Polysiloxane/Polystyrene Thermo-Responsive and Self-Healing Polymer Network via Lewis Acid-Lewis Base Pair Formation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
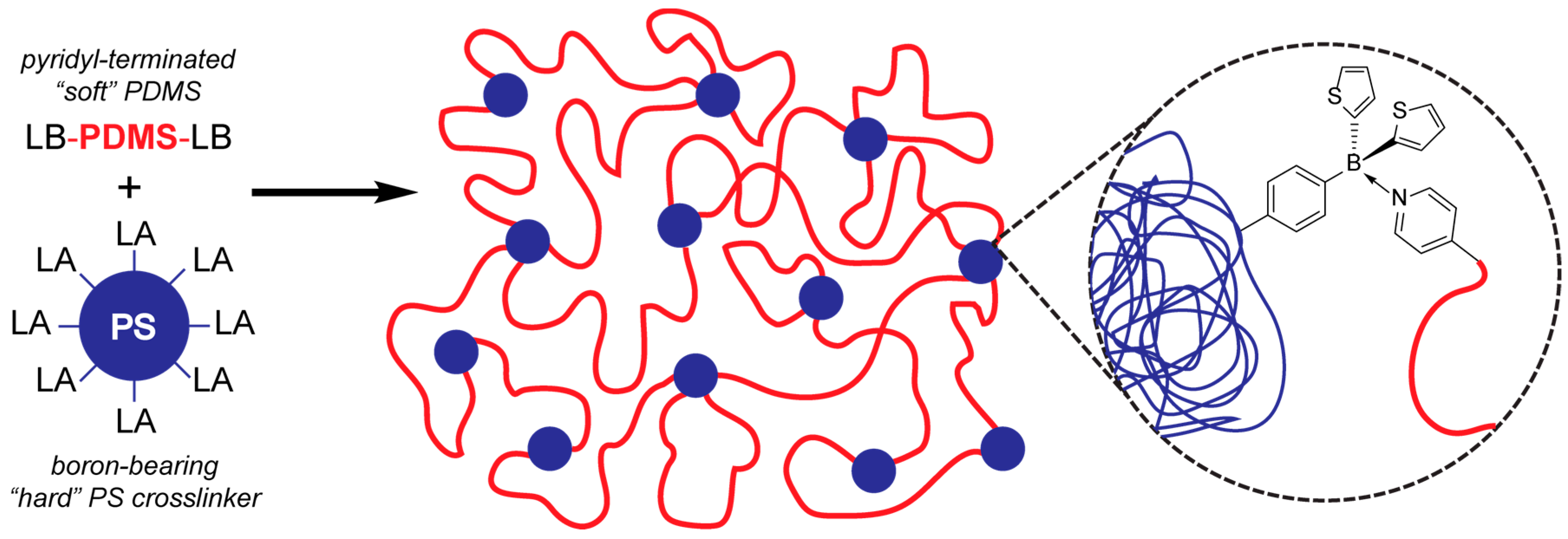
2. Results and Discussion
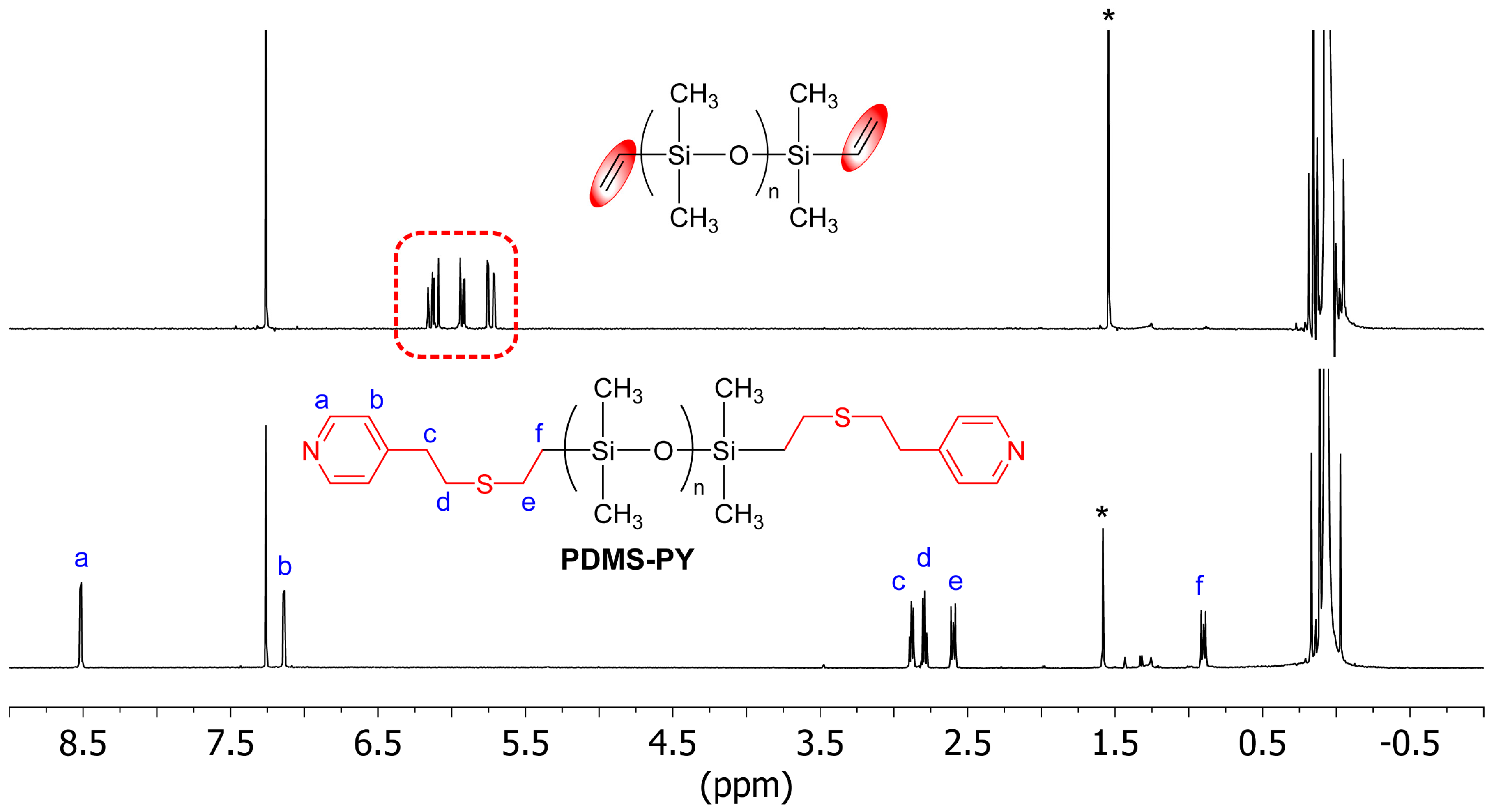
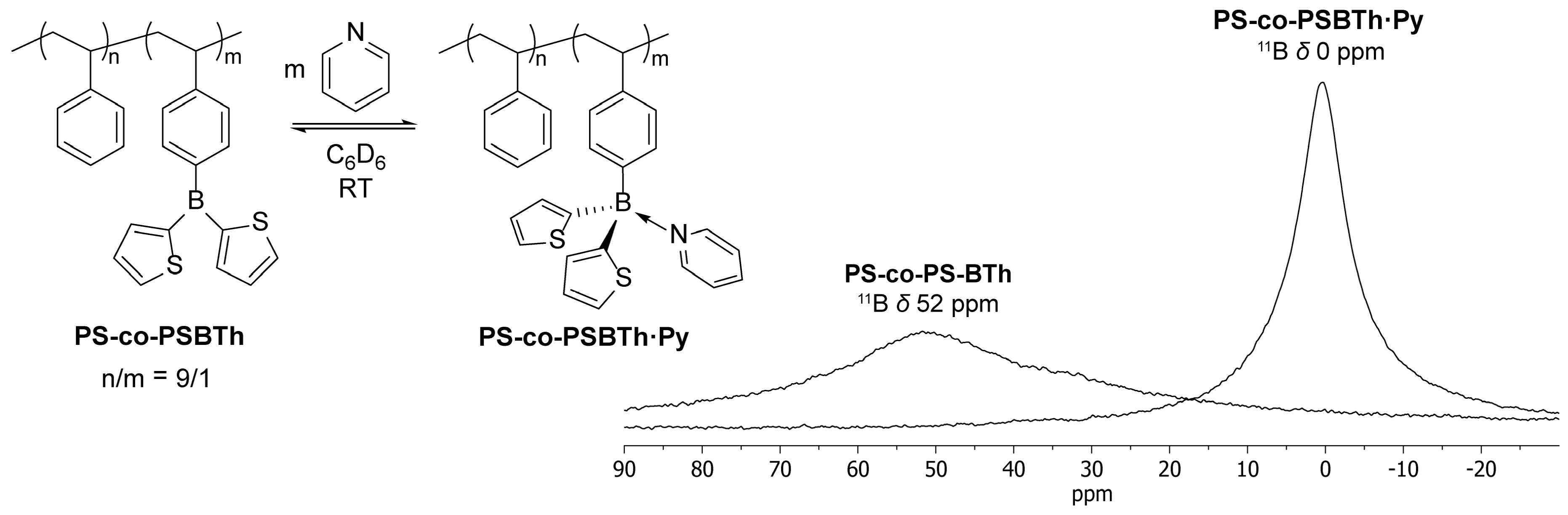
2.1. Synthesis of LA/LB-Containing Polymers and Adduct Formation
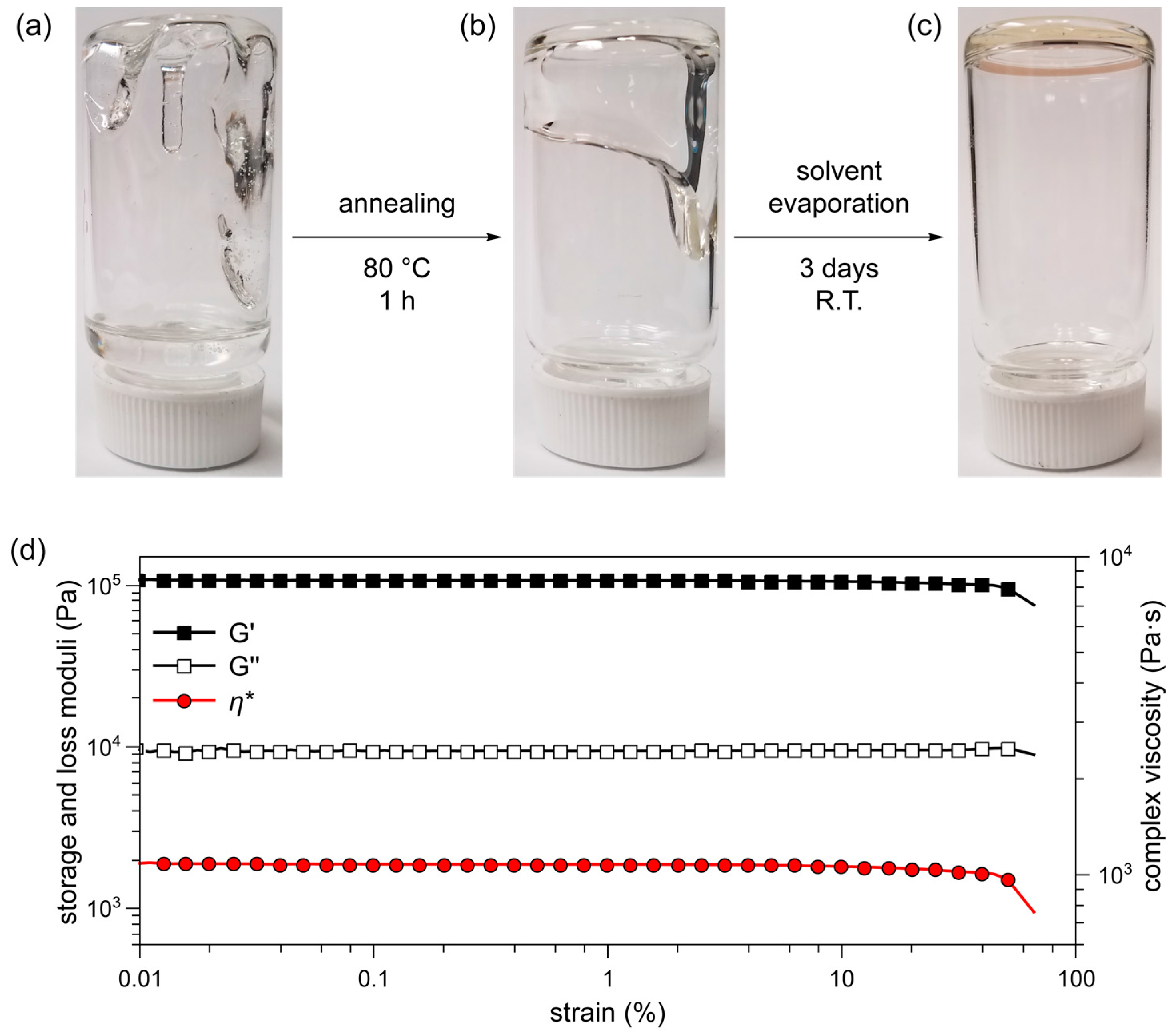
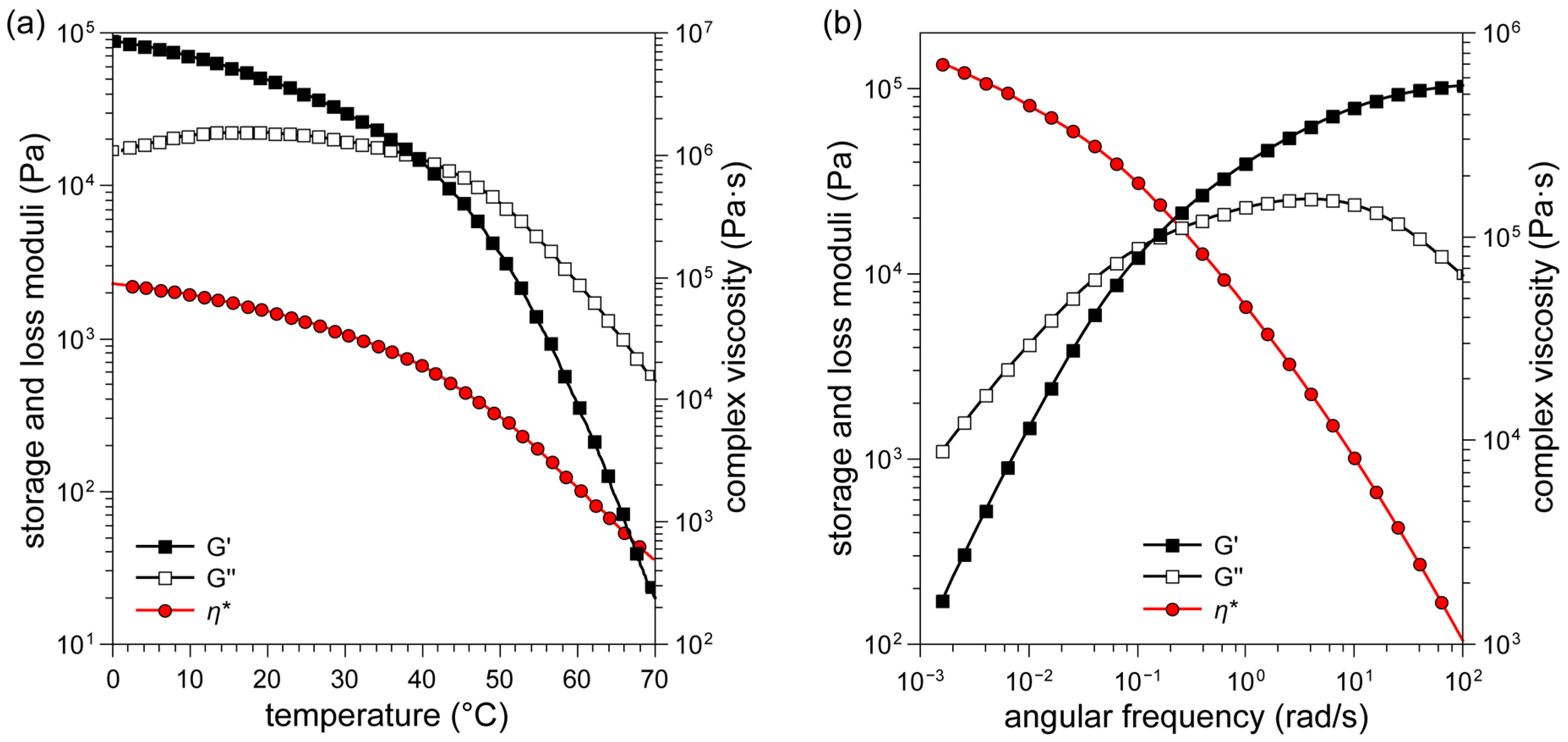
2.2. Preparation of PDMS/PS Gel Formulation and Physical Characterization
2.3. Observation of Self-Healing Properties
3. Materials and Methods
3.1. General Considerations
3.2. Syntheses and Characterizations
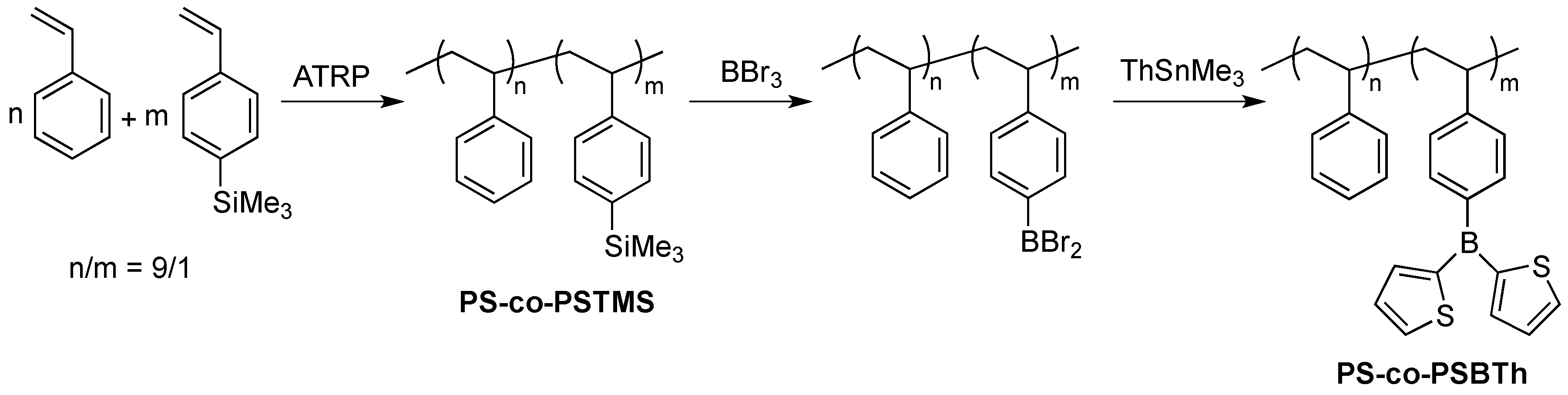
3.2.1. Synthesis of Poly(styrene-co-4-trimethylsilylstyrene), PS-co-PSTMS
3.2.2. Synthesis of Poly(styrene-co-4-(di-2-thienylboryl)styrene), PS-co-PSBTh
3.2.3. Reaction of PS-co-PSBTh with Pyridine
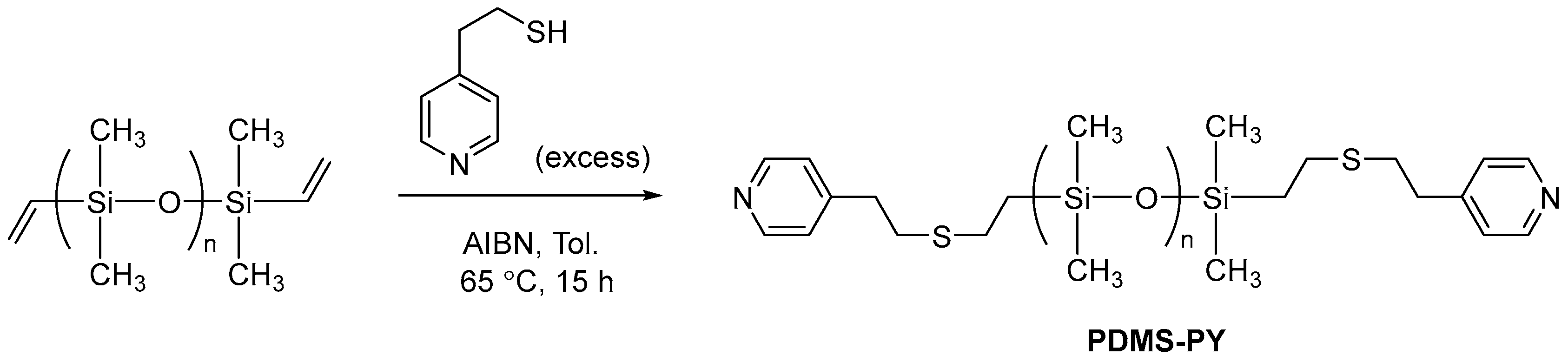
3.2.4. Synthesis of Pyridine-Terminated Telechelic PDMS, PDMS-PY
3.2.5. Procedure for the Preparation of Gel Formulations
3.2.6. Rheology Measurements
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Piepenbrock, M.-O.M.; Lloyd, G.O.; Clarke, N.; Steed, J.W. Metal- and anion-binding supramolecular gels. Chem. Rev. 2010, 110, 1960–2004. [Google Scholar] [CrossRef] [PubMed]
- Aida, T.; Meijer, E.W.; Stupp, S.I. Functional supramolecular polymers. Science 2012, 335, 813–817. [Google Scholar] [CrossRef] [PubMed]
- Voorhaar, L.; Hoogenboom, R. Supramolecular polymer networks: Hydrogels and bulk materials. Chem. Soc. Rev. 2016, 45, 4013–4031. [Google Scholar] [CrossRef] [PubMed]
