
Safe Synthesis of Alkylhydroxy and Alkylamino Nitramines


Abstract
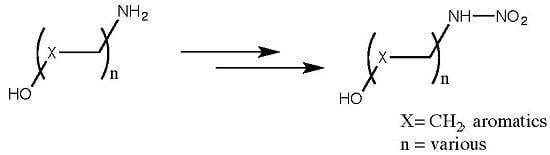
:
1. Introduction
2. Results and Discussion
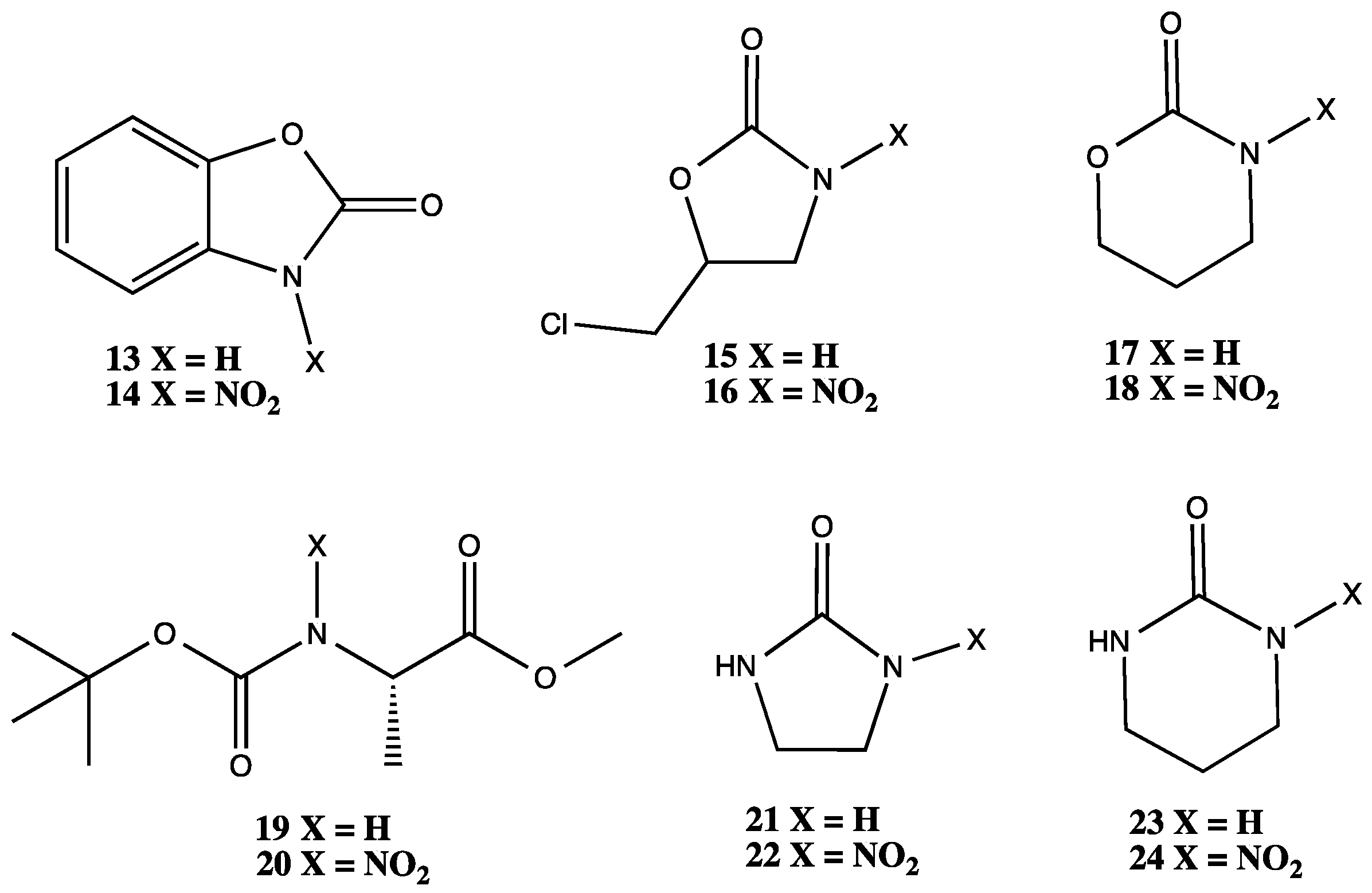
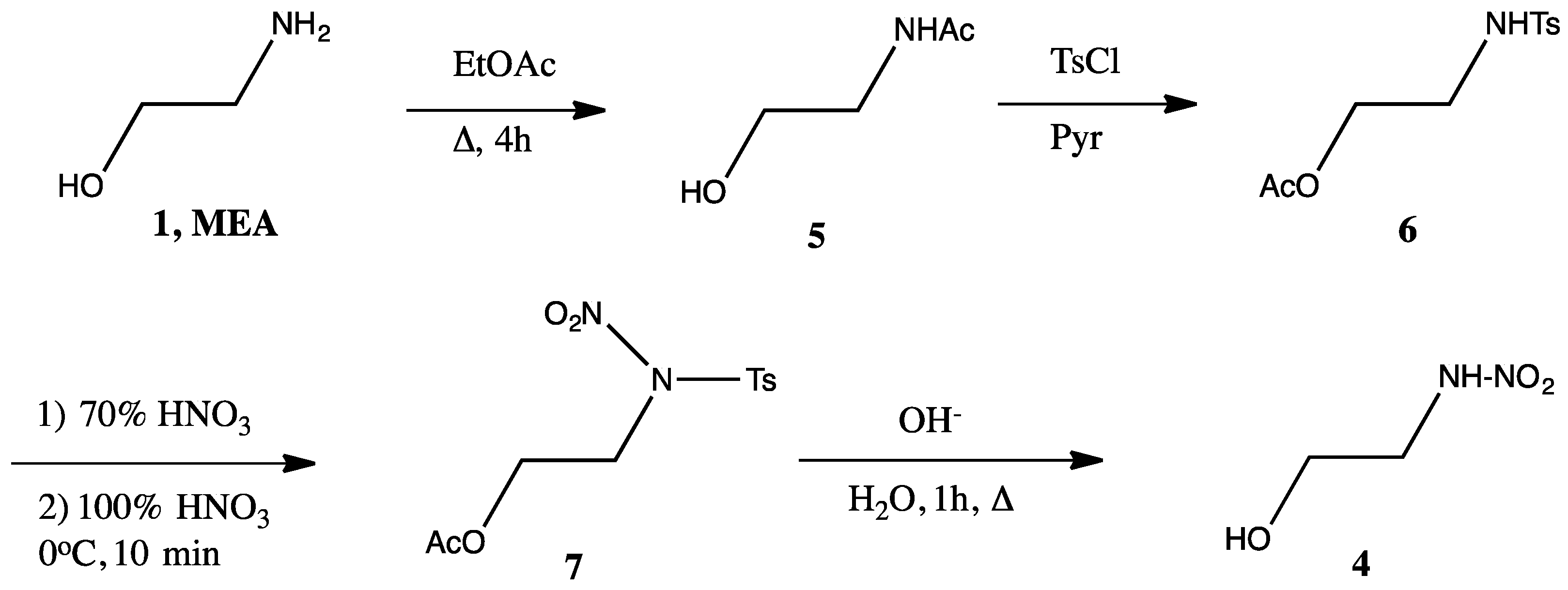
2.1. MEA Nitramine
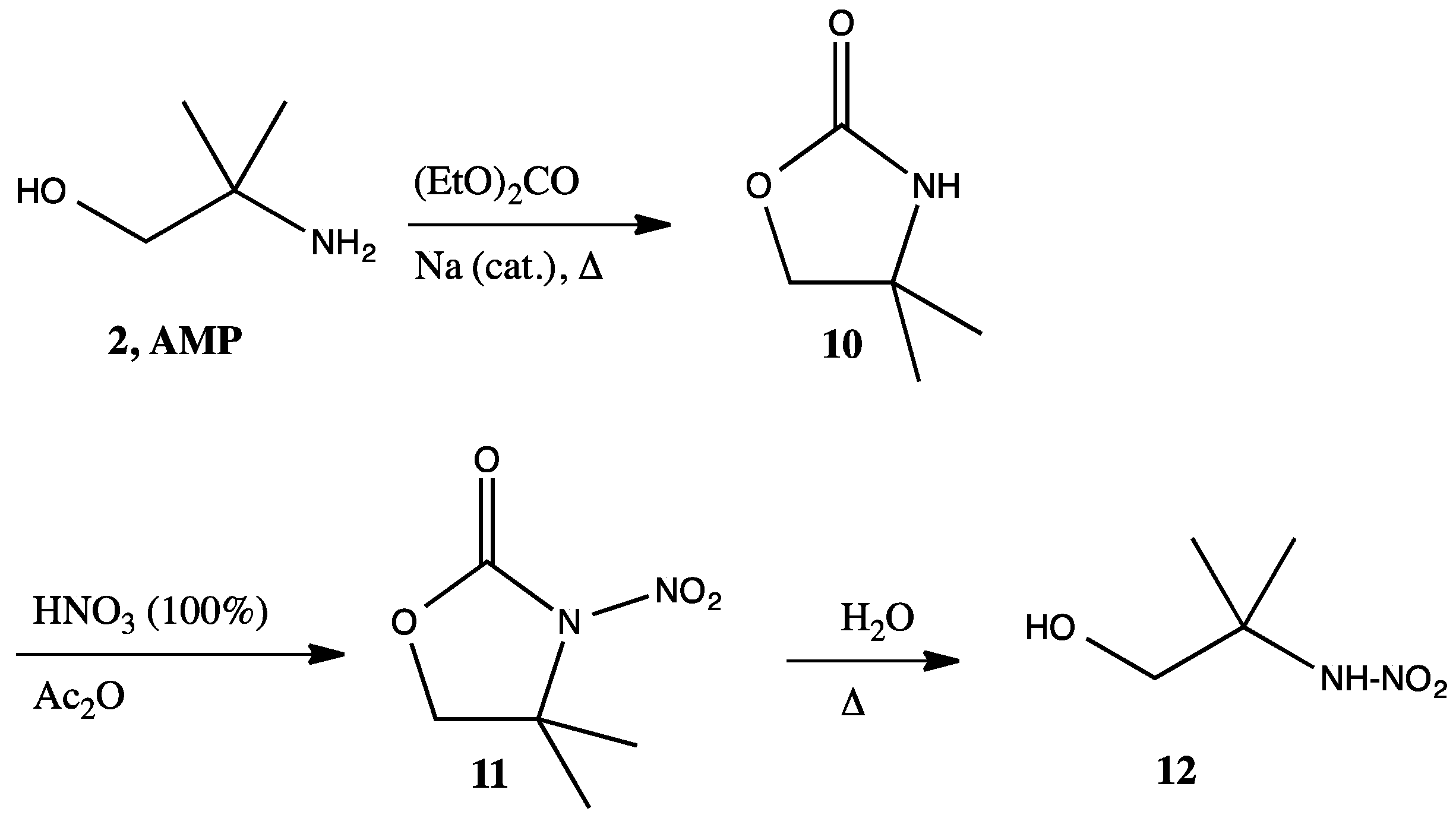
2.2. AMP Nitramine
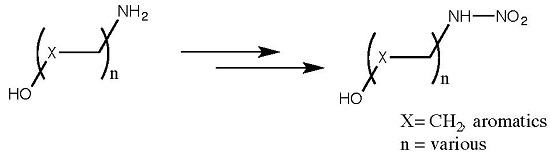
2.3. Scope and Limitations
3. Materials and Methods
3.1. General Nitration Procedures
3.1.1. Method A
3.1.2. Method B
3.1.3. Method C
3.2. Spectral Data
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Walters, M.S.; Edgar, T.F.; Rochelle, G.T. Regulatory Control of Amine Scrubbing for CO2 Capture from Power Plants. Ind. Eng. Chem. Res. 2016, 55, 4646–4657. [Google Scholar] [CrossRef]
- Rochelle, G.T. Amine Scrubbing for CO2 Capture. Science 2009, 325, 1652–1654. [Google Scholar] [CrossRef] [PubMed]
- Dai, N.; Shah, A.D.; Hu, L.; Plewa, M.J.; McKague, B.; Mitch, W.A. Measurement of Nitrosamine and Nitramine Formation from NOx Reactions with Amines during Amine-Based Carbon Dioxide Capture for Postcombustion Carbon Sequestration. Environ. Sci. Technol. 2012, 46, 9793–9801. [Google Scholar] [CrossRef] [PubMed]
- Rochelle, G.T.; Fine, N.A. Absorption of Nitrogen Oxides in Aqueous Amines. Energy Procedia 2014, 63, 830–847. [Google Scholar]
- Helgesen, L.I.; Gjernes, E. A way of qualifying Amine Based Capture Technologies with respect to Health and Environmental Properties. Energy Procedia 2016, 86, 239–251. [Google Scholar] [CrossRef]
