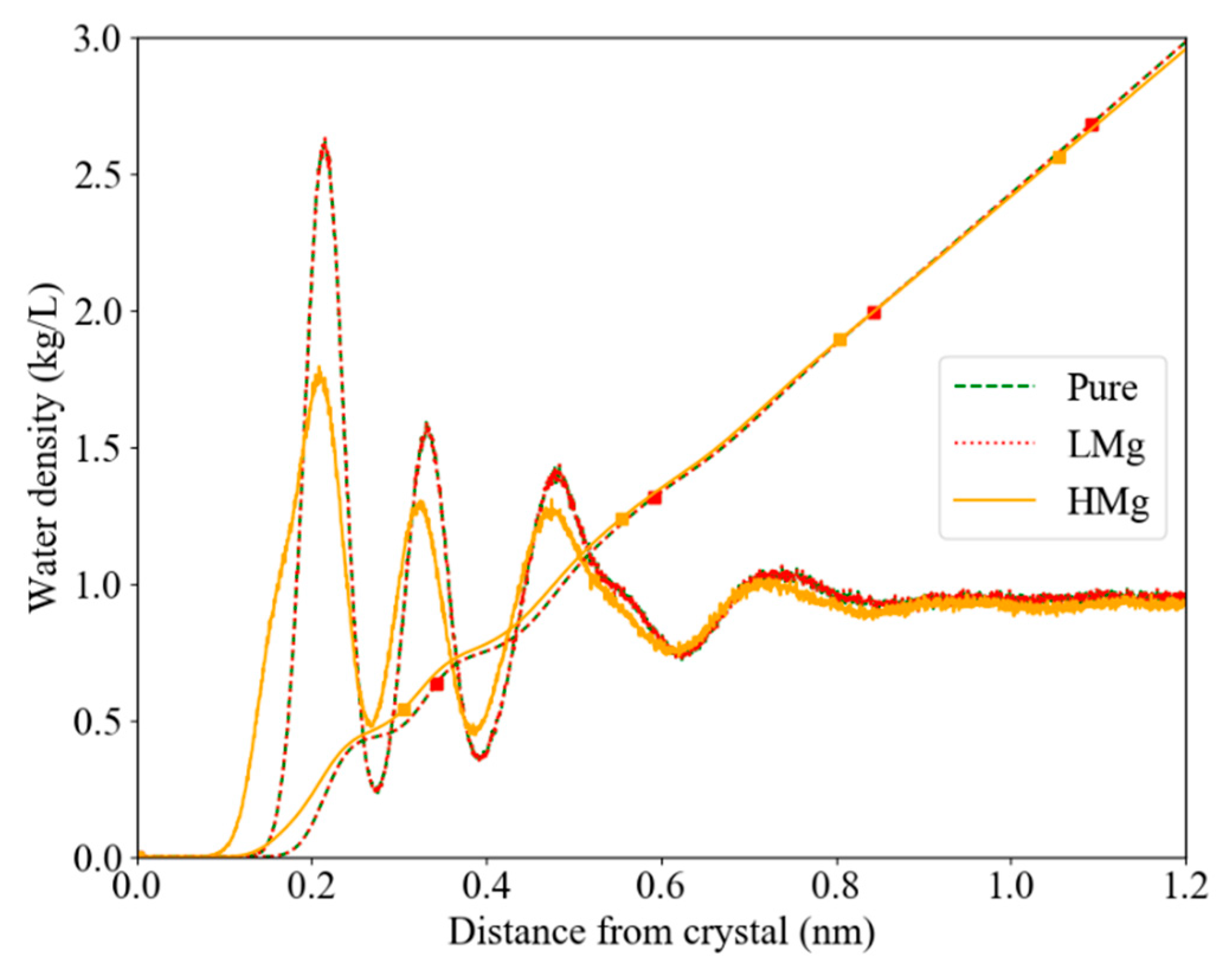
3.1. Water Density, Structure and Dynamics in the Interface
3.2. Surface Energies and Structural Relaxation
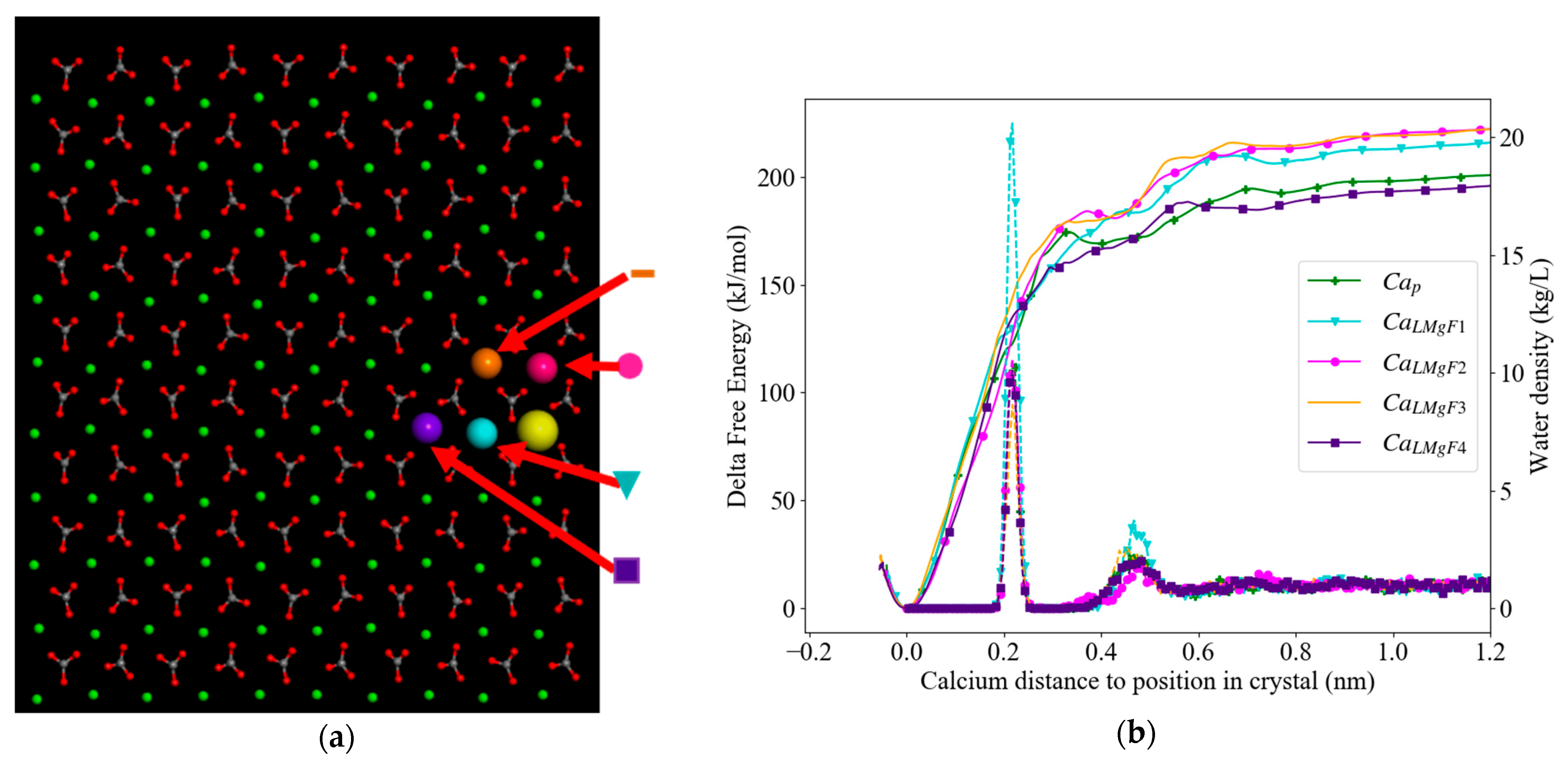
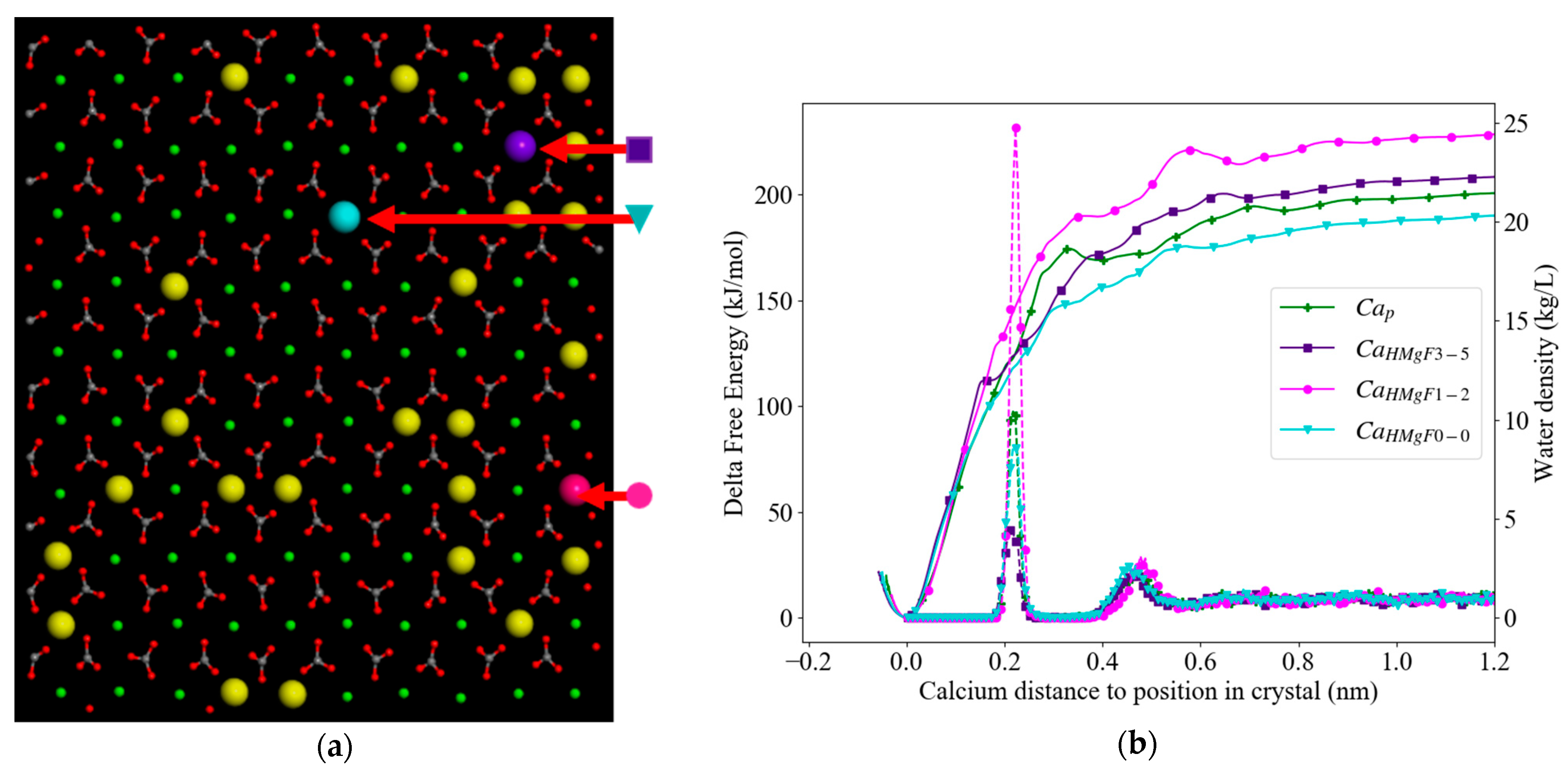
3.3. Free Energy Profiles
3.3.1. Low Mg2+ Calcite
3.3.2. High Mg2+ Calcite
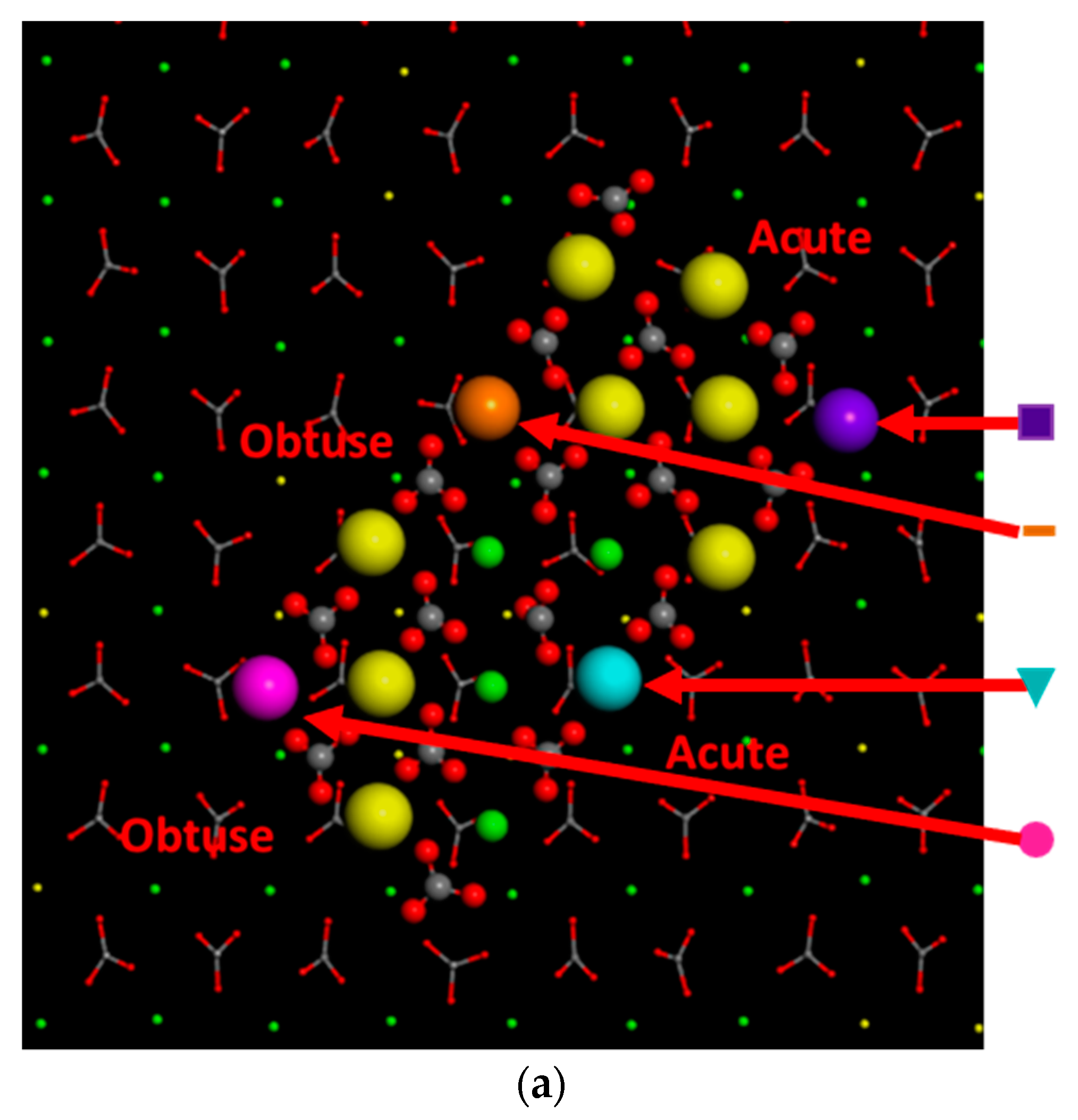
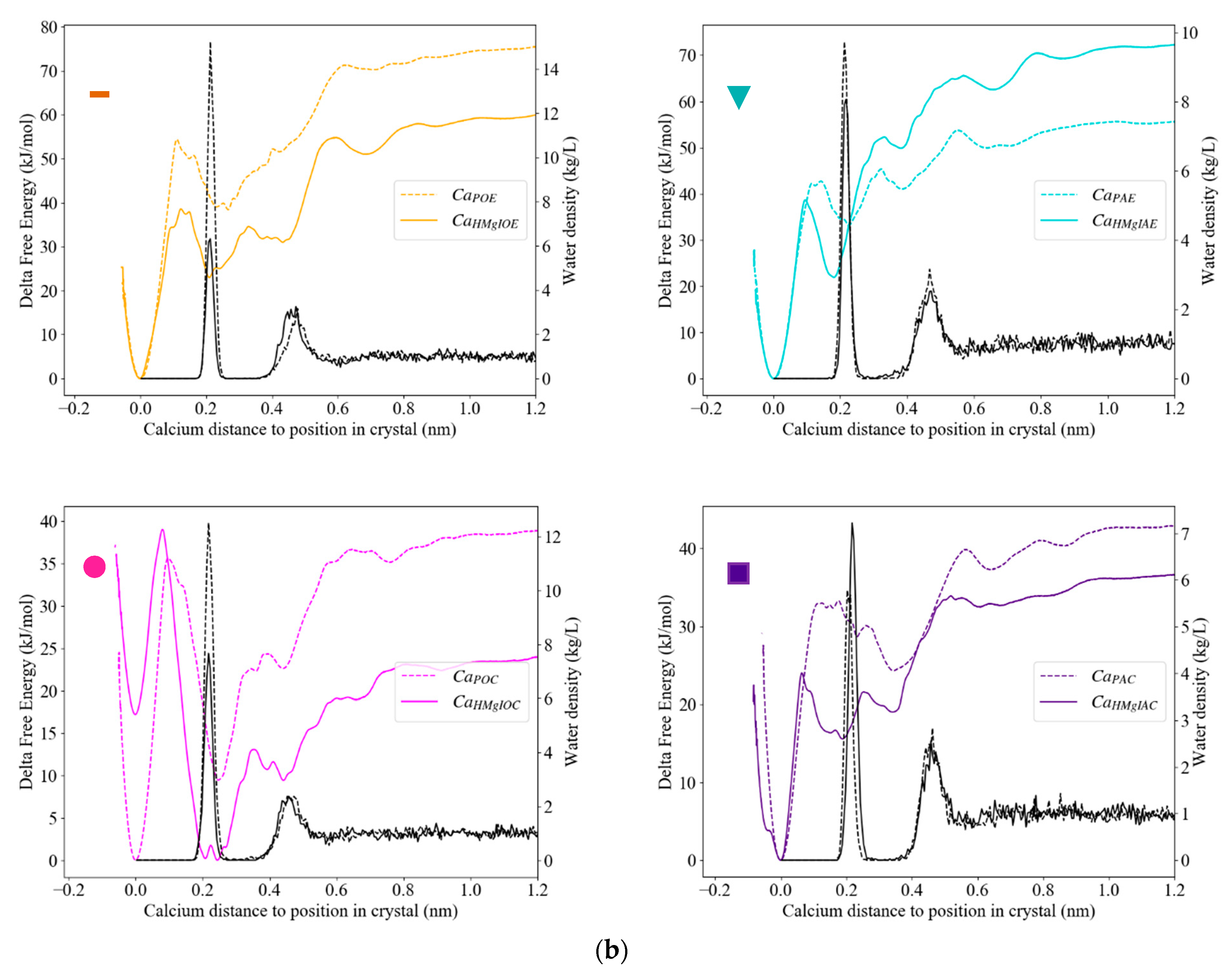
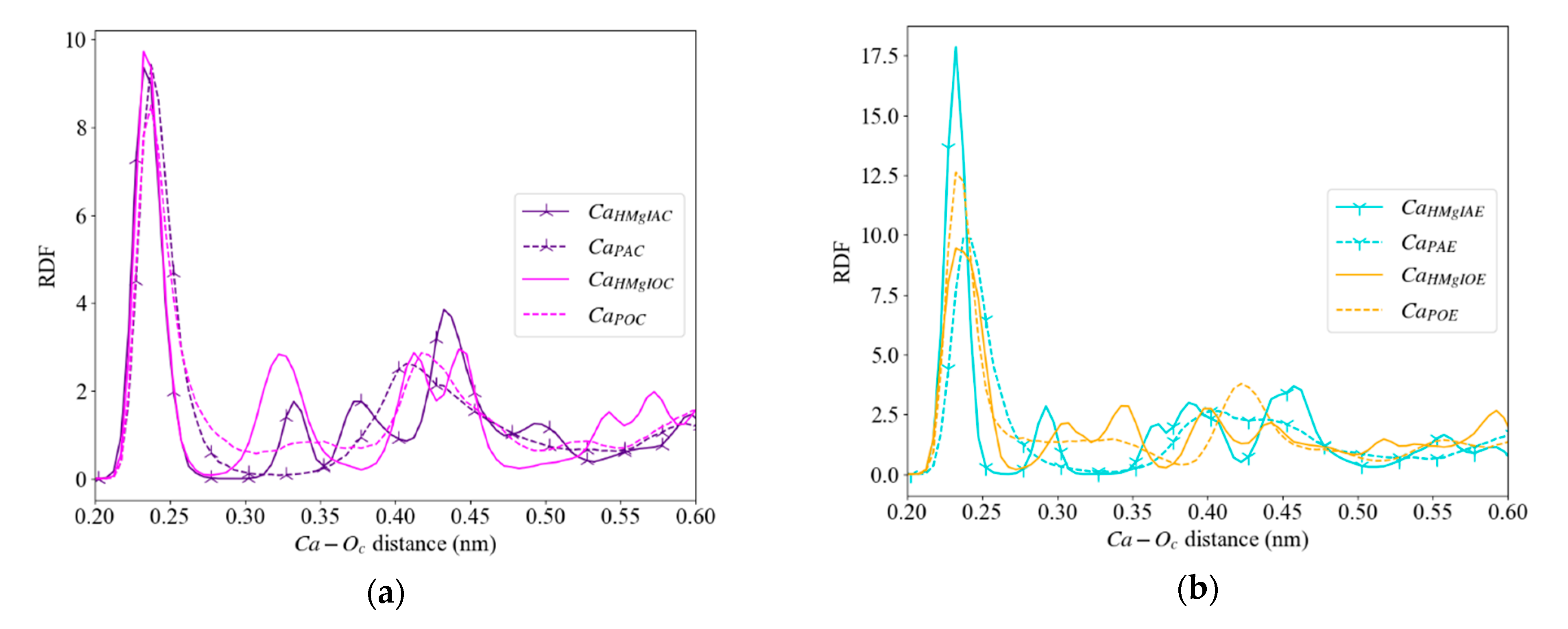
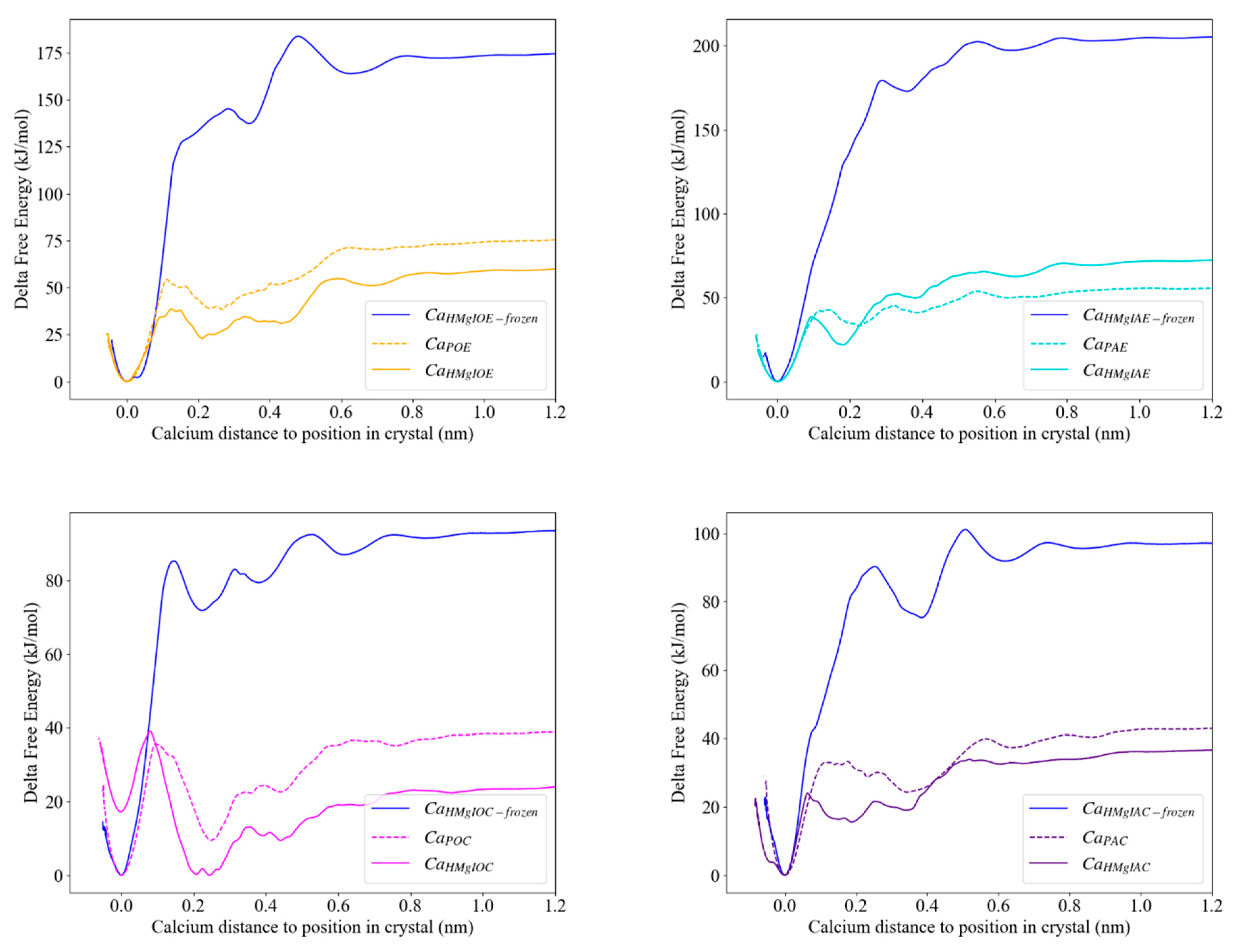
3.3.3. Calcite with an Island
3.4. Impact of Surface Carbonate on Energy Profiles
4. Discussion
4.1. Influence of Magnesium on Calcite Solubility and Dissolution
4.1.1. Interfacial Water Structure
4.1.2. Surface Energies and Structural Relaxation
4.1.3. Local Variability in Surface Free Energies
4.2. Implications for the Influence of Magnesium on Calcite Growth
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ahr, W.M. Geology of Carbonate Reservoirs: The Identification, Description, and Characterization of Hydrocarbon Reservoirs in Carbonate Rocks; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2008; ISBN 9780470164914. [Google Scholar]
- Molins, S.; Trebotich, D.; Yang, L.; Ajo-Franklin, J.B.; Ligocki, T.J.; Shen, C.; Steefel, C.I. Pore-scale controls on calcite dissolution rates from flow-through laboratory and numerical experiments. Environ. Sci. Technol. 2014, 48, 7453–7460. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, P.; Raoof, A.; Iliev, O.; Wolthers, M. Evolution of pore-shape and its impact on pore conductivity during CO2 injection in calcite: Single pore simulations and microfluidic experiments. Adv. Water Resour. 2020, 136, 103480. [Google Scholar] [CrossRef]
- Molins, S.; Soulaine, C.; Prasianakis, N.I.; Abbasi, A.; Poncet, P.; Ladd, A.J.C.; Starchenko, V.; Roman, S.; Trebotich, D.; Tchelepi, H.A.; et al. Simulation of mineral dissolution at the pore scale with evolving fluid-solid interfaces: Review of approaches and benchmark problem set. Comput. Geosci. 2020, 1–34. [Google Scholar] [CrossRef] [Green Version]
- Cao, B.; Stack, A.G.; Steefel, C.I.; DePaolo, D.J.; Lammers, L.N.; Hu, Y. Investigating calcite growth rates using a quartz crystal microbalance with dissipation (QCM-D). Geochim. Cosmochim. Acta 2018, 222, 269–283. [Google Scholar] [CrossRef] [Green Version]