- Goor, O.J.G.M.; Hendrikse, S.I.S.; Dankers, P.Y.W.; Meijer, E.W. From supramolecular polymers to multi-component biomaterials. Chem. Soc. Rev. 2017, 46, 6621–6637. [Google Scholar] [CrossRef] [PubMed]
- Kajita, T.; Noro, A.; Matsushita, Y. Design and properties of supramolecular elastomers. Polymer 2017, 128, 297–310. [Google Scholar] [CrossRef]
- Yan, X.; Wang, F.; Zheng, B.; Huang, F. Stimuli-responsive supramolecular polymeric materials. Chem. Soc. Rev. 2012, 41, 6042–6065. [Google Scholar] [CrossRef] [PubMed]
- Herbert, K.M.; Schrettl, S.; Rowan, S.J.; Weder, C. 50th anniversary perspective: Solid-state multistimuli, multiresponsive polymeric materials. Macromolecules 2017, 50, 8845–8870. [Google Scholar] [CrossRef]
- Calvino, C.; Neumann, L.; Weder, C.; Schrettl, S. Approaches to polymeric mechanochromic materials. J. Polym. Sci. Polym. Chem. 2017, 55, 640–652. [Google Scholar] [CrossRef]
- Herbst, F.; Seiffert, S.; Binder, W.H. Dynamic supramolecular poly(isobutylene)s for self-healing materials. Polym. Chem. 2012, 3, 3084–3092. [Google Scholar] [CrossRef]
- Roy, N.; Tomović, Ž.; Buhler, E.; Lehn, J.-M. An easily accessible self-healing transparent film based on a 2D supramolecular network of hydrogen-bonding interactions between polymeric chains. Chem. Eur. J. 2016, 22, 13513–13520. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-X.; Long, Y.-Y.; Pan, L.; Men, Y.-F.; Li, Y.