Adding an Artificial Tail—Anchor to a Peptide-Based HIV-1 Fusion Inhibitor for Improvement of Its Potency and Resistance Profile

Abstract
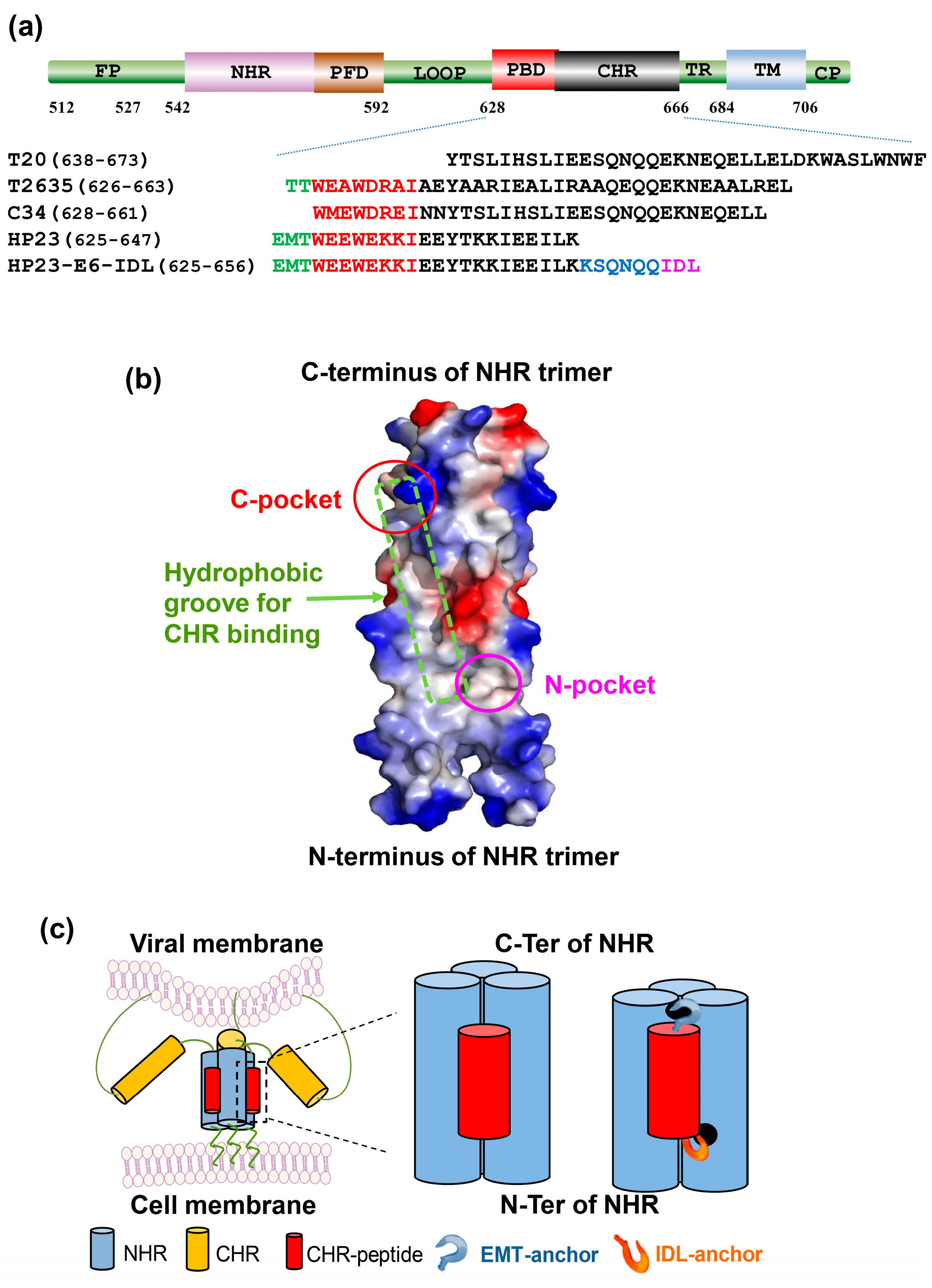
:1. Introduction
2. Results
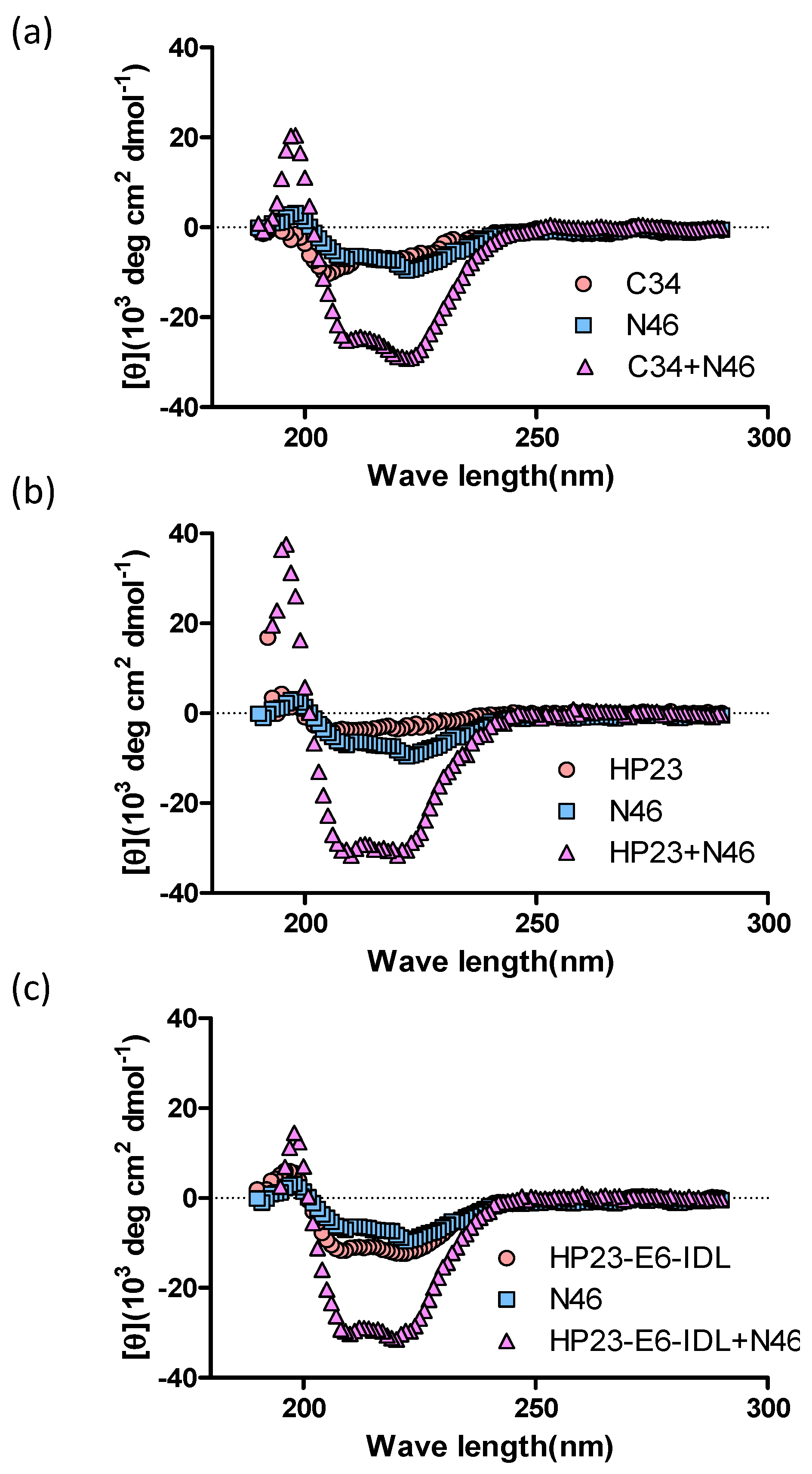
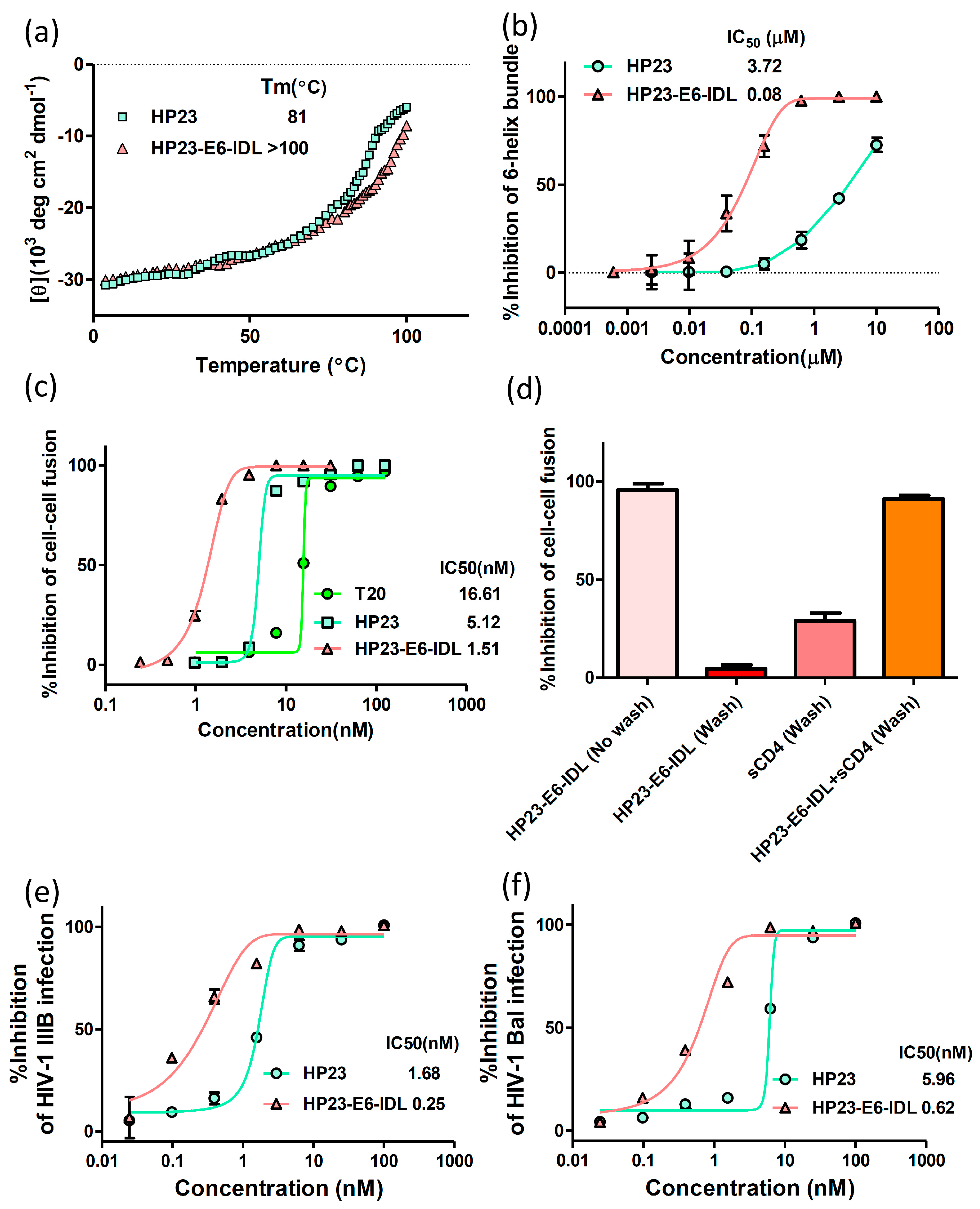
2.1. HP23-E6-IDL Formed Stable 6-HB With N46
2.2. HP23-E6-IDL Inhibited HIV-1 Env-Mediated Cell–Cell Fusion
2.3. HP23-E6-IDL Exhibited Potent Antiviral Activity Against a Broad Spectrum of HIV-1 Strains, Including Those Resistant to T20, T2635, and HP23
3. Discussion
4. Materials and Methods
4.1. Peptides
4.2. Circular Dichroism (CD) Spectroscopy
4.3. Inhibition of 6-HB Formation by Peptides In Vitro
4.4. HIV-1-Mediated Cell–Cell Fusion Assay
4.5. Inhibition of HIV-1 Infection by Peptides
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Vandamme, A.M.; Van Laethem, K.; De Clercq, E. Managing resistance to anti-HIV drugs: An important consideration for effective disease management. Drugs 1999, 57, 337–361. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Tong, P.; Yu, X.; Pan, C.; Zou, P.; Chen, Y.H.; Jiang, S. HIV-1 variants with a single-point mutation in the gp41 pocket region exhibiting different susceptibility to HIV fusion inhibitors with pocket-or membrane-binding domain. Biochim. Biophys. Acta 2012, 1818, 2950–2957. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Wang, K.; Lu, L.; Yu, F.; Cheng, M.; Jiang, S.; Liu, K.; Cai, L. Hydrophobic mutations in buried polar residues enhance HIV-1 gp41 n-terminal heptad repeat-c-terminal heptad repeat interactions and c-peptides’ anti-HIV activity. AIDS 2014, 28, 1251–1260. [Google Scholar] [CrossRef] [PubMed]
- Avila-Rios, S.; Garcia-Morales, C.; Matias-Florentino, M.; Romero-Mora, K.A.; Tapia-Trejo, D.; Quiroz-Morales, V.S.; Reyes-Gopar, H.; Ji, H.; Sandstrom, P.; Casillas-Rodriguez, J.; et al. Pretreatment HIV-drug resistance in mexico and its impact on the effectiveness of first-line antiretroviral therapy: A nationally representative 2015 who survey. Lancet HIV 2016, 3, e579–e591. [Google Scholar] [CrossRef]
