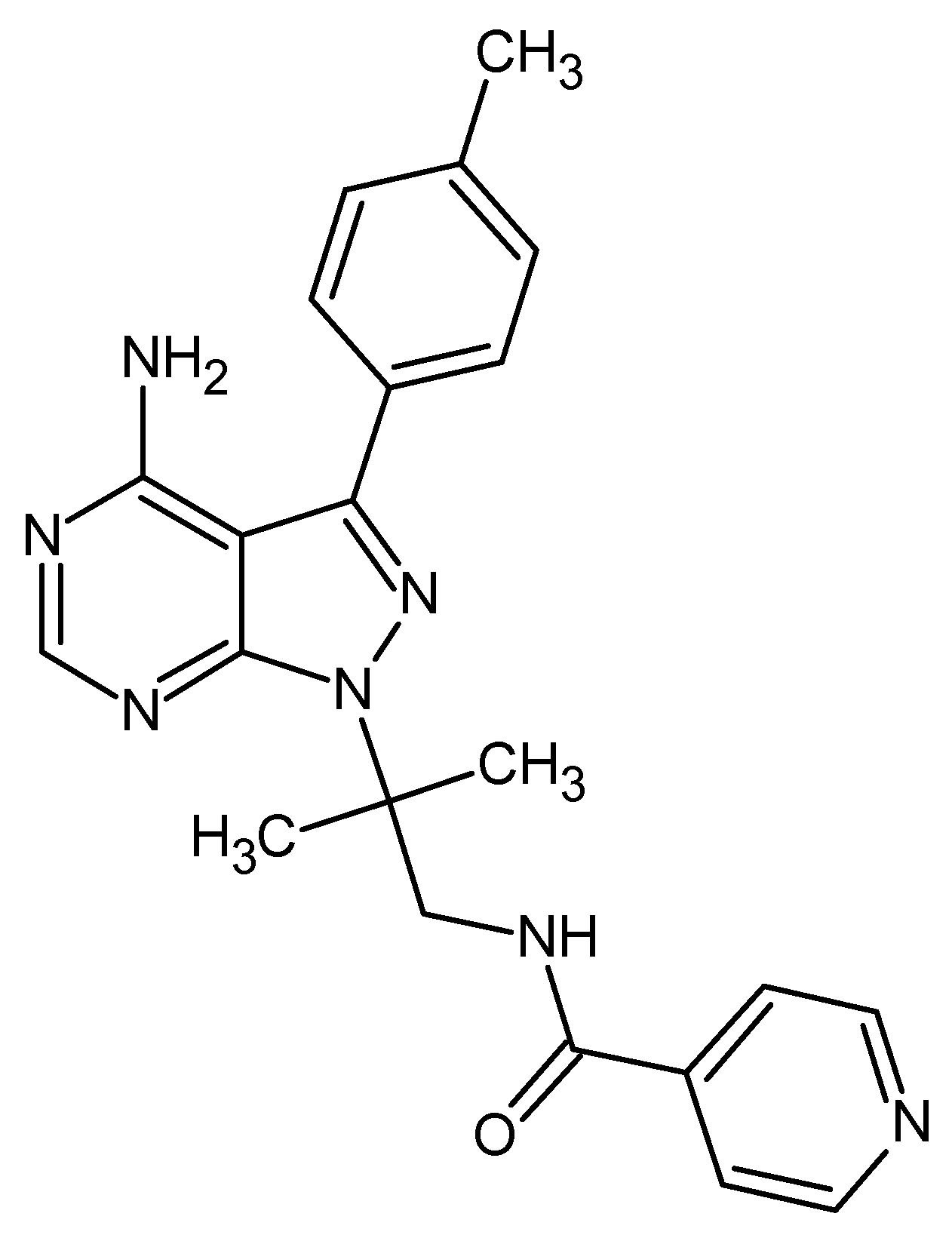
Figure 1.
2D chemical structure of compound WEHI-345.
Figure 1.
2D chemical structure of compound WEHI-345.
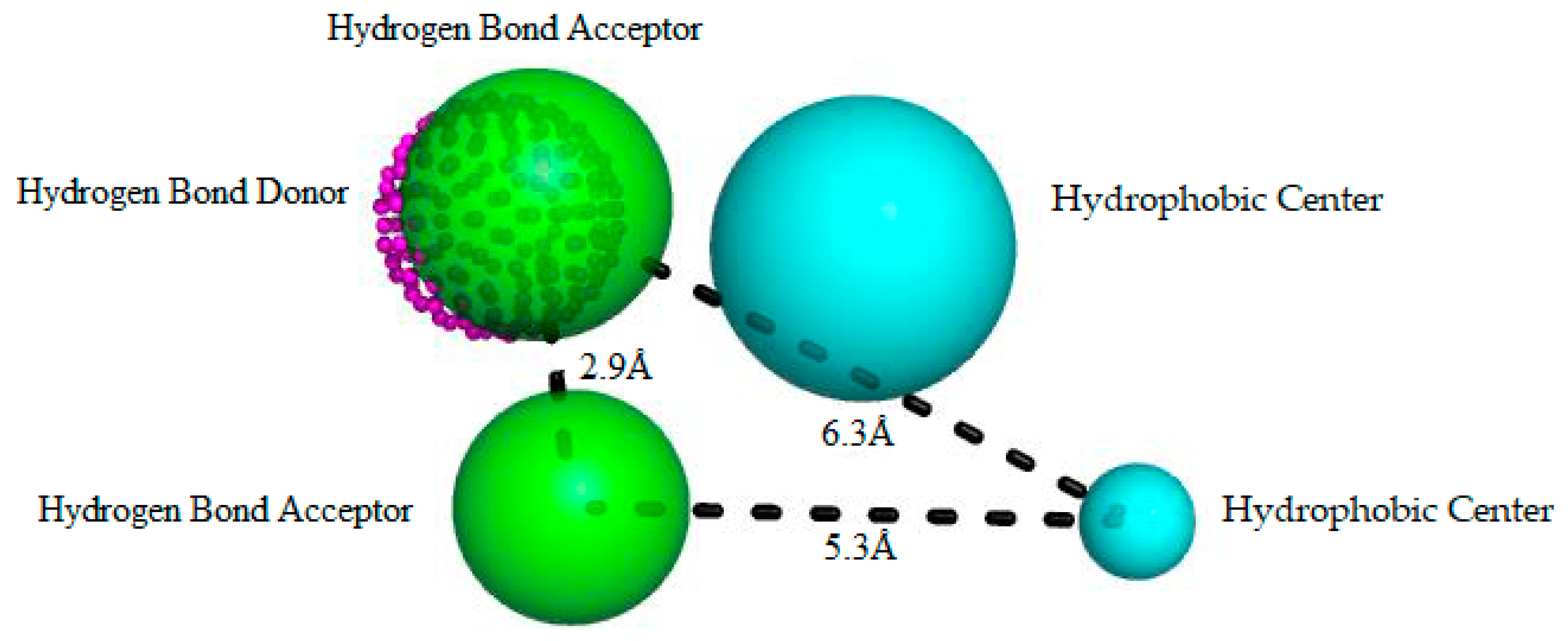
Figure 2.
Best pharmacophore model here obtained for RIPK2 inhibitors. This model has two hydrophobic centers (cyan spheres), two hydrogen bond acceptors (green spheres) and a hydrogen bond donor (magenta dotted sphere). The size of the beads varies according to the tolerance radius calculated using GALAHAD. All the distances are measured in Angstroms.
Figure 2.
Best pharmacophore model here obtained for RIPK2 inhibitors. This model has two hydrophobic centers (cyan spheres), two hydrogen bond acceptors (green spheres) and a hydrogen bond donor (magenta dotted sphere). The size of the beads varies according to the tolerance radius calculated using GALAHAD. All the distances are measured in Angstroms.
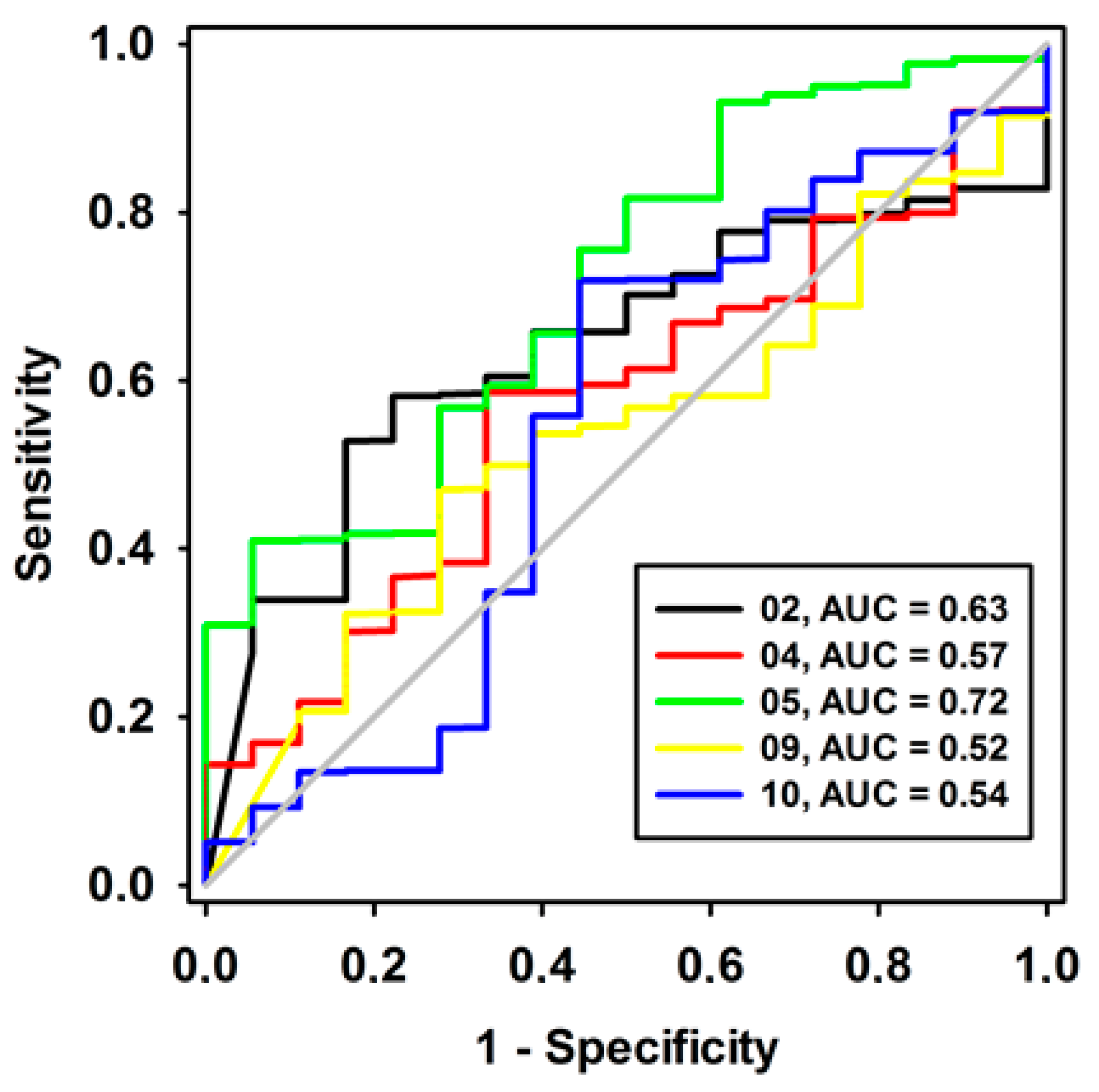
Figure 3.