- Coutris, C.; Macken, A.L.; Collins, A.R.; Yamani, N.; Brooks, S.J. Marine ecotoxicity of nitramines, transformation products of amine-based carbon capture technology. Sci. Total Environ. 2015, 527–528, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Buist, H.E.; Devito, S.; Goldbohm, R.A.; Stierum, R.H.; Venhorst, J.; Kroese, E.D. Hazard assessment of nitrosamine and nitramine by-products of amine-based CCS: Alternative approaches. Regul. Toxicol. Pharmacol. 2015, 71, 601–623. [Google Scholar] [CrossRef] [PubMed]
- Gundersen, C.B.; Andersen, T.; Lindahl, S.; Linke, D.; Vogt, R.D. Bacterial response from exposure to selected aliphatic nitramines. Energy Procedia 2014, 63, 791–800. [Google Scholar] [CrossRef]
- Nielsen, C.J.; Herrmann, H.; Weller, C. Atmospheric chemistry and environmental impact of the use of amines in carbon capture and storage (CCS). Chem. Soc. Rev. 2012, 41, 6684–6704. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, C.J.; D’Anna, B.; Dye, C.; Graus, M.; Karl, M.; King, S.; Maguto, M.M.; Müller, M.; Schmidbauer, N.; Stenstrøm, Y.; et al. Atmospheric chemistry of 2-aminoethanol (MEA). Energy Procedia 2001, 4, 2245–2252. [Google Scholar] [CrossRef]
- Wagner, E.D.; Osiol, J.; Mitch, W.A.; Plewa, M.J. Comparative in vitro toxicity of nitrosamines and nitramines associated with aminebased carbon capture and storage. Environ. Sci. Technol. 2014, 48, 8203–8211. [Google Scholar] [CrossRef] [PubMed]