- Li, T.; Qian, F.; Yuan, T.; Xu, W.; Zhu, L.; Huang, J.; Wang, H.; Zhu, Y.; Wang, Y.; Li, X.; et al. Drug resistance mutation profiles of the drug-naive and first-line regimen-treated HIV-1-infected population of suzhou, china. Virol. Sin. 2017, 32, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Phillips, A.N.; Cambiano, V.; Miners, A.; Revill, P.; Pillay, D.; Lundgren, J.D.; Bennett, D.; Raizes, E.; Nakagawa, F.; De Luca, A.; et al. Effectiveness and cost-effectiveness of potential responses to future high levels of transmitted HIV drug resistance in antiretroviral drug-naive populations beginning treatment: Modelling study and economic analysis. Lancet HIV 2014, 1, e85–e93. [Google Scholar] [CrossRef]
- Boerma, R.S.; Sigaloff, K.C.; Akanmu, A.S.; Inzaule, S.; Boele van Hensbroek, M.; Rinke de Wit, T.F.; Calis, J.C. Alarming increase in pretreatment HIV drug resistance in children living in sub-saharan africa: A systematic review and meta-analysis. J. Antimicrob. Chemother. 2017, 72, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Weissenhorn, W.; Dessen, A.; Harrison, S.C.; Skehel, J.J.; Wiley, D.C. Atomic structure of the ectodomain from HIV-1 gp41. Nature 1997, 387, 426–428. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Kim, P.S. A trimeric structural subdomain of the HIV-1 transmembrane glycoprotein. J. Biomol. Struct. Dyn. 1997, 15, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Lalezari, J.P.; Henry, K.; O’Hearn, M.; Montaner, J.S.; Piliero, P.J.; Trottier, B.; Walmsley, S.; Cohen, C.; Kuritzkes, D.R.; Eron, J.J., Jr.; et al. Enfuvirtide, an HIV-1 fusion inhibitor, for drug-resistant HIV infection in north and south america. N. Engl. J. Med. 2003, 348, 2175–2185. [Google Scholar] [CrossRef] [PubMed]
- Tilton, J.C.; Wilen, C.B.; Didigu, C.A.; Sinha, R.; Harrison, J.E.; Agrawal-Gamse, C.; Henning, E.A.; Bushman, F.D.; Martin, J.N.; Deeks, S.G.; et al. A maraviroc-resistant HIV-1 with narrow cross-resistance to other ccr5 antagonists depends on both n-terminal and extracellular loop domains of drug-bound ccr5. J. Virol. 2010, 84, 10863–10876. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, C.E.; Sanders, R.W.; Deng, Y.; Jurriaans, S.; Lange, J.M.; Lu, M.; Berkhout, B. Emergence of a drug-dependent human immunodeficiency virus type 1 variant during therapy with the t20 fusion inhibitor. J. Virol. 2004, 78, 12428–12437. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, M.L.; Cammack, N. Resistance to enfuvirtide, the first HIV fusion inhibitor. J. Antimicrob. Chemother. 