ROC curves of pharmacophore models here investigated, containing low strain energy. The diagonal line represents a model that would not be better than a randomic one (AUC < 0.5).
Figure 3.
ROC curves of pharmacophore models here investigated, containing low strain energy. The diagonal line represents a model that would not be better than a randomic one (AUC < 0.5).
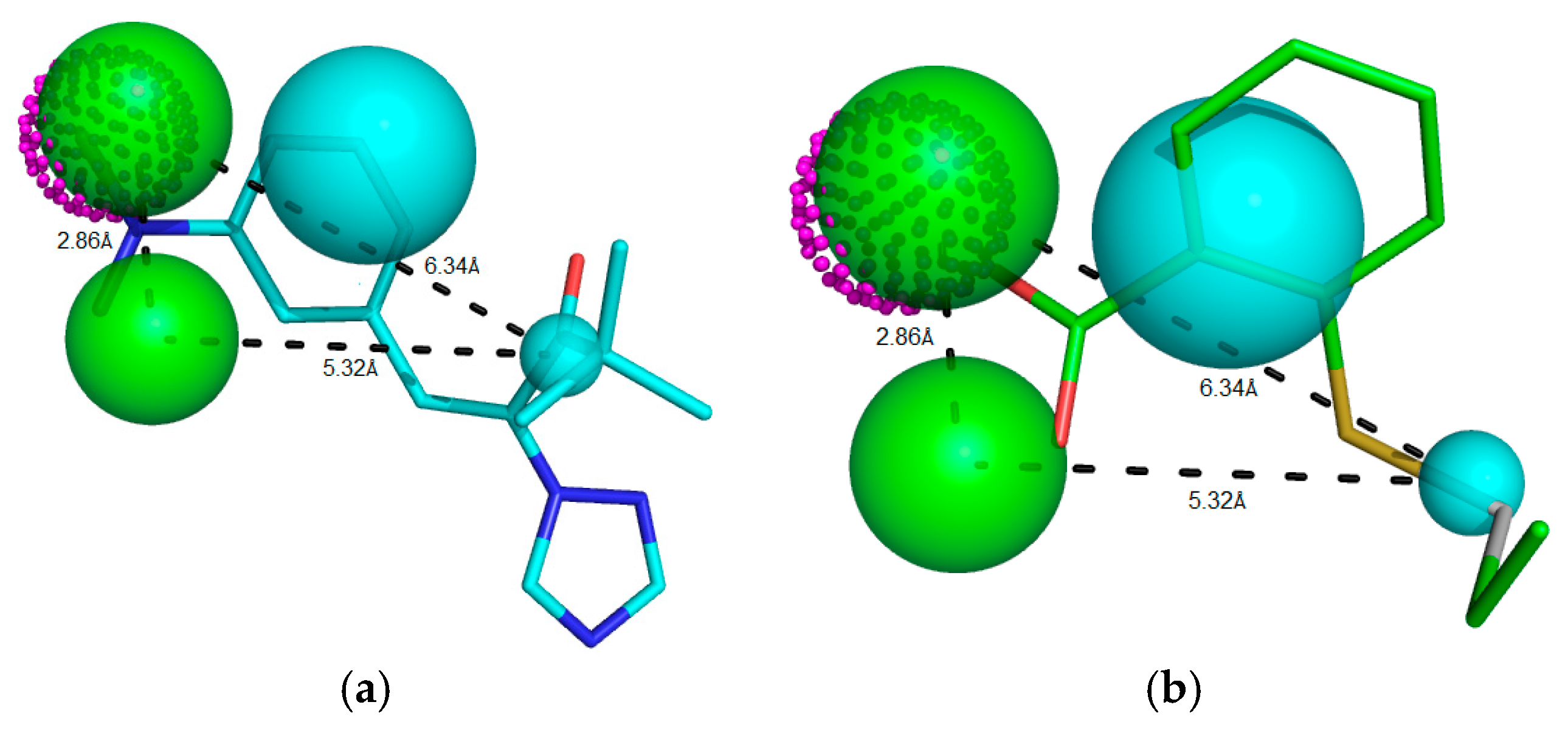
Figure 4.
Potent RIPK2 inhibitor (a) IC50 = 0.90 μM, Mol_qry value = 51.35; and (b) weak RIPK2 inhibitor IC50 = 1.40 μM, Mol_qry value = 04.10 superimposed to the pharmacophoric Model 05, where green beads/spheres represents Hbond acceptor groups, magenta doted sphere represents Hbond donor groups, while hydrophobic ones are shown in cyan spheres. The size of the beads varies according to the tolerance radius calculated using GALAHAD. All the distances are measured in angstroms.
Figure 4.
Potent RIPK2 inhibitor (a) IC50 = 0.90 μM, Mol_qry value = 51.35; and (b) weak RIPK2 inhibitor IC50 = 1.40 μM, Mol_qry value = 04.10 superimposed to the pharmacophoric Model 05, where green beads/spheres represents Hbond acceptor groups, magenta doted sphere represents Hbond donor groups, while hydrophobic ones are shown in cyan spheres. The size of the beads varies according to the tolerance radius calculated using GALAHAD. All the distances are measured in angstroms.
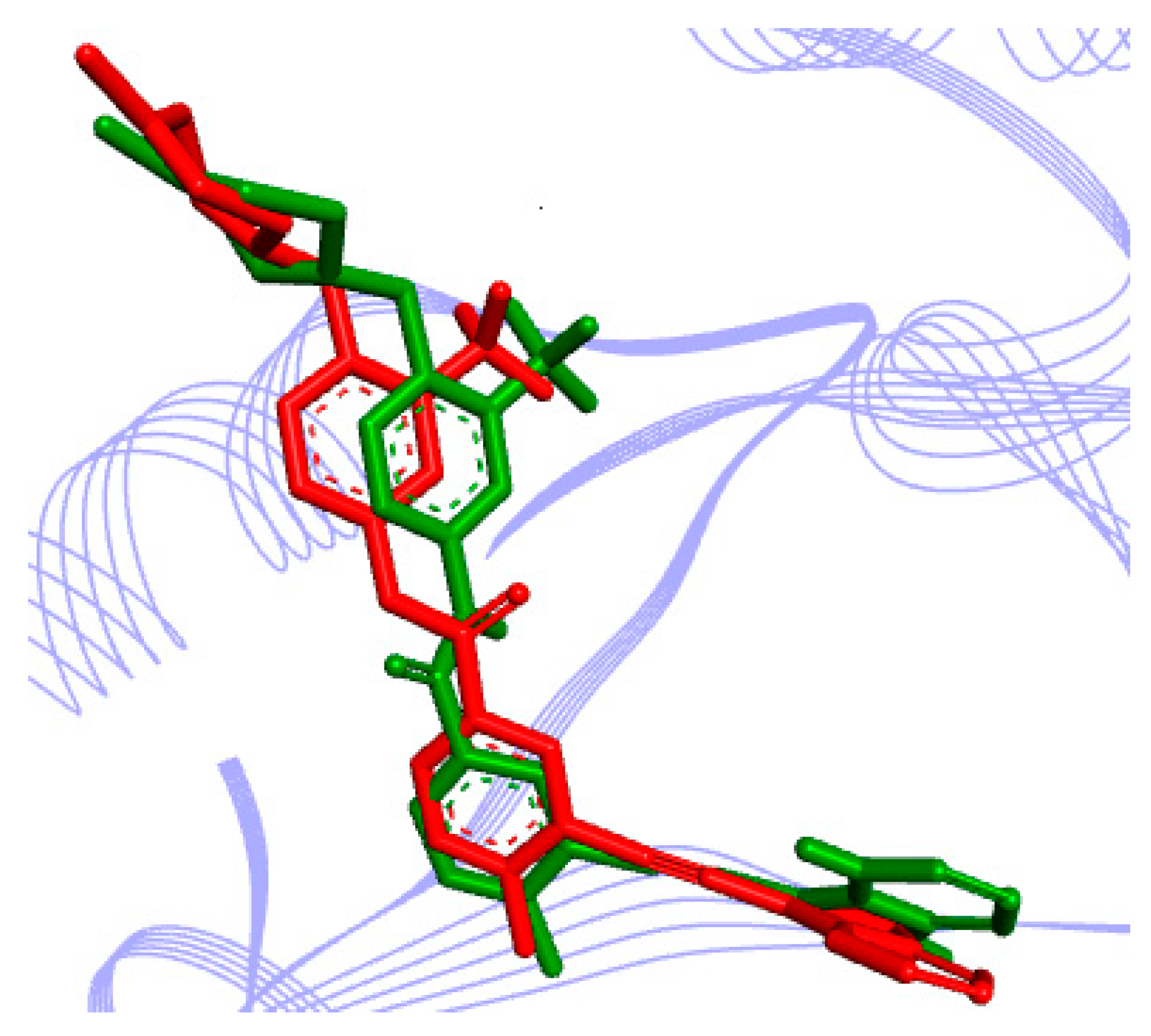
Figure 5.
