Chiral Peptide Nucleic Acids with a Substituent in the N-(2-Aminoethy)glycine Backbone


Abstract
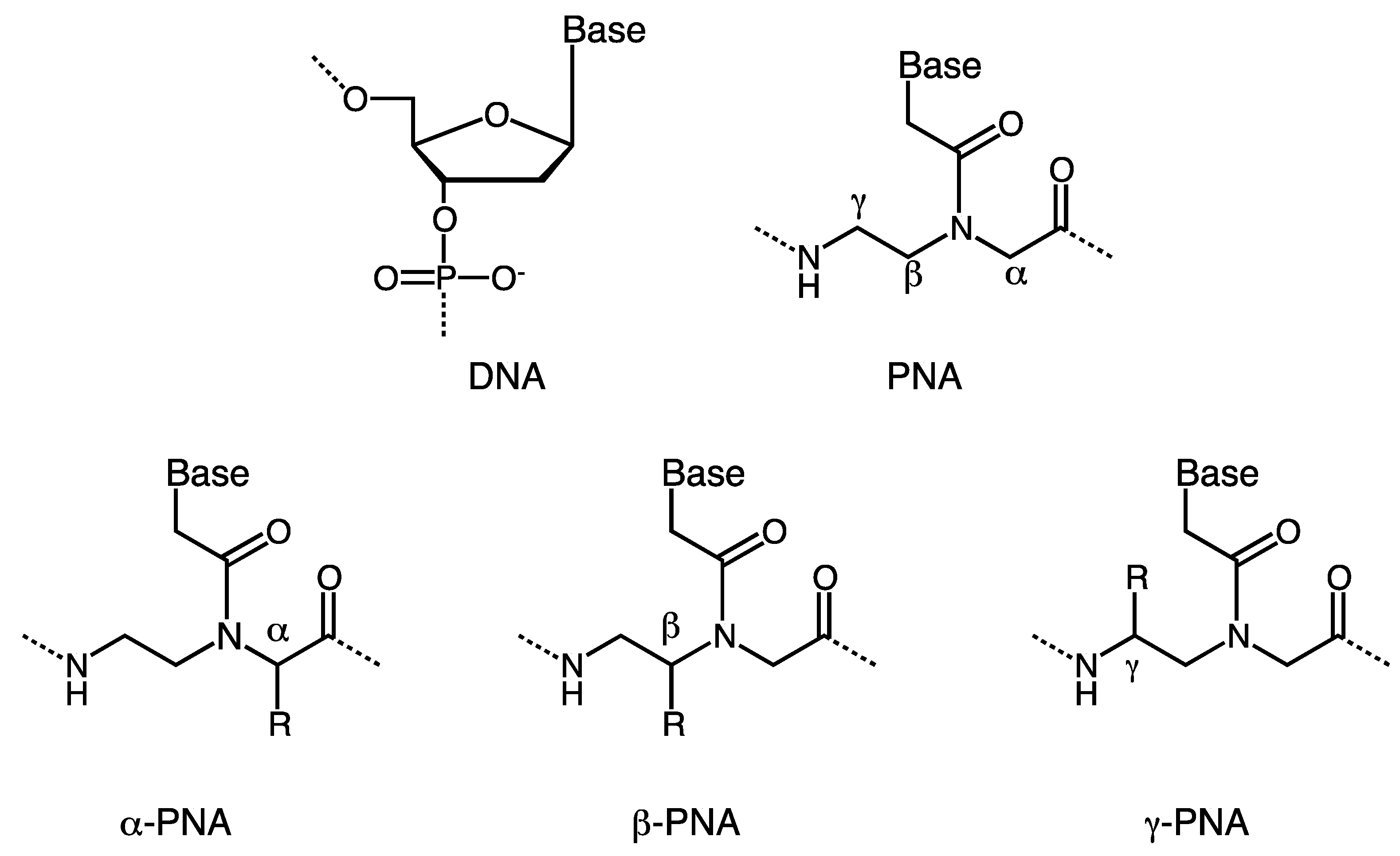
:1. Introduction

2. α-PNA
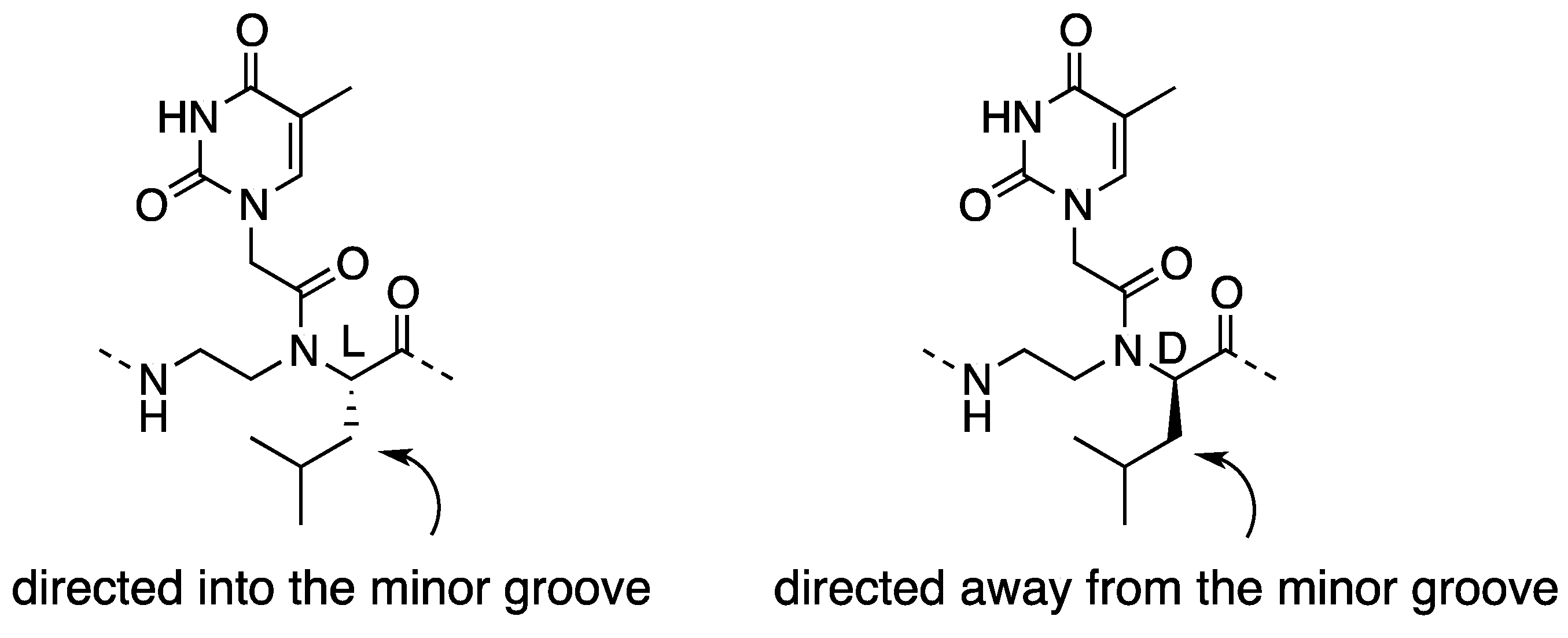
2.1. Synthesis and Properties of α-PNAs


| PNA a | Tm (°C) | Tm (°C) |
|---|---|---|
| perfect match | single mismatch | |
| 5'-AGTGATCTAC-3' | 5'-AGTGGTCTAC-3' | |
| H-GTAGATCACT-NH2 | 50 | 40 |
| H-GTAGATCACT-NH2 | 43 | - |
 | ||
| Sequence | Tm (°C) | Tm | |
|---|---|---|---|
 | H-GCATGTTTGA-LLys-NH2 | 43 | |
