
DHFR Inhibitors: Reading the Past for Discovering Novel Anticancer Agents

 , , ,
, , , 
Abstract
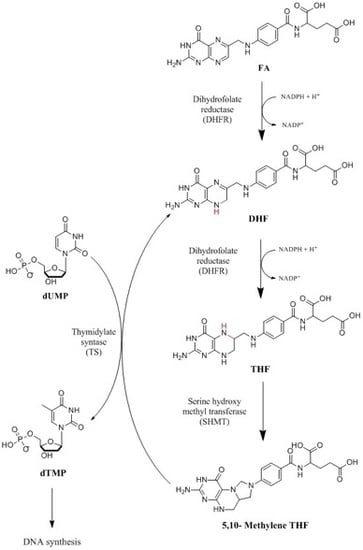
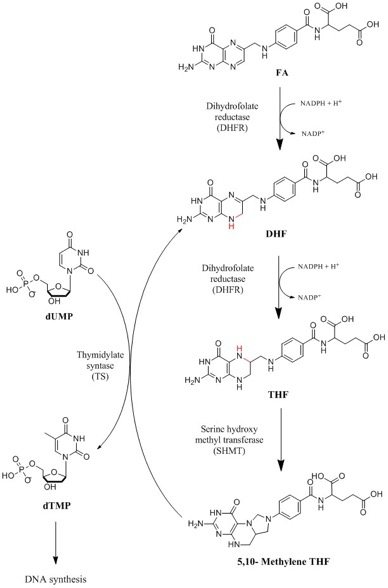
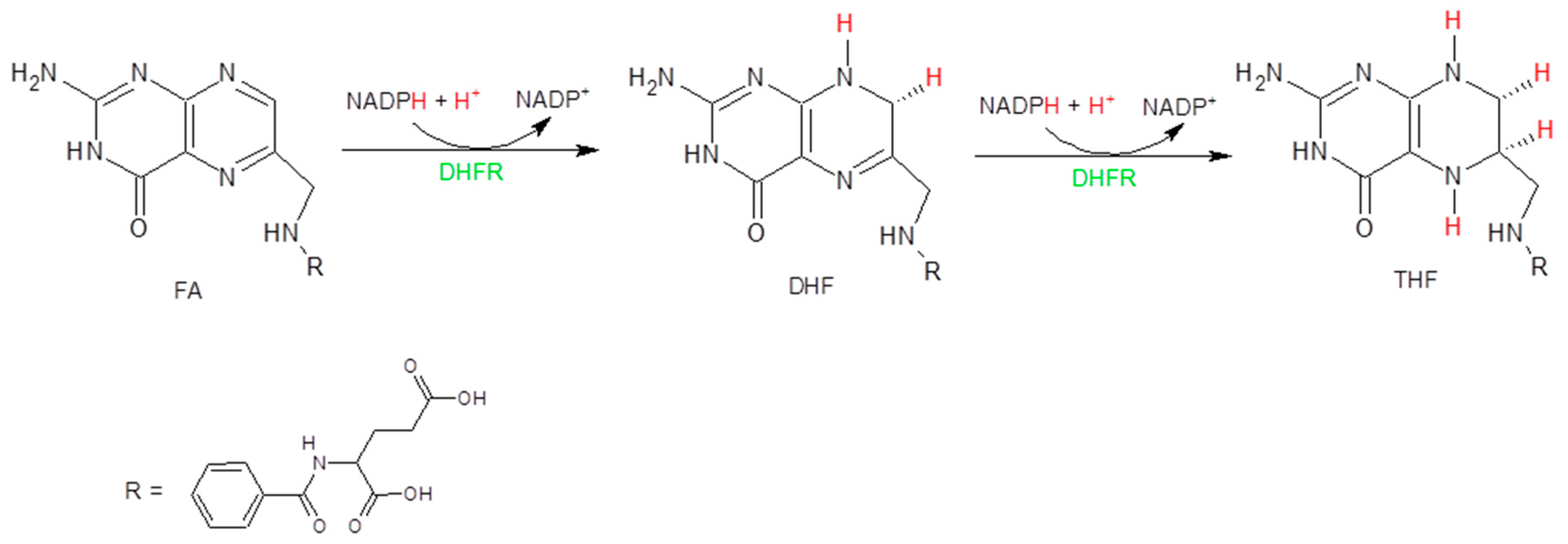
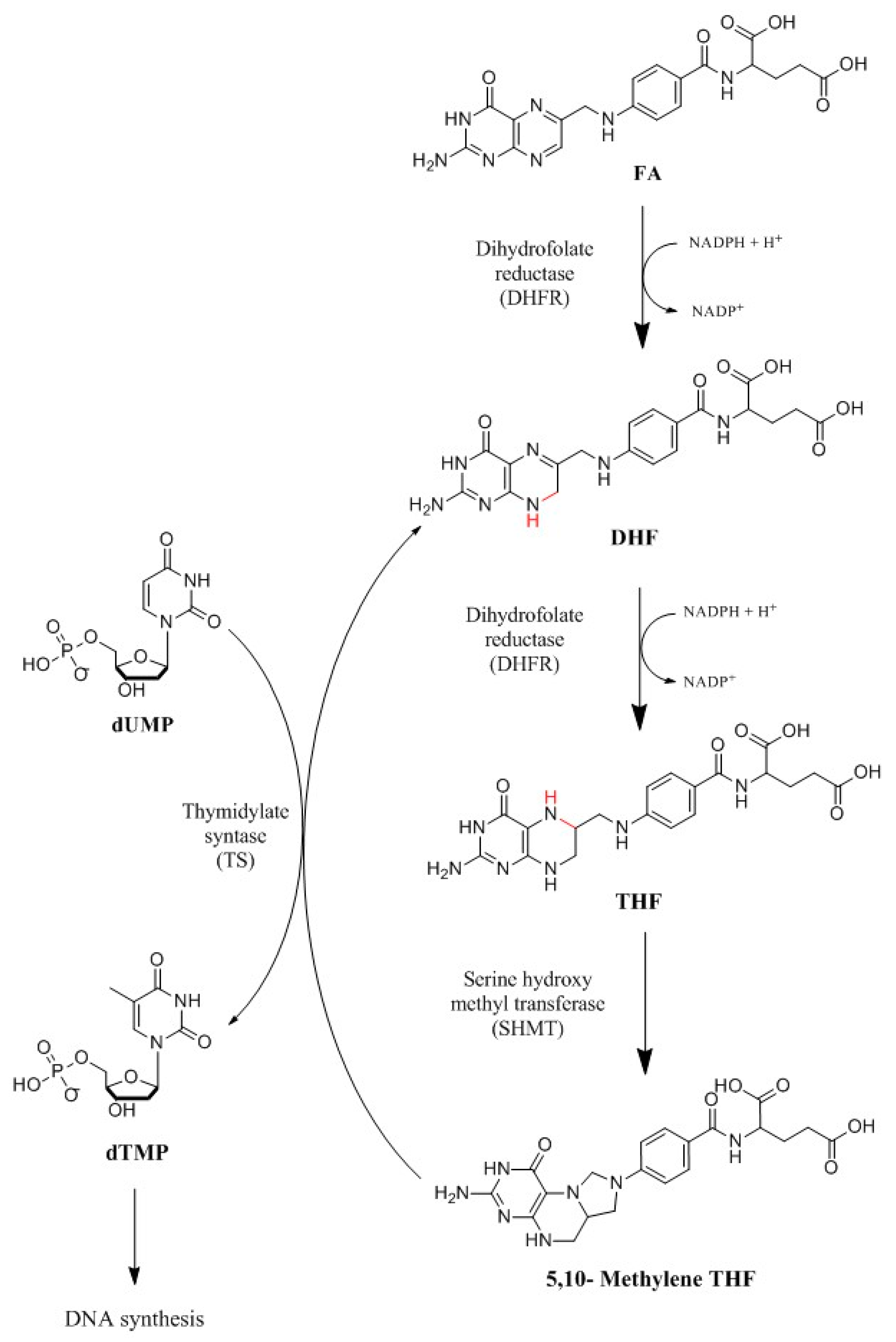
:
1. Introduction
2. Physiological Role and Structure of DHFR
3. Relevance of DHFR Inhibitors in Cancer Therapy
4. Inhibitors of Bovine DHFR
5. Inhibitors of Human DHFR under Preclinical Investigation
6. New Strategies in DHFR Drug Discovery
7. Conclusions
Funding
Conflicts of Interest
Abbreviations
| FA | Folic acid |
| THF | tetrahydrofolate |
| DHFR | dihydrofolate reductase enzyme |
| hDHFR | human DHFR |
| bDHFR | bovine DHFR |
| rhDHFR | recombinant human DHFR |
| hTS | human thymidylate synthase |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| dUMP | dihydrofolate synthase (DHFS), deoxyuridine monophosphate |
| dTMP | deoxythymidine monophosphate |
| 5,10-Methylene THF | N5-N10-methylenetetrahydrofolate |
| TrxR | thioredoxin reductases |
| FRs | folate receptors |
| ct-DNA | calf thymus-DNA |
| RNR | ribonucleotide reductase |
| TS | thymidylate synthase |
| TP | thymidylated phosphorylase |
| MTX | methotrexate |
| ANN | artificial neural network |
| PDB | Protein Data Bank |
| Asp127 | Aspartate 127 |
| Glu30 | Glutamate 30 |
| Phe31 | Phenylalanine |
| ELISA assay | enzyme-linked immunosorbent assay |
| FDA | U.S. Food and Drug Administration |
| EMA | European Medicines Agency |
| ADMET | absorption, distribution, metabolism, excretion, toxicity |
| OVCAR-3 | ovarian cancer cell line |
| MDA-MB-435 | melanoma cell line |
| NSCLC | non-small-cell lung carcinoma |
| HCC | hepatocellular carcinoma |
| A549 | adenocarcinomic human alveolar basal epithelial cells |
| SK-OV-3 | human ovary cancer cell line |
| HCT15; HCT116 | human colon adenocarcinoma colorectal adenocarcinoma |
| K562 | human chronic myeloid leukemia cell |
| HeLa | human of immortal cervical cancer cell |
| KB | keratin-forming tumor cell line HeLa |
| MCF-7; MDA-MB-231; MDA-MB-468 | human breast cancer cell line |
| DU145 | human prostate cancer cell line |
| HMEC | normal human mammary epithelial cells |
| HL-60 | human caucasian promyelocytic leukemia cell line |
| NCI-H1299; NCI-H522 | human non-small cell lung carcinoma cell line |
| HepG2 | human liver cancer cell line |
| SK-n-SH | human neuroblastoma |
| A17 | amacrine cells in mammalian retina |
| SAR | structure activity relationships |
References
- Schweitzer, B.I.; Dicker, A.P.; Bertino, J.R. Dihydrofolate reductase as a therapeutic target. FASEB J. 1990, 4, 2441–2452. [Google Scholar] [CrossRef]