Result of validation for a known potent RIPK2 inhibitor, ponatinib, inside the enzyme active site (derived from PDB ID 4C8B), obtained with the AutoDock software: in red, the crystallographic pose; in green, the top-ranked docking pose. Inhibitor is represented in stick, whereas the RIPK2 active site is represented as a Ribbons diagram (in lines).
Figure 5.
Result of validation for a known potent RIPK2 inhibitor, ponatinib, inside the enzyme active site (derived from PDB ID 4C8B), obtained with the AutoDock software: in red, the crystallographic pose; in green, the top-ranked docking pose. Inhibitor is represented in stick, whereas the RIPK2 active site is represented as a Ribbons diagram (in lines).
Figure 6.
Binding affinity provided by the AutoDock/Vina software for the potent RIPK2 inihibitor and here used as a template—ponatinib, as well as for the control compound—WEHI-345 and the compounds obtained using a virtual screening approach, regarding RIPK2.
Figure 6.
Binding affinity provided by the AutoDock/Vina software for the potent RIPK2 inihibitor and here used as a template—ponatinib, as well as for the control compound—WEHI-345 and the compounds obtained using a virtual screening approach, regarding RIPK2.
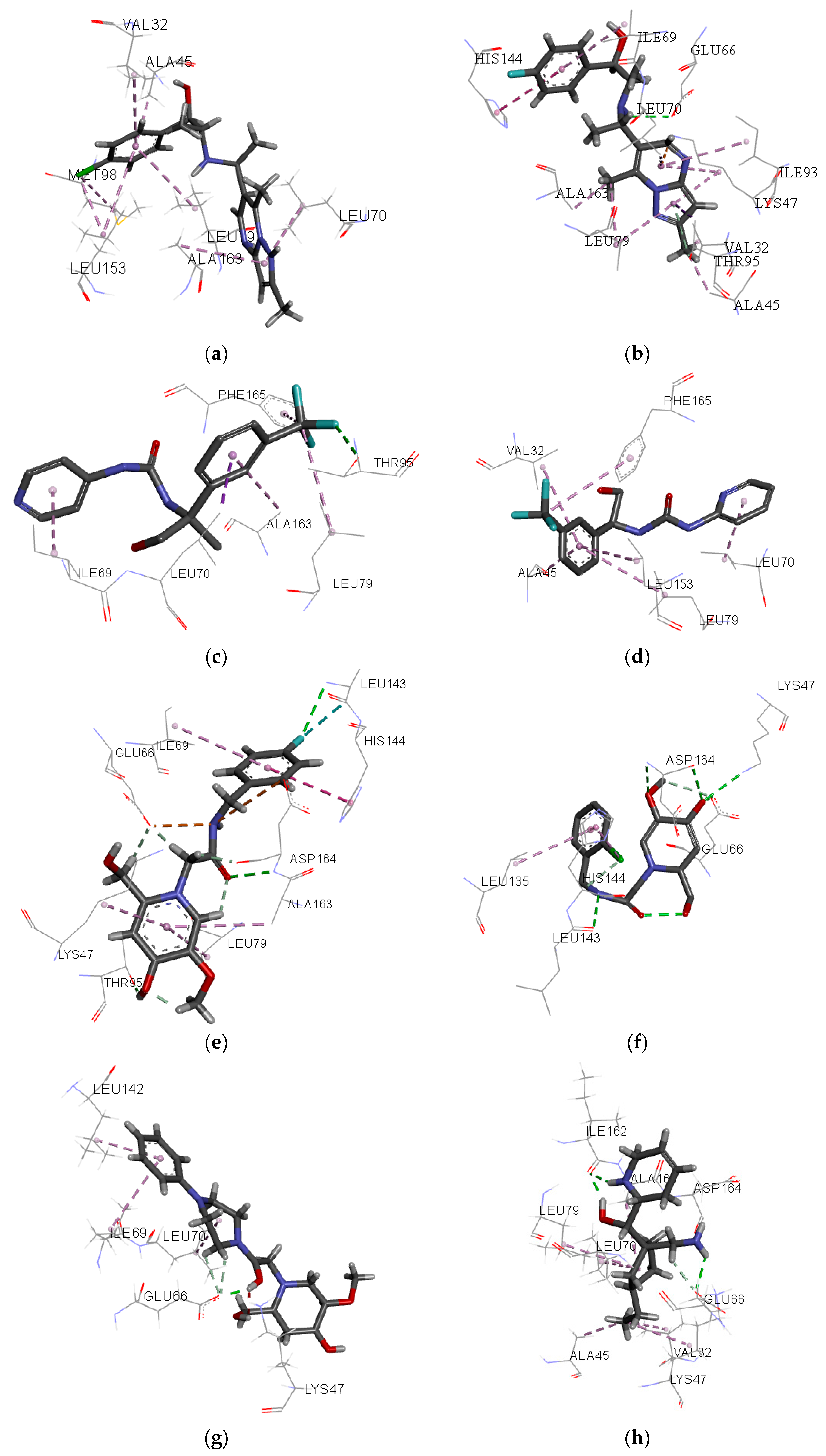
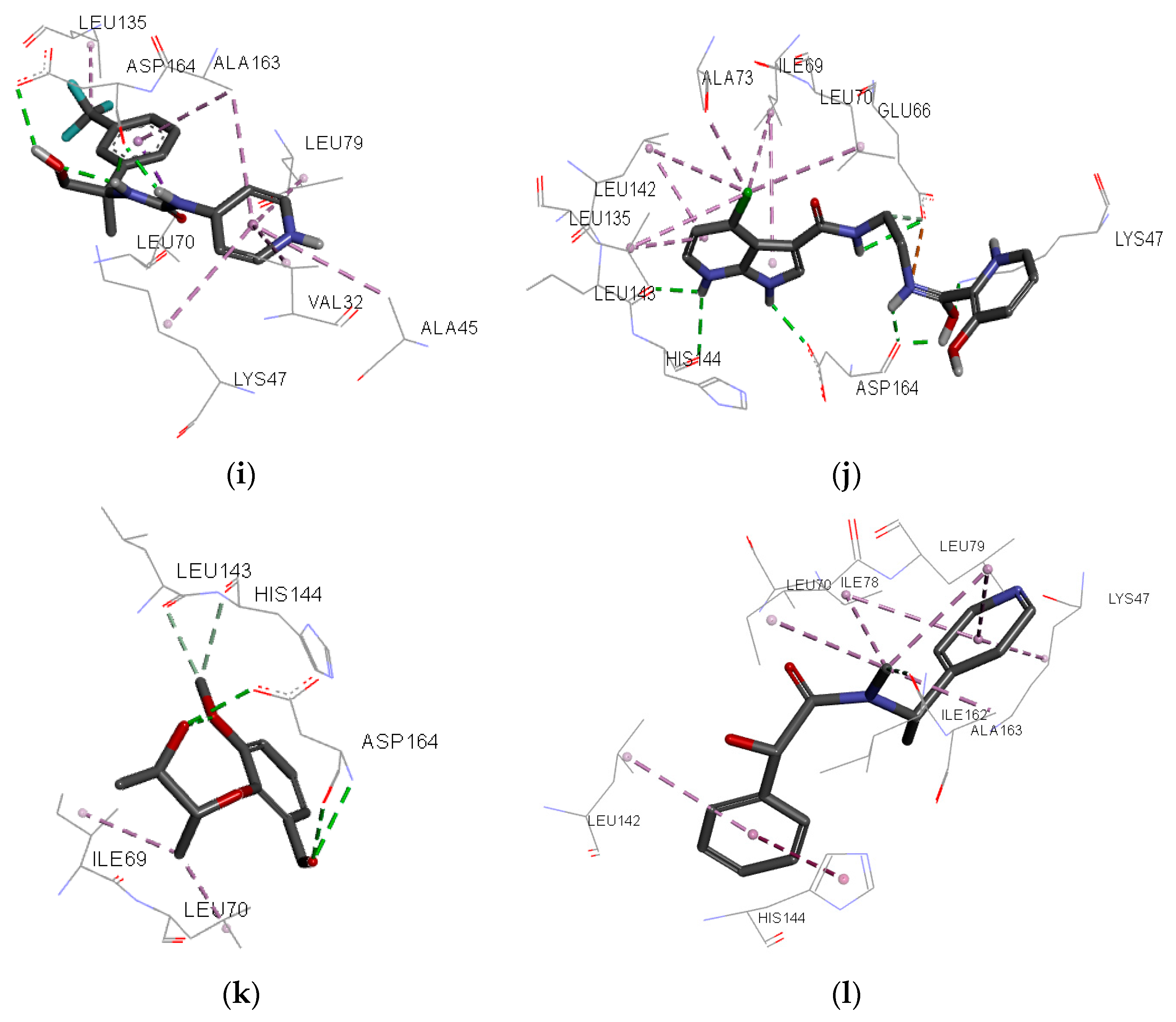
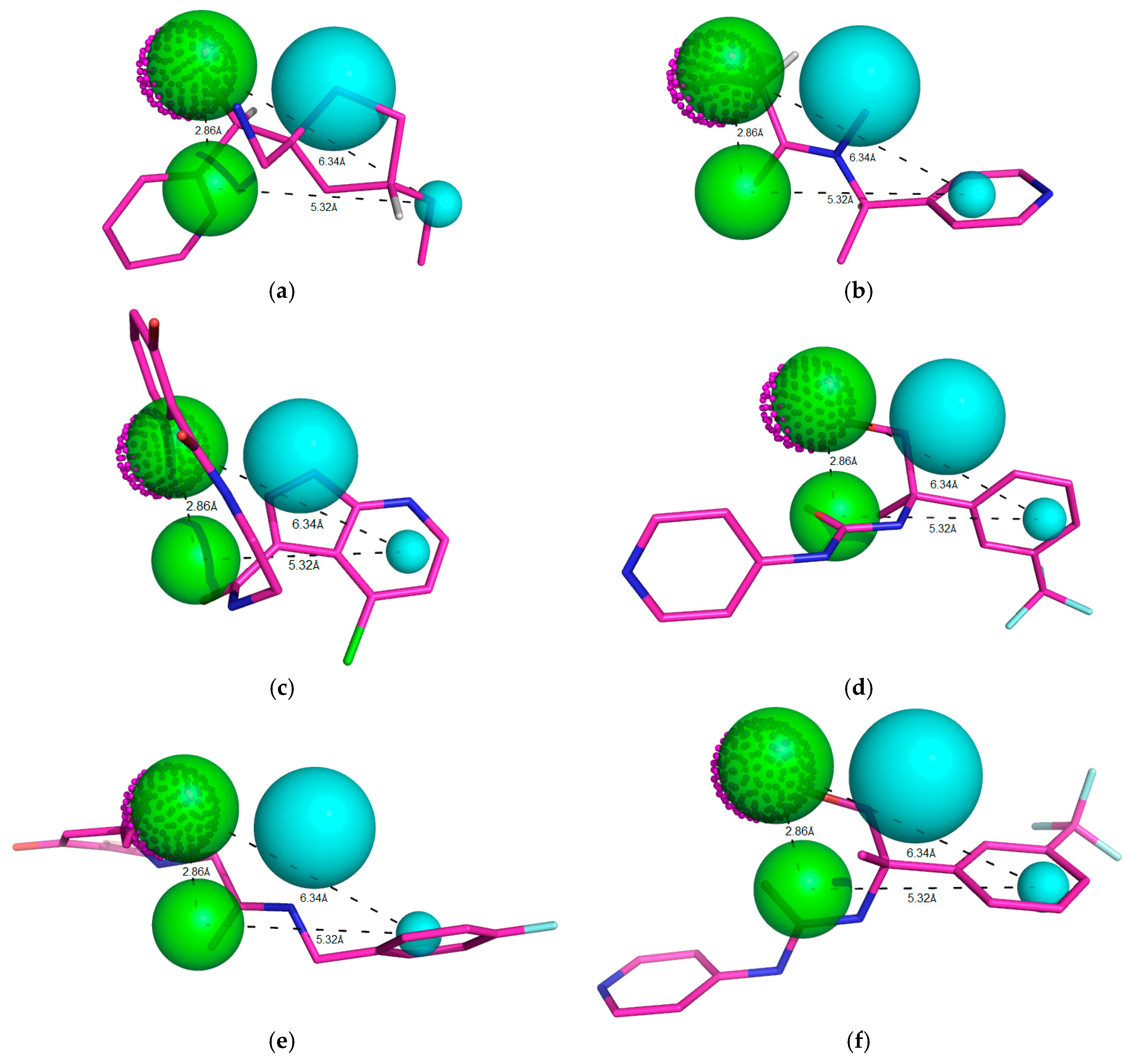
Figure 7.