-S. Spontaneously healable thermoplastic elastomers achieved through one-pot living ring-opening metathesis copolymerization of well-designed bulky monomers. ACS Appl. Mater. Interfaces 2016, 8, 12445–12455. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Viveros, J.A.; Wrue, M.H.; Anthamatten, M. Shape-memory effects in polymer networks containing reversibly associating side-groups. Adv. Mater. 2007, 19, 2851–2855. [Google Scholar] [CrossRef]
- Binder, W.H.; Petraru, L.; Roth, T.; Groh, P.W.; Pálfi, V.; Keki, S.; Ivan, B. Magnetic and temperature-sensitive release gels from supramolecular polymers. Adv. Funct. Mater. 2007, 17, 1317–1326. [Google Scholar] [CrossRef]
- Seiffert, S.; Sprakel, J. Physical chemistry of supramolecular polymer networks. Chem. Soc. Rev. 2012, 41, 909–930. [Google Scholar] [CrossRef] [PubMed]
- Sijbesma, R.P.; Beijer, F.H.; Brunsveld, L.; Folmer, B.J.B.; Hirschberg, J.H.K.K.; Lange, R.F.M.; Lowe, J.K.L.; Meijer, E.W. Reversible polymers formed from self-complementary monomers using quadruple hydrogen bonding. Science 1997, 278, 1601. [Google Scholar] [CrossRef] [PubMed]
- Folmer, B.J.B.; Sijbesma, R.P.; Versteegen, R.M.; van der Rijt, J.A.J.; Meijer, E.W. Supramolecular polymer materials: Chain extension of telechelic polymers using a reactive hydrogen-bonding synthon. Adv. Mater. 2000, 12, 874–878. [Google Scholar] [CrossRef]
- Sivakova, S.; Bohnsack, D.A.; Mackay, M.E.; Suwanmala, P.; Rowan, S.J. Utilization of a combination of weak hydrogen-bonding interactions and phase segregation to yield highly thermosensitive supramolecular polymers. J. Am. Chem. Soc. 2005, 127, 18202–18211. [Google Scholar] [CrossRef] [PubMed]