- Bailey, L.B. Folate in Health and Disease, 2nd ed.; CRC Press: Boca Raton, FL, USA; Taylor & Francis Group: Abingdon, UK, 2017; ISBN 9781138111882. Available online: https://www.crcpress.com/Folate-in-Health-and-Disease/Bailey/p/book/9781138111882 (accessed on 20 January 2019).
- Srinivasan, B.; Tonddast-Navaei, S.; Roy, A.; Zhou, H.; Skolnick, J. Chemical Space of Escherichia coli Dihydrofolate Reductase Inhibitors: New Approaches for Discovering Novel Drugs for Old Bugs. Med. Res. Rev. 2019, 39, 684–705. [Google Scholar] [CrossRef]
- Baird, J.K. Effectiveness of Antimalarial Drugs. N. Engl. J. Med. 2005, 352, 1565–1577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fidock, D.A.; Rosenthal, P.J.; Croft, S.L.; Brun, R.; Nwaka, S. Antimalarial drug discovery: efficacy models for compound screening. Nat. Rev. Drug Discov. 2004, 3, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Alsaad, N.; van Altena, R.; Pranger, A.D.; van Soolingen, D.; de Lange, W.C.M.; van der Werf, T.S.; Kosterink, J.G.W.; Alffenaar, J.-W.C. Evaluation of co-trimoxazole in the treatment of multidrug-resistant tuberculosis. Eur. Respir. J. 2013, 42, 504–512. [Google Scholar] [CrossRef]
- Boeree, M.J.; Sauvageot, D.; Banda, H.T.; Harries, A.D.; Zijlstra, E.E. Efficacy and safety of two dosages of cotrimoxazole as preventive treatment for HIV-infected Malawian adults with new smear-positive tuberculosis. Trop. Med. Int. Health. 2005, 10, 723–733. [Google Scholar] [CrossRef] [Green Version]
- Huennekens, F.M. The methotrexate story: A paradigm for development of cancer chemotherapeutic agents. Adv. Enzyme Regul. 1994, 34, 397–419. [Google Scholar] [CrossRef]
- Takimoto, C.H.; Allegra, C.J. New antifolates in clinical development. Oncology 1995, 9, 649–656, 659 DISC 660, 662, 665. Available online: https://www.cancernetwork.com/oncology-journal/new-antifolates-clinical-development (accessed on 20 January 2019). [PubMed]
- Banerjee, R.; Dey, M.; Maity, S.; Bagchi, S.; Vora, A.; Shakil, U.; Goswami, R. Preventive role of Curcumin against hepatotoxic effects of Methotrexate and Cyclophosphamide. J. Chem. Pharm. Sci. 2016, 4, 38–42. Available online: https://www.jchps.com/specialissues/2016%20Special%20Issue%204/0870916.pdf (accessed on 20 January 2019).
- Marar, T.; Singh, K.; Bhori, M.; Dhanesha, M. Impact of antioxidant supplementation on toxicity of methotrexate-an in vitro study on erythrocytes using vitamin E. Asian J. Pharm. Clin. Res. 2015, 8, 339–343. Available online: https://innovareacademics.in/journals/index.php/ajpcr/article/view/5828 (accessed on 20 January 2019).
