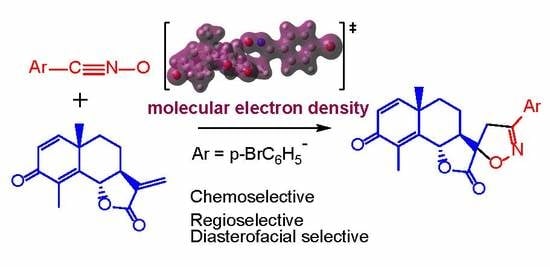
A Molecular Electron Density Theory Study of the Chemoselectivity, Regioselectivity, and Diastereofacial Selectivity in the Synthesis of an Anticancer Spiroisoxazoline derived from α-Santonin



Abstract
:
1. Introduction
2. Computational Methods
3. Results and Discussion
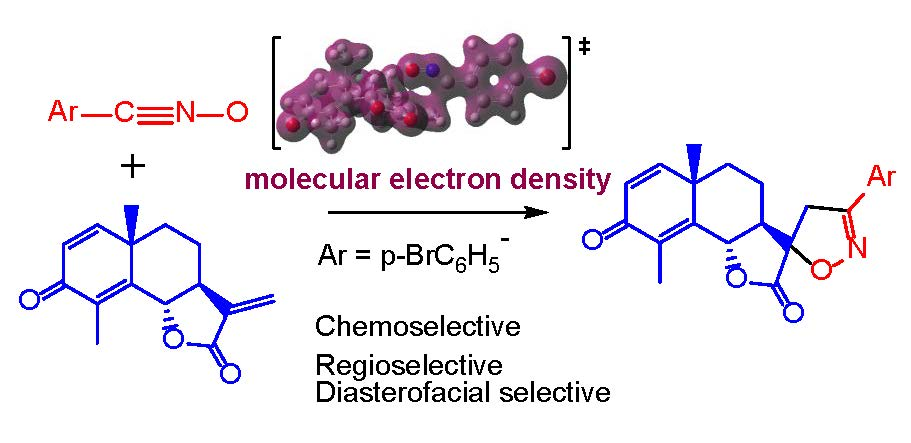
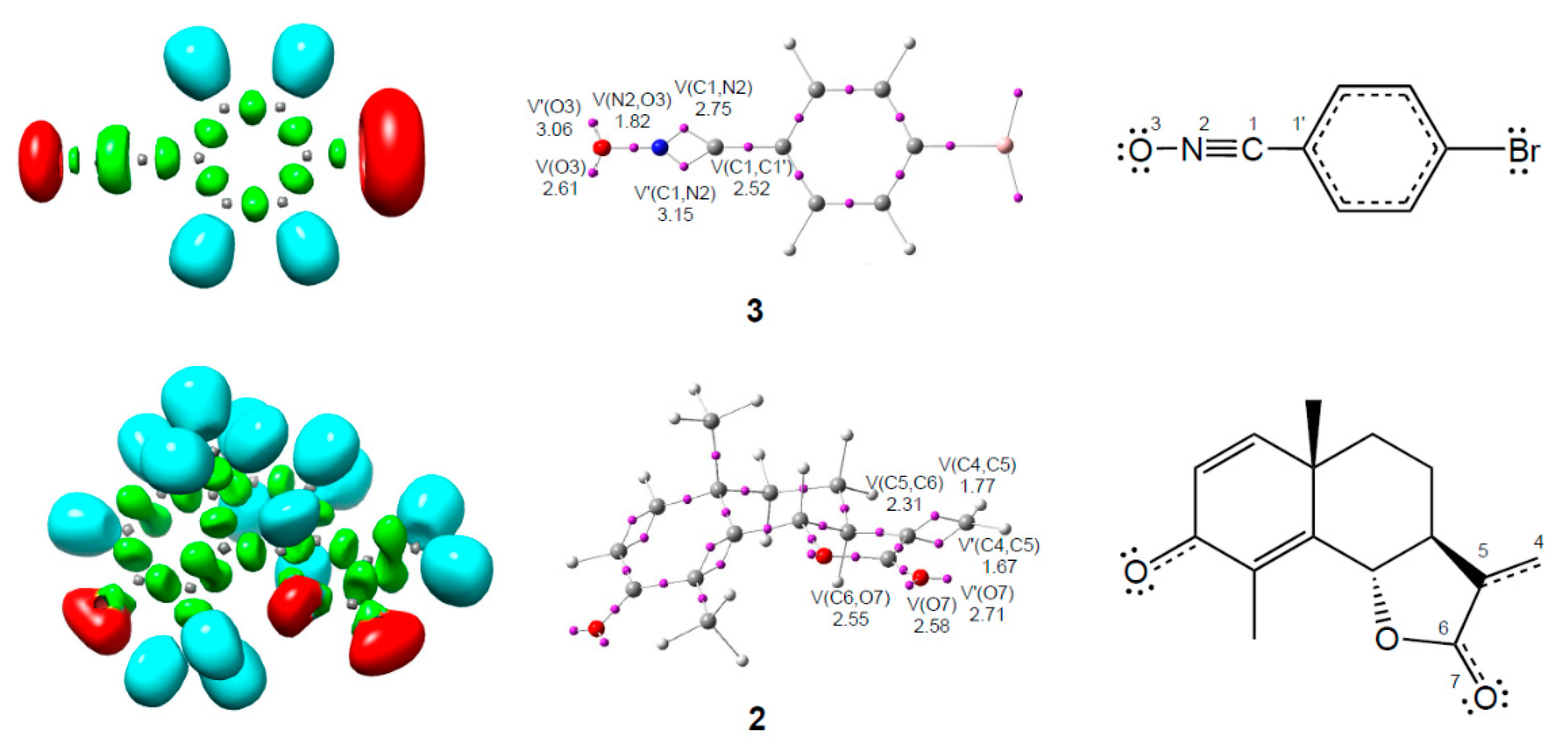
3.1. ELF Topological Analysis of α-Santonin Derivative 2 and p-Bromophenyl Nitrile Oxide 3
3.2. Analysis of the CDFT Reactivity Indices of the Reagents
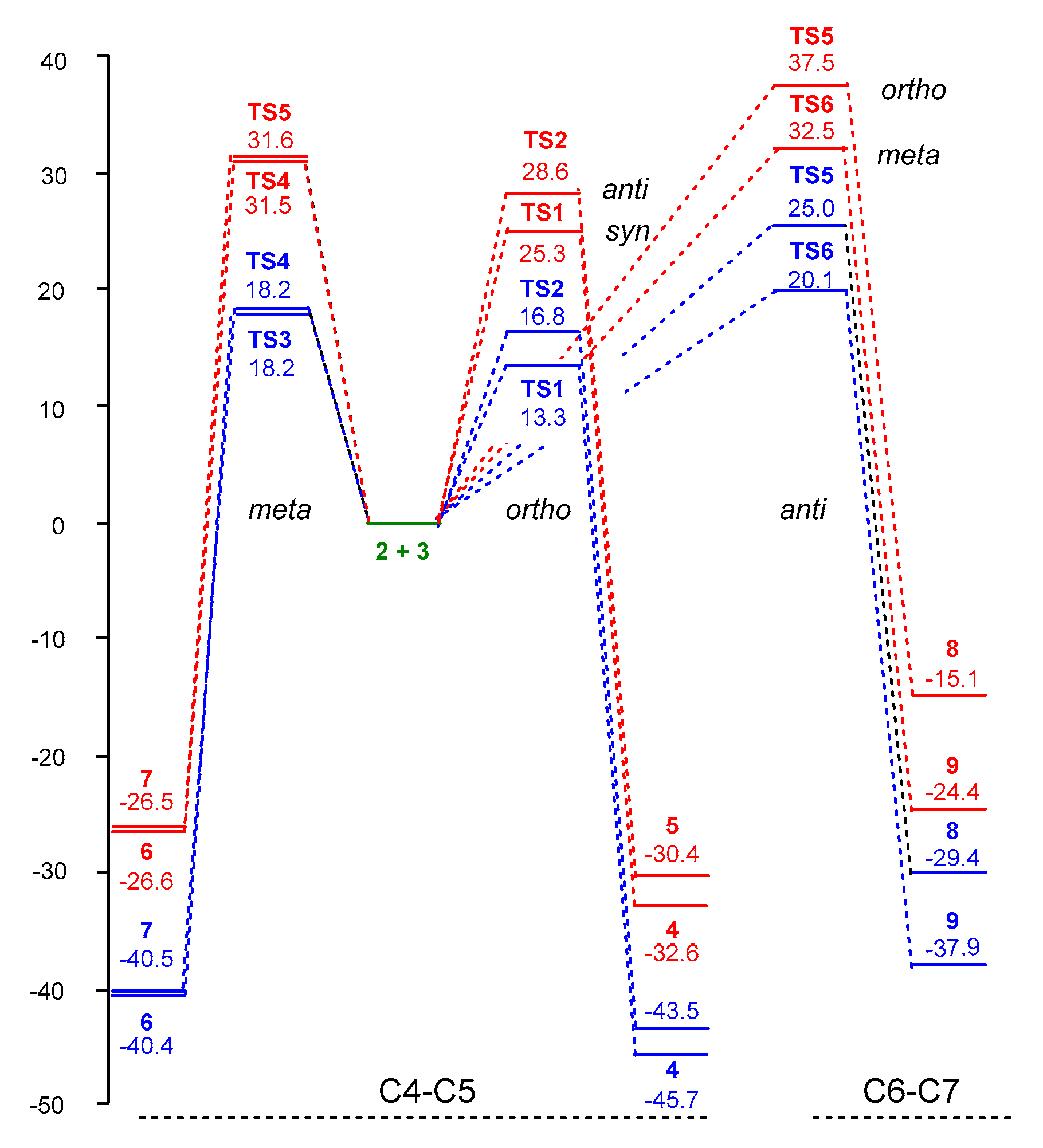