2004, 54, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Nameki, D.; Kodama, E.; Ikeuchi, M.; Mabuchi, N.; Otaka, A.; Tamamura, H.; Ohno, M.; Fujii, N.; Matsuoka, M. Mutations conferring resistance to human immunodeficiency virus type 1 fusion inhibitors are restricted by gp41 and rev-responsive element functions. J. Virol. 2005, 79, 764–770. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Pozniak, A.; Wildfire, A.; Stanfield-Oakley, S.A.; Mosier, S.M.; Ratcliffe, D.; Workman, J.; Joall, A.; Myers, R.; Smit, E.; et al. Emergence and evolution of enfuvirtide resistance following long-term therapy involves heptad repeat 2 mutations within gp41. Antimicrob. Agents Chemother. 2005, 49, 1113–1119. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, C.; Marfil, S.; Garcia, E.; Martinez-Picado, J.; Bonjoch, A.; Bofill, M.; Moreno, S.; Ribera, E.; Domingo, P.; Clotet, B.; et al. Genetic evolution of gp41 reveals a highly exclusive relationship between codons 36, 38 and 43 in gp41 under long-term enfuvirtide-containing salvage regimen. AIDS 2006, 20, 2075–2080. [Google Scholar] [CrossRef] [PubMed]
- Ray, N.; Harrison, J.E.; Blackburn, L.A.; Martin, J.N.; Deeks, S.G.; Doms, R.W. Clinical resistance to enfuvirtide does not affect susceptibility of human immunodeficiency virus type 1 to other classes of entry inhibitors. J. Virol. 2007, 81, 3240–3250. [Google Scholar] [CrossRef] [PubMed]
- Svicher, V.; Aquaro, S.; D’Arrigo, R.; Artese, A.; Dimonte, S.; Alcaro, S.; Santoro, M.M.; Di Perri, G.; Caputo, S.L.; Bellagamba, R.; et al. Specific enfuvirtide-associated mutational pathways in HIV-1 gp41 are significantly correlated with an increase in cd4(+) cell count, despite virological failure. J. Infect. Dis. 2008, 197, 1408–1418. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Xiao, Y.; Song, H.; Liang, Q.; Ju, D.; Chen, X.; Lu, H.; Jing, W.; Jiang, S.; Zhang, L. Design and evaluation of sifuvirtide, a novel HIV-1 fusion inhibitor. J. Biol. Chem. 2008, 283, 11126–11134. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, J.J.; Wilson, K.L.; Davison, D.K.; Freel, S.A.; Seedorff, J.E.; Wring, S.A.; Tvermoes, N.A.; Matthews, T.J.; Greenberg, M.L.; Delmedico, M.K. Design of helical, oligomeric HIV-1 fusion inhibitor peptides with potent activity against enfuvirtide-resistant virus. Proc. Natl. Acad. Sci. USA 2007, 104, 12772–12777. [Google Scholar] [CrossRef] [PubMed]