- Padmanabhan, S.; Tripathi, D.N.; Vikram, A.; Ramarao, P.; Jena, G.B. Methotrexate-induced cytotoxicity and genotoxicity in germ cells of mice: intervention of folic and folinic acid. Mutat. Res. 2009, 673, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Galassi, R.; Oumarou, C.S.; Burini, A.; Dolmella, A.; Micozzi, D.; Vincenzetti, S.; Pucciarelli, S. A study on the inhibition of dihydrofolate reductase (DHFR) from Escherichia coli by gold(i) phosphane compounds. X-ray crystal structures of (4,5-dichloro-1H-imidazolate-1-yl)-triphenylphosphane-gold(i) and (4,5-dicyano-1H-imidazolate-1-yl)-triphenylphosphane-gold(i). Dalton Trans. 2015, 44, 3043–3056. [Google Scholar] [CrossRef]
- Hao, M.; Zhao, W.; Zhang, L.; Wang, H.; Yang, X. Low folate levels are associated with methylation-mediated transcriptional repression of miR-203 and miR-375 during cervical carcinogenesis. Oncol. Lett. 2016, 11, 3863–3869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blount, B.C.; Mack, M.M.; Wehr, C.M.; MacGregor, J.T.; Hiatt, R.A.; Wang, G.; Wickramasinghe, S.N.; Everson, R.B.; Ames, B.N. Folate deficiency causes uracil misincorporation into human DNA and chromosome breakage: implications for cancer and neuronal damage. Proc. Natl. Acad. Sci. USA 1997, 94, 3290–3295. [Google Scholar] [CrossRef] [PubMed]
- Mhashal, A.R.; Pshetitsky, Y.; Cheatum, C.M.; Kohen, A.; Thomas Major, D. Evolutionary Effects on Bound Substrate pKa in Dihydrofolate Reductase. J. Am. Chem. Soc. 2018, 140, 16650–16660. [Google Scholar] [CrossRef]
- Mhashal, A.R.; Vardi-Kilshtain, A.; Kohen, A.; Major, D.T. The role of the Met20 loop in the hydride transfer in Escherichia coli dihydrofolate reductase. J. Biol. Chem. 2017, 292, 14229–14239. [Google Scholar] [CrossRef]
- Brut, M.; Estève, A.; Landa, G.; Renvez, G.; Djafari Rouhani, M.; Vaisset, M. Atomic Scale Determination of Enzyme Flexibility and Active Site Stability through Static Modes: Case of Dihydrofolate Reductase. J. Phys. Chem. B 2011, 115, 1616–1622. [Google Scholar] [CrossRef]
- Arora, K.; Brooks, C.L., III. Functionally Important Conformations of the Met20 Loop in Dihydrofolate Reductase are Populated by Rapid Thermal Fluctuations. J. Am. Chem. Soc. 2009, 131, 5642–5647. [Google Scholar] [CrossRef] [Green Version]
- Khavrutskii, I.V.; Price, D.J.; Lee, J.; Brooks, C.L. Conformational change of the methionine 20 loop of Escherichia coli dihydrofolate reductase modulates pKa of the bound dihydrofolate. Protein Sci. 2007, 16, 1087–1100. [Google Scholar] [CrossRef]
- Klon, A.E.; Héroux, A.; Ross, L.J.; Pathak, V.; Johnson, C.A.; Piper, J.R.; Borhani, D.W. Atomic structures of human dihydrofolate reductase complexed with NADPH and two lipophilic antifolates at 1.09 a and 1.05 a resolution. J. Mol. Biol. 2002, 320, 677–693. [Google Scholar] [CrossRef]