- Hellevang, H.; Miri, R.; Haile, B.G. New insights into the mechanisms controlling the rate of crystal growth. Cryst. Growth Des. 2014, 14, 6451–6458. [Google Scholar] [CrossRef]
- Kurganskaya, I.; Luttge, A. Kinetic Monte Carlo Approach to Study Carbonate Dissolution. J. Phys. Chem. C 2016, 120, 6482–6492. [Google Scholar] [CrossRef]
- Wolthers, M.; Nehrke, G.; Gustafsson, J.P.; Van Cappellen, P. Calcite growth kinetics: Modeling the effect of solution stoichiometry. Geochim. Cosmochim. Acta 2012, 77, 121–134. [Google Scholar] [CrossRef] [Green Version]
- Sand, K.K.; Tobler, D.J.; Dobberschütz, S.; Larsen, K.K.; Makovicky, E.; Andersson, M.P.; Wolthers, M.; Stipp, S.L.S. Calcite Growth Kinetics: Dependence on Saturation Index, Ca2+:CO32− Activity Ratio, and Surface Atomic Structure. Cryst. Growth Des. 2016, 16, 3602–3612. [Google Scholar] [CrossRef]
- Larsen, K.; Bechgaard, K.; Stipp, S.L.S. Modelling spiral growth at dislocations and determination of critical step lengths from pyramid geometries on calcite {1014} surfaces. Geochim. Cosmochim. Acta 2010, 74, 558–567. [Google Scholar] [CrossRef]
- Bracco, J.N.; Grantham, M.C.; Stack, A.G. Calcite Growth Rates as a Function of Aqueous Calcium-to-Carbonate Ratio, Saturation Index, and Inhibitor Concentration: Insight into the Mechanism of Reaction and Poisoning by Strontium. Cryst. Growth Des. 2012, 12, 3540–3548. [Google Scholar] [CrossRef]
- Kirch, A.; Mutisya, S.M.; Sánchez, V.M.; De Almeida, J.M.; Miranda, C.R. Fresh Molecular Look at Calcite-Brine Nanoconfined Interfaces. J. Phys. Chem. C 2018, 122, 6117–6127. [Google Scholar] [CrossRef] [Green Version]
- Lardge, J.S.; Duffy, D.M.; Gillan, M.J.; Watkins, M. Ab initio simulations of the interaction between water and defects on the calcite (101 4) surface. J. Phys. Chem. C 2010, 114, 2664–2668. [Google Scholar] [CrossRef]
- Fenter, P.; Kerisit, S.; Raiteri, P.; Gale, J.D. Is the Calcite–Water Interface Understood? Direct Comparisons of Molecular Dynamics Simulations with Specular X-ray Reflectivity Data. J. Phys. Chem. C 2013, 117, 5028–5042. [Google Scholar] [CrossRef]
- Harstad, A.O.; Stipp, S.L.S. Calcite dissolution: Effects of trace cations naturally present in Iceland spar calcites. Geochim. Cosmochim. Acta 2007, 71, 56–70. [Google Scholar] [CrossRef]