| H-GCATGLTTTGA-LLys-NH2 | 39 | −4 | |
| H-GCATGDTTTGA-LLys-NH2 | 41 | −2 | |
| H-GCADTGDTTDTGA-LLys-NH2 | 47 | +4 | |
| H-GDCADTGDTTDTGDA-LLys-NH2 | 50 | +7 |



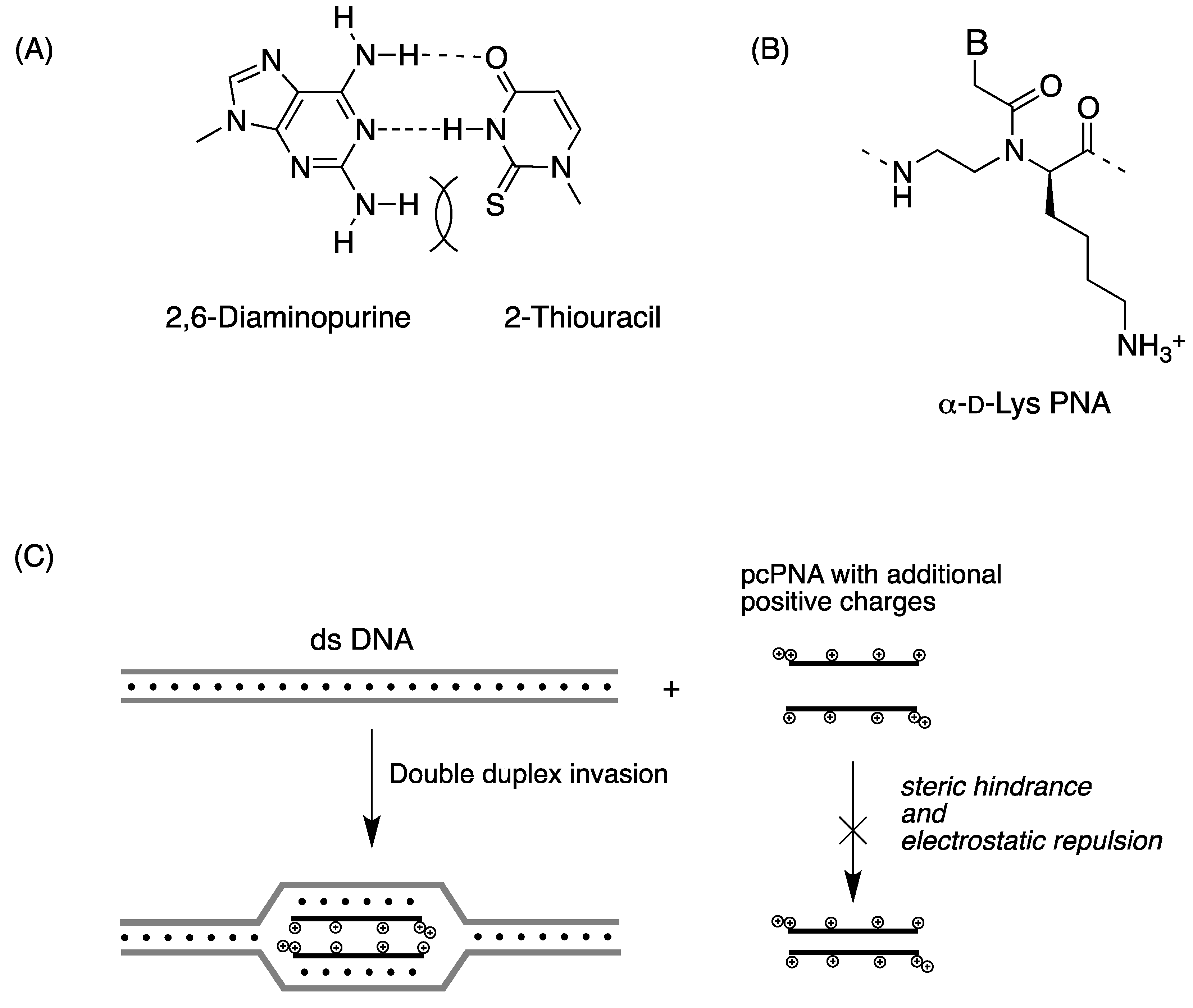
2.2. Double-Duplex Invasion

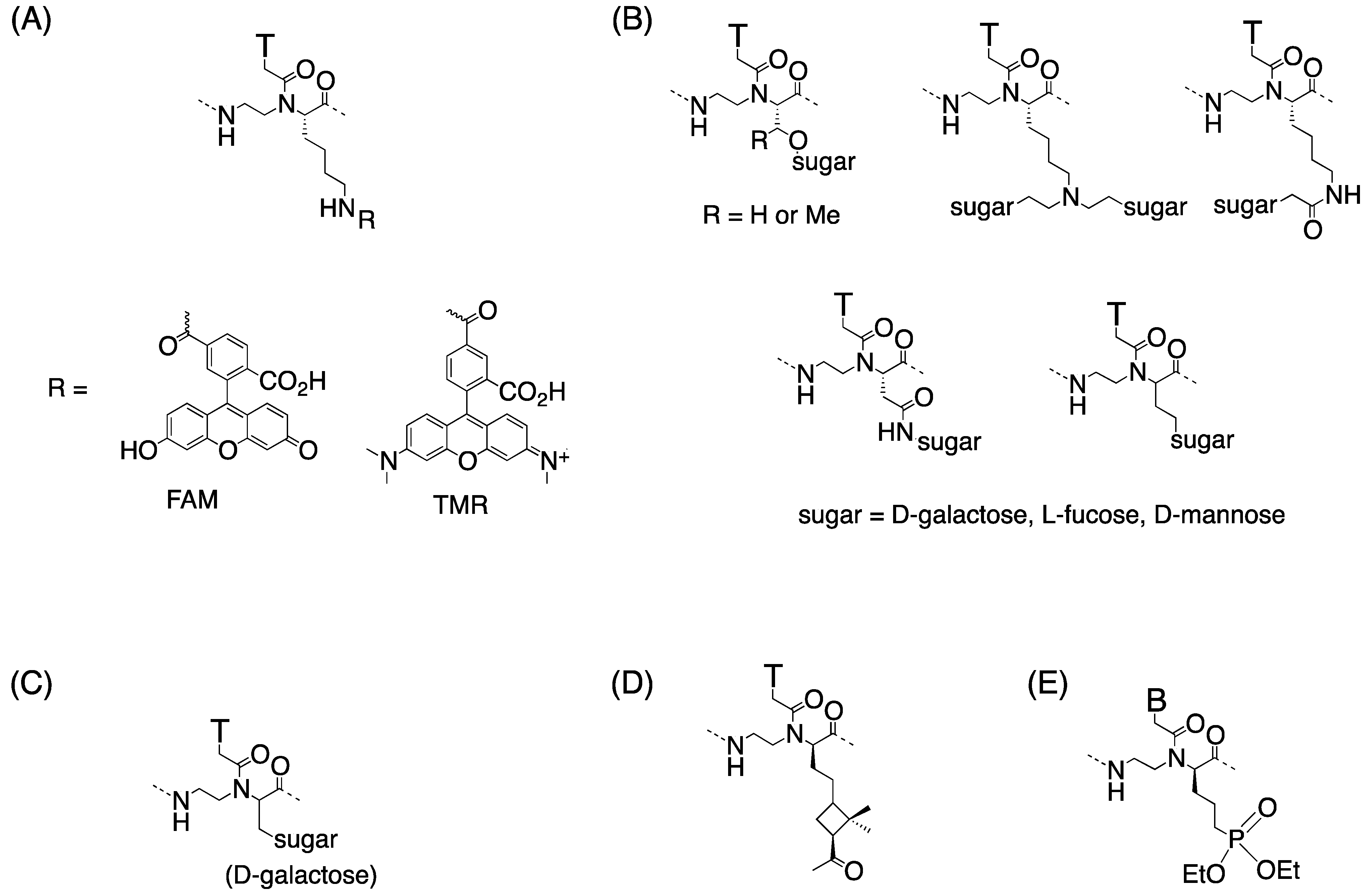
2.3. Cellular Uptake of α-PNAs

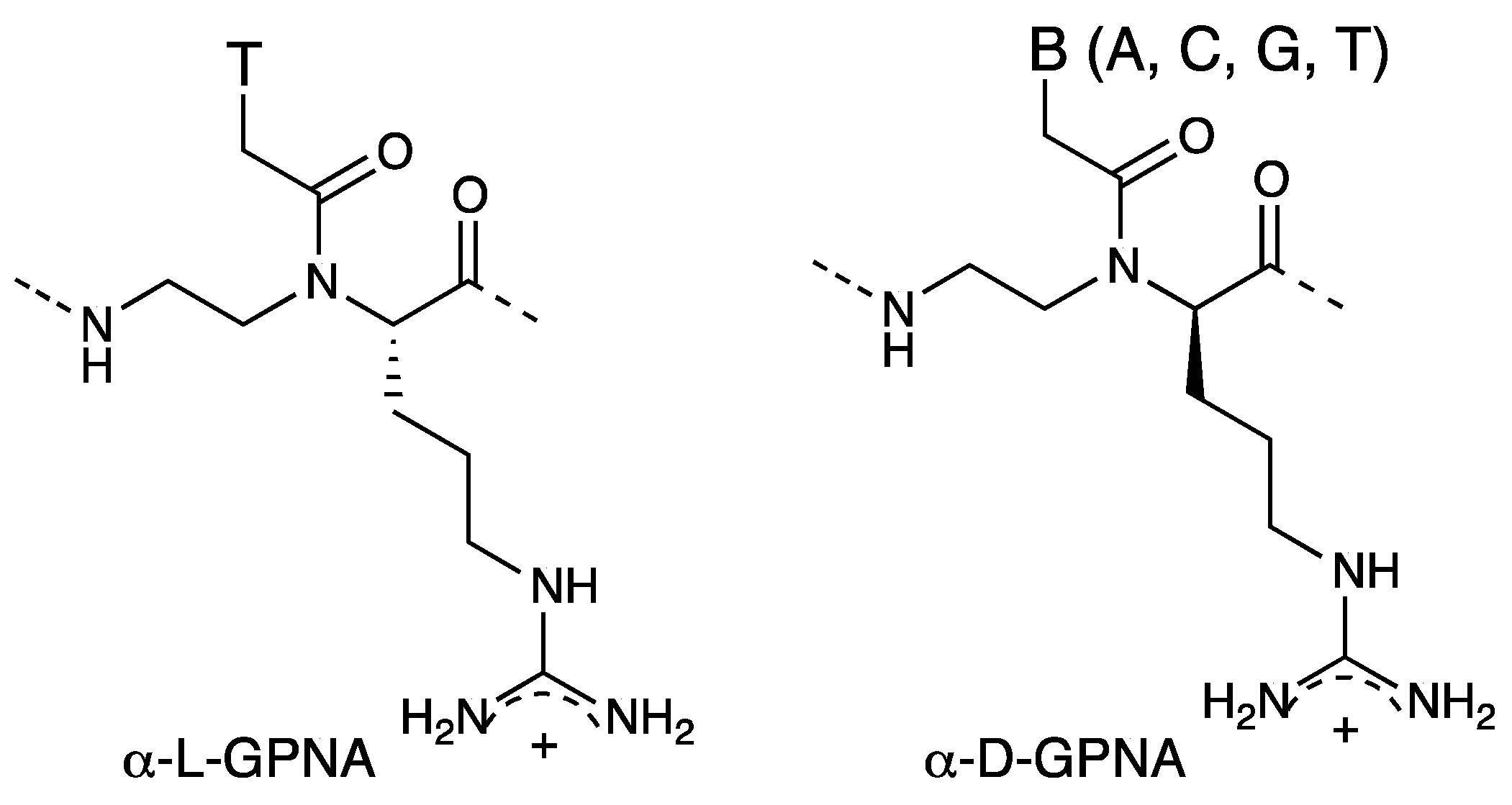
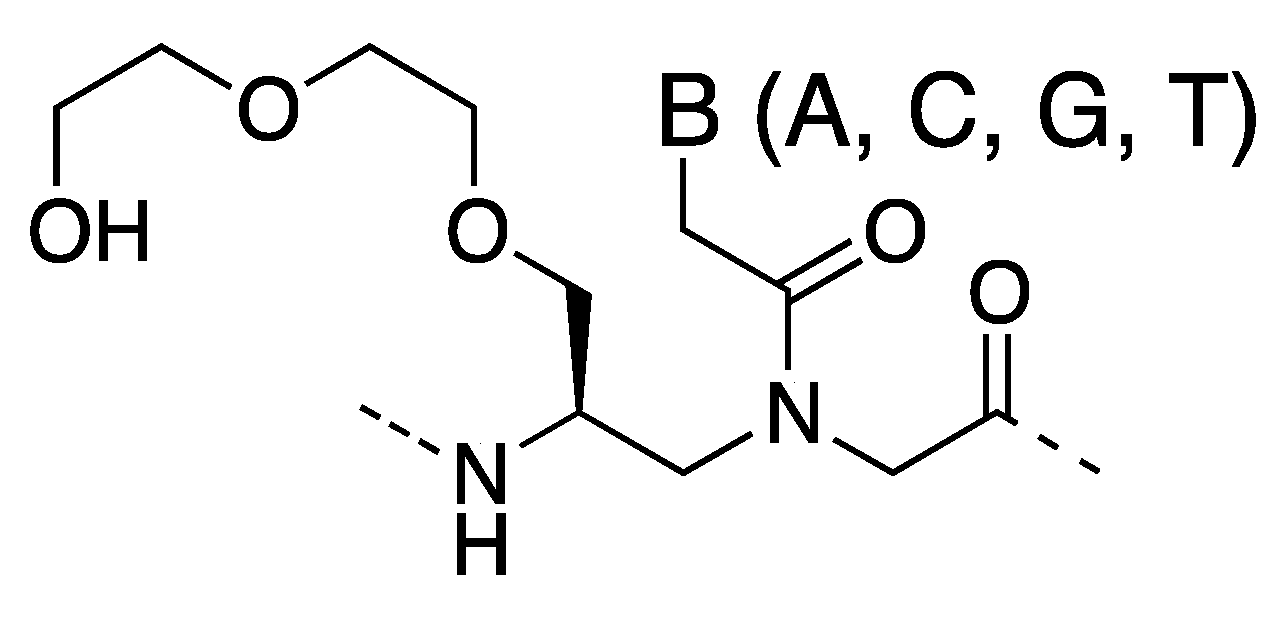
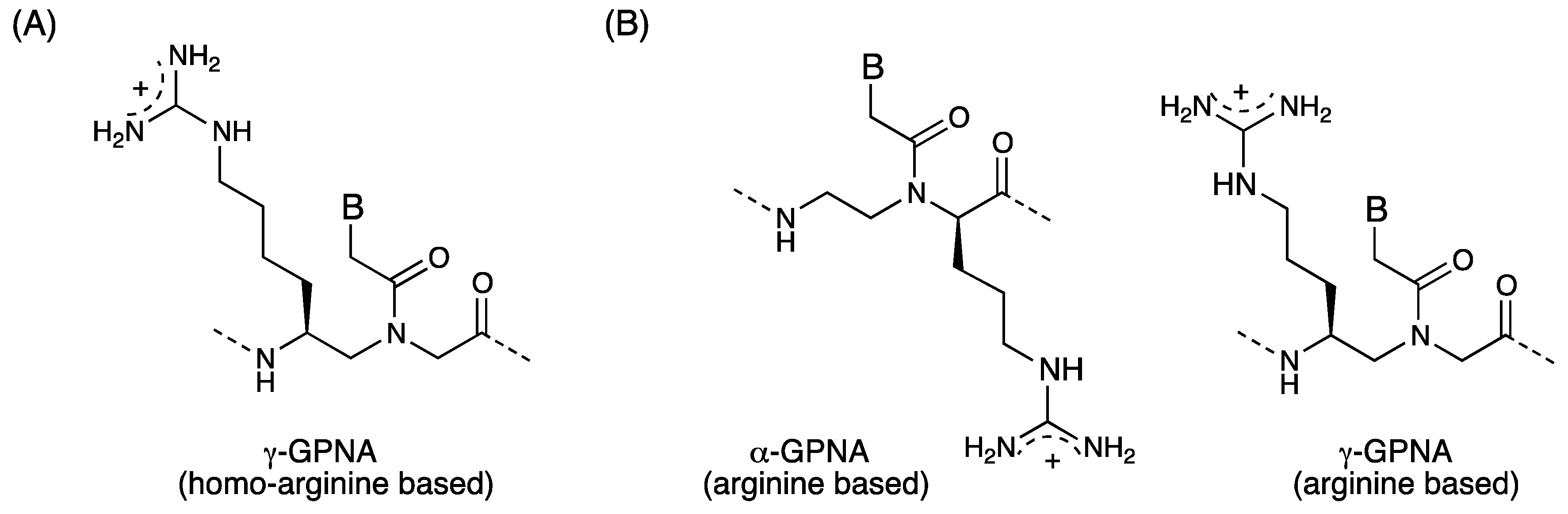
3. γ-PNA
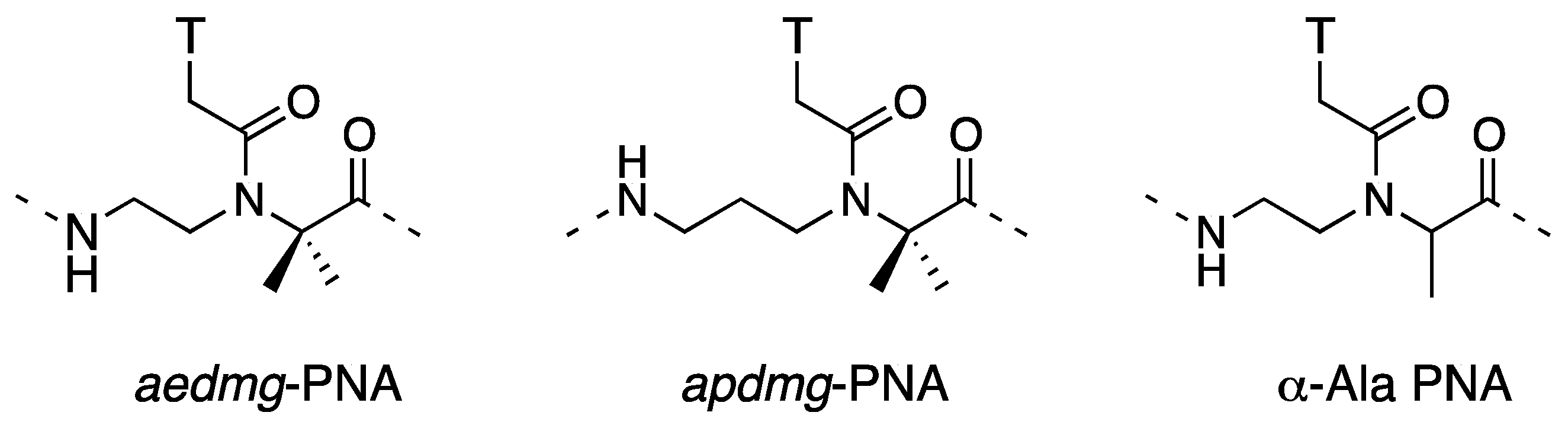
3.1. Synthesis and Properties of γ-PNAs