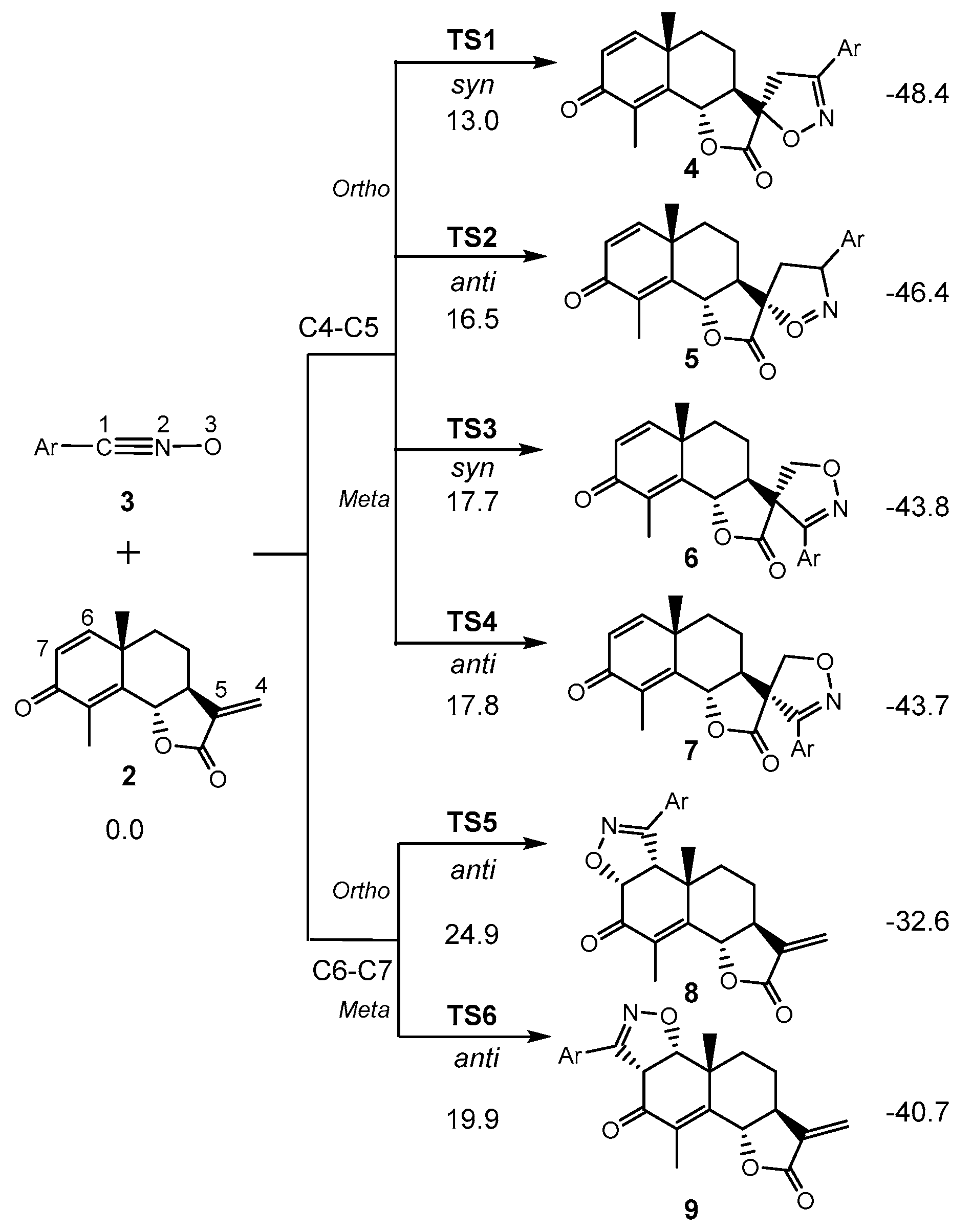
3.3. Study of the Energy Profile Associated with the 32CA Reaction between α-Santonin Derivative 2 and p-Bromophenyl Nitrile Oxide 3
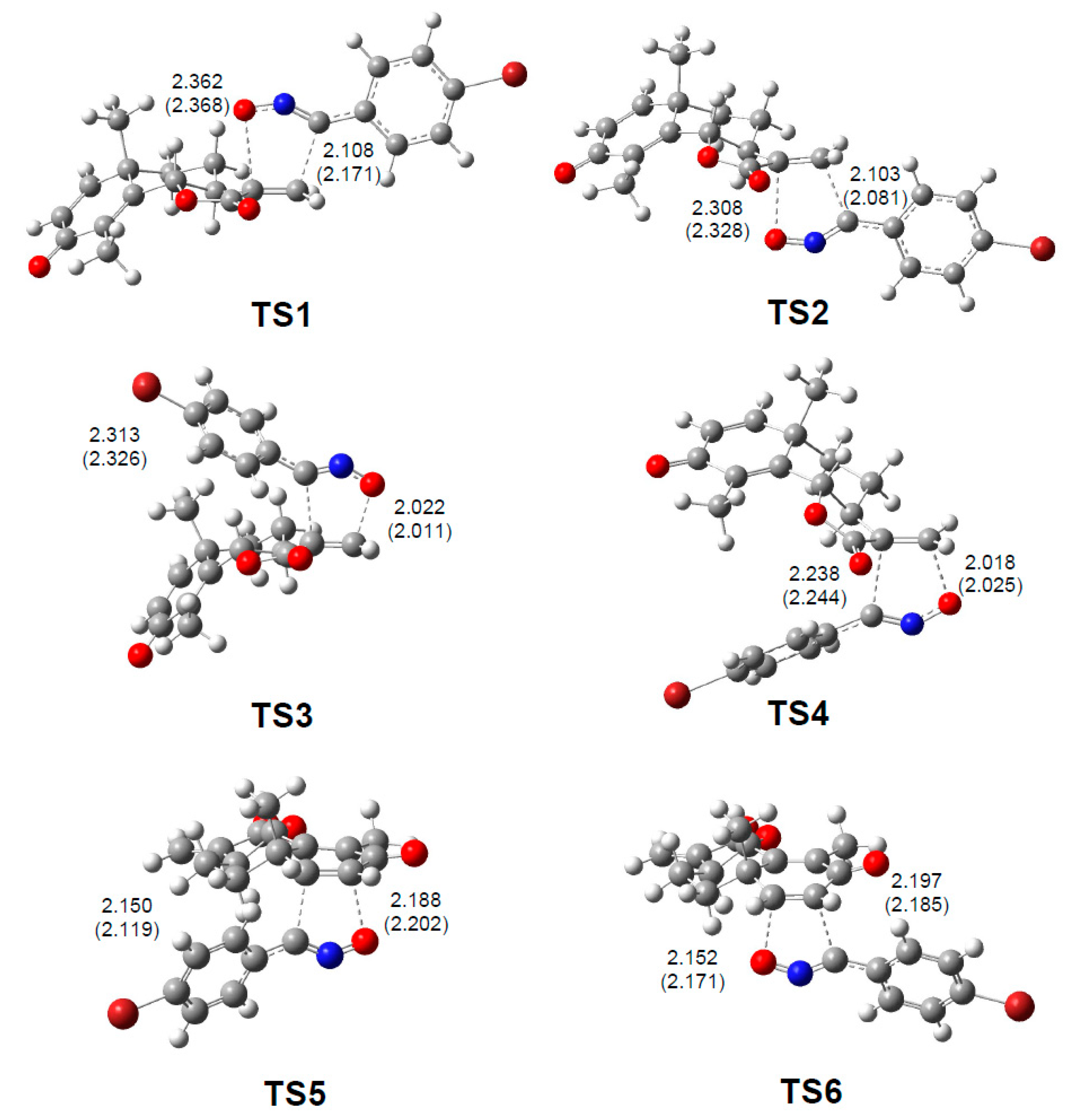
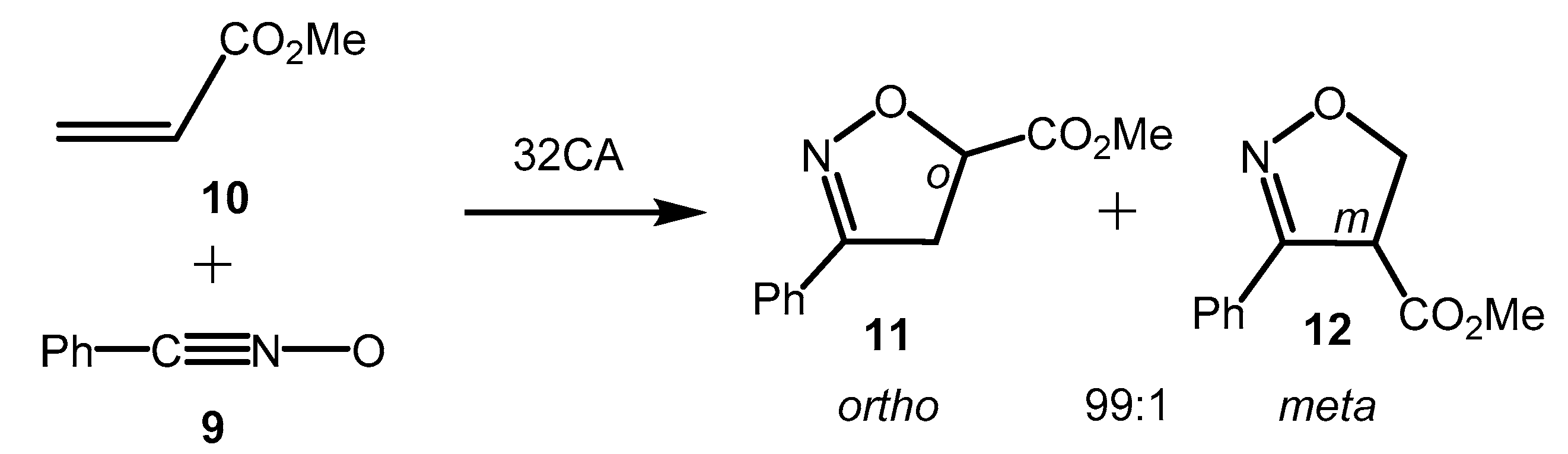
3.4. Origin of the Ortho Regioselectivity along the 32CA Reaction of α-Santonin Derivative 2 with p-Bromophenyl Nitrile Oxide 3
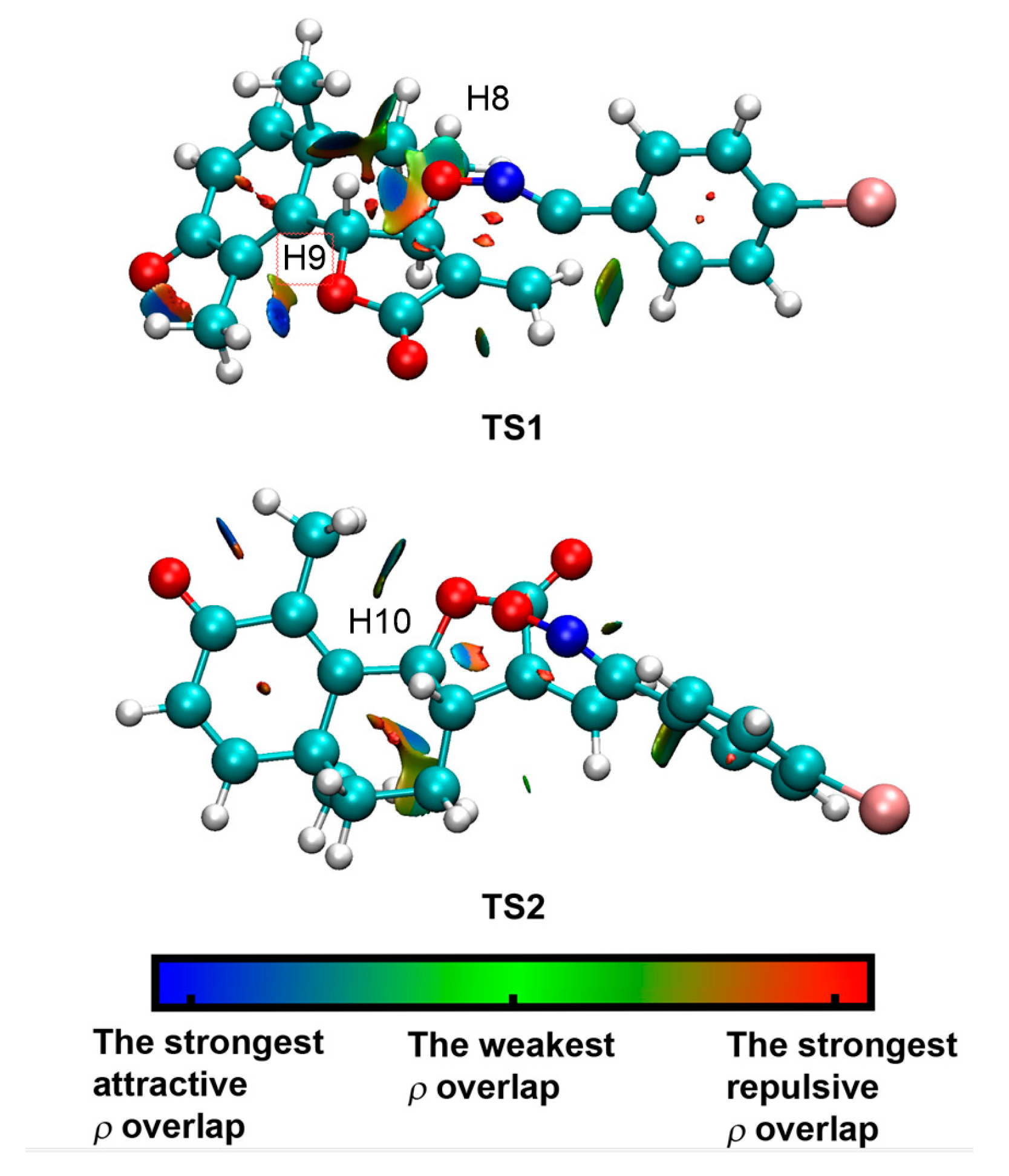
3.5. Origin of the syn Diastereofacial Selectivity along the 32CA Reaction of α-Santonin Derivative 2 with p-Bromophenyl Nitrile Oxide 3
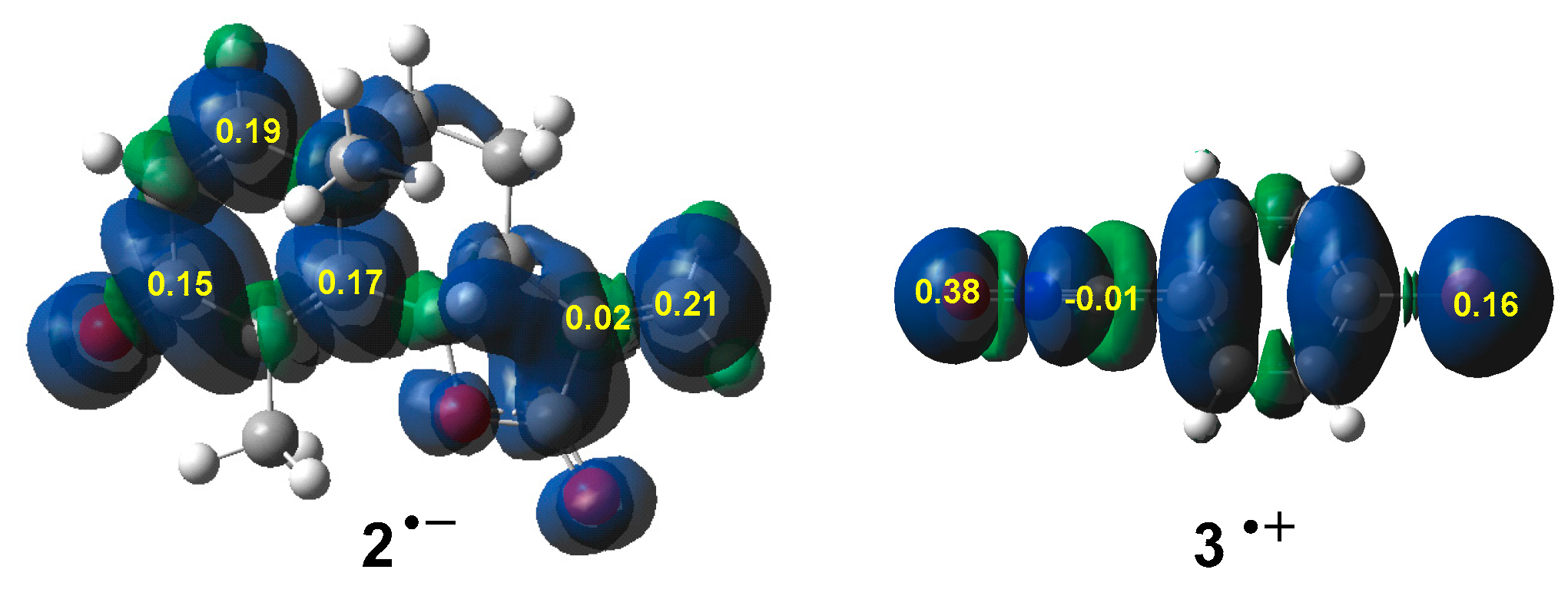