- Müller, G.; Tietz, R. Recent dolomitization of quaternary biocalcarenites from fuerteventura (Canary Islands). Contrib. to Mineral. Petrol. 1966, 13, 89–96. [Google Scholar] [CrossRef]
- Long, X.; Ma, Y.; Qi, L. Biogenic and synthetic high magnesium calcite—A review. J. Struct. Biol. 2014, 185, 1–14. [Google Scholar] [CrossRef]
- Lenders, J.J.M.; Dey, A.; Bomans, P.H.H.; Spielmann, J.; Hendrix, M.M.R.M.; De With, G.; Meldrum, F.C.; Harder, S.; Sommerdijk, N.A.J.M. High-magnesian calcite mesocrystals: A coordination chemistry approach. J. Am. Chem. Soc. 2012, 134, 1367–1373. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Hamm, L.M.; Giuffre, A.J.; Echigo, T.; Rimstidt, J.D.; De Yoreo, J.J.; Grotzinger, J.; Dove, P.M. Revisiting geochemical controls on patterns of carbonate deposition through the lens of multiple pathways to mineralization. Faraday Discuss. 2012, 159, 371–386. [Google Scholar] [CrossRef] [Green Version]
- Stanienda, K.J. Carbonate phases rich in magnesium in the Triassic limestones of the eastern part of the Germanic Basin. Carbonates Evaporites 2016, 31, 387–405. [Google Scholar] [CrossRef] [Green Version]
- De Leeuw, N.H.; Parker, S.C. Surface–water interactions in the dolomite problem. Phys. Chem. Chem. Phys. 2001, 3, 3217–3221. [Google Scholar] [CrossRef]
- De Leeuw, N.H. Molecular dynamics simulations of the growth inhibiting effect of Fe2+, Mg2+, Cd2+, and Sr2+ on calcite crystal growth. J. Phys. Chem. B 2002, 106, 5241–5249. [Google Scholar] [CrossRef]
- Paquette, J.; Reeder, R.J. Relationship between surface structure, growth mechanism, and trace element incorporation in calcite. Geochim. Cosmochim. Acta 1995, 59, 735–749. [Google Scholar] [CrossRef]
- Wang, Q. A Computational Study of Calcium Carbonate. Ph.D. Thesis, UCL (University College London), London, UK, 2011. [Google Scholar]
- De Leeuw, N.H. Surface structures, stabilities, and growth of magnesian calcites: A computational investigation from the perspective of dolomite formation. Am. Miner. 2002, 87, 679–689. [Google Scholar] [CrossRef]
- Brady, P.V.; Krumhansl, J.L.; Papenguth, H.W. Surface complexation clues to dolomite growth. Geochim. Cosmochim. Acta 1996, 60, 727–731. [Google Scholar] [CrossRef]
- Busenberg, E.; Plummer, N. The kinetics of dissolution of dolomite in CO2-H2O systems at 1.5 to 65 °C and 0 to 1 atm PCO2. Am. J. Sci. 1982, 282, 45–78. [Google Scholar] [CrossRef]