- Boehr, D.D.; Dyson, H.J.; Wright, P.E. Conformational Relaxation following Hydride Transfer Plays a Limiting Role in Dihydrofolate Reductase Catalysis. Biochemistry 2008, 47, 9227–9233. [Google Scholar] [CrossRef] [PubMed]
- Sawaya, M.R.; Kraut, J. Loop and Subdomain Movements in the Mechanism of Escherichia coli Dihydrofolate Reductase: Crystallographic Evidence. Biochemistry 1997, 36, 586–603. [Google Scholar] [CrossRef]
- Farber, S.; Diamond, L.K.; Mercer, R.D.; Sylvester, R.F.; Wolff, J.A. Temporary Remissions in Acute Leukemia in Children Produced by Folic Acid Antagonist, 4-Aminopteroyl-Glutamic Acid (Aminopterin). N. Engl. J. Med. 1948, 238, 787–793. [Google Scholar] [CrossRef]
- Wang, M.; Yang, J.; Yuan, M.; Xue, L.; Li, H.; Tian, C.; Wang, X.; Liu, J.; Zhang, Z. Synthesis and antiproliferative activity of a series of novel 6-substituted pyrido[3,2-d]pyrimidines as potential nonclassical lipophilic antifolates targeting dihydrofolate reductase. Eur. J. Med. Chem. 2017, 128, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Berman, E.M.; Werbel, L.M. The renewed potential for folate antagonists in contemporary cancer chemotherapy. J. Med. Chem. 1991, 34, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Hutchings, B.L.; Mowat, J.H.; Oleson, J.J.; Stokstad, E.L.R.; Booths, J.H.; Waller, C.W.; Angler, R.B.; Semb, I.; SubbaRow, Y. Pteroylaspartic acid, an antagonist for pteroylglutamic acid. J. Biol. Chem. 1947, 170, 323–328. [Google Scholar]
- Gubner, R.; August, S.; Ginsberg, V. Therapeutic suppression of tissue reactivity. 2. Effect of aminopterin in rheumatoid arthritis and psoriasis. Am. J. Med. Sci. 1951, 221, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Kremer, J.M. Toward a better understanding of methotrexate. Arthritis Rheumatol. 2004, 50, 1370–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chabner, B.A.; Longo, D.L. Cancer Chemotherapy and Biotherapy. Principles and Practice, 5th ed.; Wolters Kluwer, Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2011; ISBN 978-1-60547-431-1, 1-60547-431-2. [Google Scholar]
- De Oliveira, C.P.; Büttenbender, S.L.; Prado, W.A.; Beckenkamp, A.; Asbahr, A.C.; Buffon, A.; Guterres, S.S.; Pohlmann, A.R. Enhanced and Selective Antiproliferative Activity of Methotrexate-Functionalized-Nanocapsules to Human Breast Cancer Cells (MCF-7). Nanomaterials 2018, 8, 24. [Google Scholar] [CrossRef]
- Kaye, S.B. New antimetabolites in cancer chemotherapy and their clinical impact. Br. J. Cancer 1998, 78, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G. Targeting cancer metabolism: A therapeutic window opens. Nat. Rev. Drug Discov. 2011, 10, 671–684. [Google Scholar] [CrossRef] [PubMed]
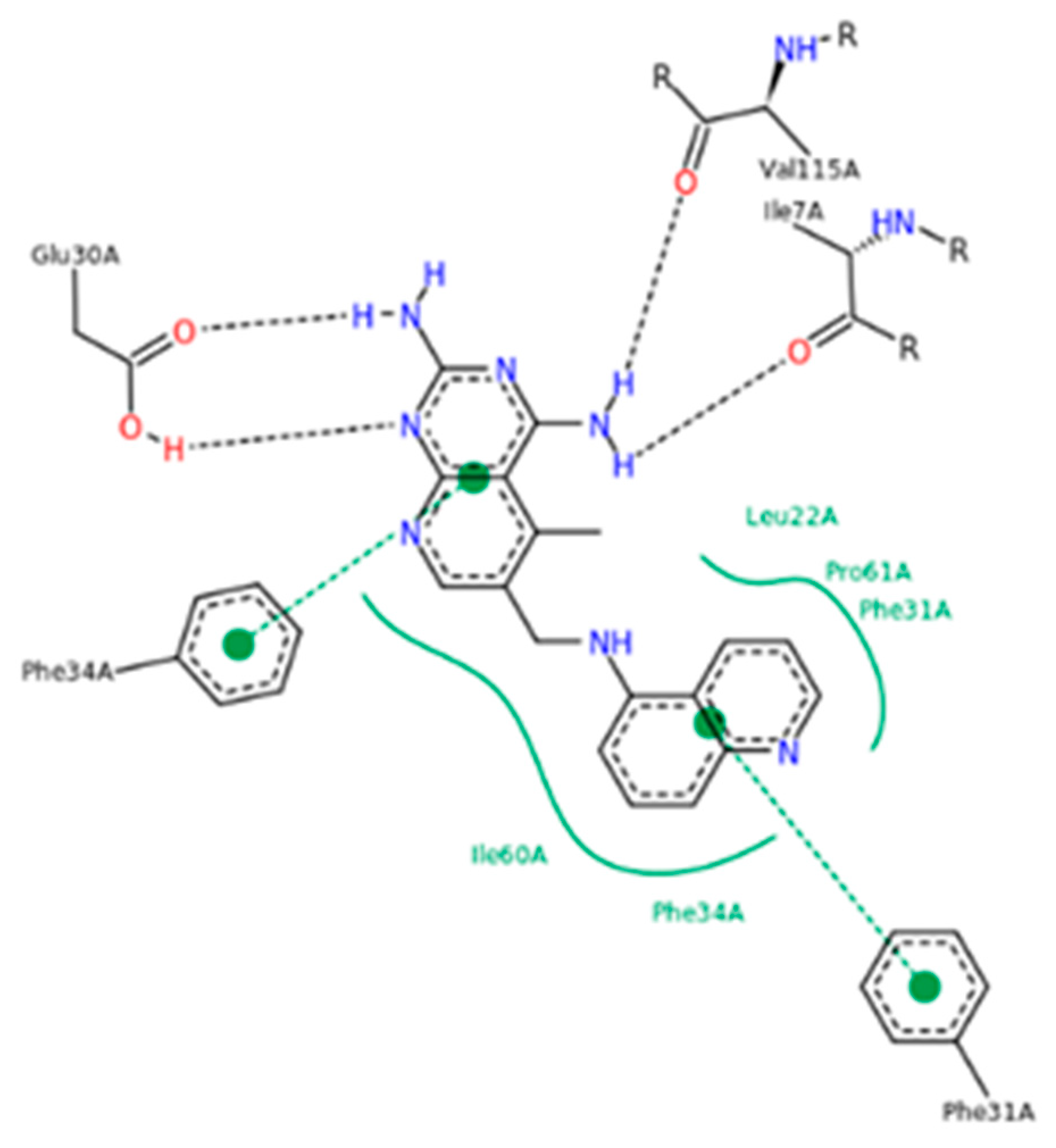
