Aleuritolic Acid Impaired Autophagic Flux and Induced Apoptosis in Hepatocellular Carcinoma HepG2 Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
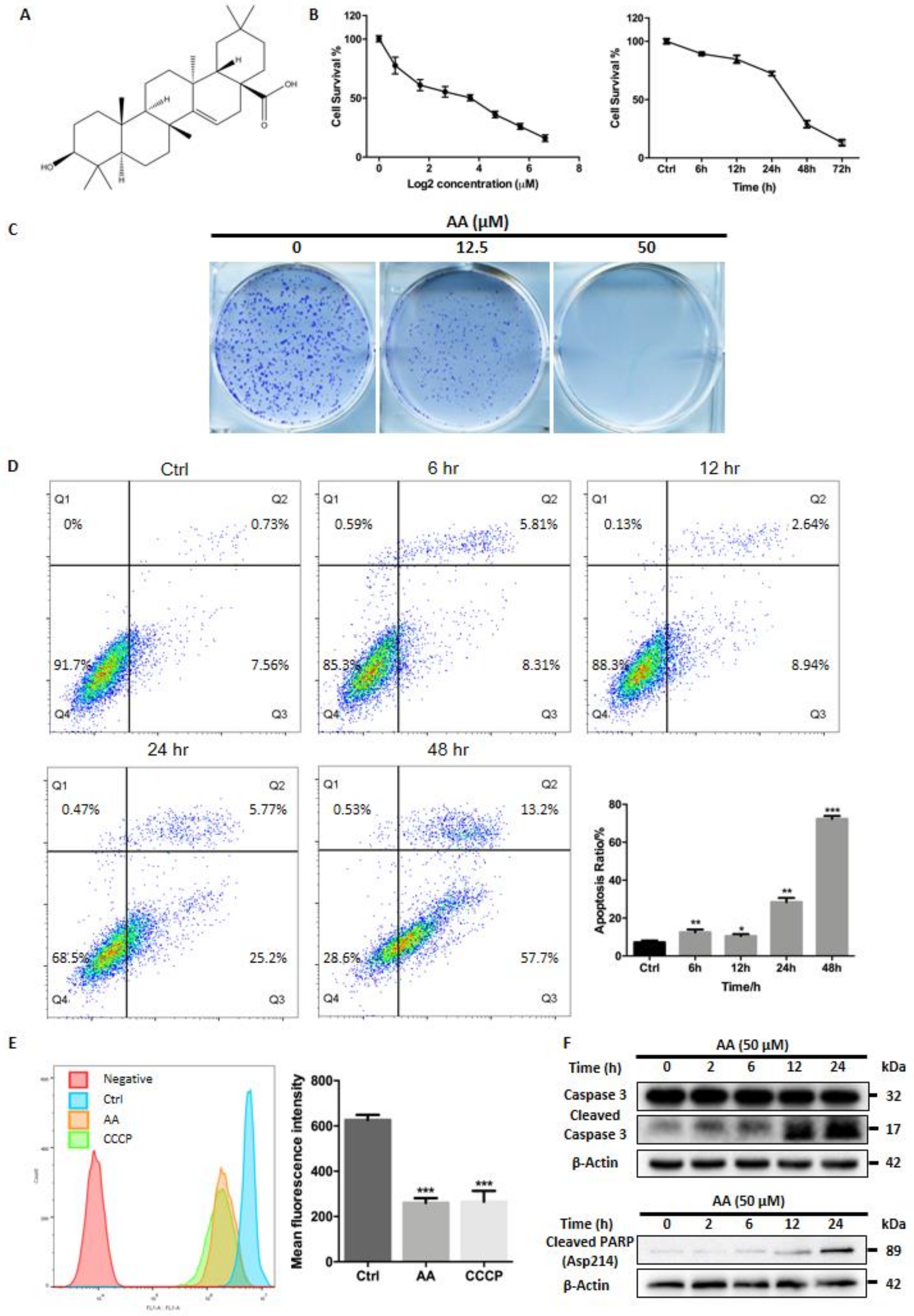
2.1. Cytotoxic Activity of AA In Vitro
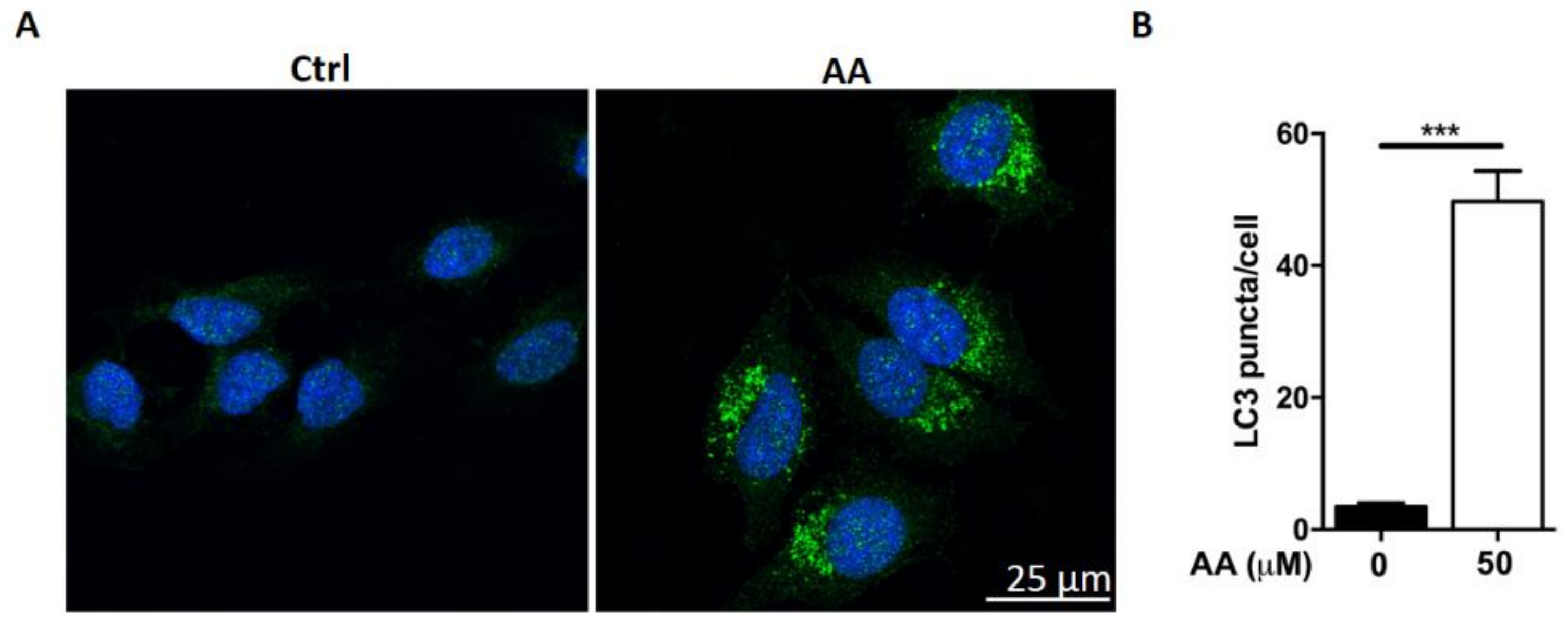
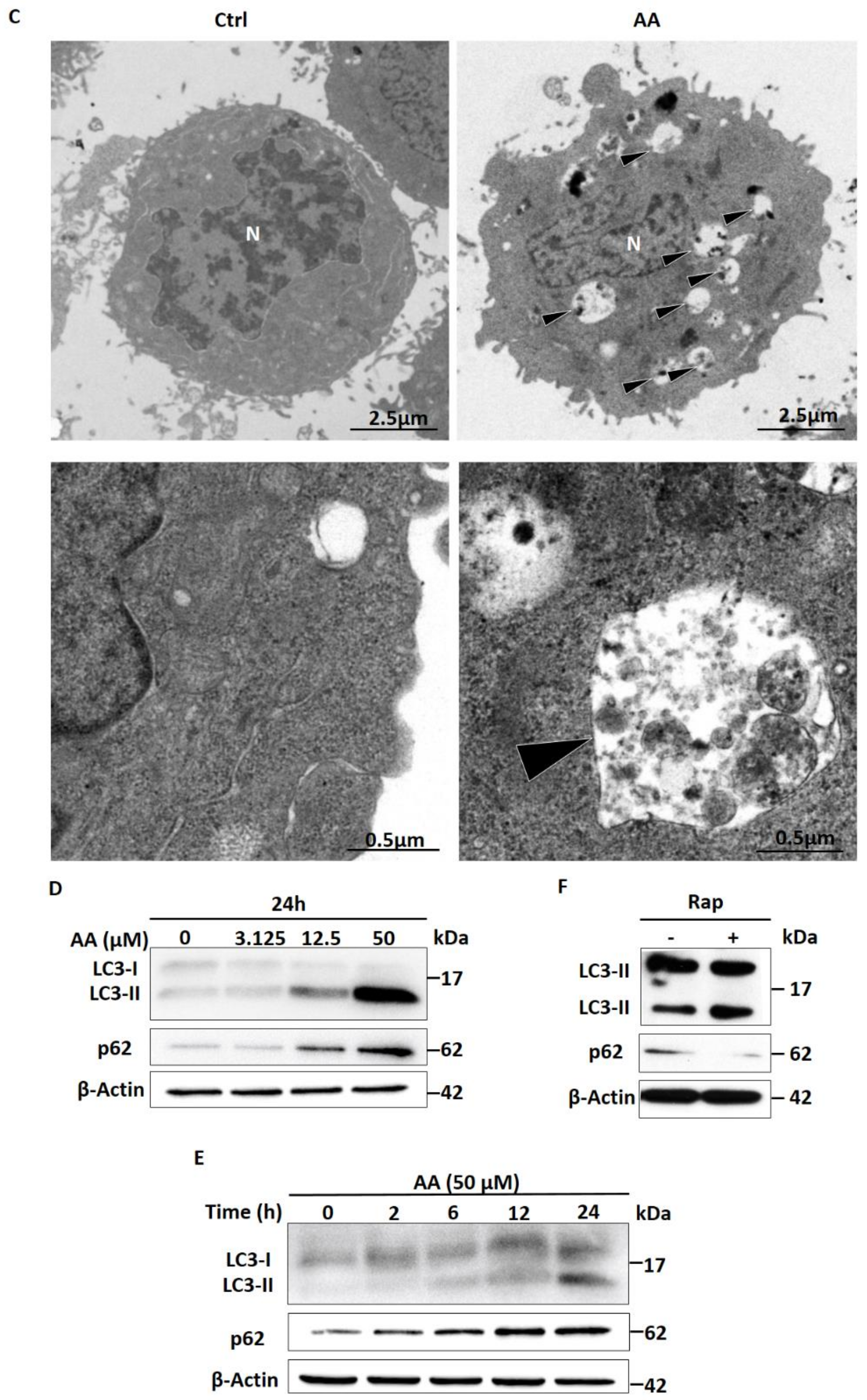
2.2. Treatment with AA Impairs Autophagic Flux in HepG2 Cells
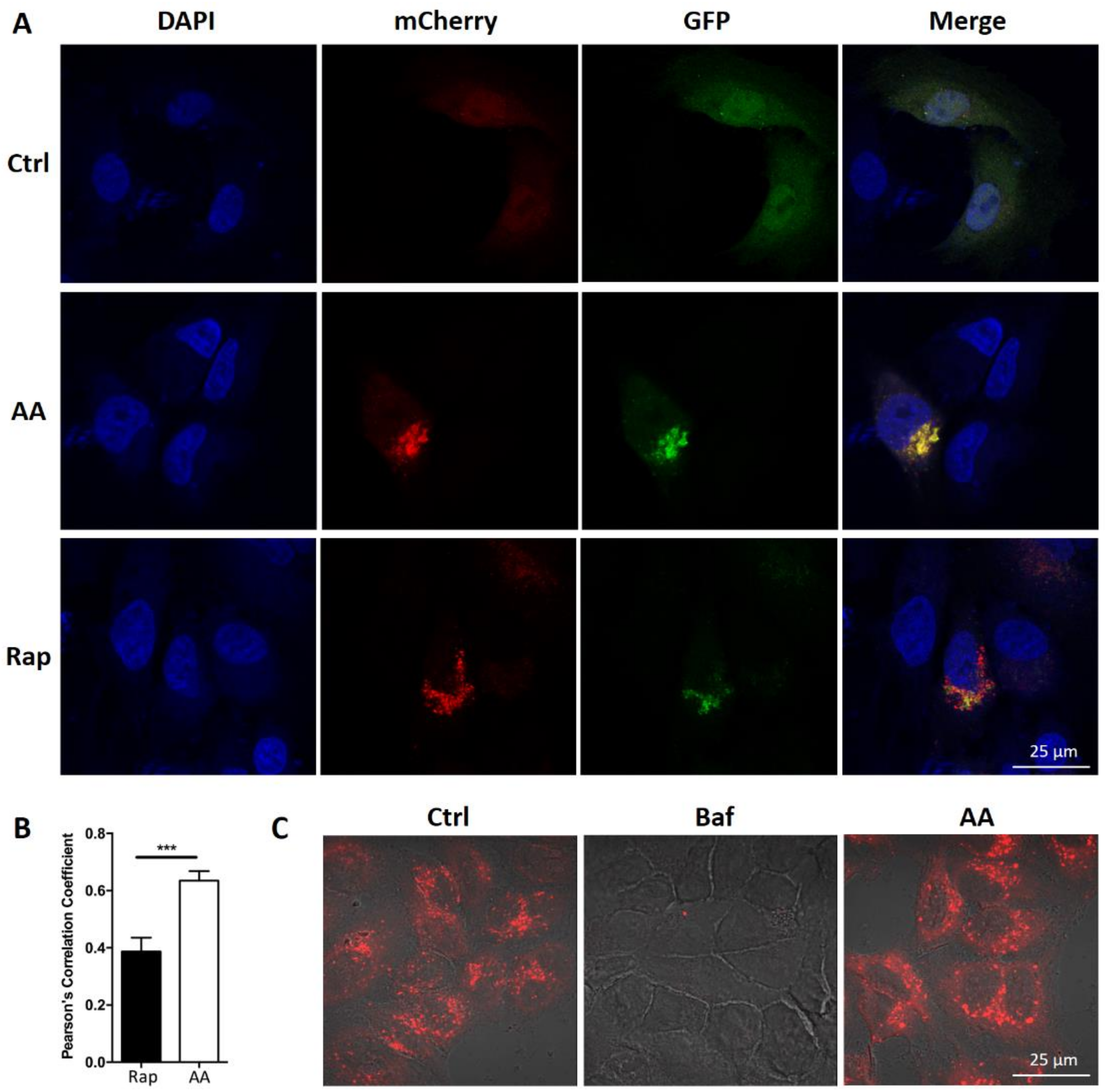
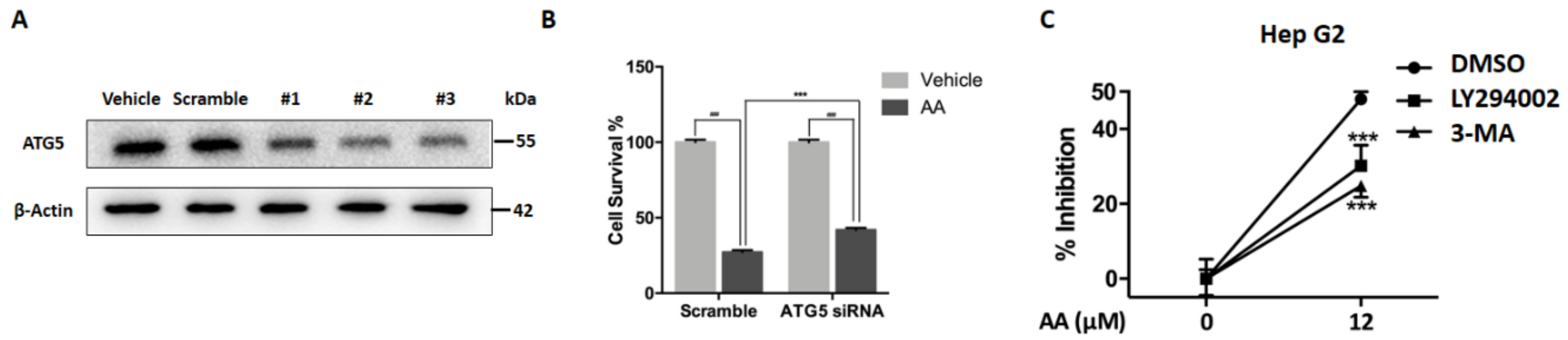
2.3. Impaired Autophagic Flux Contributes to AA Induced HepG2 Cell Death
3. Materials and Methods
3.1. Reagents and Antibodies
3.2. Cell Culture
3.3. Cell Viability Assay
3.4. Mitochondrial Membrane Potential Detection
3.5. Annexin V/Propidium Iodide Apoptosis Assay
3.6. Transmission Electron Microscopy (TEM)
3.7. LC3 Staining
3.8. Lysotracker Staining
3.9. Transfection
3.10. Western Blotting
4. Discussion
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Boonyarathanakornkit, L.; Che, C.-T.; Fong, H.H.; Farnsworth, N.R. Constituents of croton crassifolius roots. Planta Med. 1988, 54, 61–63. [Google Scholar] [CrossRef] [PubMed]