- Pokrovsky, O.S.; Schott, J. Kinetics and mechanism of dolomite dissolution in neutral to alkaline solutions revisited. Am. J. Sci. 2001, 301, 597–626. [Google Scholar] [CrossRef]
- Subhas, A.V.; Rollins, N.E.; Berelson, W.M.; Erez, J.; Ziveri, P.; Langer, G.; Adkins, J.F. The dissolution behavior of biogenic calcites in seawater and a possible role for magnesium and organic carbon. Mar. Chem. 2018, 205, 100–112. [Google Scholar] [CrossRef]
- Ruiz-Agudo, E.; Putnis, C.V. Direct observations of mineral fluid reactions using atomic force microscopy: The specific example of calcite. Miner. Mag. 2012, 76, 227–253. [Google Scholar] [CrossRef]
- Astilleros, J.M.; Fernández-Díaz, L.; Putnis, A. The role of magnesium in the growth of calcite: An AFM study. Chem. Geol. 2010, 271, 52–58. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Guo, J.; Wu, S.; Chen, F.; Yang, Y. Divalent heavy metals and uranyl cations incorporated in calcite change its dissolution process. Sci. Rep. 2020, 10, 16864. [Google Scholar] [CrossRef]
- Xu, M.; Higgins, S.R. Effects of magnesium ions on near-equilibrium calcite dissolution: Step kinetics and morphology. Geochim. Cosmochim. Acta 2011, 75, 719–733. [Google Scholar] [CrossRef]
- Di Tommaso, D.; Ruiz-Agudo, E.; De Leeuw, N.H.; Putnis, A.; Putnis, C.V. Modelling the effects of salt solutions on the hydration of calcium ions. Phys. Chem. Chem. Phys. 2014, 16, 7772–7785. [Google Scholar] [CrossRef] [Green Version]
- Wasylenki, L.E.; Dove, P.M.; Wilson, D.S.; De Yoreo, J.J. Nanoscale effects of strontium on calcite growth: An in situ AFM study in the absence of vital effects. Geochim. Cosmochim. Acta 2005, 69, 3017–3027. [Google Scholar] [CrossRef]
- Wolthers, M.; Di Tommaso, D.; Du, Z.; De Leeuw, N.H. Variations in calcite growth kinetics with surface topography: Molecular dynamics simulations and process-based growth kinetics modelling. CrystEngComm 2013, 15, 5506. [Google Scholar] [CrossRef] [Green Version]
- De La Pierre, M.; Raiteri, P.; Gale, J.D. Structure and Dynamics of Water at Step Edges on the Calcite {1014} Surface. Cryst. Growth Des. 2016, 16, 5907–5914. [Google Scholar] [CrossRef]
- De La Pierre, M.; Raiteri, P.; Stack, A.G.; Gale, J.D. Uncovering the Atomistic Mechanism for Calcite Step Growth. Angew. Chem. Int. Ed. 2017, 56, 8464–8467. [Google Scholar] [CrossRef] [Green Version]