| Backbone Modification | Tm (°C) | PNA sequence | Reference |
|---|---|---|---|
| Gly | 47 | a | [70] |
| L-Ala | 51 | a | |
| L-Val | 51 | a | |
| L-Ile | 51 | a | |
| L-Phe | 51 | a | |
| Gly | 44 | a | [68] |
| L-Ser | 48 | a | |
| Gly | 49.7 | b | [62] |
| L-Lys | 51.4 | b | |
| D-Lys | 36.4 | b | |
| Gly | 50 | c | [52] |
| L-Lys | 56 | c | |
| D-Lys | 32 | C |
3.2. Duplex Invasion of γ-PNAs

| PNA sequences | Tm (°C) | Duplex invasion | Reference |
|---|---|---|---|
| PNA1: H-LLys-GACCACAGAT-LLys-NH2 | 59 | - | [75] |
| PNA2: H-LLys-GACCACAGAT-LLys-NH2 | ~90 | - | |
| PNA3: H-LLys-GACCACAGAT-LLys(Acr)-LLys-NH2 | ~90 | + | |
| PNA4: H-LLys-GAXCACAGAT-LLys-NH2 | n.d. | + | [76,77] |
| PNA5: H-LLys-GAXCAXAGAT-LLys-NH2 | n.d. | + | |
| PNA6: H-LLys-GAXXAXAGAT-LLys-NH2 | n.d. | + | |
| PNA7: H-LLys-GACCACAGATCTAAG-LLys-NH2 | >95 | + | [78] |
| PNA8: H-LLys-TATGAGACCACAGATCTAAG-LLys-NH2 | >95 | + |

3.3. Cell Internalization of γ-PNAs and Related Modified PNAs



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