- Flora of China Editorial Committee. Flora of China; Institutum Academiae Science Press: Beijing, China, 2004; Volume 40, pp. 135–137. [Google Scholar]
- Huang, W.; Wang, J.; Liang, Y.; Ge, W.; Wang, G.; Li, Y.; Chung, H.Y. Potent anti-angiogenic component in croton crassifolius and its mechanism of action. J. Ethnopharmacol. 2015, 175, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.-L.; Yao, G.-D.; Wang, Y.-X.; Gao, P.-Y.; Wang, D.; Li, L.-Z.; Lin, B.; Huang, X.-X.; Song, S.-J. Cytotoxic clerodane diterpenoids from croton crassifolius. Bioorg. Med. Chem. Lett. 2017, 27, 1237–1242. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of autophagy in cancer. Nat. Rev. Cancer 2007, 7, 961. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.; Karp, C.M.; Beaudoin, B.; Vuong, N.; Chen, G.; Chen, H.-Y.; Bray, K.; Reddy, A.; Bhanot, G.; Gelinas, C. Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009, 137, 1062–1075. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Rodriguez, A.; Mayoral, R.; Agra, N.; Valdecantos, M.; Pardo, V.; Miquilena-Colina, M.; Vargas-Castrillón, J.; Iacono, O.L.; Corazzari, M.; Fimia, G. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of nafld. Cell Death Dis. 2014, 5, e1179. [Google Scholar] [CrossRef] [PubMed]
- Mauvezin, C.; Neufeld, T.P. Bafilomycin a1 disrupts autophagic flux by inhibiting both v-atpase-dependent acidification and ca-p60a/serca-dependent autophagosome-lysosome fusion. Autophagy 2015, 11, 1437–1438. [Google Scholar] [CrossRef] [PubMed]
- Chude, C.I.; Amaravadi, R.K. Targeting autophagy in cancer: Update on clinical trials and novel inhibitors. Int. J. Mol. Sci. 2017, 18, 1279. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Hu, S.-E.; Tan, S.-H.; Cao, R.; Chen, Y.; Xia, D.; Zhu, X.; Yang, X.-F.; Ong, C.-N.; Shen, H.-M. Andrographolide sensitizes cisplatin-induced apoptosis via suppression of autophagosome-lysosome fusion in human cancer cells. Autophagy 2012, 8, 