- Chong, H.; Qiu, Z.; Su, Y.; Yang, L.; He, Y. Design of a highly potent HIV-1 fusion inhibitor targeting the gp41 pocket. AIDS 2015, 29, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Shan, M.; Li, L.; Lu, L.; Meng, S.; Chen, C.; He, Y.; Jiang, S.; Zhang, L. In vitro selection and characterization of HIV-1 variants with increased resistance to sifuvirtide, a novel HIV-1 fusion inhibitor. J. Biol. Chem. 2011, 286, 3277–3287. [Google Scholar] [CrossRef] [PubMed]
- Eggink, D.; Bontjer, I.; Langedijk, J.P.; Berkhout, B.; Sanders, R.W. Resistance of human immunodeficiency virus type 1 to a third-generation fusion inhibitor requires multiple mutations in gp41 and is accompanied by a dramatic loss of gp41 function. J. Virol. 2011, 85, 10785–10797. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Chong, H.; Qiu, Z.; Xiong, S.; He, Y. Mechanism of HIV-1 resistance to short-peptide fusion inhibitors targeting the gp41 pocket. J. Virol. 2015, 89, 5801–5811. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Lu, L.; Cai, L.; Tong, P.; Tan, S.; Zou, P.; Meng, F.; Chen, Y.H.; Jiang, S. Mutations of gln64 in the HIV-1 gp41 n-terminal heptad repeat render viruses resistant to peptide HIV fusion inhibitors targeting the gp41 pocket. J. Virol. 2012, 86, 589–593. [Google Scholar] [CrossRef] [PubMed]
- Qi, Q.; Wang, Q.; Chen, W.; Du, L.; Dimitrov, D.S.; Lu, L.; Jiang, S. HIV-1 gp41-targeting fusion inhibitory peptides enhance the gp120-targeting protein-mediated inactivation of HIV-1 virions. Emerg. Microbes Infect. 2017, 6, e59. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Zhu, Y.; Ye, S.; Qi, Q.; Xia, S.; Ma, Z.; Yu, F.; Wang, Q.; Zhang, R.; Jiang, S.; et al. Creating an artificial tail anchor as a novel strategy to enhance the potency of peptide-based HIV fusion inhibitors. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, C.; Berkhout, B. Mechanistic studies of a t20-dependent human immunodeficiency virus type 1 variant. J. Virol. 2008, 82, 7735–7740. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Melby, T.; DeMasi, R.; Ravindran, P.; Heilek-Snyder, G. Genotypic changes in human immunodeficiency virus type 1 envelope glycoproteins on treatment with the fusion inhibitor enfuvirtide and their influence on changes in drug susceptibility in vitro. J. Clin. Virol. 