3.6. ELF Topological Analysis of the C–C and C–O Bond Formation along the 32CA Reaction of α-Santonin Derivative 2 with p-Bromophenyl Nitrile Oxide 3
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Borad, M.A.; Bhoi, M.N.; Prajapati, N.P.; Patel, H.D. Review of synthesis of spiro heterocyclic compounds from isatin. Synth. Commun. 2014, 44, 897–922. [Google Scholar] [CrossRef]
- Molvi, I.K.; Haque, N.; Awen, B.Z.S.; Zameeruddin, M. Synthesis of Spiro Compounds as Medicinal Agents: New Opportunities for Drug Design and Discovery. Part I: A Review. Word J. Pharm. Pharm. Sci. 2014, 3, 536–563. [Google Scholar] [CrossRef]
- Goyard, D.; Kónya, B.; Chajistamatiou, A.S.; Chrysina, E.D.; Leroy, J.; Balzarin, S.; Tournier, M.; Tousch, D.; Petit, P.; Duret, C.; et al. Glucose-derived spiro-isoxazolines are anti-hyperglycemic agents against type 2 diabetes through glycogen phosphorylase inhibition. Eur. J. Med. Chem. 2016, 108, 444–454. [Google Scholar] [CrossRef] [PubMed]
- Maiuolo, L.; Merino, P.; Algieri, V.; Nardi, M.; Di Gioia, M.L.; Russo, B.; Delso, I.M.; Tallarida, A.; De Nino, A. Nitrones and nucleobase-containing spiro-isoxazolidines derived from isatin and indanone: Solvent-free microwave-assisted