- Nair, K.P.; Breedveld, V.; Weck, M. Complementary hydrogen-bonded thermoreversible polymer networks with tunable properties. Macromolecules 2008, 41, 3429–3438. [Google Scholar] [CrossRef]
- Söntjens, S.H.M.; Renken, R.A.E.; van Gemert, G.M.L.; Engels, T.A.P.; Bosman, A.W.; Janssen, H.M.; Govaert, L.E.; Baaijens, F.P.T. Thermoplastic elastomers based on strong and well-defined hydrogen-bonding interactions. Macromolecules 2008, 41, 5703–5708. [Google Scholar] [CrossRef]
- Fawcett, A.S.; Brook, M.A. Thermoplastic silicone elastomers through self-association of pendant coumarin groups. Macromolecules 2014, 47, 1656–1663. [Google Scholar] [CrossRef]
- Tan, C.S.Y.; Agmon, G.; Liu, J.; Hoogland, D.; Janecek, E.-R.; Appel, E.A.; Scherman, O.A. Distinguishing relaxation dynamics in transiently crosslinked polymeric networks. Polym. Chem. 2017, 8, 5336–5343. [Google Scholar] [CrossRef]
- Yount, W.C.; Juwarker, H.; Craig, S.L. Orthogonal control of dissociation dynamics relative to thermodynamics in a main-chain reversible polymer. J. Am. Chem. Soc. 2003, 125, 15302–15303. [Google Scholar] [CrossRef] [PubMed]
- Yount, W.C.; Loveless, D.M.; Craig, S.L. Strong means slow: Dynamic contributions to the bulk mechanical properties of supramolecular networks. Angew. Chem. Int. Ed. 2005, 44, 2746–2748. [Google Scholar] [CrossRef] [PubMed]
- Yount, W.C.; Loveless, D.M.; Craig, S.L. Small-molecule dynamics and mechanisms underlying the macroscopic mechanical properties of coordinatively cross-linked polymer networks. J. Am. Chem. Soc. 2005, 127, 14488–14496. [Google Scholar] [CrossRef] [PubMed]