Individual net of interactions and contacts between RIPK2 (from PDB ID 4C8B) and the following compounds: (a) ZINC90174766; (b) ZINC91725665; (c) ZINC69349685; (d) ZINC69431616; (e) ZINC12230819; (f) ZINC12230826; (g) ZINC12230756; (h) ZINC91781108; (i) ZINC69349687; (j) ZINC89571615; (k) ZINC87131463; (l) ZINC81021663, thus calculated using the AutoDock/Vina software.
Figure 7.
Individual net of interactions and contacts between RIPK2 (from PDB ID 4C8B) and the following compounds: (a) ZINC90174766; (b) ZINC91725665; (c) ZINC69349685; (d) ZINC69431616; (e) ZINC12230819; (f) ZINC12230826; (g) ZINC12230756; (h) ZINC91781108; (i) ZINC69349687; (j) ZINC89571615; (k) ZINC87131463; (l) ZINC81021663, thus calculated using the AutoDock/Vina software.
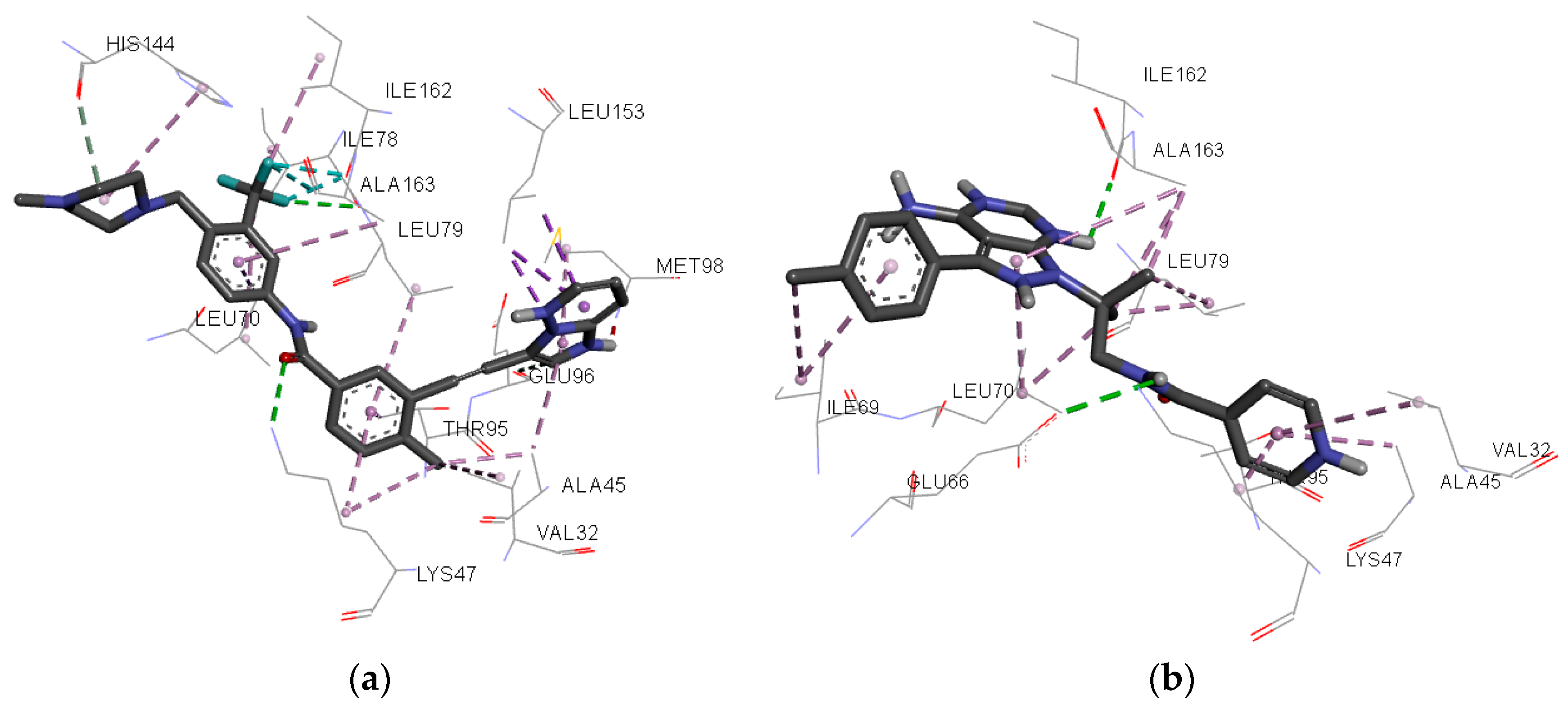
Figure 8.
Individual net of interactions and contacts between RIPK2 (from PDB ID 4C8B) and (a) Ponatinib and (b) WEHI-345, thus calculated using the AutoDock/Vina software.
Figure 8.
Individual net of interactions and contacts between RIPK2 (from PDB ID 4C8B) and (a) Ponatinib and (b) WEHI-345, thus calculated using the AutoDock/Vina software.
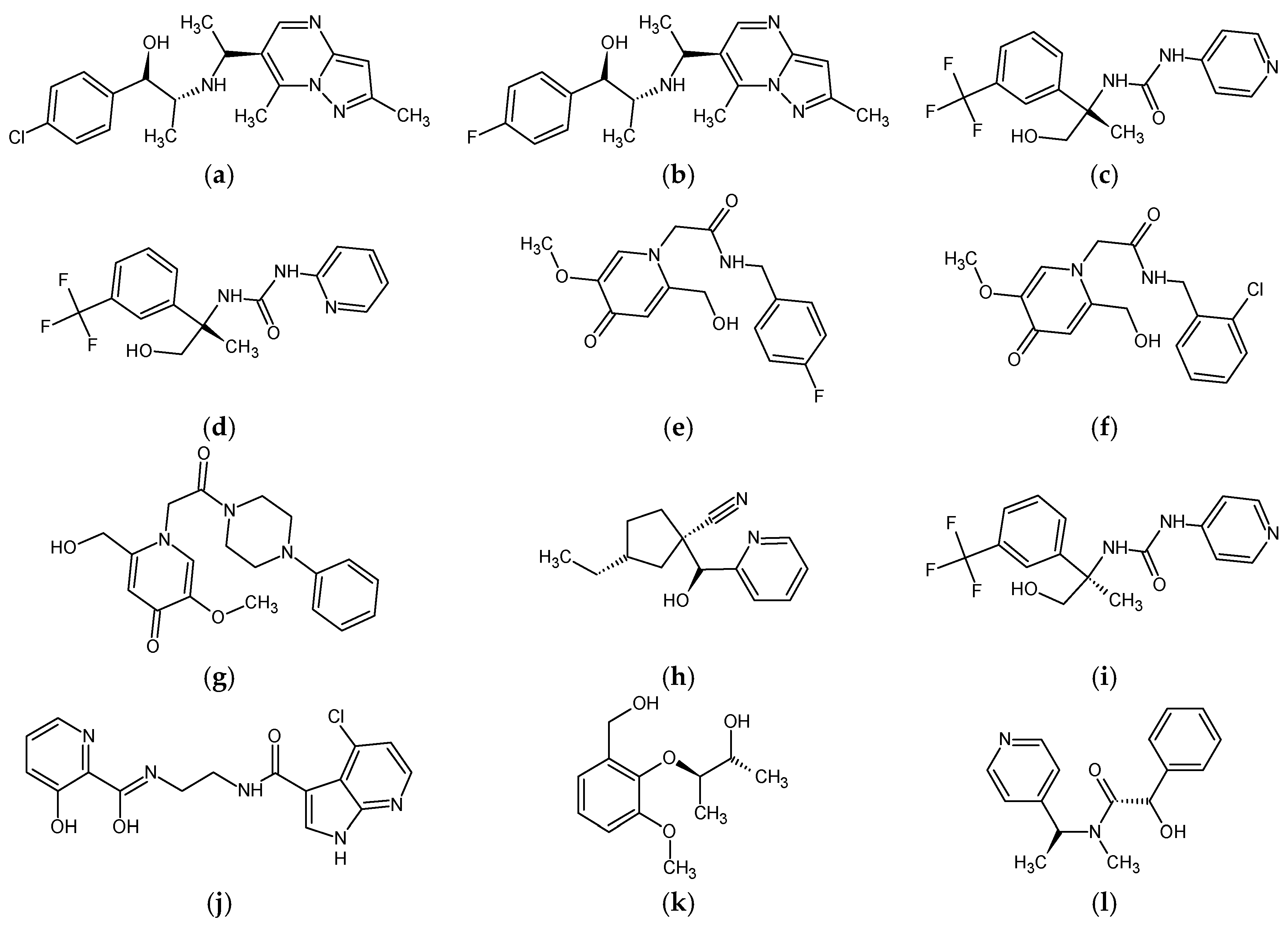
Figure 9.