2006, 36, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Tolstrup, M.; Selzer-Plon, J.; Laursen, A.L.; Bertelsen, L.; Gerstoft, J.; Duch, M.; Pedersen, F.S.; Ostergaard, L. Full fusion competence rescue of the enfuvirtide resistant HIV-1 gp41 genotype (43d) by a prevalent polymorphism (137k). AIDS 2007, 21, 519–521. [Google Scholar] [CrossRef] [PubMed]
- Binley, J.M.; Sanders, R.W.; Clas, B.; Schuelke, N.; Master, A.; Guo, Y.; Kajumo, F.; Anselma, D.J.; Maddon, P.J.; Olson, W.C.; et al. A recombinant human immunodeficiency virus type 1 envelope glycoprotein complex stabilized by an intermolecular disulfide bond between the gp120 and gp41 subunits is an antigenic mimic of the trimeric virion-associated structure. J. Virol. 2000, 74, 627–643. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Bergeron, L.; Helseth, E.; Thali, M.; Repke, H.; Sodroski, J. Effects of amino acid changes in the extracellular domain of the human immunodeficiency virus type 1 gp41 envelope glycoprotein. J. Virol. 1993, 67, 2747–2755. [Google Scholar] [PubMed]
- Abrahamyan, L.G.; Markosyan, R.M.; Moore, J.P.; Cohen, F.S.; Melikyan, G.B. Human immunodeficiency virus type 1 env with an intersubunit disulfide bond engages coreceptors but requires bond reduction after engagement to induce fusion. J. Virol. 2003, 77, 5829–5836. [Google Scholar] [CrossRef] [PubMed]
- Welch, B.D.; VanDemark, A.P.; Heroux, A.; Hill, C.P.; Kay, M.S. Potent d-peptide inhibitors of hiv-1 entry. Proc. Natl. Acad. Sci. USA 2007, 104, 16828–16833. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Curreli, F.; Zhang, X.; Bhattacharya, S.; Waheed, A.A.; Cooper, A.; Cowburn, D.; Freed, E.O.; Debnath, A.K. Antiviral activity of alpha-helical stapled peptides designed from the hiv-1 capsid dimerization domain. Retrovirology 2011, 8, 28. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Chang, X.; Wang, Y.; Gao, G.F.; Shao, Y.; Ma, L.; Li, X. Glycosylated enfuvirtide: A long-lasting glycopeptide with potent anti-hiv activity. J. Med. Chem. 2015, 58, 1372–1379. [Google Scholar] [CrossRef] [PubMed]