stereoselective synthesis and theoretical calculations. RSC Adv. 2017, 7, 48980–48988. [Google Scholar] [CrossRef]
- Das, P.; Omollo, A.O.; Sitole, L.J.; McClendon, E.; Valente, E.J.; Raucher, D.; Walker, L.R.; Hamme, A.T., II. Synthesis and investigation of novel spiro-isoxazolines as anti-cancer agents. Tetrahedron Lett. 2015, 56, 1794–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, D.M.; Qazi, N.A.; Sawant, S.D.; Bandey, A.H.; Srinivas, J.; Shankar, M.; Singh, S.; Verma, K.M.; Chashoo, G.; Saxena, A.; et al. Design and synthesis of spiro derivatives of parthenin as novel anti-cancer agents. Eur. J. Med. Chem. 2011, 46, 3210–3217. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Song, S.; Shu, S.; Miao, Z.; Zhang, A.; Ding, C. Novel spirobicyclic artemisinin analogues (artemalogues): Synthesis and antitumor activities. Eur. J. Med. Chem. 2015, 103, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Zaki, M.; Oukhrib, A.; Akssira, M.; Berteina-Raboin, S. Synthesis of novel spiro-isoxazoline and spiro-isoxazolidine derivatives of tomentosin. RSC Adv. 2017, 7, 6523–6529. [Google Scholar] [CrossRef] [Green Version]
- Merck Index, 11th ed.; Merck and Co.: Rahway, NJ, USA, 1989; p. 1327.