2D chemical structures of 12 predicted molecules selected with molecular coupling: (a) ZINC90174766; (b) ZINC91725665; (c) ZINC69349685; (d) ZINC69431616; (e) ZINC12230819; (f) ZINC12230826; (g) ZINC12230756; (h) ZINC91881108; (i) ZINC69349687; (j) ZINC89571615; (k) ZINC87131463 and (l) ZINC81021663.
Figure 9.
2D chemical structures of 12 predicted molecules selected with molecular coupling: (a) ZINC90174766; (b) ZINC91725665; (c) ZINC69349685; (d) ZINC69431616; (e) ZINC12230819; (f) ZINC12230826; (g) ZINC12230756; (h) ZINC91881108; (i) ZINC69349687; (j) ZINC89571615; (k) ZINC87131463 and (l) ZINC81021663.
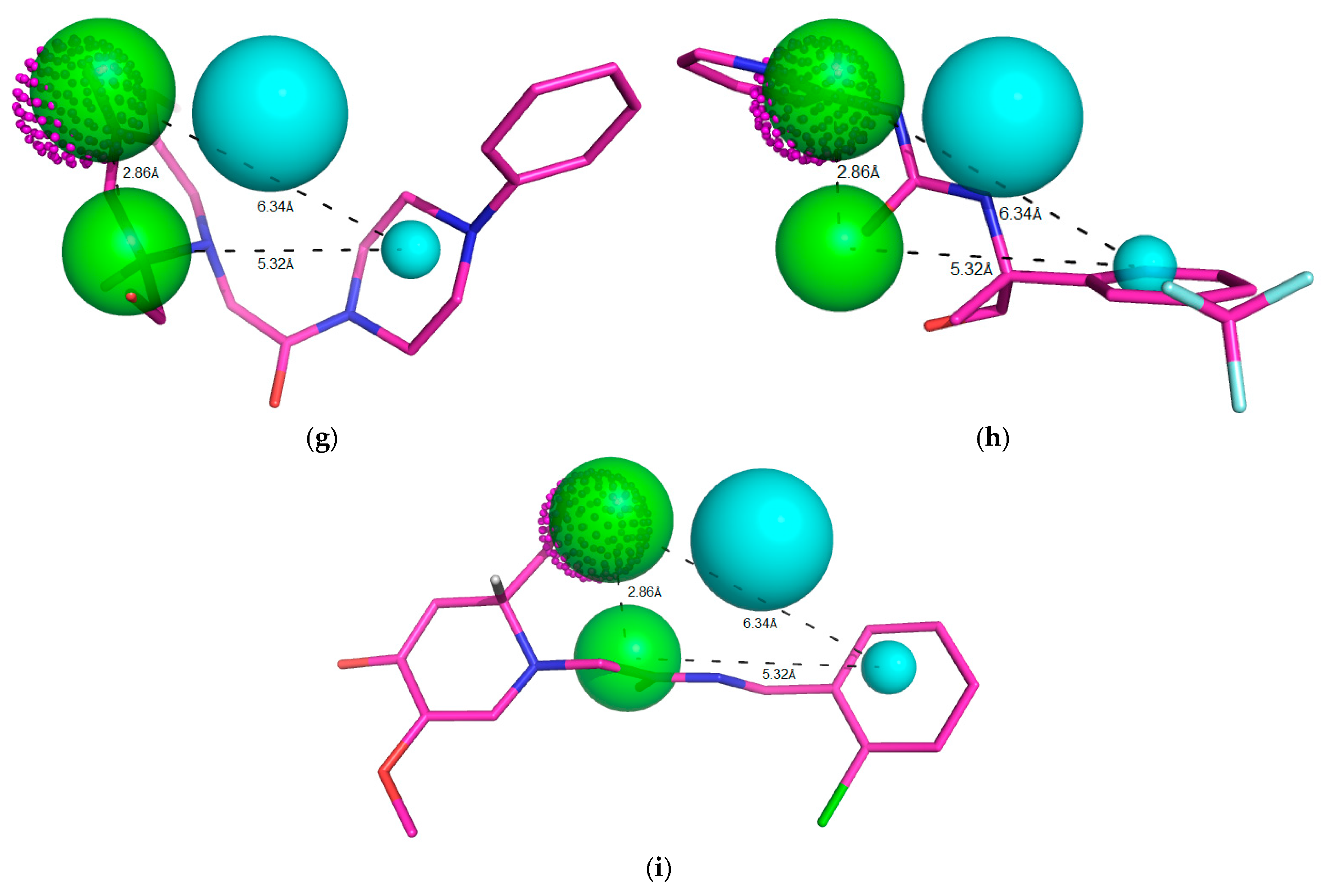
Figure 10.
Representation of the ten compounds that fit to the pharmacophoric model, with QFIT > 0. Green beads/spheres represent H-bond acceptor groups, magenta dotted spheres represent H-bond donor groups, while hydrophobic centers are shown in cyan spheres. Size of the beads varies according to the tolerance radius calculated using GALAHAD. All the distances are measured in angstroms (a) ZINC91881108; (b) ZINC81021663; (c) ZINC89571615; (d) ZINC69349687; (e) ZINC12230819; (f) ZINC69349685; (g) ZINC12230756; (h) ZINC69431616 and (i) ZINC12230826.
Figure 10.
Representation of the ten compounds that fit to the pharmacophoric model, with QFIT > 0. Green beads/spheres represent H-bond acceptor groups, magenta dotted spheres represent H-bond donor groups, while hydrophobic centers are shown in cyan spheres. Size of the beads varies according to the tolerance radius calculated using GALAHAD. All the distances are measured in angstroms (a) ZINC91881108; (b) ZINC81021663; (c) ZINC89571615; (d) ZINC69349687; (e) ZINC12230819; (f) ZINC69349685; (g) ZINC12230756; (h) ZINC69431616 and (i) ZINC12230826.
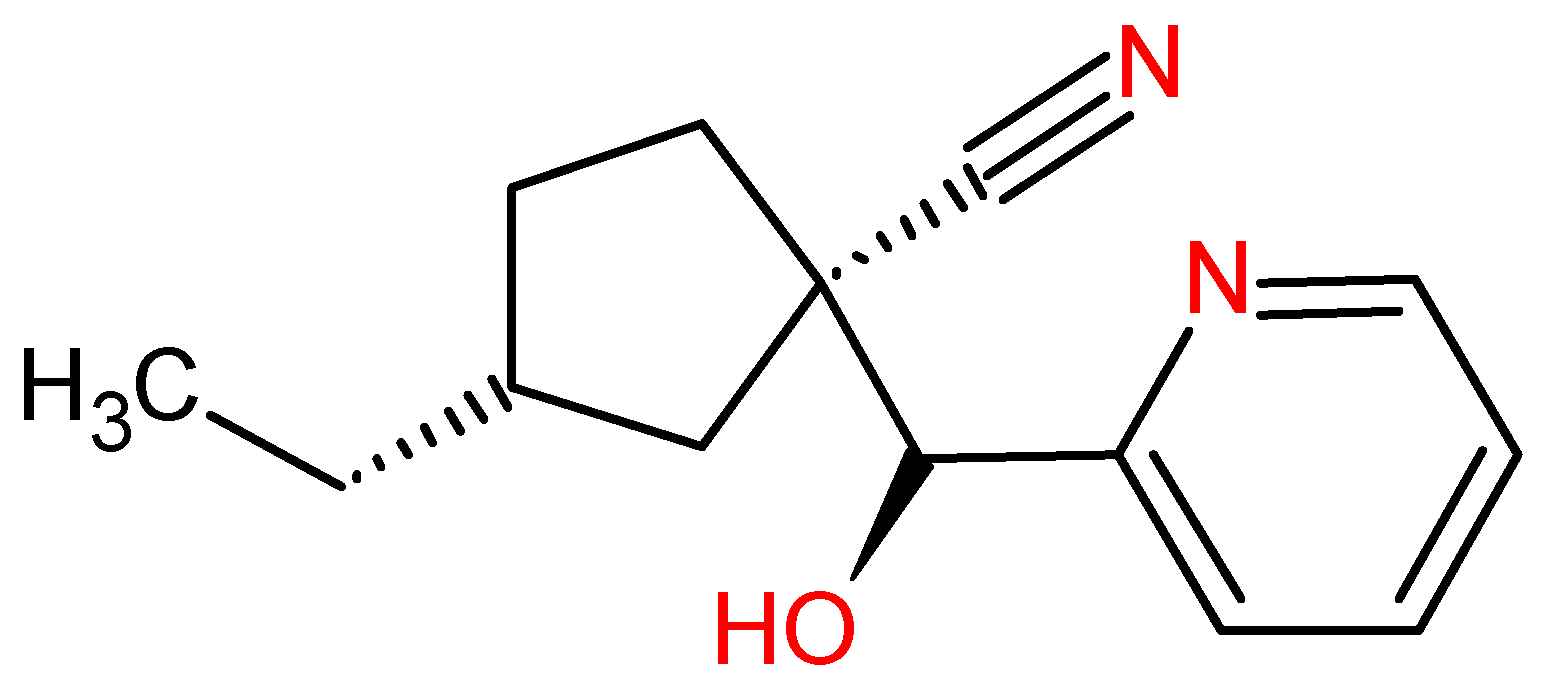
Figure 11.
Promising molecule ZINC91881108 obtained after virtual screening.
Figure 11.
Promising molecule ZINC91881108 obtained after virtual screening.