- Al-Rashood, S.T.; Hassan, G.S.; El-Messery, S.M.; Nagi, M.N.; Habib, E.S.E.; Al-Omary, F.A.M.; El-Subbagh, H.I. Synthesis, biological evaluation and molecular modeling study of 2-(1,3,4-thiadiazolyl-thio and 4-methyl-thiazolyl-thio)-quinazolin-4-ones as a new class of DHFR inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 4557–4567. [Google Scholar] [CrossRef] [PubMed]
- Drug.com. FDA Approves Folotyn (pralatrexate) for Treatment of Peripheral T-Cell Lymphoma. Available online: https://www.drugs.com/newdrugs/fda-approves-folotyn-pralatrexate-peripheral-t-cell-lymphoma-1666.html (accessed on 14 March 2019).
- Hagner, N.; Joerger, M. Cancer chemotherapy: targeting folic acid synthesis. Cancer Manag. Res. 2010, 2, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Hassan, G.S.; El-Messery, S.M.; Al-Omary, F.A.M.; Al-Rashood, S.T.; Shabayek, M.I.; Abulfadl, Y.S.; Habib, E.S.E.; El-Hallouty, S.M.; Fayad, W.; Mohamed, K.M.; et al. Nonclassical antifolates, part 4. 5-(2-Aminothiazol-4-yl)-4-phenyl-4H-1,2,4-triazole-3-thiols as a new class of DHFR inhibitors: Synthesis, biological evaluation and molecular modeling study. Eur. J. Med. Chem. 2013, 66, 135–145. [Google Scholar] [CrossRef] [PubMed]
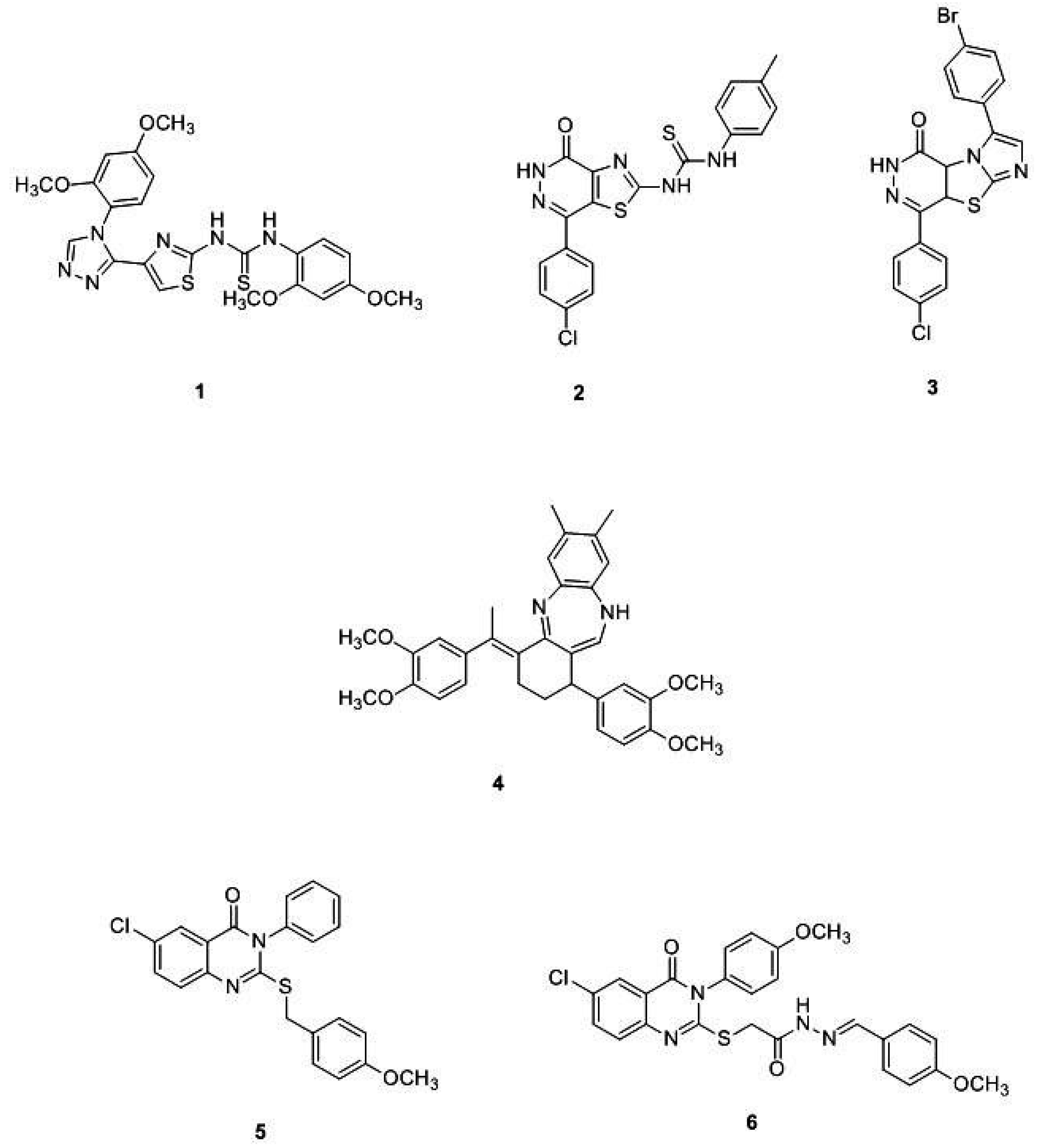
- Ewida, M.A.; Abou El Ella, D.A.; Lasheen, D.S.; Ewida, H.A.; El-Gazzar, Y.I.; El-Subbagh, H.I. Thiazolo[4,5-d]pyridazine analogues as a new class of dihydrofolate reductase (DHFR) inhibitors: Synthesis, biological evaluation and molecular modeling study. Bioorg. Chem. 2017, 74, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Ewida, M.A.; Abou El Ella, D.A.; Lasheen, D.S.; Ewida, H.A.; El-Gazzar, Y.I.; El-Subbagh, H.I. Imidazo[2′,1′:2,3]thiazolo[4,5-d]pyridazinone as a new scaffold of DHFR inhibitors: Synthesis, biological evaluation and molecular modeling study. Bioorg. Chem. 2018, 80, 11–23. [Google Scholar] [CrossRef]
- El-Subbagh, H.I.; Hassan, G.S.; El-Messery, S.M.; Al-Rashood, S.T.; Al-Omary, F.A.M.; Abulfadl, Y.S.; Shabayek, M.I. Nonclassical antifolates, part 5. Benzodiazepine analogs as a new class of DHFR inhibitors: Synthesis, antitumor testing and molecular modeling study. Eur. J. Med. Chem. 2014, 74, 234–245. [Google Scholar] [CrossRef]
- El-Messery, S.M.; Hassan, G.S.; Nagi, M.N.; Habib, E.S.E.; Al-Rashood, S.T.; El-Subbagh, H.I. Synthesis, biological evaluation and molecular modeling study of some new methoxylated 2-benzylthio-quinazoline-4(3H)-ones as nonclassical antifolates. Bioorg. Med. Chem. Lett. 2016, 26, 4815–4823. [Google Scholar] [CrossRef]
- El-Gazzar, Y.I.; Georgey, H.H.; El-Messery, S.M.; Ewida, H.A.; Hassan, G.S.; Raafat, M.M.; Ewida, M.A.; El-Subbagh, H.I. Synthesis, biological evaluation and molecular modeling study of new (1,2,4-triazole or 1,3,4-thiadiazole)-methylthio-derivatives of quinazolin-4(3H)-one as DHFR inhibitors. Bioorg. Chem. 2017, 72, 282–292. [Google Scholar] [CrossRef]