- Khazir, J.; Singh, P.P.; Reddy, D.M.; Hyder, I.; Shafi, S.; Sawant, S.D.; Chashoo, G.; Mahajan, A.; Alam, M.S.; Saxena, A.K. Synthesis and anticancer activity of novel spiro-isoxazoline and spiro-isoxazolidine derivatives of α-santonin. Eur. J. Med. Chem. 2013, 63, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R. Molecular electron density theory: A modern view of reactivity in organic chemistry. Molecules 2016, 21, 1319. [Google Scholar] [CrossRef] [PubMed]
- Ríos-Gutiérrez, M.; Domingo, L.R. Unravelling the mysteries of the [3 + 2] cycloaddition reactions. Eur. J. Org. Chem. 2019, 267–282. [Google Scholar] [CrossRef]
- Ndassa, I.M.; Adjieufack, A.I.; Ketcha, J.M.; Berski, S.; Ríos-Gutiérrez, M.; Domingo, L.R. Understanding the reactivity and regioselectivity of [3 + 2] cycloaddition reactions between substituted nitrile oxides and methyl acrylate. A molecular electron density theory study. Int. J. Quantum Chem. 2017, 117, 25451. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. Hybrid meta density functional theory methods for thermochemistry, thermochemical kinetics, and noncovalent Interactions: The MPW1B95 and MPWB1K models and comparative assessments for hydrogen bonding and van der Waals interactions. J. Phys. Chem. A 2004, 108, 6908–6918. [Google Scholar] [CrossRef]
- Hehre, M.J.; Radom, L.; Schleyer, P.v.R.; Pople, J. Ab initio Molecular Orbital Theory; Wiley: New York, NY, USA, 1986. [Google Scholar]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. A Molecular electron density theory study of the reactivity and selectivities in [3 + 2] cycloaddition reactions of C,N-dialkyl nitrones with ethylene derivatives. J. Org. Chem. 2018, 83, 2182–2197. [Google Scholar] [CrossRef] [PubMed]
- Schlegel, H.B. Optimization of equilibrium geometries and transition structures. J. Comput. Chem. 1982, 3, 214–218. [Google Scholar] [CrossRef]
- Schlegel, H.B. Modern Electronic Structure Theory; Yarkony, D.R., Ed.; World Scientific Publishing: Singapore, 1994. [Google Scholar]
- Fukui, K. Formulation of the reaction coordinate. J. Phys. Chem. 1970, 74, 4161–4163. [Google Scholar] [CrossRef]
- González, C.; Schlegel, H.B. Reaction path following in mass-weighted internal coordinates. J. Phys. Chem. 1990, 94, 5523–5527. [Google Scholar] [CrossRef]
- González, C.; Schlegel, H.B. Improved algorithms for reaction path following: Higher-order implicit algorithms. J. Chem. Phys. 1991, 95, 5853–5860. [Google Scholar] [CrossRef]
- Tomasi, J.; Persico, M. Molecular interactions in solution: And overview of methods based on continuous distributions of the solvent. Chem. Rev. 1994, 94, 2027–2094. [Google Scholar] [CrossRef]
- Simkin, B.I.A.; Sheikhet, I.I. Quantum chemical and statistical theory of solutions—Computational approach; Ellis Horwood: London, UK, 1995. [Google Scholar]
- Cossi, M.; Barone, V.; Cammi, R.; Tomasi, J. Ab initio study of solvated molecules: A new implementation of the polarizable continuum model. Chem. Phys. Lett. 1996, 255, 327–335. [Google Scholar] [CrossRef]
- Cances, E.; Mennucci, B.; Tomasi, J. A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to isotropic and anisotropic dielectrics. J. Chem. Phys. 1997, 107, 3032–3041. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M.; Tomasi, J. Geometry optimization of molecular structures in solution by the polarizable continuum model. J. Comput. Chem. 1998, 19, 404–417. [Google Scholar] [CrossRef]
- Domingo, L.R. A new C-C bond formation model based on the quantum chemical topology of electron density. RSC Adv. 2014, 4, 32415–32428. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Geerlings, P.; De Proft, F.; Langenaeker, W. Conceptual density functional theory. Chem. Rev. 2003, 103, 1793–1873. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the conceptual density functional indices to organic chemistry reactivity. Molecules 2016, 21, 748. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R.; Pérez, P.; Sáez, J.A. Understanding the local reactivity in polar organic reactions through electrophilic and nucleophilic Parr functions. RSC Adv. 2013, 3, 1486–1494. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular-systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Noury, S.; Krokidis, X.; Fuster, F.; Silvi, B. Computational tools for the electron localization function topological analysis. Comput. Chem. 1999, 23, 597–604. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, J.; Yang, A.W. Revealing noncovalent interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [PubMed]
- Contreras-Garcia, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D.N.; Yang, W. NCIPLOT: A program for plotting noncovalent interaction regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Blanco, M.A.; Martín Pendás, A.; Francisco, E. Interacting Quantum Atoms: A Correlated Energy Decomposition Scheme Based on the Quantum Theory of Atoms in Molecules. J. Chem. Theory Comput. 2005, 1, 1096–1109. [Google Scholar] [CrossRef] [PubMed]
- Martín Pendás, A.; Francisco, E. Promolden: A QTAIM/IQA code, (available from the authors upon request by writing to [email protected]).