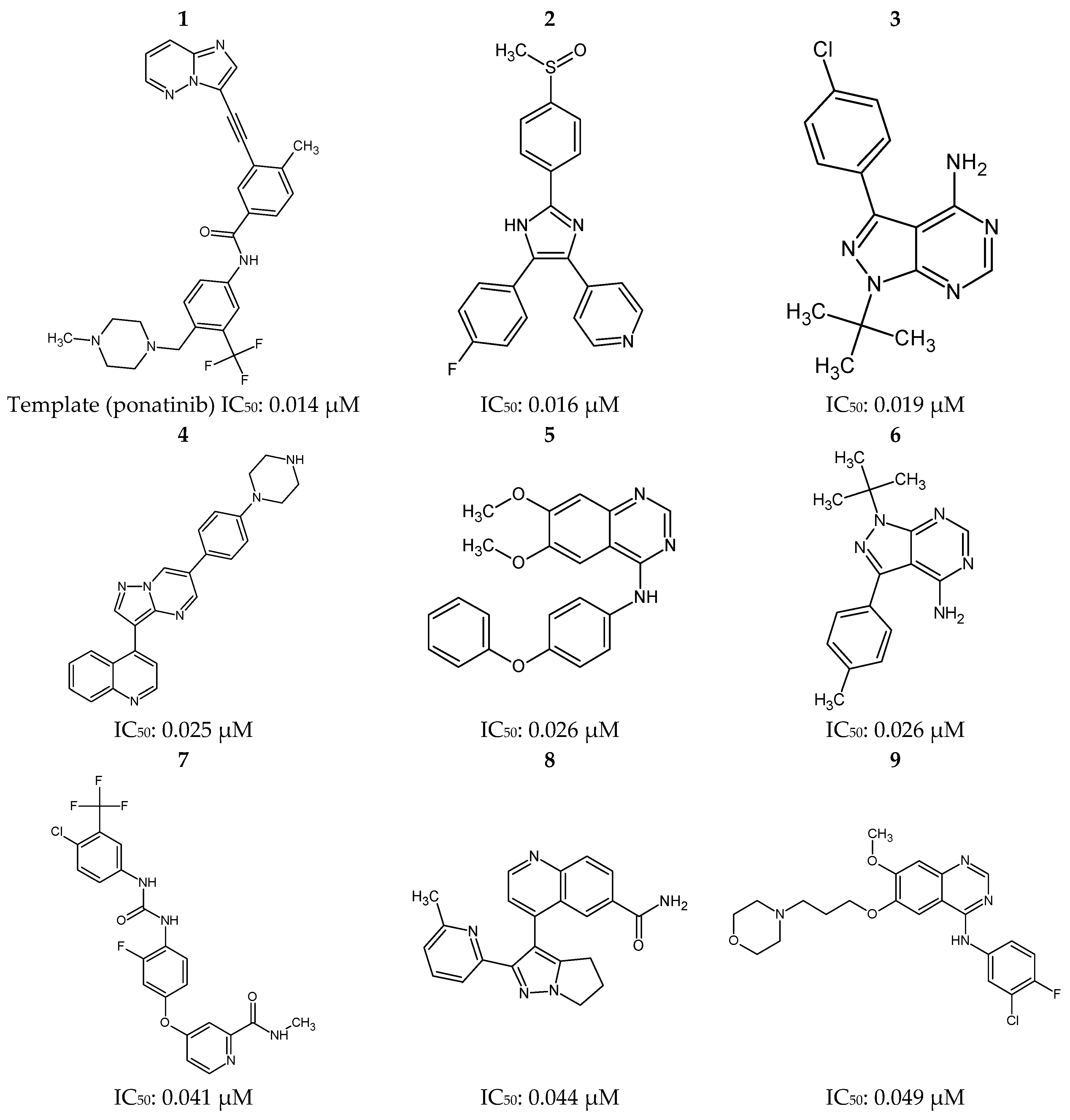
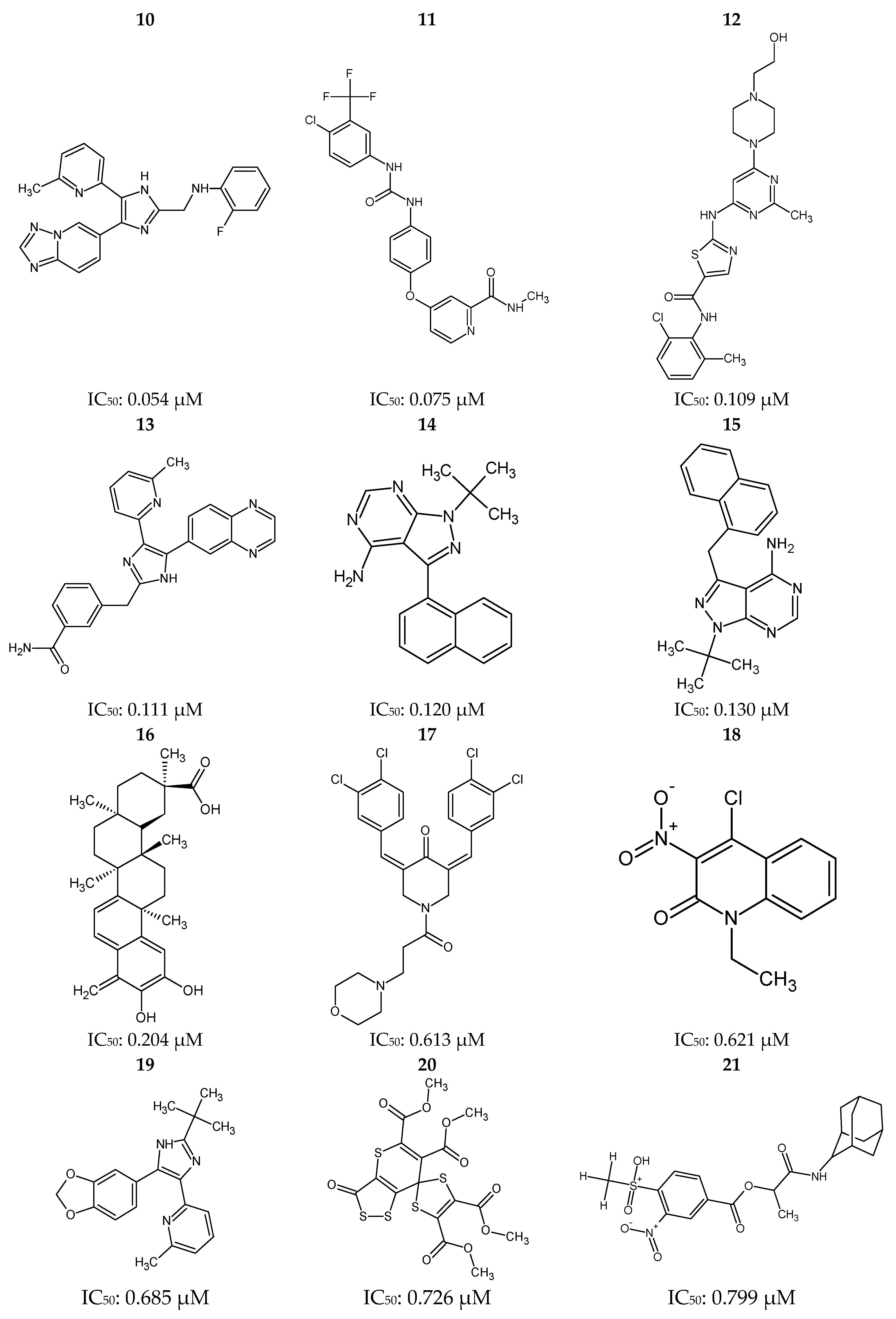
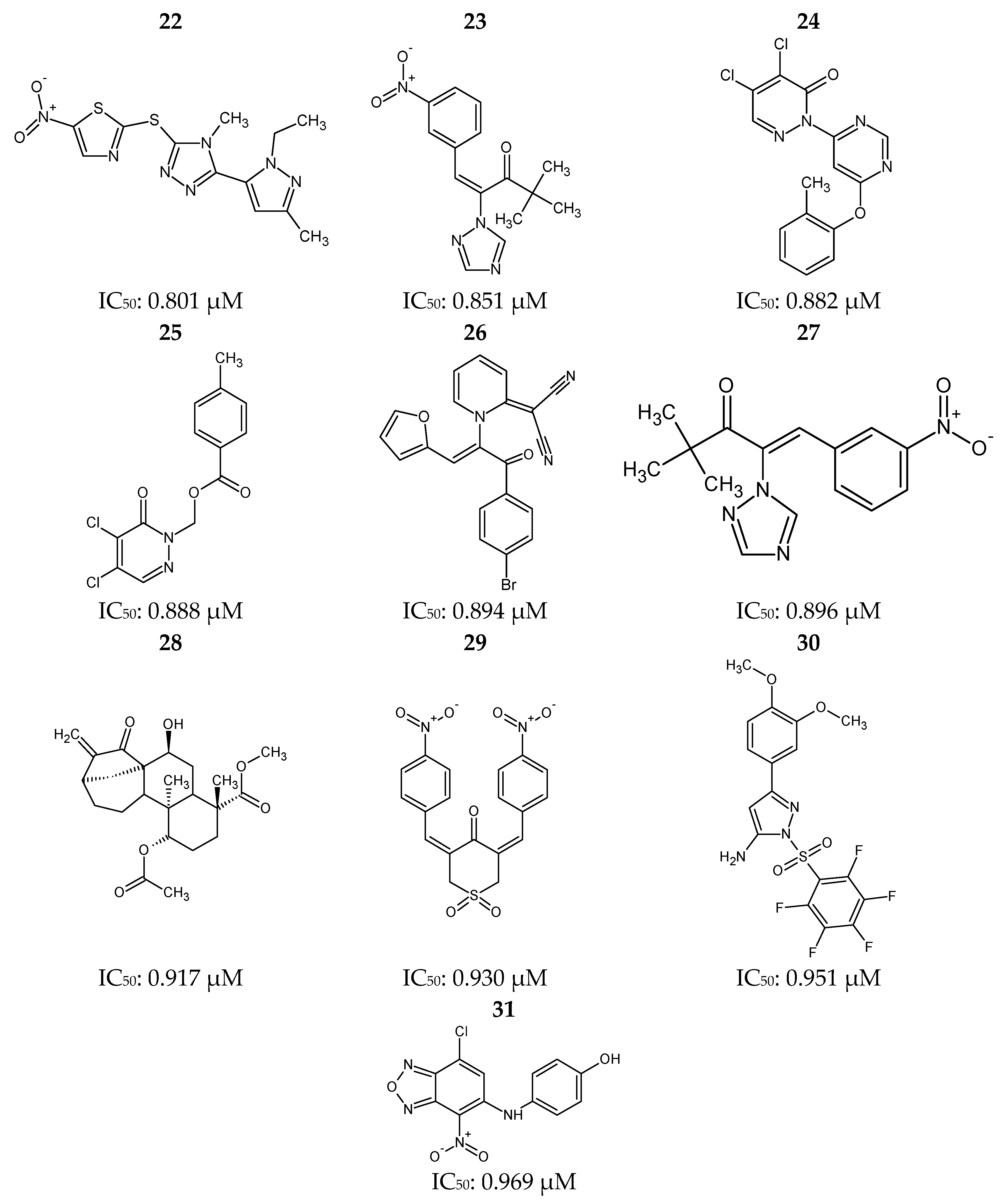
Figure 12.