- Spagnoli, D.; Kerisit, S.; Parker, S.C. Atomistic simulation of the free energies of dissolution of ions from flat and stepped calcite surfaces. J. Cryst. Growth 2006, 294, 103–110. [Google Scholar] [CrossRef]
- De Leeuw, N.H.; Parker, S.C.; Harding, J.H. Molecular dynamics simulation of crystal dissolution from calcite steps. Phys. Rev. B 1999, 60, 13792–13799. [Google Scholar] [CrossRef]
- Mutisya, S.M.; Kirch, A.; De Almeida, J.M.; Sánchez, V.M.; Miranda, C.R. Molecular Dynamics Simulations of Water Confined in Calcite Slit Pores: An NMR Spin Relaxation and Hydrogen Bond Analysis. J. Phys. Chem. C 2017, 121, 6674–6684. [Google Scholar] [CrossRef]
- Spagnoli, D.; Cooke, D.J.; Kerisit, S.; Parker, S.C. Molecular dynamics simulations of the interaction between the surfaces of polar solids and aqueous solutions. J. Mater. Chem. 2006, 16, 1997–2006. [Google Scholar] [CrossRef]
- Koleini, M.M.; Mehraban, M.F.; Ayatollahi, S. Effects of low salinity water on calcite/brine interface: A molecular dynamics simulation study. Colloids Surf. A Physicochem. Eng. Asp. 2018, 537, 61–68. [Google Scholar] [CrossRef]
- Kerisit, S.; Parker, S.C. Free energy of adsorption of water and metal ions on the calcite surface. J. Am. Chem. Soc. 2004, 126, 10152–10161. [Google Scholar] [CrossRef]
- Liang, Y.; Lea, A.S.; Baer, D.R.; Engelhard, M.H. Structure of the cleaved CaCo3 surface in an aqueous environment. Surf. Sci. 1996, 351, 172–182. [Google Scholar] [CrossRef]
- Kerisit, S.; Parker, S.C.; Harding, J.H. Atomistic simulation of the dissociative adsorption of water on calcite surfaces. J. Phys. Chem. B 2003, 107, 7676–7682. [Google Scholar] [CrossRef]
- Andersson, M.P.; Dobberschütz, S.; Sand, K.K.; Tobler, D.J.; De Yoreo, J.J.; Stipp, S.L.S. A Microkinetic Model of Calcite Step Growth. Angew. Chemie Int. Ed. 2016, 55, 11086–11090. [Google Scholar] [CrossRef]
- Xu, J.; Yan, C.; Zhang, F.; Konishi, H.; Xu, H.; Teng, H.H. Testing the cation-hydration effect on the crystallization of Ca-Mg-CO3 systems. Proc. Natl. Acad. Sci. USA 2013, 110, 17750–17755. [Google Scholar] [CrossRef] [Green Version]
- Davis, K.J.; Dove, P.M.; De Yoreo, J.J. The role of Mg2+ as an impurity in calcite growth. Science 2000, 290, 1134–1137. [Google Scholar] [CrossRef]