- Cody, V.; Mao, Q.; Queener, S.F. Recombinant bovine dihydrofolate reductase produced by mutagenesis and nested PCR of murine dihydrofolate reductase cDNA. Protein Expr. Purif. 2008, 62, 104–110. [Google Scholar] [CrossRef]
- Sahu, M.; Nerkar, A.G. In silico screening, synthesis and in vitro evaluation of some quinazolinone derivatives as dihydrofolate reductase inhibitors for anticancer activity: Part-I. Int. J. Pharm. Pharm. Sci. 2014, 6, 193–199. Available online: http://www.ijppsjournal.com/Vol6Issue5/9201.pdf (accessed on 20 January 2019).
- Sahu, M.; Nerkar, A.G. In silico design, synthesis and pharmacological screening of some quinazolinone metal complexes as dihydrofolate reductase inhibitors for anticancer activity: Part-II. Int. J. Pharm. Pharm. Sci. 2014, 6, 509–514. Available online: http://www.ijppsjournal.com/Vol6Issue5/9425.pdf (accessed on 20 January 2019).
- Al-Omary, F.A.M.; Hassan, G.S.; El-Messery, S.M.; Nagi, M.N.; Habib, E.S.E.; El-Subbagh, H.I. Nonclassical antifolates, part 3: Synthesis, biological evaluation and molecular modeling study of some new 2-heteroarylthio-quinazolin-4-ones. Eur. J. Med. Chem. 2013, 63, 33–45. [Google Scholar] [CrossRef]
- Chen, J.; Kassenbrock, A.; Li, B.X.; Xiao, X. Discovery of a potent anti-tumor agent through regioselective mono-N-acylation of 7H-pyrrolo[3,2-f]quinazoline-1,3-diamine. Med. Chem. Commun. 2013, 4, 1275–1282. [Google Scholar] [CrossRef] [PubMed]
- Du, X.H.; Zhuang, W.C. Neural Network Model for Predicting Anticancer Activity of Pyridopyrimidines Derivatives. Adv. Mat. Res. 2014, 905, 96–100. [Google Scholar] [CrossRef]
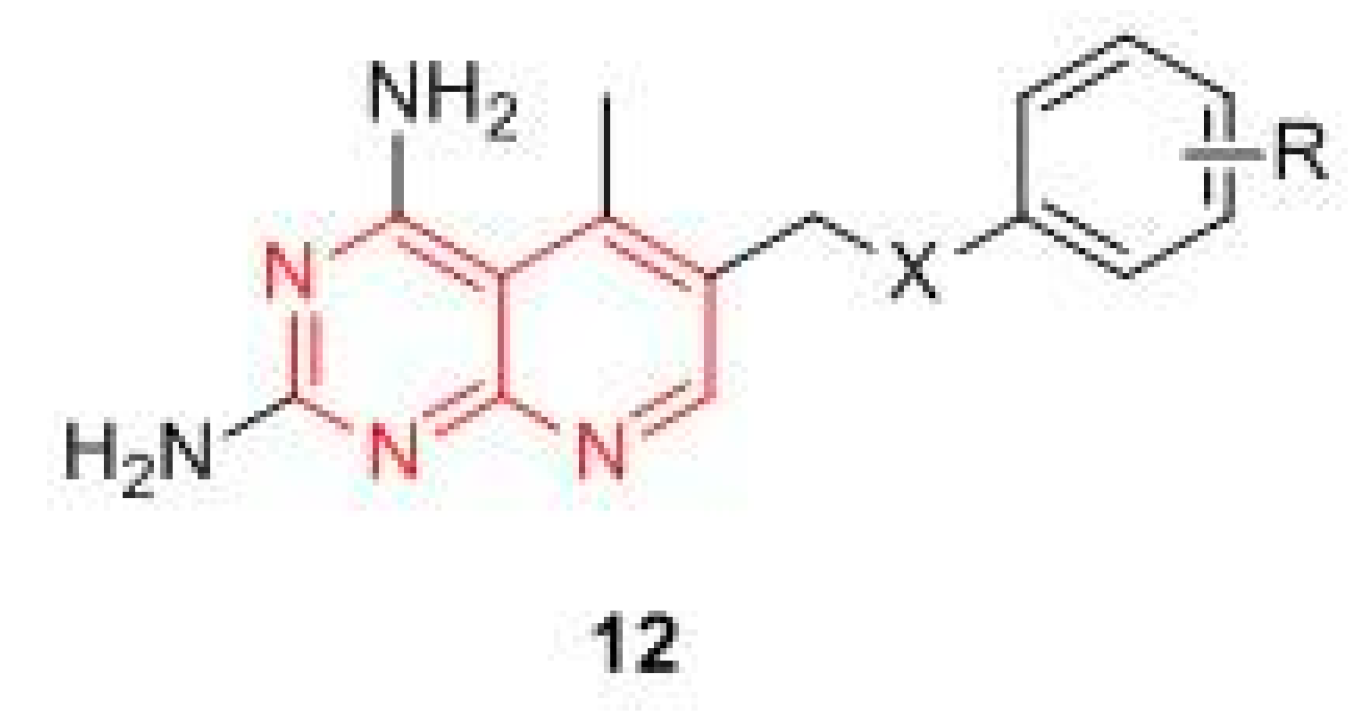
- Yang, J.; Wang, M.; Li, X.; Fan, N.; Xue, L.; Li, H.; Tian, C.; Wang, X.; Liu, J.; Zhang, Z. Syntheses and antiproliferative evaluation of 6-thienyl, 6-polyphenyl aryl and 6-naphthyl derivatives of 2,4-diaminopyrido[3,2-d]pyramidine as non-classical antifolate targeting DHFR. Chem. Res. Chin. Univ. 2017, 33, 559–568. [Google Scholar] [CrossRef]
- Li, H.; Fang, F.; Liu, Y.; Xue, L.; Wang, M.; Guo, Y.; Wang, X.; Tian, C.; Liu, J.; Zhang, Z. Inhibitors of dihydrofolate reductase as antitumor agents: design, synthesis and biological evaluation of a series of novel nonclassical 6-substituted pyrido[3,2-d]pyrimidines with a three- to five-carbon bridge. Bioorg. Med. Chem. 2018, 26, 2674–2685. [Google Scholar] [CrossRef] [PubMed]
- Rapolu, S.; Alla, M.; Ganji, R.J.; Saddanapu, V.; Kishor, C.; Bommena, V.R.; Addlagatta, A. Synthesis, cytotoxicity and hDHFR inhibition studies of 2H-pyrido[1,2-a]pyrimidin-2-ones. Med. Chem. Commun. 2013, 4, 817–821. [Google Scholar] [CrossRef]