- Lewis, G.N. Valence and the Structure of Atoms and Molecules; Chemical Catalog Co.: New York, NY, USA, 1923. [Google Scholar]
- Parr, R.G.; Yang, W. Density Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Parr, R.G.; Szentpaly, L.v.; Liu, S. Electrophilicity index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Domingo, L.R.; Chamorro, E.; Pérez, P. Understanding the reactivity of captodative ethylenes in polar cycloaddition reactions. A theoretical study. J. Org. Chem. 2008, 73, 4615–4624. [Google Scholar] [CrossRef] [PubMed]
- Aurell, M.J.; Domingo, L.R.; Pérez, P.; Contreras, R. A theoretical study on the regioselectivity of 1,3-dipolar cycloadditions using DFT-based reactivity indexes. Tetrahedron 2004, 60, 11503–11509. [Google Scholar] [CrossRef]
- Khorief Nacereddine, A.; Djerourou, A.; Ríos-Gutiérrez, M.; Domingo, L.R. Non-classical CH…O hydrogen-bond determining the regio- and stereoselectivity in the [3 + 2] cycloaddition reaction of (Z) C-phenyl-N-methylnitrone with dimethyl 2-benzylidenecyclopropane-1,1-dicarboxylate. A topological electron-density study. RSC Adv. 2015, 5, 99299–99311. [Google Scholar] [CrossRef]
- Adjieufack, A.I.; Ndassa, I.M.; Ketcha Mbadcam, J.; Ríos-Gutiérrez, M.; Domingo, L.R. Steric interactions controlling the syn diastereofacial selectivity in the [3 + 2] cycloaddition reaction between acetonitrile axide and 7-oxanorborn-5-en-2-ones. A Molecular Electron Density Theory study. J. Phys. Org. Chem. 2017, 30, 3710. [Google Scholar] [CrossRef]
- Suárez, D.; Díaz, N.; Francisco, E.; Martín Pendás, A. Application of the interacting quantum atoms approach to the S66 and ionic-hydrogen-bond datasets for noncovalent Interactions. ChemPhysChem 2018, 19, 973–987. [Google Scholar] [CrossRef] [PubMed]
- Martin Pendás, A.; Casals-Sainz, J.L.; Francisco, E. On Electrostatics, Covalency, and Chemical Dashes: Physical Interactions versus Chemical Bonds. Chem. Eur. J. 2019, 25, 309–314. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
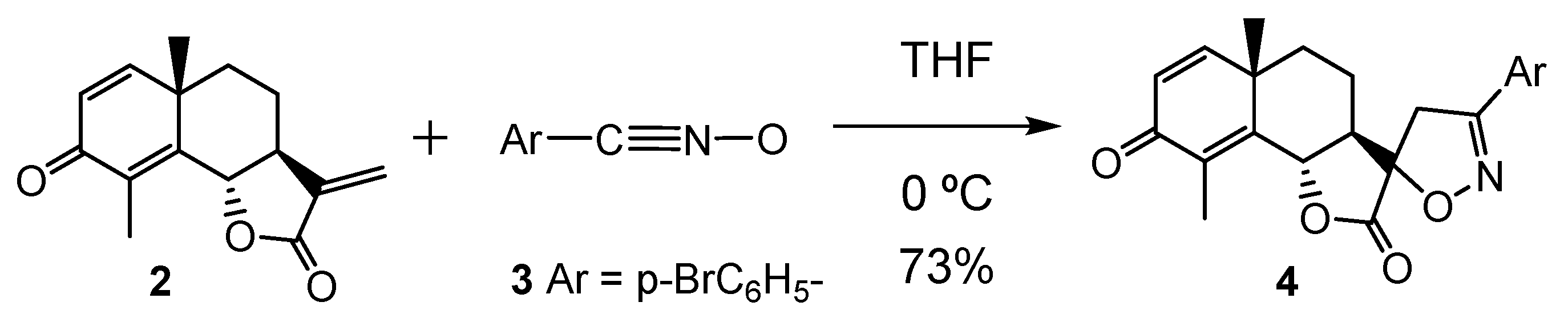{kind=link}
{kind=link}
{kind=link}
| μ | η | ω | N | |
|---|---|---|---|---|
| α-santonin derivative 2 | −4.16 | 4.90 | 1.76 | 2.51 |
| p-bromophenyl nitrile oxide 3 | −4.03 | 4.81 | 1.69 | 2.69 |
| 3-methylene lactone 16 | −4.29 | 5.99 | 1.54 | 1.84 |
| simplest nitrile oxide 13 | −3.40 | 7.94 | 0.73 | 1.75 |
| Eint | Ecl | Exc | |
|---|---|---|---|
| O3–H8 | −4.4 | −1.6 | −2.8 |
| O3–H9 | −8.1 | −3.1 | −5.0 |
| O3–H10 | −5.7 | −2.0 | −3.6 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Domingo, L.R.; Ríos-Gutiérrez, M.; Acharjee, N. A Molecular Electron Density Theory Study of the Chemoselectivity, Regioselectivity, and Diastereofacial Selectivity in the Synthesis of an Anticancer Spiroisoxazoline derived from α-Santonin. Molecules 2019, 24, 832. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24050832
Domingo LR, Ríos-Gutiérrez M, Acharjee N. A Molecular Electron Density Theory Study of the Chemoselectivity, Regioselectivity, and Diastereofacial Selectivity in the Synthesis of an Anticancer Spiroisoxazoline derived from α-Santonin. Molecules. 2019; 24(5):832. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24050832
Chicago/Turabian StyleDomingo, Luis R., Mar Ríos-Gutiérrez, and Nivedita Acharjee. 2019. "A Molecular Electron Density Theory Study of the Chemoselectivity, Regioselectivity, and Diastereofacial Selectivity in the Synthesis of an Anticancer Spiroisoxazoline derived from α-Santonin" Molecules 24, no. 5: 832. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24050832