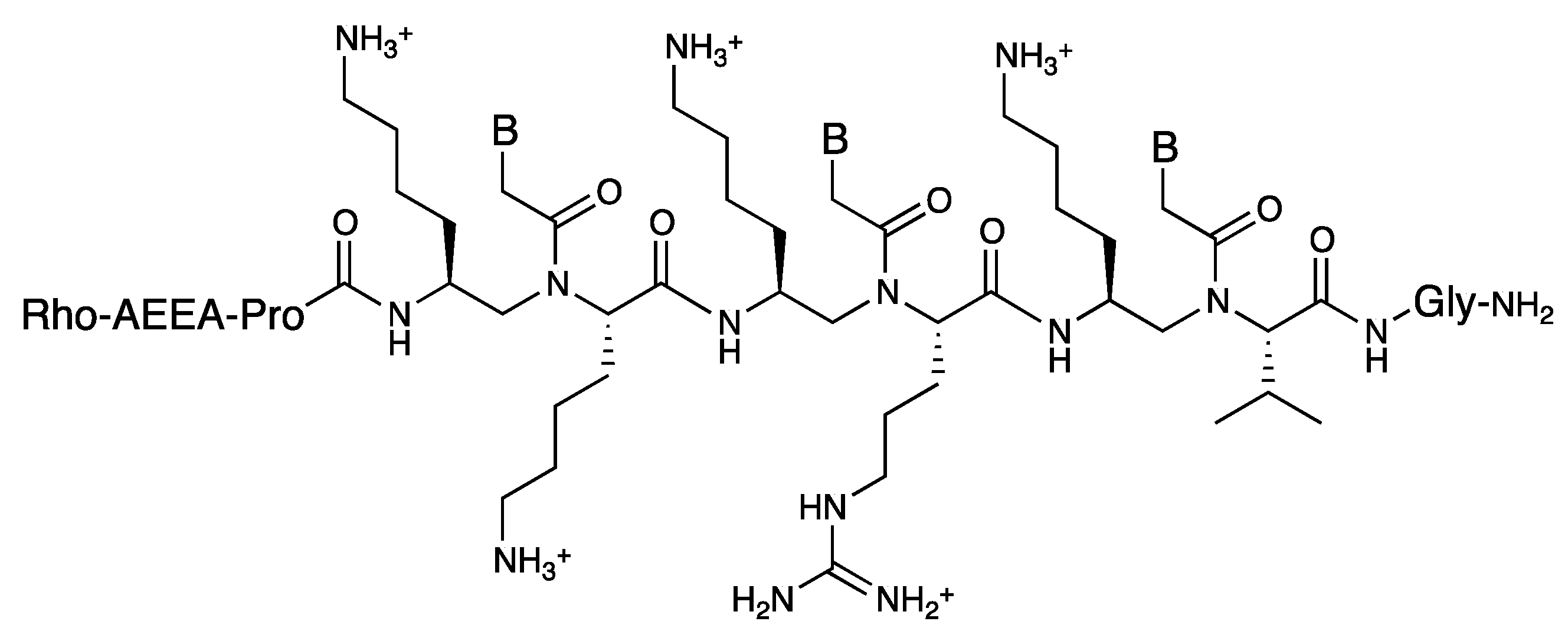{kind=link}
{kind=link}
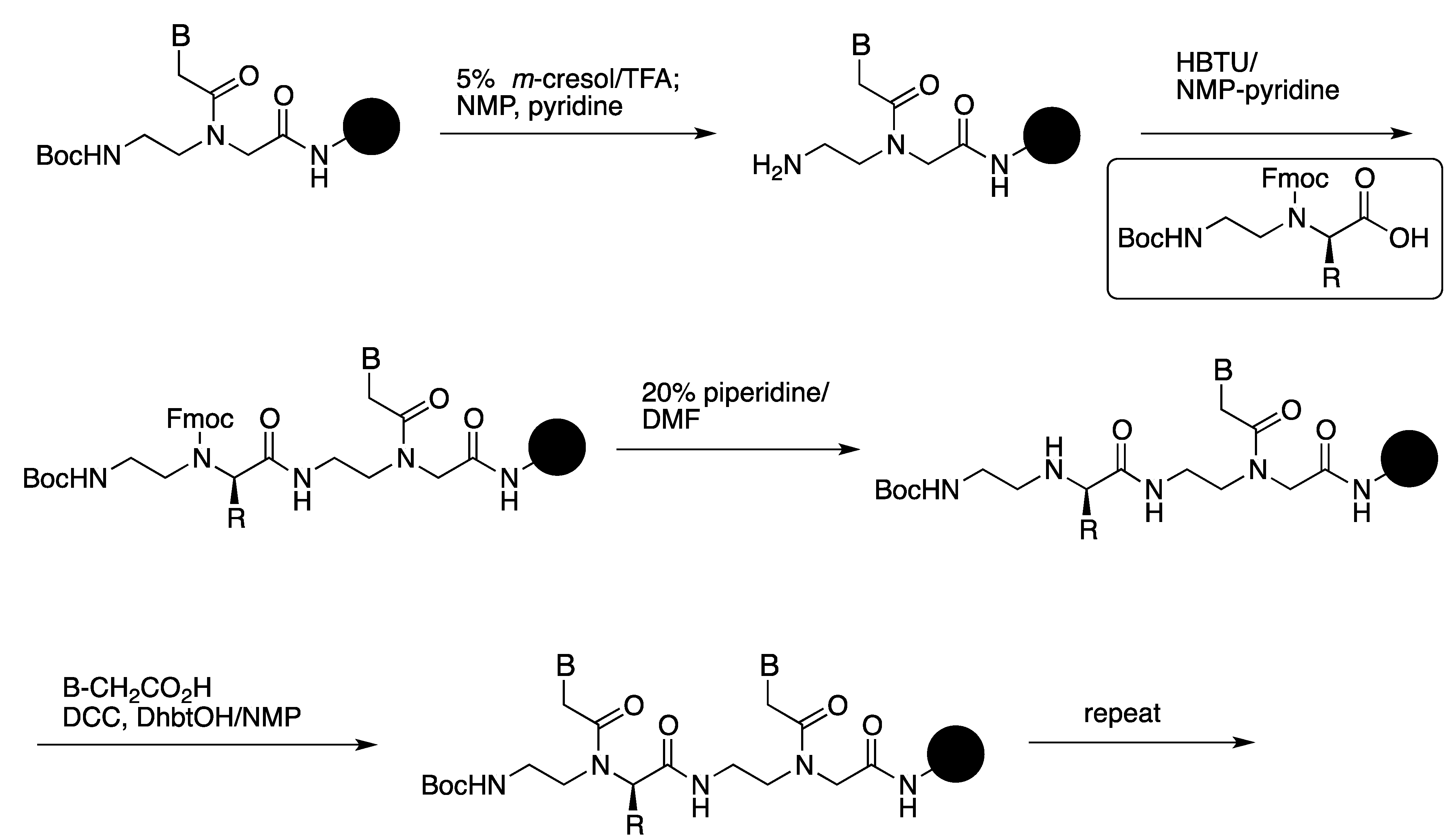{kind=link}
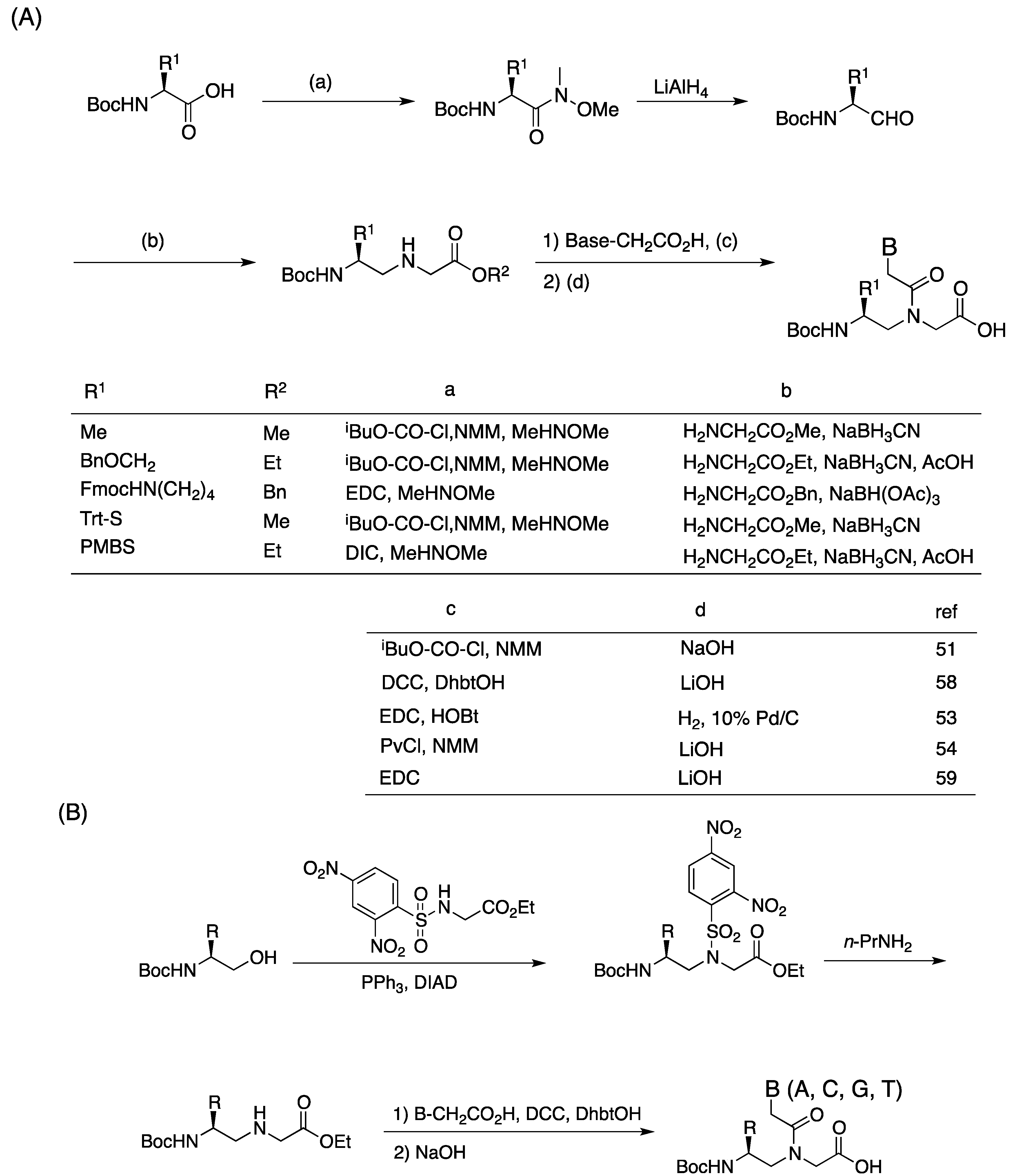{kind=link}
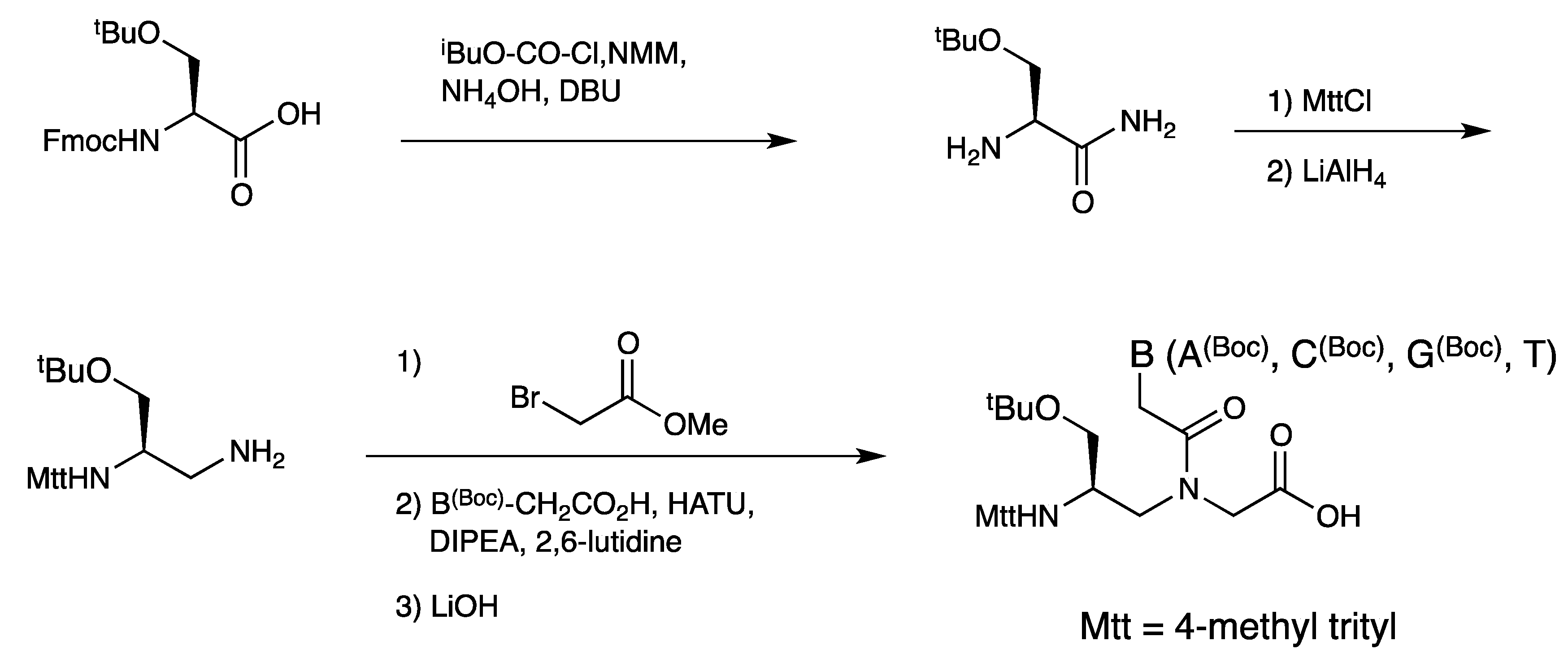{kind=link}
{kind=link}
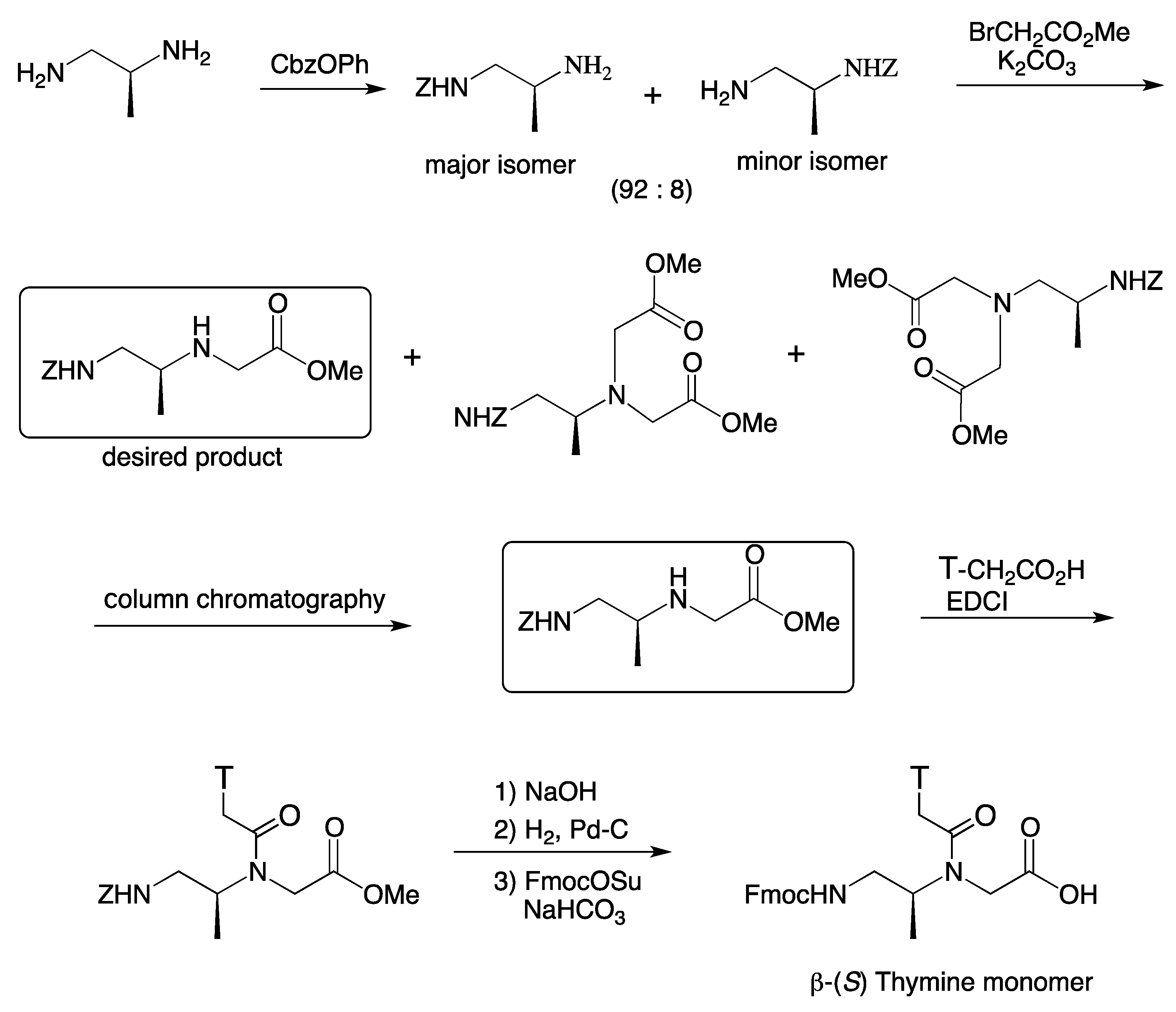
4. β-PNA

| PNA sequence | Tm (°C) |
|---|---|
| H-GTAGATCACT-LLys-NH2 | 51.4 |
| H-G TSAGATSCACTS-LLys-NH2 | 51.0 |
| H-G TRAGATRCACTR-DLys-NH2 | n.d. |
 | |
5. Conclusions
Acknowledgments
- Sample Availability: Not available.