- Aboudzadeh, M.A.; Muñoz, M.E.; Santamaría, A.; Fernández-Berridi, M.J.; Irusta, L.; Mecerreyes, D. Synthesis and rheological behavior of supramolecular ionic networks based on citric acid and aliphatic diamines. Macromolecules 2012, 45, 7599–7606. [Google Scholar] [CrossRef]
- Qin, Y.; Jäkle, F. Formation of polymeric Lewis acid–Lewis base complexes with well-defined organoboron polymers. J. Inorg. Organomet. Polym. Mater. 2007, 17, 149–157. [Google Scholar] [CrossRef]
- Doshi, A.; Jäkle, F. Polystyrene-supported borane complexes PS-BH2. Main Group Chem. 2006, 5, 309–318. [Google Scholar] [CrossRef]
- Cheng, F.; Jäkle, F. Boron-containing polymers as versatile building blocks for functional nanostructured materials. Polym. Chem. 2011, 2, 2122–2132. [Google Scholar] [CrossRef]
- Jäkle, F. Recent advances in the synthesis and applications of organoborane polymers. In Synthesis and Application of Organoboron Compounds; Fernández, E., Whiting, A., Eds.; Springer International Publishing: Cham, Switzerland, 2015; pp. 297–325. [Google Scholar]
- Korich, A.L.; Iovine, P.M. Boroxine chemistry and applications: A perspective. Dalton Trans. 2010, 39, 1423–1431. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Cui, C.; Jäkle, F. Silylated initiators for the efficient preparation of borane-end-functionalized polymers via atrp. Macromolecules 2007, 40, 1413–1420. [Google Scholar] [CrossRef]
- Cheng, F.; Bonder, E.M.; Jäkle, F. Electron-Deficient Triarylborane Block Copolymers: Synthesis by Controlled Free Radical Polymerization and Application in the Detection of Fluoride Ions. J. Am. Chem. Soc. 2013, 135, 17286–17289. [Google Scholar] [CrossRef] [PubMed]