- Dai, N.; Mitch, W.A. Effects of flue gas compositions on nitrosamine and nitramine formation in postcombustion CO2 capture systems. Environ. Sci. Technol. 2014, 48, 7519–7526. [Google Scholar] [CrossRef] [PubMed]
- Pitts, J.N.; Grosjean, D.; Vancauwenberghe, K.; Schmid, J.P.; Fitz, D.R. Photo-oxidation of aliphatic-amines under simulated atmospheric conditions–formation of nitrosamines, nitramines, amides, and photo-chemical oxidant. Environ. Sci. Technol. 1978, 12, 946–953. [Google Scholar] [CrossRef]
- Beckstead, M.W.; Puduppakkam, K.; Thakre, P.; Yang, V. Modeling of combustion and ignition of solid-propellant ingredients. Prog. Energy Combust. Sci. 2007, 33, 497–551. [Google Scholar] [CrossRef]
- Sikder, A.K.; Maddala, G.; Agrawal, J.P.; Singh, H. Important aspects of behaviour of organic energetic compounds: A review. J. Hazard. Mater. 2001, 84, 1–26. [Google Scholar] [CrossRef]
- Buckle, D.R.; Pinto, I.L. 4.09—Functions Bearing Two Nitrogens. In Comprehensive Organic Functional Group Transformations; Katritzky, A.R., Meth-Cohn, O., Rees, C.W., Eds.; Elsevier Science: Oxford, UK, 1995; pp. 403–449. ISBN 9780080447056. [Google Scholar]
- Agrawal, J.P.; Hodgson, R.D. Organic Chemistry of Explosives; Wiley: New York, NY, USA, 2007; ISBN 978-0-470-02967-1. [Google Scholar]
- Matyáš, R.; Pachman, J. Primary Explosives; Springer: Berlin, Germany, 2013; pp. 325–328. ISBN 978-3-642-28435-9. [Google Scholar]
- Jianming, Y.; Lv, J.; Yu, Q.; Li, F.; Liu, B.; Wang, W. Method for Preparing N-Nitroamino Alcohol. CN 101973908 A, 16 February 2011. [Google Scholar]