338–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- N’Diaye, E.N.; Kajihara, K.K.; Hsieh, I.; Morisaki, H.; Debnath, J.; Brown, E.J. Plic proteins or ubiquilins regulate autophagy-dependent cell survival during nutrient starvation. EMBO Rep. 2009, 10, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Yoshida, T.; Tsujioka, M.; Arakawa, S. Autophagic cell death and cancer. Int. J. Mol. Sci. 2014, 15, 3145–3153. [Google Scholar] [CrossRef] [PubMed]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Button, R.W.; Luo, S. The formation of autophagosomes during lysosomal defect: A new source of cytotoxicity. Autophagy 2017, 13, 1797–1798. [Google Scholar] [CrossRef] [PubMed]
- Lamark, T.; Kirkin, V.; Dikic, I.; Johansen, T. Nbr1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle 2009, 8, 1986–1990. [Google Scholar] [CrossRef] [PubMed]
- Zientara-Rytter, K.; Subramani, S. Autophagic degradation of peroxisomes in mammals. Biochem. Soc. Trans. 2016, 44, 431–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Sample Availability: Samples of the compounds Aleuritolic acid are available from the authors. |





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yi, H.; Wang, K.; Du, B.; He, L.; HO, H.; Qiu, M.; Zou, Y.; Li, Q.; Jin, J.; Zhan, Y.; et al. Aleuritolic Acid Impaired Autophagic Flux and Induced Apoptosis in Hepatocellular Carcinoma HepG2 Cells. Molecules 2018, 23, 1338. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23061338
Yi H, Wang K, Du B, He L, HO H, Qiu M, Zou Y, Li Q, Jin J, Zhan Y, et al. Aleuritolic Acid Impaired Autophagic Flux and Induced Apoptosis in Hepatocellular Carcinoma HepG2 Cells. Molecules. 2018; 23(6):1338. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23061338
Chicago/Turabian StyleYi, Hua, Kun Wang, Biaoyan Du, Lina He, Hiuting HO, Maosong Qiu, Yidan Zou, Qiao Li, Junfeng Jin, Yujuan Zhan, and et al. 2018. "Aleuritolic Acid Impaired Autophagic Flux and Induced Apoptosis in Hepatocellular Carcinoma HepG2 Cells" Molecules 23, no. 6: 1338. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules23061338