- Christinat, N.; Croisier, E.; Scopelliti, R.; Cascella, M.; Röthlisberger, U.; Severin, K. Formation of boronate ester polymers with efficient intrastrand charge-transfer transitions by three-component reactions. Eur. J. Inorg. Chem. 2007, 2007, 5177–5181. [Google Scholar] [CrossRef]
- Icli, B.; Solari, E.; Kilbas, B.; Scopelliti, R.; Severin, K. Multicomponent assembly of macrocycles and polymers by coordination of pyridyl ligands to 1,4-bis(benzodioxaborole)benzene. Chem. Eur. J. 2012, 18, 14867–14874. [Google Scholar] [CrossRef] [PubMed]
- Luisier, N.; Schenk, K.; Severin, K. A four-component organogel based on orthogonal chemical interactions. Chem. Commun. 2014, 50, 10233–10236. [Google Scholar] [CrossRef] [PubMed]
- Luisier, N.; Scopelliti, R.; Severin, K. Supramolecular gels based on boronate esters and imidazolyl donors. Soft Matter 2016, 12, 588–593. [Google Scholar] [CrossRef] [PubMed]
- Dodge, L.; Chen, Y.; Brook, M.A. Silicone boronates reversibly crosslink using Lewis acid–Lewis base amine complexes. Chem. Eur. J. 2014, 20, 9349–9356. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Nudelman, F.; Matthes, R.R.; Shaver, M.P. Frustrated Lewis pair polymers as responsive self-healing gels. J. Am. Chem. Soc. 2017, 139, 14232–14236. [Google Scholar] [CrossRef] [PubMed]
- Marić, M.; Macosko, C.W. Block copolymer compatibilizers for polystyrene/poly(dimethylsiloxane) blends. J. Polym. Sci. B Polym. Phys. 2002, 40, 346–357. [Google Scholar] [CrossRef]