- Gangjee, A. Pyrimidine Compounds and Pyrimido Indole Compounds and Methods of Use. U.S. Patent WO2016022890A1, 11 February 2016. Available online: https://worldwide.espacenet.com/publicationDetails/biblio?CC=WO&NR=2016022890A1&KC=A1&FT=D# (accessed on 20 January 2019).
- Zhou, X.; Lin, K.; Ma, X.; Chui, W.K.; Zhou, W. Design, synthesis, docking studies and biological evaluation of novel dihydro-1,3,5-triazines as human DHFR inhibitors. Eur. J. Med. Chem. 2017, 125, 1279–1288. [Google Scholar] [CrossRef]
- Bashandy, M.S.; Al-Harbi, S.A. Synthesis, Antimicrobial and Antihuman Liver Cancer Activities of Novel Sulfonamides Incorporating Benzofuran, Pyrazole, Pyrimidine, 1,4-Diazepine and Pyridine Moieties Prepared from (E)-4-(3-(Dimethylamino)acryloyl)-N-ethyl-N-methylbenzenesulfonamide. Heterocycles 2015, 91, 1905. [Google Scholar] [CrossRef]
- Debbabi, K.F.; Bashandy, M.S.; Al-Harbi, S.A.; Aljuhani, E.H.; Al-Saidi, H.M. Synthesis and molecular docking against dihydrofolate reductase of novel pyridin-N-ethyl-N-methylbenzenesulfonamides as efficient anticancer and antimicrobial agents. J. Mol. Struct. 2017, 1131, 124–135. [Google Scholar] [CrossRef]
- Shen, B. A New Golden Age of Natural Products Drug Discovery. Cell 2015, 163, 1297–1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakao, Y.; Fusetani, N. Enzyme Inhibitors from Marine Invertebrates. J. Nat. Prod. 2007, 70, 689–710. [Google Scholar] [CrossRef] [PubMed]
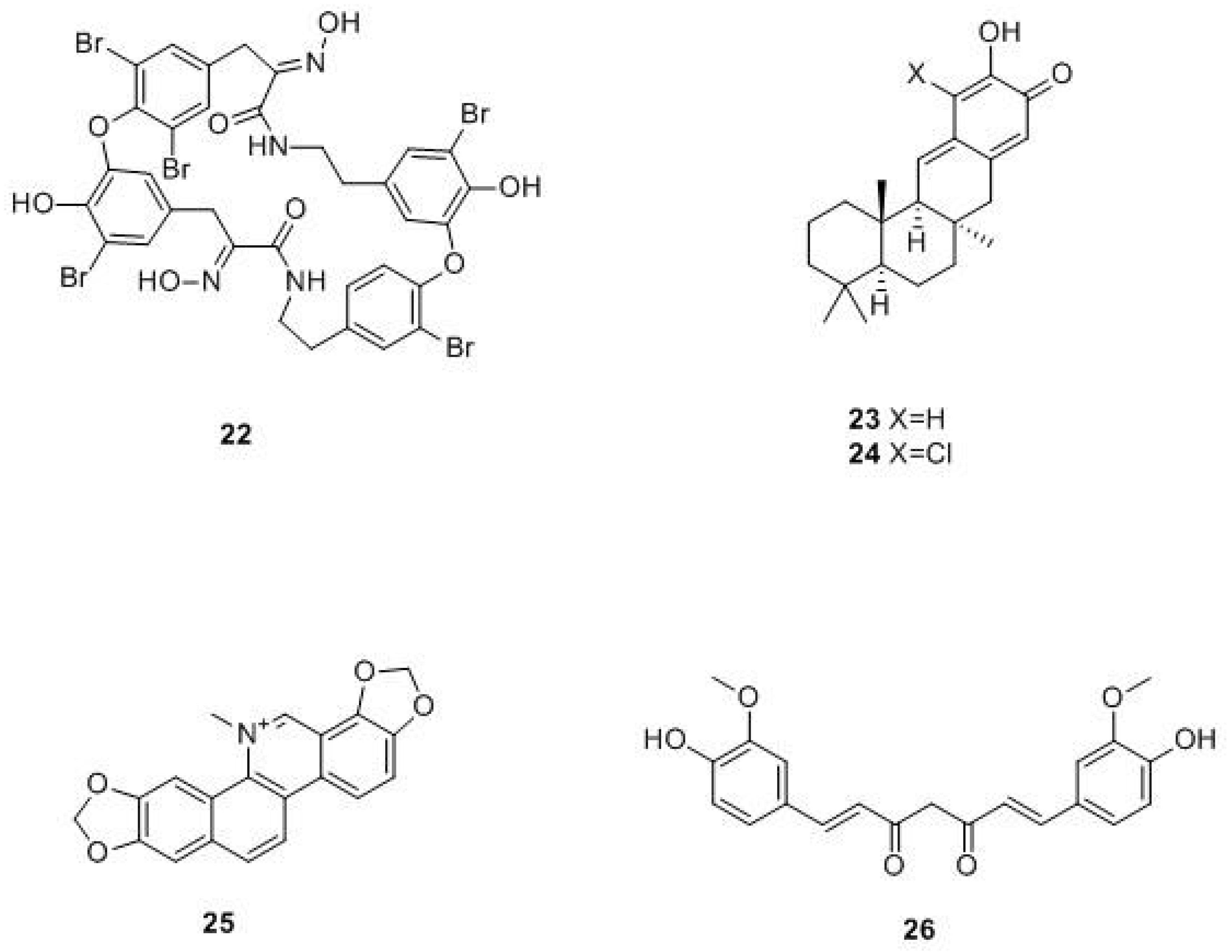
- Kalogris, C.; Garulli, C.; Pietrella, L.; Gambini, V.; Pucciarelli, S.; Lucci, C.; Tilio, M.; Zabaleta, M.E.; Bartolacci, C.; Andreani, C.; et al. Sanguinarine suppresses basal-like breast cancer growth through dihydrofolate reductase inhibition. Biochem. Pharm. 2014, 90, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Albalawi, M.A.D.; Bashir, N.A.O.; Tawfik, A. Anticancer and Antifolate Activities of Extracts of Six Saudi Arabian Wild Plants Used in Folk Medicine. J. Life Sci. 2015, 9, 334–340. [Google Scholar] [CrossRef]
- Hobani, Y.; Jerah, A.; Bidwai, A. A comparative molecular docking study of curcumin and methotrexate to dihydrofolate reductase. Bioinformation 2017, 13, 63–66. [Google Scholar] [CrossRef]
- Arooj, M.; Sakkiah, S.; Cao, G.; Lee, K.W. An Innovative Strategy for Dual Inhibitor Design and Its Application in Dual Inhibition of Human Thymidylate Synthase and Dihydrofolate Reductase Enzymes. PLoS ONE 2013, 8, e60470. [Google Scholar] [CrossRef]
- Tian, C.; Wang, M.; Han, Z.; Fang, F.; Zhang, Z.; Wang, X.; Liu, J. Design, synthesis and biological evaluation of novel 6-substituted pyrrolo [3,2-d] pyrimidine analogues as antifolate antitumor agents. Eur. J. Med. Chem. 2017, 138, 630–643. [Google Scholar] [CrossRef]
- Shavet, P.S. Structural optimization of indole based compounds for highly promising anti-cancer activities: Structure activity relationship studies and identification of lead molecules. Eur. J. Med. Chem. 2014, 74, 440–450. [Google Scholar] [CrossRef]