- Barrott, J.; Denton, I.N.; Lamberton, A.H. Nitramines and nitramides. Part IV. The acid-catalysed decomposition of primary nitramines. J. Chem. Soc. 1953, 1998–2005. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Hesek, D.; Lee, M.; Oliver, A.G.; Mobashery, S. Sulfonylation-induced N- to O-acetyl migration in 2-acetamidoethanol derivatives. J. Org. Chem. 2010, 75, 3515–3517. [Google Scholar] [CrossRef] [PubMed]
- Maguta, M.M.; Aursnes, M.; Bunkan, A.J.C.; Edelen, K.; Mikoviny, T.; Nielsen, C.J.; Stenstrom, Y.; Tang, Y.; Wisthaler, A. Atmospheric fate of nitramines: An experimental and theoretical study of the OH reactions with CH3NHNO2 and (CH3)2NNO2. J. Phys. Chem. 2014, 118, 3450–3462. [Google Scholar] [CrossRef] [PubMed]
- Franchimont, A.P.N.; Lublin, A. Nitroamino-alcohols. Rec. Trav. Chim. 1902, 21, 45–55. [Google Scholar]
- Bordwell, F.G.; Garbisch, E.W. Nitrations with acetyl nitrate. I. The nature of the nitrating agent and the mechanism of reaction with simple alkenes. J. Am. Chem. Soc. 1960, 82, 3588–3598. [Google Scholar] [CrossRef]
- White, E.H.; Chen, M.C.; Dolak, L.A. N-nitroamides and N-nitrocarbamates. III. Rotational isomerism, steric effects, and physical properties at low temperatures. J. Org. Chem. 1966, 31, 3038–3046. [Google Scholar] [CrossRef]
- Menke, J.B. Nitration with nitrates. Recl. Travaux Chim. Pays-Bas 1925, 44, 141–149. [Google Scholar] [CrossRef]