References
- Nielsen, P.E.; Egholm, M.; Berg, R.H.; Buchardt, O. Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science 1991, 254, 1497–1500. [Google Scholar]
- Egholm, M.; Buchardt, O.; Christensen, L.; Behrens, C.; Freier, S.M.; Driver, D.A.; Berg, R.H.; Kim, S.K.; Norden, B.; Nielsen, P.E. PNA hybridizes to complementary oligonucleotides obeying the Watson–Crick hydrogen-bonding rules. Nature 1993, 365, 566–568. [Google Scholar]
- Jensen, K.K.; Orum, H.; Nielsen, P.E.; Norden, B. Kinetics for hybridization of peptide nucleic acids (PNA) with DNA and RNA studied with the BIAcore technique. Biochemistry 1997, 36, 5072–5077. [Google Scholar] [CrossRef]
- Ratilainen, T.; Holmen, A.; Tuite, E.; Nielsen, P.E.; Norden, B. Thermodynamics of sequence-specific binding of PNA to DNA. Biochemistry 2000, 39, 7781–7791. [Google Scholar] [CrossRef]
- Demidov, V.V.; Potaman, V.N.; Frank-Kamenetskii, M.D.; Egholm, M.; Buchardt, O.; Sonnichsen, S.H.; Nielsen, P.E. Stability of peptide nucleic acids in human serum and cellular extracts. Biochem. Pharmacol. 1994, 48, 1310–1313. [Google Scholar] [CrossRef]
- Hamilton, S.E.; Iyer, M.; Norton, J.C.; Corey, D.R. Specific and nonspecific inhibition of transcription by DNA, PNA, and phosphorothioate promoter analog duplexes. Bioorg. Med. Chem. Lett. 1996, 6, 2897–2900. [Google Scholar] [CrossRef]
- Cutrona, G.; Carpaneto, E.M.; Ulivi, M.; Roncella, S.; Landt, O.; Ferrarini, M.; Boffa, L.C. Effects in live cells of a c-myc anti-gene PNA linked to a nuclear localization signal. Nat. Biotechnol. 2000, 18, 300–303. [Google Scholar] [CrossRef]
- Boffa, L.C.; Cutrona, G.; Cilli, M.; Mati, S.; Damonte, G.; Mariani, M.R.; Millo, E.; Moroni, M.; Roncella, S.; Fedeli, F.; Ferrarini, M. Inhibition of Burkitt's lymphoma cells growth in SCID mice by a PNA specific for a regulatory sequence of the translocated c-myc. Cancer Gene Ther. 2007, 14, 220–226. [Google Scholar] [CrossRef]
- Tonelli, R.; Purgato, S.; Camerin, C.; Fronza, R.; Bologna, F.; Alboresi, S.; Franzoni, M.; Corradini, R.; Sforza, S.; Faccini, A.; Shohet, J.M.; Marchelli, R.; Pession, A. Antigene peptide nucleic acid specifically inhibits MYCN expression in human neuroblastoma cells leading to cell growth inhibition and apoptosis. Mol. Cancer Ther. 2005, 4, 779–786. [Google Scholar] [CrossRef]
- Cogoi, S.; Codognotto, A.; Rapozzi, V.; Meeuwenoord, N.; van der Marel, G.; Xodo, L.E. Transcription inhibition of oncogenic KRAS by a mutation-selective peptide nucleic acid conjugated to the PKKKRKV nuclear localization signal peptide. Biochemistry 2005, 44, 10510–10519. [Google Scholar] [CrossRef]
- Janowski, B.A.; Kaihatsu, K.; Huffman, K.E.; Schwartz, J.C.; Ram, R.; Hardy, D.; Mendelson, C.R.; Corey, D.R. Inhibiting transcription of chromosomal DNA with antigene peptide nucleic acids. Nat. Chem. Biol. 2005, 1, 210–215. [Google Scholar] [CrossRef]
- Kumar, V.A.; Ganesh, K.N. Conformationally constrained PNA analogues: Structural evolution toward DNA/RNA binding selectivity. Acc. Chem. Res. 2005, 38, 404–412. [Google Scholar] [CrossRef]
- Corradini, R.; Sforza, S.; Tedeschi, T.; Totsingan, F.; Manicardi, A.; Marchelli, R. Peptide nucleic acids with a structurally biased backbone. Updated review and emerging challenges. Curr. Top. Med. Chem. 2011, 11, 1535–1554. [Google Scholar] [CrossRef]
- Dueholm, K.L.; Petersen, K.H.; Jensen, D.K.; Egholm, M.; Nielsen, P.E.; Buchardt, O. Peptide nucleic acid (PNA) with a chiral backbone based on alanine. Bioorg. Med. Chem. Lett. 1994, 4, 1077–1080. [Google Scholar] [CrossRef]
- Haaima, G.; Lohse, A.; Buchardt, O.; Nielsen, P.E. Peptide nucleic acids (PNAs) containing thymine monomers derived from chiral amino acids: Hybridization and solubility properties of D-lysine PNA. Angew. Chem. Int. Ed. Engl. 1996, 35, 1939–1942. [Google Scholar] [CrossRef]
- Püschl, A.; Sforza, S.; Haaima, G.; Dahl, O.; Nielsen, P.E. Peptide nucleic acids (PNAs) with a functional backbone. Tetrahedron Lett. 1998, 39, 4707–4710. [Google Scholar] [CrossRef]
- Gupta, P.; Muse, O.; Rozners, E. Recognition of double-stranded RNA by guanidine-modified peptide nucleic acids. Biochemistry 2012, 51, 63–73. [Google Scholar]
- Balaji, B.S.; Gallazzi, F.; Jia, F.; Lewis, M.R. An efficient, convenient solid-phase synthesis of amino acid-modified peptide nucleic acid monomers and oligomers. Bioconjug. Chem. 2006, 17, 551–558. [Google Scholar] [CrossRef]
- Corradini, R.; Sforza, S.; Dossena, A.; Palla, G.; Rocchi, R.; Filira, F.; Nastri, F.; Marchelli, R. Epimerization of peptide nucleic acids analogs during solid-phase synthesis: optimization of the coupling conditions for increasing the optical purity. J. Chem. Soc. Perkin Trans. 1 2001, 2690–2696. [Google Scholar]
- Tedeschi, T.; Corradini, R.; Marchelli, R.; Pushl, A.; Nielsen, P.E. Racemization of chiral PNAs during solid-phase synthesis: Effect of the coupling conditions on enantiomeric purity. Tetrahedron: Asymmetry 2002, 13, 1629–1636. [Google Scholar]
- Richter, L.S.; Zuckermann, R.N. Synthesis of peptide nucleic acids (PNA) by submonomer solid-phase synthesis. Bioorg. Med. Chem. Lett. 1995, 5, 1159–1163. [Google Scholar] [CrossRef]
- Viirre, R.D.; Hudson, R.H.E. Optimization of a solid-phase synthesis of a PNA monomer. Org. Lett. 2001, 3, 3931–3934. [Google Scholar] [CrossRef]
- Sforza, S.; Tedeschi, T.; Corradini, R.; Ciavardelli, D.; Dossena, A.; Marchelli, R. Fast, solid-phase synthesis of chiral peptide nucleic acids with a high optical purity by a submonomeric strategy. Eur. J. Org. Chem. 2003, 2003, 1056–1063. [Google Scholar]
- Sforza, S.; Haaima, G.; Marchelli, R.; Nielsen, P.E. Chiral peptide nucleic acids (PNAs): Helix handedness and DNA recognition. Eur. J. Org. Chem. 1999, 1999, 197–204. [Google Scholar] [CrossRef]
- Sforza, S.; Corradini, R.; Ghirardi, S.; Dossena, A.; Marchelli, R. DNA binding of a D-lysine-based chiral PNA: Direction control and mismatch recognition. Eur. J. Org. Chem. 2000, 2000, 2905–2913. [Google Scholar] [CrossRef]
- Menchise, V.; de Simone, G.; Tedeschi, T.; Corradini, R.; Sforza, S.; Marchelli, R.; Capasso, D.; Saviano, M.; Pedone, C. Insights into peptide nucleic acid (PNA) structural features: The crystal structure of a D-lysine-based chiral PNA-DNA duplex. Proc. Natl. Acad. Sci. USA 2003, 100, 12021–12026. [Google Scholar]
- Dose, C.; Seitz, O. Single nucleotide specific detection of DNA by native chemical ligation of fluorescence labeled PNA-probes. Bioorg. Med. Chem. 2008, 16, 65–77. [Google Scholar] [CrossRef]
- Zhou, P.; Dragulescu-Andrasi, A.; Bhattacharya, B.; O’Keefe, H.; Vatta, P.; Hyldig-Nielsen, J.J.; Ly, D.H. Synthesis of cell-permeable peptide nucleic acids and characterization of their hybridization and uptake properties. Bioorg. Med. Chem. Lett. 2006, 16, 4931–4935. [Google Scholar] [CrossRef]
- Rossi, S.; Calabretta, A.; Tedeschi, T.; Sforza, S.; Arcioni, S.; Baldoni, L.; Corradini, R.; Marchelli, R. Selective recognition of DNA from olive leaves and olive oil by PNA and modified-PNA microarrays. Artificial DNA PNA XNA 2012, 3, 63–72. [Google Scholar]
- Hamzavi, R.; Dolle, F.; Tavitian, B.; Dahl, O.; Nielsen, P.E. Modulation of the pharmacokinetic properties of PNA: Preparation of galactosyl, mannosyl, fucosyl, N-acetylgalactosaminyl, and N-acetylglucosaminyl derivatives of aminoethylglycine peptide nucleic acid monomers and their incorporation into PNA oligomers. Bioconjug. Chem. 2003, 14, 941–954. [Google Scholar] [CrossRef]
- Hamzavi, R.; Meyer, C.; Metzler-Nolte, N. Synthesis of a C-linked glycosylated thymine-based PNA monomer and its incorporation into a PNA oligomer. Org. Biomol. Chem. 2005, 4, 3648–3651. [Google Scholar]