- Pasut, G.; Veronese, F.M. Pegylation for improving the effectiveness of therapeutic biomolecules. Drugs Today (Barc.) 2009, 45, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, A.; Viard, M.; Unger, L.; Blumenthal, R.; Shai, Y. Sphingopeptides: Dihydrosphingosine-based fusion inhibitors against wild-type and enfuvirtide-resistant hiv-1. FASEB J. 2012, 26, 4628–4636. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, Y.; Martin, M.A. Of mice, macaques, and men: Broadly neutralizing antibody immunotherapy for HIV-1. Cell Host Microbe 2017, 22, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, S.; Sasaki, M.; Tanaka, T.; Inoue, M.; Ophinni, Y.; Kotaki, T.; Kameoka, M. A 2–4-amino-acid deletion in the v5 region of HIV-1 env gp120 confers viral resistance to the broadly neutralizing human monoclonal antibody, vrc01. AIDS Res. Hum. Retrovir. 2017. [Google Scholar] [CrossRef] [PubMed]
- Dingens, A.S.; Haddox, H.K.; Overbaugh, J.; Bloom, J.D. Comprehensive mapping of HIV-1 escape from a broadly neutralizing antibody. Cell Host Microbe 2017, 21, 659–660. [Google Scholar] [CrossRef] [PubMed]
- Wheatley, A.K.; Kent, S.J. Twist in the tail: Escape from HIV neutralising antibodies at a single site confers broad susceptibility to others. EBioMedicine 2016, 12, 14–15. [Google Scholar] [CrossRef] [PubMed]
- Nie, J.; Zhao, J.; Chen, Q.; Huang, W.; Wang, Y. Three amino acid residues in the envelope of human immunodeficiency virus type 1 crf07_bc regulate viral neutralization susceptibility to the human monoclonal neutralizing antibody igg1b12. Virol. Sin. 2014, 29, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zony, C.; Chen, P.; Chen, B.K. Reduced potency and incomplete neutralization of broadly neutralizing antibodies against cell-to-cell transmission of HIV-1 with transmitted founder envs. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Qi, Q.; Wang, Q.; Chen, W.; Yu, F.; Du, L.; Dimitrov, D.S.; Lu, L.; Jiang, S. Anti-HIV antibody and drug combinations exhibit synergistic activity against drug-resistant HIV-1 strains. J. Infect. 2017, 75, 68–71. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Su, S.; Qin, L.; Wang, Q.; Shi, L.; Ma, Z.; Tang, J.; Jiang, S.; Lu, L.; Ye, S.; et al. Rational improvement of gp41-targeting HIV-1 fusion inhibitors: An innovatively designed ile-asp-leu tail with alternative conformations. Sci. Rep. 2016, 6, 31983. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Zhu, Y.; Wang, Q.; Ye, L.; Dai, Y.; Su, S.; Yu, F.; Ying, T.; Yang, C.; Jiang, S.; et al. An immunogen containing four tandem 10e8 epitope repeats with exposed key residues induces antibodies that neutralize HIV-1 and activates an adcc reporter gene. Emerg. Microbes Infect. 2016, 5, e65. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Zhu, Y.; Ye, S.; Wang, Q.; Xu, W.; Su, S.; Sun, Z.; Yu, F.; Liu, Q.; Wang, C.; et al. Improved pharmacological and structural properties of HIV fusion inhibitor ap3 over enfuvirtide: Highlighting advantages of artificial peptide strategy. Sci. Rep. 2015, 5, 13028. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Lin, K.; Lu, M. A conformation-specific monoclonal antibody reacting with fusion-active gp41 from the human immunodeficiency virus type 1 envelope glycoprotein. J. Virol. 1998, 72, 10213–10217. [Google Scholar] [PubMed]
- Wang, Q.; Bi, W.; Zhu, X.; Li, H.; Qi, Q.; Yu, F.; Lu, L.; Jiang, S. Nonneutralizing antibodies induced by the HIV-1 gp41 nhr domain gain neutralizing activity in the presence of the HIV fusion inhibitor enfuvirtide: A potential therapeutic vaccine strategy. J. Virol. 2015, 89, 6960–6964. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Wang, Q.; Xu, W.; Yu, F.; Hua, C.; Zhu, Y.; Jiang, S.; Lu, L. A novel HIV-1 gp41 tripartite model for rational design of HIV-1 fusion inhibitors with improved antiviral activity. AIDS 2017, 31, 885–894. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wang, Q.; Li, Y.; Yu, F.; Liu, Q.; Qin, B.; Xie, L.; Lu, L.; Jiang, S. A nanoparticle-encapsulated non-nucleoside reverse-transcriptase inhibitor with enhanced anti-HIV-1 activity and prolonged circulation time in plasma. Curr. Pharm. Des. 2015, 21, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Tong, P.; Lu, Z.; Chen, X.; Wang, Q.; Yu, F.; Zou, P.; Yu, X.; Li, Y.; Lu, L.; Chen, Y.H.; et al. An engineered HIV-1 gp41 trimeric coiled coil with increased stability and anti-HIV-1 activity: Implication for developing anti-HIV microbicides. J. Antimicrob. Chemother. 2013, 68, 2533–2544. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |




{kind=link}
{kind=link}
{kind=link}
| Viruses | IC50 (nM) | |
|---|---|---|
| HP23 | HP23-E6-IDL | |
| 92UG029 (A, X4) | 1.6 ± 0.3 | 0.8 ± 0.4 |
| KER2018 (A, R5) | 1.9 ± 0.2 | 0.8 ± 0.1 |
| KNH1135 (A, R5) | 2.2 ± 0.3 | 0.5 ± 0.2 |