Ponatinib (1) and 30 most potent RIPK2 inhibitors (2–31) obtained from the Protein Data Bank (PDB) and the BindingDB database.
Figure 12.
Ponatinib (1) and 30 most potent RIPK2 inhibitors (2–31) obtained from the Protein Data Bank (PDB) and the BindingDB database.
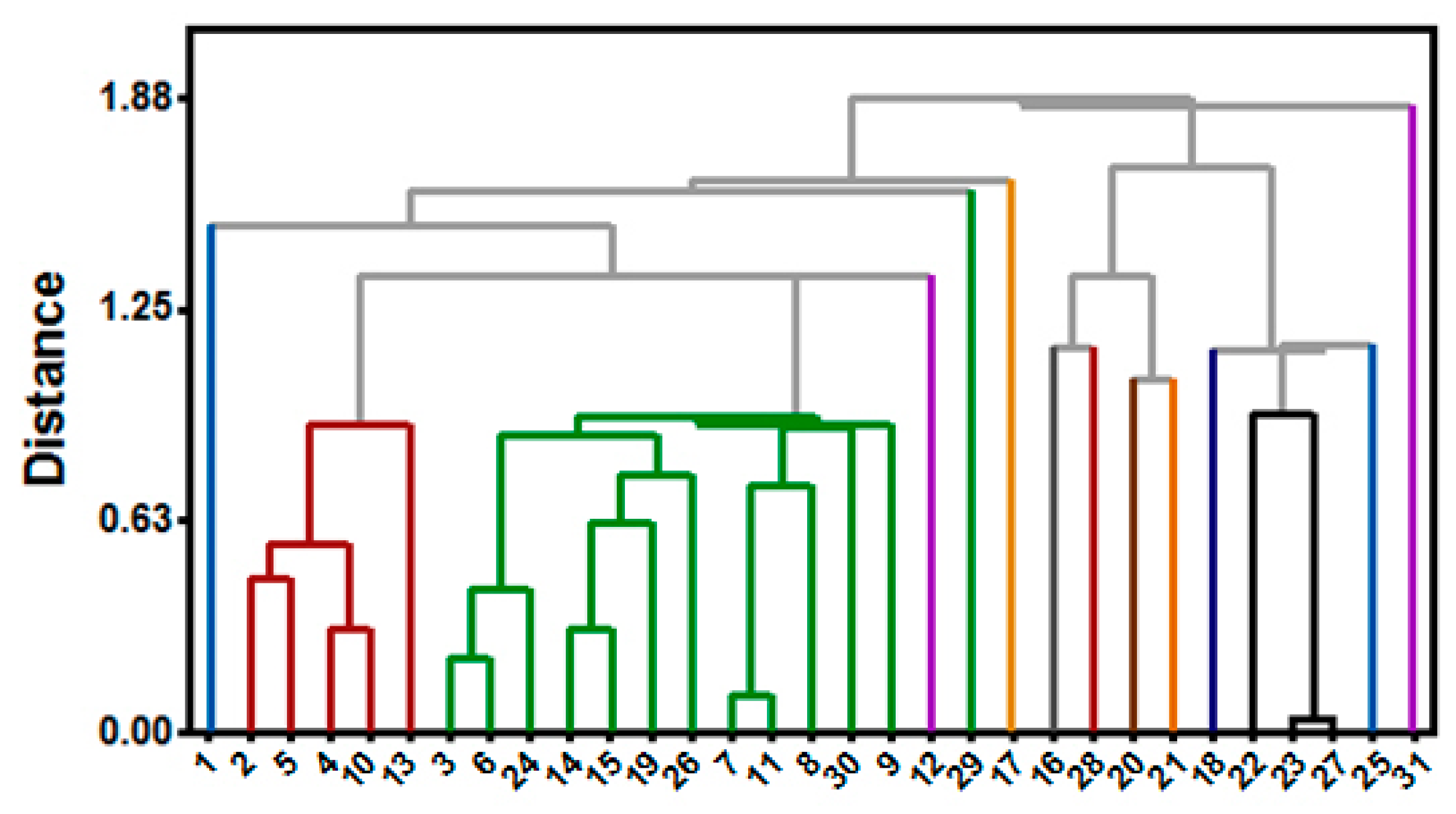
Figure 13.
Dendrogram used to investigate chemical similarity of RIPK2 inhibitors.
Figure 13.
Dendrogram used to investigate chemical similarity of RIPK2 inhibitors.
Table 1.
Features of each pharmacophoric model generated using GALAHAD.
Table 1.
Features of each pharmacophoric model generated using GALAHAD.
| Model | Specificity | N_Hits | Features | Pareto | Energy (kcal/mol) | Sterics | HBond | Mol_QRY |
|---|
| 01 | 2.399 | 03 | 07 | 00 | 1215.88 | 541.80 | 57.00 | 3.29 |
| 02 | 3.069 | 01 | 09 | 00 | 90.97 | 532.10 | 54.00 | 2.22 |
| 03 | 3.875 | 00 | 06 | 00 | 303, 52.40 | 537.80 | 55.30 | 5.78 |
| 04 | 3.676 | 01 | 05 | 00 | 31.35 | 529.70 | 55.30 | 0.58 |
| 05 | 2.993 | 03 | 05 | 00 | 30.33 | 472.30 | 54.60 | 2.25 |
| 06 | 1.900 | 03 | 04 | 00 | 148.83 | 533.80 | 52.90 | 2.67 |
| 07 | 5.526 | 00 | 08 | 00 | 187, 147.79 | 547.20 | 56.80 | 0.53 |
| 08 | 3.939 | 01 | 06 | 00 | 4453, 411.50 | 549.30 | 56.50 | 1.67 |
| 09 | 3.068 | 03 | 05 | 00 | 45.49 | 536.90 | 54.60 | 0.22 |
| 10 | 3.082 | 04 | 04 | 00 | 72.56 | 514.90 | 55.20 | 0.80 |
Table 2.
Prediction of biological activity of substances (PASS).
Table 2.
Prediction of biological activity of substances (PASS).
| Compound (ZINC Code) | Biological Activity | Pa | Pi |
|---|
| ZINC69349685 | Autoimmune | 0.84 | 0.005 |
| Anti-inflammatory | 0.538 | 0.046 |
| Treatment of rheumatoid arthritis | 0.434 | 0.019 |
| Kinase Inhibitor | 0.316 | 0.128 |
| ZINC69349687 | Autoimmune | 0.784 | 0.005 |
| Anti-inflamatória | 0.538 | 0.046 |
| Treatment of rheumatoid arthritis | 0.434 | 0.019 |
| Kinase Inhibitor | 0.316 | 0.128 |
| ZINC69431616 | Autoimmune | 0.790 | 0.005 |
| Anti-inflammatory | 0.573 | 0.038 |
| Treatment of rheumatoid arthritis | 0.436 | 0.019 |
| Kinase Inhibitor | 0.133 | 0.104 |
| ZINC69431621 | Autoimmune | 0.790 | 0.005 |
| Anti-inflammatory | 0.573 | 0.038 |
| Treatment of rheumatoid arthritis | 0.436 | 0.019 |
| Kinase Inhibitor | 0.133 | 0.104 |
| ZINC91072217 | Kinase Inhibitor | 0.572 | 0.023 |
| Anti-inflammatory | 0.259 | 0.203 |
Table 3.
Interactions between RIPK2 (PDB 4C8B) and ponatinib or WEHI-345, obtained using the AutoDock/Vina software are shown.
Table 3.
Interactions between RIPK2 (PDB 4C8B) and ponatinib or WEHI-345, obtained using the AutoDock/Vina software are shown.
| Compound | Amino Acid | Type | Distance (Å) | Binding Affinity (in kcal/mol) |
|---|
| Ponatinib | LYS47 | Hydrogen Bond | 3.09 | −13.30 |
| HIS144 | Hydrogen Bond | 3.12 |
| ALA45 | Alkyl | 3.33 |
| VAL32 | Alkyl | 4.98 |
| LYS47 | Alkyl | 4.80 |
| LEU70 | Alkyl | 5.42 |
| HIS144 | π-Alkyl | 4.90 |
| LYS47 | π-Alkyl | 4.23 |
| LEU79 | π-Alkyl | 5.47 |
| ALA45 | π-Alkyl | 4.03 |
| ALA163 | π-Alkyl | 5.04 |
| WEHI-345 | ILE162 | Hydrogen Bond | 2.43 | −7.50 |
| GLU66 | Hydrogen Bond | 3.06 |
| THR95 | π-Donor | 4.03 |
| ALA163 | Alkyl | 4.17 |
| ALA163 | Alkyl | 3.09 |
| LEU70 | Alkyl | 3.60 |
| LEU79 | Alkyl | 3.87 |
| LEU79 | Alkyl | 5.08 |
| ILE69 | Alkyl | 4.98 |
| LEU70 | π-Alkyl | 4.600 |
| ALA163 | π-Alkyl | 5.19 |
| VAL32 | π-Alkyl | 4.50 |
| ALA45 | π-Alkyl | 4.48 |
| LYS47 | π-Alkyl | 3.82 |
| ILE69 | π-Alkyl | 4.63 |
Table 4.