- Aguado, G.P.; Rúa, F.; Branchadell, V.; Nielsen, P.E.; Ortuño, R.M. Cyclobutyl-carbonyl substituted PNA: Synthesis and study of a novel PNA derivative. Tetrahedron: Asymmetry 2006, 17, 2499–2503. [Google Scholar]
- Dorn, S.; Aghaallaei, N.; Jung, G.; Bajoghli, B.; Werner, B.; Bock, H.; Lindhorst, T.; Czerny, T. Side chain modified peptide nucleic acids (PNA) for knock-down of six3 in medaka embryos. BMC Biotechnol. 2012, 12. [Google Scholar] [CrossRef]
- Dilek, I.; Madrid, M.; Singh, R.; Urrea, C.P.; Armitage, B.A. Effect of PNA backbone modifications on cyanine dye binding to PNA-DNA duplexes investigated by optical spectroscopy and molecular dynamics simulations. J. Am. Chem. Soc. 2005, 127, 3339–3345. [Google Scholar]
- Gourishankar, A.; Ganesh, K.N. (α,α-dimethyl)glycyl (dmg) PNAs. Achiral PNA analogs that form stronger hybrids with cDNA relative to isosequential RNA. Artificial DNA PNA XNA 2012, 3, 5–13. [Google Scholar] [CrossRef]
- Lohse, J.; Dahl, O.; Nielsen, P.E. Double duplex invasion by peptide nucleic acid: A general principle for sequence-specific targeting of double-stranded DNA. Proc. Natl. Acad. Sci. USA 1999, 96, 11804–11808. [Google Scholar]
- Ishizuka, T.; Yoshida, J.; Yamamoto, Y.; Sumaoka, J.; Tedeschi, T.; Corradini, R.; Sforza, S.; Komiyama, M. Chiral introduction of positive charges to PNA for double-duplex invasion to versatile sequences. Nucleic Acids Res. 2008, 36, 1464–1471. [Google Scholar] [CrossRef]
- Koppelhus, U.; Nielsen, P.E. Cellular delivery of peptide nucleic acid (PNA). Adv. Drug Deliv. Rev. 2003, 55, 267–280. [Google Scholar] [CrossRef]
- Nielsen, P.E. Addressing the challenges of cellular delivery and bioavailability of peptide nucleic acids (PNA). Q. Rev. Biophys. 2005, 38, 345–350. [Google Scholar] [CrossRef]
- Shiraishi, T.; Pankratova, S.; Nielsen, P.E. Calcium ions effectively enhance the effect of antisense peptide nucleic acids conjugated to cationic Tat and oligoarginine Peptides. Chem. Biol. 2005, 12, 923–929. [Google Scholar] [CrossRef]
- Hu, J.; Corey, D.R. Inhibiting gene expression with peptide nucleic acid (PNA)-peptide conjugates that target chromosomal DNA. Biochemistry 2007, 46, 7581–7589. [Google Scholar] [CrossRef]
- Koppelhus, U.; Awasthi, S.K.; Zachar, V.; Holst, H.U.; Ebbesen, P.; Nielsen, P.E. Cell-dependent differential cellular uptake of PNA, peptides, and PNA-peptide conjugates. Antisense Nucleic Acid Drug Dev. 2002, 12, 51–63. [Google Scholar] [CrossRef]
- Zhou, P.; Wang, M.; Du, L.; Fisher, G.W.; Waggoner, A.; Ly, D.H. Novel binding and efficient cellular uptake of guanidine-based peptide nucleic acids (GPNA). J. Am. Chem. Soc. 2003, 125, 6878–6879. [Google Scholar] [CrossRef]
- Nagahara, H.; Vocero-Akbini, A.M.; Snyder, E.L.; Ho, A.; Latham, D.G.; Lissy, N.A.; Becker-Hapak, M.; Ezhevsky, S.; Dowdy, S.F. Transduction of full-length TAT fusion proteins into mammalian cells: TAT-p27Kip1 induces cell migration. Nat. Med. 1998, 4, 1449–1452. [Google Scholar] [CrossRef]
- Futaki, S.; Suzuki, T.; Ohashi, W.; Yagami, T.; Tanaka, S.; Ueda, K.; Sugiura, Y. Arginine-rich peptides. J. Biol. Chem. 2001, 276, 5836–5840. [Google Scholar]
- Wender, P.A.; Mitchell, D.J.; Pattabiraman, K.; Pelkey, E.T.; Steinman, L.; Rothbard, J.B. The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: Peptoid molecular transporters. Proc. Natl. Acad. Sci. USA 2000, 97, 13003–13008. [Google Scholar]
- Dragulescu-Andrasi, A.; Zhou, P.; He, G.; Ly, D.H. Cell-permeable GPNA with appropriate backbone stereochemistry and spacing binds sequence-specifically to RNA. Chem. Commun. 2005, 41, 244–246. [Google Scholar] [CrossRef]
- Rothbard, J.B.; Jessop, T.C.; Lewis, R.S.; Murray, B.A.; Wender, P.A. Role of membrane potential and hydrogen bonding in the mechanism of translocation of guanidinium-rich peptides into cells. J. Am. Chem. Soc. 2004, 126, 9506–9507. [Google Scholar] [CrossRef]
- Dragulescu-Andrasi, A.; Rapireddy, S.; He, G.; Bhattacharya, B.; Hyldig-Nielsen, J.J.; Zon, G.; Ly, D.H. Cell-permeable peptide nucleic acid designed to bind to the 5'-untranslated region of E-cadherin transcript induces potent and sequence-specific antisense effects. J. Am. Chem. Soc. 2006, 128, 16104–16112. [Google Scholar]
- Doyle, D.F.; Braasch, D.A.; Simmons, C.G.; Janowski, B.A.; Corey, D.R. Inhibition of gene expression inside cells by peptide nucleic acids: effect of mRNA target sequence, mismatched bases, and PNA length. Biochemistry 2001, 40, 53–64. [Google Scholar]
- Kosynkina, L.; Wang, W.; Liang, T.C. A Convenient synthesis of chiral peptide nucleic acid (PNA) monomers. Tetrahedron Lett. 1994, 35, 5173–5176. [Google Scholar] [CrossRef]
- Tedeschi, T.; Sforza, S.; Corradini, R.; Marchelli, R. Synthesis of new chiral PNAs bearing a dipeptide-mimic monomer with two lysine-derived stereogenic centres. Tetrahedron Lett. 2005, 46, 8395–8399. [Google Scholar] [CrossRef]
- Englund, E.A.; Appella, D.H. Synthesis of γ-substituted peptide nucleic acids: A new place to attach fluorophores without affecting DNA binding. Org. Lett. 2005, 7, 3465–3467. [Google Scholar] [CrossRef]
- Dose, C.; Seitz, O. Convergent Synthesis of Peptide Nucleic Acids by Native Chemical Ligation. Org. Lett. 2005, 7, 4365–4368. [Google Scholar] [CrossRef]
- Ficht, S.; Dose, C.; Seitz, O. As fast and selective as enzymatic ligations: Unpaired nucleobases increase the selectivity of DNA-controlled native chemical PNA ligation. ChemBioChem 2005, 6, 2098–2103. [Google Scholar] [CrossRef]
- Stanfield, C.F.; Parker, J.E.; Kanellis, P. Preparation of protected amino aldehydes. J. Org. Chem. 1981, 46, 4797–4798. [Google Scholar] [CrossRef]
- Rittle, K.E.; Homnick, C.F.; Ponticello, G.S.; Evans, B.E. A synthesis of statine utilizing an oxidative route to chiral α-amino aldehydes. J. Org. Chem. 1982, 47, 3016–3018. [Google Scholar] [CrossRef]
- Dragulescu-Andrasi, A.; Rapireddy, S.; Frezza, B.M.; Gayathri, C.; Gil, R.R.; Ly, D.H. A simple γ-backbone modification preorganizes peptide nucleic acid into a helical structure. J. Am. Chem. Soc. 2006, 128, 10258–10267. [Google Scholar]
- De Koning, M.C.; Petersen, L.; Weterings, J.J.; Overhand, M.; van der Marel, G.A.; Filippov, D.V. Synthesis of thiol-modified peptide nucleic acids designed for post-assembly conjugation reactions. Tetrahedron 2006, 62, 3248–3258. [Google Scholar]
- Sahu, B.; Chenna, V.; Lathrop, K.L.; Thomas, S.M.; Zon, G.; Livak, K.J.; Ly, D.H. Synthesis of conformationallypreorganized and cell-permeable guanidine-based γ-peptide nucleic acids (γGPNAs). J. Org. Chem. 2009, 74, 1509–1516. [Google Scholar] [CrossRef]
- Sahu, B.; Sacui, I.; Rapireddy, S.; Zanotti, K.J.; Bahal, R.; Armitage, B.A.; Ly, D.H. Synthesis and characterization of conformationally preorganized, (R)-diethylene glycol-containing γ-peptide nucleic acids with superior hybridization properties and water solubility. J. Org. Chem. 2011, 76, 5614–5627. [Google Scholar] [CrossRef]
- Falkiewicz, B.; Kolodziejczyk, A.S.; Liberek, B.; Wisniewski, K. Synthesis of achiral and chiralpeptide nucleic acid (PNA) monomers using Mitsunobu reaction. Tetrahedron 2001, 57, 7909–7917. [Google Scholar] [CrossRef]
- Chouikhi, D.; Ciobanu, M.; Zambaldo, C.; Duplan, V.; Barluenga, S.; Winssinger, N. Expanding the scope of PNA-encoded synthesis (PES): Mtt-protected PNA fully orthogonal to fmoc chemistry and a broad array of robust diversity-generating reactions. Chem. Eur. J. 2012, 18, 12698–12704. [Google Scholar] [CrossRef]
- Englund, E.A.; Appella, D.H. γ-Substituted peptide nucleic acids constructed from L-lysine are a versatile scaffold for multifunctional display. Angew.Chem. Int. Ed. 2007, 46, 1414–1418. [Google Scholar] [CrossRef]