- Nielsen, L.C.; De Yoreo, J.J.; DePaolo, D.J. General model for calcite growth kinetics in the presence of impurity ions. Geochim. Cosmochim. Acta 2013, 115, 100–114. [Google Scholar] [CrossRef] [Green Version]
- Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef] [Green Version]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [Green Version]
- Koskamp, J.A.; Ruiz-Hernandez, S.E.; Di Tommaso, D.; Elena, A.M.; De Leeuw, N.H.; Wolthers, M. Reconsidering Calcium Dehydration as the Rate-Determining Step in Calcium Mineral Growth. J. Phys. Chem. C 2019, 123, 26895–26903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Leeuw, N.H.; Parker, S.C. Molecular-dynamics simulation of MgO surfaces in liquid water using a shell-model potential for water. Phys. Rev. B Condens. Matter Mater. Phys. 1998, 58, 13901–13908. [Google Scholar] [CrossRef]
- Reischl, B.; Raiteri, P.; Gale, J.D.; Rohl, A.L. Atomistic Simulation of Atomic Force Microscopy Imaging of Hydration Layers on Calcite, Dolomite, and Magnesite Surfaces. J. Phys. Chem. C 2019, 123, 14985–14992. [Google Scholar] [CrossRef] [Green Version]
- Reischl, B.; Watkins, M.; Foster, A.S. Free energy approaches for modeling atomic force microscopy in liquids. J. Chem. Theory Comput. 2013, 9, 600–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raiteri, P.; Demichelis, R.; Gale, J.D. Thermodynamically Consistent Force Field for Molecular Dynamics Simulations of Alkaline-Earth Carbonates and Their Aqueous Speciation. J. Phys. Chem. C 2015, 119, 24447–24458. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Tepper, H.L.; Voth, G.A. Flexible simple point-charge water model with improved liquid-state properties. J. Chem. Phys. 2006, 124, 024503. [Google Scholar] [CrossRef] [PubMed]
- Tribello, G.A.; Bonomi, M.; Branduardi, D.; Camilloni, C.; Bussi, G. PLUMED 2: New feathers for an old bird. Comput. Phys. Commun. 2014, 185, 604–613. [Google Scholar] [CrossRef] [Green Version]
- Grossfield, A. “WHAM: The weighted histogram analysis method” version 2.0.9. Available online: http://membrane.urmc.rochester.edu/wordpress/?page_id=126 (accessed on 10 April 2021).
- Smith, W. Mean Square Displacement. 1996. Available online: https://www.scd.stfc.ac.uk/Pages/DL_POLY.aspx (accessed on 10 April 2021).