- Qin, Y.; Cheng, G.; Achara, O.; Parab, K.; Jäkle, F. A new route to organoboron polymers via highly selective polymer modification reactions. Macromolecules 2004, 37, 7123–7131. [Google Scholar] [CrossRef]
- Qin, Y.; Cheng, G.; Sundararaman, A.; Jäkle, F. Well-defined boron-containing polymeric Lewis acids. J. Am. Chem. Soc. 2002, 124, 12672–12673. [Google Scholar] [CrossRef] [PubMed]
- Nose, T. Coexistence curves of polystyrene/poly(dimethylsiloxane) blends. Polymer 1995, 36, 2243–2248. [Google Scholar] [CrossRef]
- Chuai, C.; Li, S.; Almdal, K.; Alstrup, J.; Lyngaae-Jørgensen, J. Influence of diblock copolymer on the morphology and properties of polystyrene/poly(dimethylsiloxane) blends. J. Appl. Polym. Sci. 2004, 92, 2747–2757. [Google Scholar] [CrossRef]
- Van den Brande, N.; Koning, C.; Geerlings, P.; Van Lier, G.; Van Assche, G.; Van Mele, B. Partially miscible polystyrene/polymethylphenylsiloxane blends for nanocomposites. J. Therm. Anal. Calorim. 2011, 105, 775. [Google Scholar] [CrossRef]
- Dvornic, P.R.; Jovanovic, J.D.; Govedarica, M.N. On the critical molecular chain length of polydimethylsiloxane. J. Appl. Polym. Sci. 1993, 49, 1497–1507. [Google Scholar] [CrossRef]
- Kawakami, Y.; Hisada, H.; Yamashita, Y. Polystyrenes with p-oligo-siloxane, silane, germanosiloxane, germane, or stannane as p-substituents as materials for oxygen permeable membranes. J. Polym. Sci. Polym. Chem. 1988, 26, 1307–1314. [Google Scholar] [CrossRef]