- Englund, E.A.; Wang, D.; Fujigaki, H.; Sakai, H.; Micklitsch, C.M.; Ghirlando, R.; Martin-Manso, G.; Pendrak, M.L.; Roberts, D.D.; Durell, S.R.; et al. Programmable multivalent display of receptor ligands using peptide nucleic acid nanoscaffolds. Nat. Commun. 2012. [Google Scholar] [CrossRef]
- Scheibe, C.; Bujotzek, A.; Dernedde, J.; Weberb, M.; Seitz, O. DNA-programmed spatial screening of carbohydrate-lectin interactions. Chem. Sci. 2011, 2, 770–775. [Google Scholar] [CrossRef]
- Sforza, S.; Tedeschi, T.; Corradini, R.; Marchelli, R. Induction of helical handedness and DNA binding properties of peptide nucleic acids (PNAs) with two stereogenic centres. Eur. J. Org. Chem. 2007, 2007, 5879–5885. [Google Scholar]
- Avitabile, C.; Moggio, L.; Malgieri, G.; Capasso, D.; Gaetano, S.D.; Saviano, M.; Pedone, C.; Romanelli, A. γ sulphate PNA (PNA S): Highly selective DNA binding molecule showing promising antigene activity. PLoS One 2012, 7, e35774. [Google Scholar]
- Yeh, J.I.; Boris Shivachev, B.; Rapireddy, S.; Crawford, M.J.; Gil, R.R.; Du, S.; Madrid, M.; Ly, D.H. Crystal structure of chiral γPNA with complementary DNA strand: Insights into the stability and specificity of recognition and conformational preorganization. J. Am. Chem. Soc. 2010, 132, 10717–10727. [Google Scholar]
- Crawford, M.J.; Rapireddy, S.; Bahal, R.; Sacui, I.; Ly, D.H. Effect of steric constraint at the γ-backbone position on the conformations and hybridization properties of PNAs. J. Nucleic Acids 2011, 2011. [Google Scholar] [CrossRef]
- Nielsen, P.E.; Christensen, L. Strand displacement binding of a duplex-forming homopurine PNA to a homopyrimidine duplex DNA target. J. Am. Chem. Soc. 1996, 118, 2287–2288. [Google Scholar] [CrossRef]
- Zhang, X.; Ishihra, T.; Corey, D.R. Strand invasion by mixed base PNAs and a PNA-peptide chimera. Nucleic Acids Res. 2000, 28, 3332–3338. [Google Scholar] [CrossRef]
- Smolina, I.V.; Demidov, V.V.; Soldatenkov1, V.A.; Chasovskikh1, S.G.; Frank-Kamenetskii, M.D. End invasion of peptide nucleic acids (PNAs) with mixed-base composition into linear DNA duplexes. Nucleic Acids Res. 2005, 33, e146. [Google Scholar] [CrossRef]
- Avitabile, C.; Saviano, M.; D’Andrea, L.D.; Bianchi, N.; Fabbri, E.; Brognara, E.; Gambari, R.; Romanelli, A. Targeting pre-miRNA by peptide nucleic acids. Artificial DNA PNA XNA 2012, 3, 88–96. [Google Scholar]
- Rapireddy, S.; He, G.; Roy, S.; Armitage, B.A.; Ly, D.H. Strand invasion of mixed-sequence B-DNA by acridine-linked, γ-peptide nucleic acid (γ-PNA). J. Am. Chem. Soc. 2007, 129, 15596–15600. [Google Scholar] [CrossRef]
- Chenna, V.; Rapireddy, S.; Sahu, B.; Ausin, C.; Pedroso, E.; Ly, D.H. A simple cytosine to G-clamp nucleobase substitution enables chiral γ-PNAs to invade mixed-sequence double-helical B-form DNA. ChemBioChem 2008, 9, 2388–2391. [Google Scholar] [CrossRef]
- Rapireddy, S.; Bahal, R.; Ly, D.H. Strand invasion of mixed-sequence, double-helical B-DNA by γ-peptide nucleic acids containing G-clamp nucleobases under physiological conditions. Biochemistry 2011, 50, 3913–3918. [Google Scholar] [CrossRef]
- He, G.; Rapireddy, S.; Bahal, R.; Sahu, B.; Ly, D.H. Strand invasion of extended, mixed-sequence B-DNA by γPNAs. J. Am. Chem. Soc. 2009, 131, 12088–12090. [Google Scholar] [CrossRef]
- Bahal, R.; Sahu, B.; Rapireddy, S.; Lee, C.-M.; Ly, D.H. Sequence-unrestricted, Watson–Crick recognition of double helical B-DNA by (R)-MiniPEG-γPNAs. ChemBioChem 2012, 13, 56–60. [Google Scholar] [CrossRef]
- Manicardi, A.; Fabbri, E.; Tedeschi, T.; Sforza, S.; Nicoletta Bianchi, N.; Brognara, E.; Gambari, R.; Marchelli, R.; Corradini, R. Cellular uptakes, biostabilities and anti-miR-210 activities of chiralarginine-PNAs in leukaemic K562 cells. ChemBioChem 2012, 13, 1327–1337. [Google Scholar] [CrossRef]
- Manizardi, A.; Calabretta, A.; Bencivenni, M.; Tedeschi, T.; Sforza, S.; Corradini, R.; Marchelli, R. Affinity and selectivity of C2- and C5-substituted “chiral-box” PNA in solution and on microarrays. Chirality 2010, 22, E161–E171. [Google Scholar] [CrossRef]
- Sforza, S.; Tedeschi, T.; Calabretta, A.; Corradini, R.; Camerin, C.; Tonelli, R.; Pession, A.; Marchelli, R. A peptide nucleic acid embedding a pseudopeptide nuclear localization sequence in the backbone behaves as a peptide mimic. Eur. J. Org. Chem. 2010, 2010, 2441–2444. [Google Scholar] [CrossRef]
- Mitra, R.; Ganesh, K.N. PNAs grafted with (α/γ, R/S)-aminomethylene pendants: Regio and stereo specific effects on DNA binding and improved cell uptake. Chem. Commun. 2011, 47, 1198–1200. [Google Scholar] [CrossRef]
- Mitra, R.; Ganesh, K.N. Aminomethylene peptide nucleic acid (am-PNA): Synthesis, regio-/stereospecific DNA binding, and differential cell uptake of (α/γ,R/S)am-PNA analogues. J. Org. Chem. 2012, 77, 5696–5704. [Google Scholar]
- Myers, M.C.; Witschi, M.A.; Larionova, N.V.; Frank, J.M.; Haynes, R.D.; Hara, T.; Grajkowski, A.; Appella, D.H. A cyclopentane conformational restraint for a peptide nucleic acid: Design, asymmetric synthesis, and improved binding affinity to DNA and RNA. Org. Lett. 2003, 5, 2695–2698. [Google Scholar]
- Pokorski, J.K.; Witschi, M.A.; Purnell, B.L.; Appella, D.H. (S,S)-trans-Cyclopentane-constrained peptide nucleic acids. A general backbone modification that improves binding affinity and sequence specificity. J. Am. Chem. Soc. 2004, 126, 15067–15073. [Google Scholar]
- Pokorski, J.K.; Myers, M.C.; Appella, D.H. Cyclopropane PNA: Observable triplex melting in a PNA constrained with a 3-membered ring. Tetrahedron Lett. 2005, 46, 915–917. [Google Scholar] [CrossRef]
- Govindaraju, T.; Kumar, V.A.; Ganesh, K.N. cis-Cyclopentyl PNA (cpPNA) as constrained chiral PNA analogues: Stereochemical dependence of DNA/RNA hybridiation. Chem. Commun. 2004, 860–861. [Google Scholar]
- Govindaraju, T.; Kumar, V.A.; Ganesh, K.N. (SR/RS)-Cyclohexanyl PNAs: Conformationally preorganized PNA analogues with unprecedented preference for duplex formation with RNA. J. Am. Chem. Soc. 2005, 127, 4144–4145. [Google Scholar] [CrossRef]
- Lagriffoule, P.; Wittung, P.; Ericksson, M.; Jensen, D.K.; Norden, B.; Buchardt, O.; Nielsen, P.E. Peptide nucleic acids with a conformationally constrained chiralcyclohexyl-derived backbone. Chem. Eur. J. 1997, 3, 912–919. [Google Scholar] [CrossRef]
- Bregant, S.; Burlina, F.; Vaissermann, J.; Chassaing, G. Synthesis and hybridization properties of thiazolidine PNAs. Eur. J. Org. Chem. 2001, 2001, 3285–3294. [Google Scholar]
- Bregant, S.; Burlina, F.; Chassaing, G. New thiazane and thiazolidine PNA monomers: Synthesis, incorporation into PNAs and hybridization studies. Bioorg. Med. Chem. Lett. 2002, 12, 1047–1050. [Google Scholar] [CrossRef]
- Sugiyama, T.; Imamura, Y.; Demizu, Y.; Kurihara, M.; Takano, M.; Kittaka, A. β-PNA: Peptide nucleic acid (PNA) with a chiral center at the β-position of the PNA backbone. Bioorg. Med. Chem. Lett. 2011, 21, 7317–7320. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sugiyama, T.; Kittaka, A. Chiral Peptide Nucleic Acids with a Substituent in the N-(2-Aminoethy)glycine Backbone. Molecules 2013, 18, 287-310. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules18010287
Sugiyama T, Kittaka A. Chiral Peptide Nucleic Acids with a Substituent in the N-(2-Aminoethy)glycine Backbone. Molecules. 2013; 18(1):287-310. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules18010287
Chicago/Turabian StyleSugiyama, Toru, and Atsushi Kittaka. 2013. "Chiral Peptide Nucleic Acids with a Substituent in the N-(2-Aminoethy)glycine Backbone" Molecules 18, no. 1: 287-310. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules18010287