- Allen, M.P.; Tildesley, D.J. Computer Simulation of Liquids; Oxford University Press Inc.: New York, NY, USA, 1987; ISBN 0198556454. [Google Scholar]
- Chandra, A. Effects of Ion Atmosphere on Hydrogen-Bond Dynamics in Aqueous Electrolyte Solutions. Phys. Rev. Lett. 2000, 85, 768–771. [Google Scholar] [CrossRef] [PubMed]
- Bruno, M.; Massaro, F.R.; Pastero, L.; Costa, E.; Rubbo, M.; Prencipe, M.; Aquilano, D. New estimates of the free energy of calcite/water interfaces for evaluating the equilibrium shape and nucleation mechanisms. Cryst. Growth Des. 2013, 13, 1170–1179. [Google Scholar] [CrossRef]
- Sekkal, W.; Zaoui, A. Nanoscale analysis of the morphology and surface stability of calcium carbonate polymorphs. Sci. Rep. 2013, 3, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Røyne, A.; Bisschop, J.; Dysthe, D.K. Experimental investigation of surface energy and subcritical crack growth in calcite. J. Geophys. Res. 2011, 116, B04204. [Google Scholar] [CrossRef] [Green Version]
- Bracco, J.N.; Stack, A.G.; Steefel, C.I. Upscaling calcite growth rates from the mesoscale to the macroscale. Environ. Sci. Technol. 2013, 47, 7555–7562. [Google Scholar] [CrossRef] [PubMed]
- Kurganskaya, I.; Rohlfs, R.D. Atomistic to meso-scale modeling of mineral dissolution: Methods, challenges and prospects. Am. J. Sci. 2020, 320, 1–26. [Google Scholar] [CrossRef]
- Hancer, M.; Celik, M.S.; Miller, J.D. The significance of interfacial water structure in soluble salt flotation systems. J. Colloid Interface Sci. 2001, 235, 150–161. [Google Scholar] [CrossRef]
- Dewan, S.; Yeganeh, M.S.; Borguet, E. Experimental correlation between interfacial water structure and mineral reactivity. J. Phys. Chem. Lett. 2013, 4, 1977–1982. [Google Scholar] [CrossRef]
- Di Tommaso, D.; Prakash, M.; Lemaire, T.; Lewerenz, M.; de Leeuw, N.H.; Naili, S. Molecular Dynamics Simulations of Hydroxyapatite Nanopores in Contact with Electrolyte Solutions: The Effect of Nanoconfinement and Solvated Ions on the Surface Reactivity and the Structural, Dynamical, and Vibrational Properties of Water. Crystals 2017, 7, 57. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, M.I.; Schenter, G.K.; Scamehorn, C.A.; Nicholas, J.B. Structure and Dynamics of the Water/MgO Interface. J. Phys. Chem. 1996, 100, 16989–16995. [Google Scholar] [CrossRef]
- Du, Z.; De Leeuw, N.H. Molecular dynamics simulations of hydration, dissolution and nucleation processes at the α-quartz (0001) surface in liquid water. J. Chem. Soc. Dalt. Trans. 2006, 2623–2634. [Google Scholar] [CrossRef]
- Cheng, L.; Fenter, P.; Nagy, K.L.; Schlegel, M.L.; Sturchio, N.C. Molecular-Scale Density Oscillations in Water Adjacent to a Mica Surface. Phys. Rev. Lett. 2001, 87, 156103. [Google Scholar] [CrossRef]
- Forbes, R.T.; York, P.; Fawcett, V.; Shields, L. Physicochemical Properties of Salts of p-Aminosalicylic Acid. I. Correlation of Crystal Structure and Hydrate Stability. Pharm. Res. 1992, 9, 1428–1435. [Google Scholar] [CrossRef]
- Gao, Z.Y.; Sun, W.; Hu, Y.H.; Liu, X.W. Anisotropic surface broken bond properties and wettability of calcite and fluorite crystals. Trans. Nonferrous Met. Soc. China Engl. Ed. 2012, 22, 1203–1208. [Google Scholar] [CrossRef]
- Bruno, M.; Bittarello, E.; Massaro, F.R.; Aquilano, D. The effect of impurities on the structure and energy of a crystal surface: Mg impurities in calcite as a case study. CrystEngComm 2018, 20, 4556–4564. [Google Scholar] [CrossRef]
- Lardge, J.S.; Duffy, D.M.; Gillan, M.J. Investigation of the interaction of water with the calcite (10.4) surface using ab initio simulation. J. Phys. Chem. C 2009, 113, 7207–7212. [Google Scholar] [CrossRef] [Green Version]
- Parker, S.C.; Kerisit, S.; Marmier, A.; Grigoleit, S.; Watson, G.W. Modelling Inorganic Solids and Their Interfaces: A Combined Approach of Atomistic and Electronic Structure Simulation Techniques. Faraday Discuss. 2003, 124, 155–170. [Google Scholar] [CrossRef]
- Ferreira, S.O. Advanced Topics on Crystal Growth; InTech: Rijeka, Croatia, 2013. [Google Scholar]
- Elstnerová, P.; Friák, M.; Fabritius, H.O.; Lymperakis, L.; Hickel, T.; Petrov, M.; Nikolov, S.; Raabe, D.; Ziegler, A.; Hild, S.; et al. Ab initio study of thermodynamic, structural, and elastic properties of Mg-substituted crystalline calcite. Acta Biomater. 2010, 6, 4506–4512. [Google Scholar] [CrossRef]
- Morse, J.W.; Mackenzie, F.T. Geochemistry of Sedimentary Carbonates; Elsevier Science Publisher: New York, NY, USA, 1990; Volume 48, ISBN 978-0-444-88781-8. [Google Scholar]
- Langmuir, D. Aqueous Environmental Geochemistry; Prentice Hall: Upper Saddle River, NJ, USA, 1997; ISBN 0023674121. [Google Scholar]
- Robie, R.; Hemingway, B. Thermodynamic Properties of Minerals and Related Substances at 298.15 K and 1 bar (105 Pascals) Pressure and at Higher Temperatures; Government Printing Office: Washington, DC, USA, 1995. [Google Scholar]
- Busenberg, E.; Niel Plummer, L. Thermodynamics of magnesian calcite solid-solutions at 25 °C and 1 atm total pressure. Geochim. Cosmochim. Acta 1989, 53, 1189–1208. [Google Scholar] [CrossRef]
- Bischoff, W.D.; Mackenzie, F.T.; Bishop, F.C. Stabilities of synthetic magnesian calcites in aqueous solution: Comparison with biogenic materials. Geochim. Cosmochim. Acta 1987, 51, 1413–1423. [Google Scholar] [CrossRef]