- Lu, S.; Pearce, E.M.; Kwei, T.K. Novel silanol-containing polymers and their blends. Polym. Prepr. (Am. Chem. Soc. Div. Polym. Chem.) 1998, 39, 560–561. [Google Scholar] [CrossRef]
- Lindsley, C.W.; Hodges, J.C.; Filzen, G.F.; Watson, B.M.; Geyer, A.G. Rasta silanes: New silyl resins with novel macromolecular architecture via living free radical polymerization. J. Comb. Chem. 2000, 2, 550–559. [Google Scholar] [CrossRef] [PubMed]
- Rossi, R.; Carpita, A.; Ciofalo, M.; Lippolis, V. Selective and efficient syntheses of phototoxic 2,2′:5′,2″-terthiophene derivatives bearing a functional substituent in the 3′- or the 5-position. Tetrahedron 1991, 47, 8443–8460. [Google Scholar] [CrossRef]
- Bauer, L.; Gardella, L.A. Addition of thiourea to 2- and 4-vinylpyridines. J. Org. Chem. 1961, 26, 82–85. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vidal, F.; Lin, H.; Morales, C.; Jäkle, F. Polysiloxane/Polystyrene Thermo-Responsive and Self-Healing Polymer Network via Lewis Acid-Lewis Base Pair Formation. Molecules 2018, 23, 405. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23020405
Vidal F, Lin H, Morales C, Jäkle F. Polysiloxane/Polystyrene Thermo-Responsive and Self-Healing Polymer Network via Lewis Acid-Lewis Base Pair Formation. Molecules. 2018; 23(2):405. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23020405
Chicago/Turabian StyleVidal, Fernando, Huina Lin, Cecilia Morales, and Frieder Jäkle. 2018. "Polysiloxane/Polystyrene Thermo-Responsive and Self-Healing Polymer Network via Lewis Acid-Lewis Base Pair Formation" Molecules 23, no. 2: 405. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23020405