- Astakhov, M.; Stepanov, R.S.; Kruglyakova, L.A.; Kekin, Y.V. 1-Nitroimidazolidin-2-one and its hydrolysis to 1-amino-2-nitroaminoethane. Russ. J. Org. Chem. 2000, 36, 575–576. [Google Scholar]
- Sample Availability: Samples of the compounds 9, 11, 14, 16, 8, 10 and 24 are available from the authors.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Starting Material | Yields (%) | Product | ||
|---|---|---|---|---|---|
| A | B | C | |||
| 1 | 2-oxazolidone (9) | 78 | 65 | 59 | 4 |
| 2 | 4,4-dimethyloxazolidin-2-one (10) | 43 | 63 | 62 | 11 |
| 3 | benzo[d]oxazol-2(3H)-one (13) | 54 | 32 | 74 | 14 |
| 4 | 5-(chloromethyl)-oxazolidin-2-one (15) | 69 | 70 | 54 | 16 |
| 5 | 1,3-oxazinan-2-one (17) | 56 | 59 | 62 | 18 |
| 6 | Methyl (tert-butoxycarbonyl)alaninate (19) | - a | - a | 52 | 20 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antonsen, S.; Aursnes, M.; Gallantree-Smith, H.; Dye, C.; Stenstrøm, Y. Safe Synthesis of Alkylhydroxy and Alkylamino Nitramines. Molecules 2016, 21, 1738. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules21121738
Antonsen S, Aursnes M, Gallantree-Smith H, Dye C, Stenstrøm Y. Safe Synthesis of Alkylhydroxy and Alkylamino Nitramines. Molecules. 2016; 21(12):1738. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules21121738
Chicago/Turabian StyleAntonsen, Simen, Marius Aursnes, Harrison Gallantree-Smith, Christian Dye, and Yngve Stenstrøm. 2016. "Safe Synthesis of Alkylhydroxy and Alkylamino Nitramines" Molecules 21, no. 12: 1738. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules21121738