Interactions between RIPK2 (from PDB ID 4C8B) and the following compounds are shown, after docking calculations: ZINC90174766, ZINC91725665, ZINC69431616, ZINC12230819, ZINC12230826, ZINC12230756, ZINC69431621, ZINC90174764, ZINC91881108, ZINC69349687, ZINC91725663, ZINC89571615, ZINC87131463 and ZINC81021663.
Table 4.
Interactions between RIPK2 (from PDB ID 4C8B) and the following compounds are shown, after docking calculations: ZINC90174766, ZINC91725665, ZINC69431616, ZINC12230819, ZINC12230826, ZINC12230756, ZINC69431621, ZINC90174764, ZINC91881108, ZINC69349687, ZINC91725663, ZINC89571615, ZINC87131463 and ZINC81021663.
| Compound | Amino Acid | Type | Distance (Å) | Binding Affinity (in kcal/mol) |
|---|
| ZINC90174766 | LEU70 | π-Alkyl | 5.12 | −11.00 |
| ALA163 | πAlkyl | 4.77 |
| LEU79 | π-Alkyl | 5.23 |
| ALA45 | π-Alkyl | 3.71 |
| VAL32 | π-Alkyl | 5.14 |
| ZINC91725665 | VAL32 | Alkyl | 4.12 | −10.80 |
| ALA45 | Alkyl | 3.04 |
| LEU79 | Alkyl | 4.42 |
| LYS47 | π-Alkyl | 4.55 |
| ALA163 | Alkyl | 2.94 |
| GLU66 | Hydrogen Bond | 2.54 |
| ILE69 | π-Alkyl | 5.11 |
| LEU70 | π-Alkyl | 4.37 |
| LYS47 | π-Alkyl | 5.29 |
| ZINC69349685 | LEU79 | Alkyl | 5.06 | −10.40 |
| ALA163 | π-Alkyl | 5.48 |
| ILE69 | π-Alkyl | 4.56 |
| ZINC69431616 | VAL32 | π-Alkyl | 5.10 | −10.20 |
| ALA45 | π-Alkyl | 3.65 |
| LEU79 | π-Alkyl | 5.40 |
| LEU70 | π-Alkyl | 4.28 |
| ZINC12230819 | GLU66 | Hydrogen Bond | 2.71 | −9.80 |
| ILE69 | π-Alkyl | 5.44 |
| LYS47 | Hydrogen Bond | 2.79 |
| LYS47 | Hydrogen Bond | 3.03 |
| ZINC12230826 | LYS47 | Hydrogen Bond | 2.81 | −9.80 |
| LYS47 | Hydrogen Bond | 2.64 |
| GLU66 | Hydrogen Bond | 2.54 |
| GLU66 | Hydrogen Bond | 3.32 |
| HIS144 | π-Alkyl | 3.23 |
| ZINC12230756 | LYS47 | Hydrogen Bond | 2.78 | −8.50 |
| LYS47 | Hydrogen Bond | 2.33 |
| GLU66 | Hydrogen Bond | 2.43 |
| LEU70 | Alkyl | 4.82 |
| ILE69 | π-Alkyl | 5.10 |
| VAL32 | π-Alkyl | 5.04 |
| ALA45 | π-Alkyl | 4.57 |
| LYS47 | π-Alkyl | 4.55 |
| LEU79 | π-Alkyl | 5.39 |
| ZINC91881108 | ASP164 | Hydrogen Bond | 2.53 | −8.10 |
| ILE162 | Hydrogen Bond | 2.18 |
| GLU66 | Hydrogen Bond | 2.28 |
| ALA45 | Alkyl | 3.84 |
| ALA163 | Alkyl | 4.54 |
| LEU70 | Alkyl | 5.24 |
| LEU79 | Alkyl | 4.75 |
| VAL32 | Alkyl | 4.31 |
| LYS47 | Alkyl | 4.39 |
| ZINC69349687 | ALA163 | π-Alkyl | 5.08 | −8.00 |
| VAL32 | π-Alkyl | 4.78 |
| ALA45 | π-Alkyl | 5.27 |
| LYS47 | π-Alkyl | 4.84 |
| LEU79 | π-Alkyl | 5.33 |
| ALA163 | π-Alkyl | 5.45 |
| ZINC89571615 | LYS47 | Hydrogen Bond | 2.52 | −7.90 |
| GLU66 | Hydrogen Bond | 3.04 |
| HIS144 | Hydrogen Bond | 2.70 |
| GLU66 | Hydrogen Bond | 2.74 |
| LEU70 | Alkyl | 4.93 |
| ILE69 | π-Alkyl | 5.38 |
| ZINC87131463 | HIS144 | Hydrogen Bond | 3.62 | −7.90 |
| ILE69 | Alkyl | 4.17 |
| LEU70 | Alkyl | 3.73 |
| ZINC81021663 | ILE162 | Hydrogen Bond | 2.89 | −7.80 |
| LYS47 | Alkyl | 5.02 |
| LEU70 | Alkyl | 5.06 |
| LEU79 | Alkyl | 5.20 |
| ALA163 | Alkyl | 4.00 |
| LEU70 | Alkyl | 4.26 |
| LEU79 | Alkyl | 4.69 |
Table 5.
Similarity analyses for the molecular overlap of the compounds for 100ste, 100elt, 60est and 40elt, 50est/elt, relative to ponatinib.
Table 5.
Similarity analyses for the molecular overlap of the compounds for 100ste, 100elt, 60est and 40elt, 50est/elt, relative to ponatinib.
| Similarity of Overlay (%) |
|---|
| Compound | 100ste | 100elt | 60est/40elt | 40est/60elt | 50est/elt |
|---|
| ZINC90174766 | 0.7169 | 0.3917 | 0.4757 | 0.3669 | 0.4202 |
| ZINC91725665 | 0.7128 | 0.4167 | 0.4802 | 0.3666 | 0.4234 |
| ZINC91881108 | 0.4865 | 0.5520 | 0.4701 | 0.4775 | 0.4740 |
Table 6.
Similarity analyses for the molecular overlap of the compounds for 100ste, 100elt, 60est and 40elt, 50est/elt, relative to WEHI-345.
Table 6.
Similarity analyses for the molecular overlap of the compounds for 100ste, 100elt, 60est and 40elt, 50est/elt, relative to WEHI-345.
| Similarity of Overlay (%) |
|---|
| Compound | 100ste | 100elt | 60est/40elt | 40est/60elt | 50est/elt |
|---|
| ZINC12230819 | 0.7824 | 0.3623 | 0.4717 | 0.3390 | 0.4036 |
| ZINC91881108 | 0.6491 | 0.5645 | 0.4611 | 0.4131 | 0.4361 |
| ZINC81021663 | 0.6802 | 0.3527 | 0.5687 | 0.5192 | 0.5438 |
Table 7.
Compounds with QFIT > 0, which were analyzed using the CHEMGPS-NP server.
Table 7.
Compounds with QFIT > 0, which were analyzed using the CHEMGPS-NP server.
| Compound | QFIT Value |
|---|
| ZINC91881108 | 52.28 |
| ZINC81021663 | 31.26 |
| ZINC89571615 | 25.83 |
| ZINC69349687 | 18.61 |
| ZINC12230819 | 17.69 |
| ZINC69349685 | 15.23 |
| ZINC12230756 | 14.62 |
| ZINC69431616 | 14.13 |
| ZINC12230826 | 12.21 |
Table 8.
Pharmacokinetic properties of nine selected compounds as well as ponatinib and WEHI-345.
Table 8.
Pharmacokinetic properties of nine selected compounds as well as ponatinib and WEHI-345.
| Compound | Stars | Rule of Five | % HOA | ACP (nm/s) | AMP (nm/s) | QPlog Po/w | CNS | QPlog BBB |
|---|
| Ponatinib | 1 | 1 | 73.645 | 67.076 | 119.394 | 4.605 | 1 | −0.026 |
| WEHI-345 | 0 | 0 | 93.941 | 456.487 | 211.950 | 3.313 | −2 | −1.220 |
| ZINC69349685 | 0 | 0 | 100.00 | 1300.94 | 3123.35 | 2.563 | −1 | −0.281 |
| ZINC69431616 | 0 | 0 | 100.00 | 1695.74 | 4585.45 | 2.910 | −1 | −0.145 |
| ZINC12230819 | 0 | 0 | 80.392 | 362.468 | 467.713 | 1.305 | −2 | −1.048 |
| ZINC12230826 | 0 | 0 | 82.927 | 462.942 | 598.190 | 1.413 | −1 | −0.871 |
| ZINC12230756 | 0 | 0 | 84.316 | 561.255 | 418.589 | 1.395 | −1 | −0.809 |
| ZINC91881108 | 0 | 0 | 94.985 | 1169.73 | 586.063 | 2.242 | 0 | −0.577 |
| ZINC69349687 | 0 | 0 | 100.00 | 1297.59 | 3147.44 | 2.566 | −1 | −0.282 |
| ZINC89571615 | 0 | 0 | 83.225 | 229.628 | 131.490 | 2.395 | −2 | −1.455 |
| ZINC81021663 | 0 | 0 | 89.971 | 946.013 | 748.108 | 1.667 | 0 | −0.526 |
Table 9.
Prediction of synthetic accessibility of ponatinib, WEHI-345 and the selected compound ZINC91881108.
Table 9.
Prediction of synthetic accessibility of ponatinib, WEHI-345 and the selected compound ZINC91881108.
| Compound | Accessibility |
|---|
| Ponatinib | 5.10 |
| WEHI-345 | 4.86 |
| ZINC91881108 | 5.01 |
Table 10.
3D Coordinates and pharmacophoric features of the best-ranked pharmacophoric model.
Table 10.
3D Coordinates and pharmacophoric features of the best-ranked pharmacophoric model.
| Pharmacophoric Feature | X | Y | Z | Radius (in Ǻ) |
|---|
| Hydrogen Bond Donor | 3.71 | −0.53 | 1.86 | 1.10 |
| Hydrogen Bond Acceptor | 2.47 | −1.65 | -0.38 | 1.11 |
| Hydrogen Bond Acceptor | 3.51 | −0.10 | 1.79 | 1.26 |
| Hydrophobic | −2.33 | 0.63 | −0.57 | 0.55 |
| Hydrophobic | 0.68 | 1.00 | 1.43 | 1.42 |

 ,
, 


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}