| US4GS007 (B, R5) | 3.9 ± 0.6 | 0.8 ± 0.1 |
| BK132/GS009 (B, X4) | 7.2 ± 0.5 | 0.7 ± 0.2 |
| 90US_873 (B, R5) | 3.4 ± 0.6 | 1.0 ± 0.1 |
| 93IN101 (C, R5) | 3.7 ± 0.3 | 0.6 ± 0.1 |
| 92UG024 (D, X4) | 3.5 ± 0.2 | 0.6 ± 0.1 |
| 93BR020 (F, X4/R5) | 4.5 ± 0.2 | 1.0 ± 0.2 |
| BCF02 (O, R5) | 9.4 ± 0.8 | 1.6 ± 0.2 |
| 92TH009 (A/E, R5) | 0.8 ± 0.2 | 0.1 ± 0.1 |
| NP1525 (A/E, X4/R5) | 9.5 ± 0.6 | 0.6 ± 0.2 |
| Viruses | IC50 (nM) | |
|---|---|---|
| HP23 | HP23-E6-IDL | |
| wild-type | 2.8 ± 0.2 | 1.5 ± 0.2 |
| H3C | 5.4 ± 0.4 | 0.7 ± 0.2 |
| A6V | 1.6 ± 0.5 | 0.3 ± 0.1 |
| Q66R | 2.8 ± 0.1 | 0.4 ± 0.2 |
| Q79E | 4.2 ± 0.7 | 1.3 ± 0.2 |
| K90E | 9.4 ± 0.3 | 2.2 ± 0.2 |
| N113E | 2.3 ± 0.5 | 0.7 ± 0.1 |
| N126K | 8.6 ± 0.4 | 1.6 ± 0.2 |
| K154Q | 7.4 ± 0.4 | 1.5 ± 0.2 |
| Q79E/N126K | 5.2 ± 0.4 | 0.7 ± 0.1 |
| K90E/N126K | 4.2 ± 0.3 | 0.3 ± 0.1 |
| Q66R/N113E | 3.6 ± 0.3 | 1.1 ± 0.1 |
| Viruses | IC50 (nM) | |
|---|---|---|
| HP23 | HP23-E6-IDL | |
| T20-resistant strains | ||
| WT | 3.1 ± 0.1 | 1.2 ± 0.2 |
| V38A | 2.1 ± 0.7 | 0.5 ± 0.2 |
| V38A, N42D | 2.9 ± 0.3 | 1.5 ± 0.2 |
| V38E, N42S | 6.2 ± 0.3 | 0.6 ± 0.1 |
| V38A, N42T | 1.5 ± 0.3 | 0.3 ± 0.1 |
| HP23-resistant strains | ||
| wild-type | 3.3 ± 0.3 | 0.6 ± 0.1 |
| E49K | 13.4 ± 0.7 | 1.9 ± 0.2 |
| E49K/N126K | 53.8 ± 1.2 | 2.6 ± 0.3 |
| D36G/E49K/N126K | >60 | 4.0 ± 0.4 |
| L34S/D36G/E49K/E136G | >60 | 5.2 ± 1.1 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, S.; Ma, Z.; Hua, C.; Li, W.; Lu, L.; Jiang, S. Adding an Artificial Tail—Anchor to a Peptide-Based HIV-1 Fusion Inhibitor for Improvement of Its Potency and Resistance Profile. Molecules 2017, 22, 1996. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules22111996
Su S, Ma Z, Hua C, Li W, Lu L, Jiang S. Adding an Artificial Tail—Anchor to a Peptide-Based HIV-1 Fusion Inhibitor for Improvement of Its Potency and Resistance Profile. Molecules. 2017; 22(11):1996. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules22111996
Chicago/Turabian StyleSu, Shan, Zhenxuan Ma, Chen Hua, Weihua Li, Lu Lu, and Shibo Jiang. 2017. "Adding an Artificial Tail—Anchor to a Peptide-Based HIV-1 Fusion Inhibitor for Improvement of Its Potency and Resistance Profile" Molecules 22, no. 11: 1996. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules22111996