- Lasaga, A.C.; Lüttge, A. A model for crystal dissolution. Eur. J. Miner. 2003, 15, 603–615. [Google Scholar] [CrossRef]
- Bibi, I.; Arvidson, R.; Fischer, C.; Lüttge, A. Temporal Evolution of Calcite Surface Dissolution Kinetics. Minerals 2018, 8, 256. [Google Scholar] [CrossRef] [Green Version]
- Teng, H.H.; Dove, P.M.; De Yoreo, J.J. Kinetics of calcite growth: Surface processes and relationships to macroscopic rate laws. Geochim. Cosmochim. Acta 2000, 64, 2255–2266. [Google Scholar] [CrossRef]
- Wolthers, M. How minerals dissolve. Science 2015, 349, 1288. [Google Scholar] [CrossRef] [PubMed]
- Fenter, P.; Sturchio, N.C. Calcite (104)-water interface structure, revisited. Geochim. Cosmochim. Acta 2012, 97, 58–69. [Google Scholar] [CrossRef]
- Heberling, F.; Trainor, T.P.; Lützenkirchen, J.; Eng, P.; Denecke, M.A.; Bosbach, D. Structure and reactivity of the calcite–water interface. J. Colloid Interface Sci. 2011, 354, 843–857. [Google Scholar] [CrossRef]
- Zhu, L.F.; Friák, M.; Lymperakis, L.; Titrian, H.; Aydin, U.; Janus, A.M.; Fabritius, H.O.; Ziegler, A.; Nikolov, S.; Hemzalová, P.; et al. Ab initio study of single-crystalline and polycrystalline elastic properties of Mg-substituted calcite crystals. J. Mech. Behav. Biomed. Mater. 2013, 20, 296–304. [Google Scholar] [CrossRef]
- Ruiz-Agudo, E.; Putnis, C.V.; Jiménez-López, C.; Rodriguez-Navarro, C. An atomic force microscopy study of calcite dissolution in saline solutions: The role of magnesium ions. Geochim. Cosmochim. Acta 2009, 73, 3201–3217. [Google Scholar] [CrossRef]
- Arvidson, R.S.; Collier, M.; Davis, K.J.; Vinson, M.D.; Amonette, J.E.; Luttge, A. Magnesium inhibition of calcite dissolution kinetics. Geochim. Cosmochim. Acta 2006, 70, 583–594. [Google Scholar] [CrossRef]
- Arvidson, R.S.; Davis, K.J.; Collier, M.; Amonette, J.E.; Lüttge, A. Etch Pit Morphology and Magnesium Inhibition of Calcite Dissolution. In Proceedings of the 11th Symposium on Water-Rock Interactions WRI-11, Saratoga Springs, New York; Wanty, R.B., Seal, R.R., Eds.; Balkema: New York, NY, USA, 2004; pp. 721–725. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Property | Ca2+ in Pure Calcite | Ca2+ in HMgF Calcite | Mg2+ in HMgF Calcite | CO32− in Pure Calcite | CO32− in HMgF Calcite |
|---|---|---|---|---|---|
| Water dipole angle with surface (°) | 76.13 | 75.89 | |||
| Average number of hydrogen bonds between water molecules | 0.633 | 0.628 | 0.485 | 0.942 | 0.910 |
| 0 H-bond (%) | 49.9 | 49.9 | 57.7 | 40.8 | 42.3 |
| 1 H-bond (%) | 37.8 | 38.0 | 36.3 | 34.0 | 33.9 |
| 2 H-bond (%) | 11.5 | 11.3 | 5.8 | 16.9 | 15.9 |
| 3 H-bond (%) | 0.8 | 0.7 | 0.2 | 7.1 | 6.7 |
| H-bond Lifetime | 3.1 | 3.3 | 2.5 | 3.5 | 3.9 |
| Average number of H-bond between cation—Ow–Hw–Oc | 1.01 | 1.02 | 1.37 | ||
| Water forming >1 H-bond with Oc (%) | 15.8 | 15.3 | 40.8 | ||
| Distance HW–Oc | 1.853 | 1.855 | 1.860 |
| Acronym | Surface Energies (J/m2) | ||
|---|---|---|---|
| Dry | Wet | ||
| Pure calcite | Pure | 0.63 | 0.30 |
| Low Mg2+ flat surface | LMgF | 0.64 | 0.31 |
| High Mg2+ flat surface | HMgF | 0.62 | 0.20 |
| Island pure calcite | PureI | 0.66 | 0.35 |
| Island high Mg2+ | HMgI | 0.68 | 0.29 |
| Calcium Site | Colour Code | Label | ΔGextraction(kJ/mol) |
|---|---|---|---|
| Ca2+ in pure calcite | Green (  ) ) | CaP | 204 |
| Low Mg2+ flat surface (LMgF;Figure 4) | |||
| Ca2+with 1 Mg2+ in first cation shell * | Turquoise (  ) ) | CaLMgF1 | 218 |
Pink (  ) ) | CaLMgF2 | 223 | |
Orange (  ) ) | CaLMgF3 | 222 | |
| Ca2+with 1 Mg2+ in second cation shell | Purple (  ) ) | CaLMgF4 | 197 |
| High Mg2+ flat surface (HMgF;Figure 5) | |||
| Ca2+with 3 Mg2+ in first cation shell and 2 Mg2+ in second | Purple ( ) | CaHMgF3-5 | 209 |
| Ca2+with 1 Mg2+ in first cation shell and 1 Mg2+ in second | Pink ( ) | CaHMgF1-2 | 230 |
| Ca2+with 0 Mg2+ in first cation shell and 0 Mg2+ in second | Turquoise ( ) | CaHMgF0-0 | 191 |
| Island-surface, pure calcite (PureI, Figure S7) | |||
| Ca2+ in an Acute Corner | Purple ( ) | CaPAC | 43 |
| Ca2+ in an Obtuse Corner | Pink ( ) | CaPOC | 39 |
| Ca2+ in an Acute Edge | Turquoise ( ) | CaPAE | 56 |
| Ca2+ in an Obtuse Edge | Orange ( ) | CaPOE | 76 |
| Island-surface, high Mg2+ calcite (HMgI; Figure 6) | |||
| Ca2+ in an Acute Corner | Purple ( ) | CaHMgIAC | 37; 97 |
| Ca2+ in an Obtuse Corner | Pink ( ) | CaHMgIOC | 24; 90 |
| Ca2+ in an Acute Edge | Turquoise ( ) | CaIHMgIAE | 72; 206 |
| Ca2+ in an Obtuse Edge | Orange ( ) | CaHMgIOE | 60; 175 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koskamp, J.A.; Ruiz Hernandez, S.E.; Leeuw, N.H.D.; Wolthers, M. First Steps towards Understanding the Non-Linear Impact of Mg on Calcite Solubility: A Molecular Dynamics Study. Minerals 2021, 11, 407. https://0-doi-org.brum.beds.ac.uk/10.3390/min11040407
Koskamp JA, Ruiz Hernandez SE, Leeuw NHD, Wolthers M. First Steps towards Understanding the Non-Linear Impact of Mg on Calcite Solubility: A Molecular Dynamics Study. Minerals. 2021; 11(4):407. https://0-doi-org.brum.beds.ac.uk/10.3390/min11040407
Chicago/Turabian StyleKoskamp, Janou A., Sergio E. Ruiz Hernandez, Nora H. De Leeuw, and Mariette Wolthers. 2021. "First Steps towards Understanding the Non-Linear Impact of Mg on Calcite Solubility: A Molecular Dynamics Study" Minerals 11, no. 4: 407. https://0-doi-org.brum.beds.ac.uk/10.3390/min11040407