- Singla, P.; Luxami, V.; Paul, K. Synthesis, in vitro antitumor activity, dihydrofolate reductase inhibition, DNA intercalation and structure–activity relationship studies of 1,3,5-triazine analogues. Bioorg. Med. Chem. Lett. 2016, 26, 518–523. [Google Scholar] [CrossRef]
- Ng, H.L.; Chen, S.; Chew, E.; Chui, W. Applying the designed multiple ligands approach to inhibit dihydrofolate reductase and thioredoxin reductase for anti-proliferative activity. Eur. J. Med. Chem. 2016, 115, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Ng, H.L.; Ma, X.; Chew, E.; Chui, W. Design, Synthesis, and Biological Evaluation of Coupled Bioactive Scaffolds as Potential Anticancer Agents for Dual Targeting of Dihydrofolate Reductase and Thioredoxin Reductase. J. Med. Chem. 2017, 60, 1734–1745. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.C.; Tedeschi, P.; AdeBisi Lawal, R.; Banerjee, D.; Scotto, K.; Kerrigan, J.E.; Lee, K.C.; Johnson-Farley, N.; Bertino, J.R.; Abali, E.E. Enhanced Degradation of Dihydrofolate Reductase through Inhibition of NAD Kinase by Nicotinamide Analogs. Mol. Pharmacol. 2013, 83, 339–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
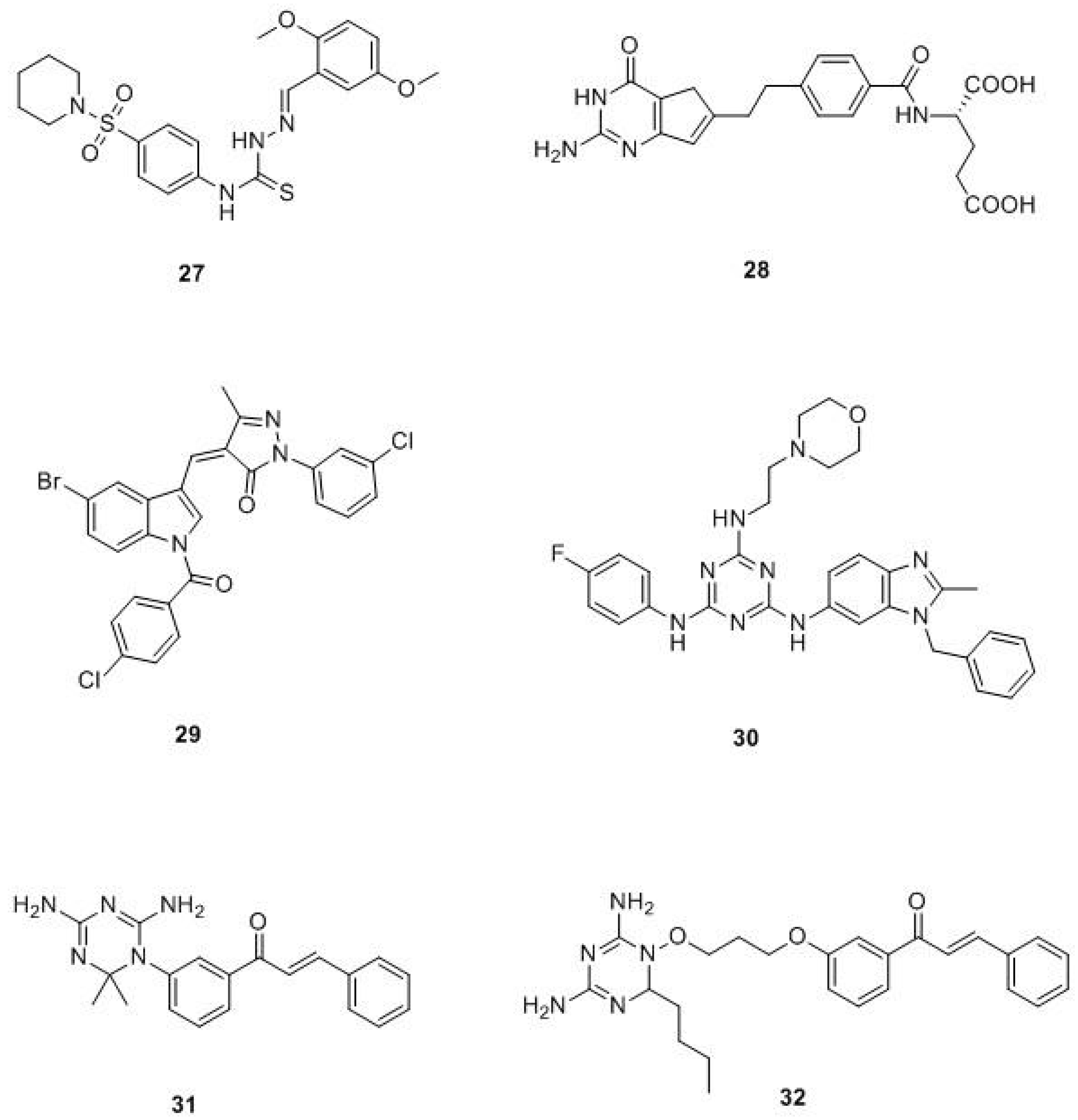{kind=link}
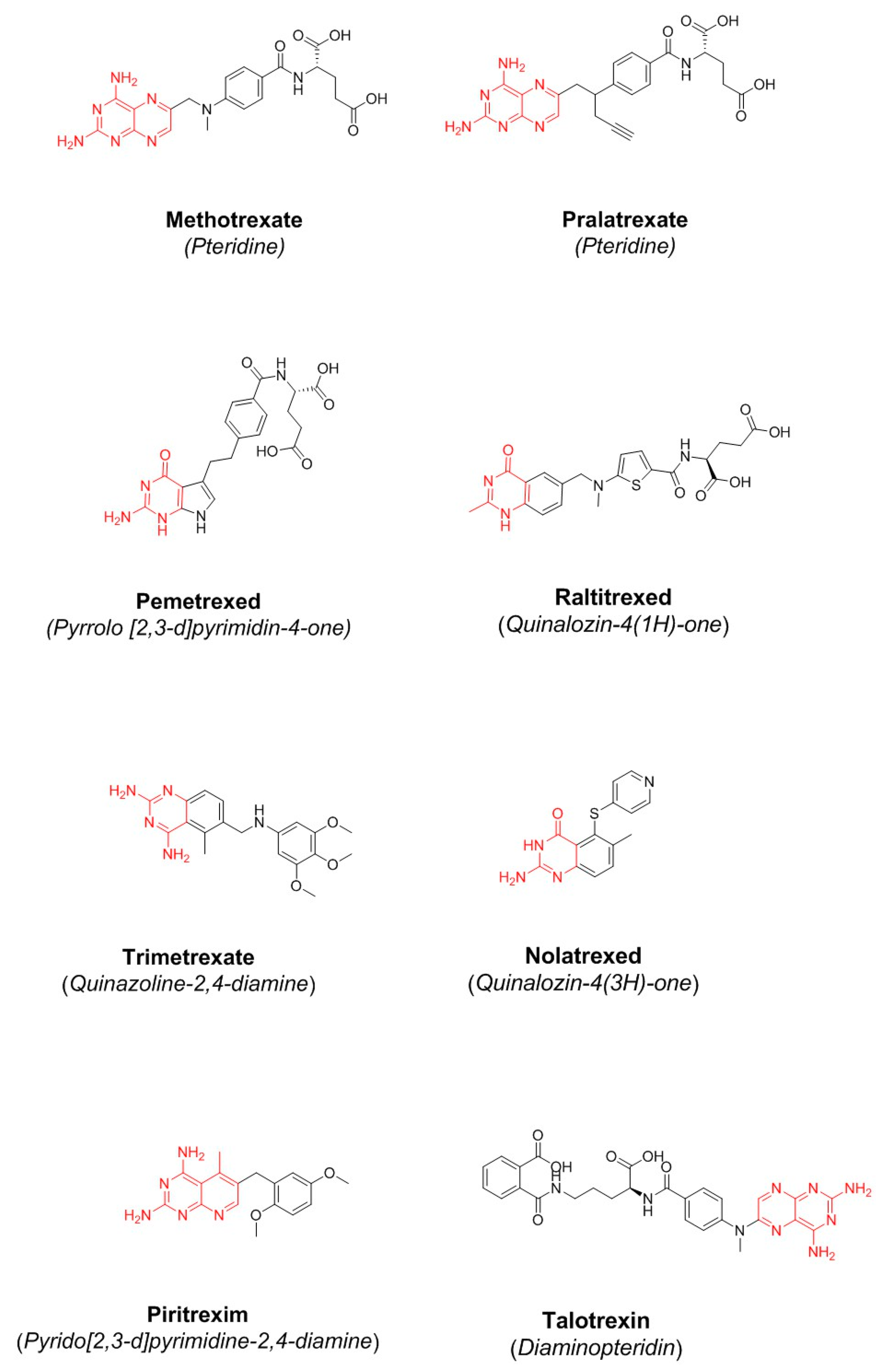
| Antifolate | Status * | Indication | Toxicity |
|---|---|---|---|
| Methotrexate | Approved by FDA and EMA in 1985 | Treatment of lymphoma, acute lymphoblastic leukemia and osteosarcoma | Symptoms of overdose include bone marrow suppression and gastrointestinal side effects |
| Raltitrexed | Approved by EMA in 1998 | Treatment of malignant colorectal cancer, but its utilization in mainly limited to patients who are intolerant to 5-fluorouracil | Gastrointestinal and hematologic side effects |
| Pemetrexed | Approved by FDA and EMA in 2001 | First-line treatment for advanced non-squamous-cell lung cancer and pleural mesothelioma in combination with cisplatin | Neutropenia, leukopenia, anemia, stomatitis and infection |
| Pralatrexate | Approved by FDA and EMA in 2009 | Treatment of relapsed or refractory peripheral T-cell lymphoma (TCL) | Mucositis |
| Antifolate | ClinicalTrials.gov RECORD ID * | Status | Toxicity |
|---|---|---|---|
| Nolatrexed | NCT00012324 | Phase 3 study in unresectable hepatocellular carcinoma (HCC) has been completed (2005) | Nausea, vomiting, stomatitis, erythematous maculopapular rash, thrombocytopenia and neutropenia |
| Piritrexim | NCT00002914 | Phase 2 study in advanced cancer of the urinary tract has been completed (2004) | Leukopenia, thrombocytopenia, mucositis |
| Talotrexin | NCT00088023 NCT00112060 NCT00129558 NCT00458744 | It has been suspended in phase 1 in the treatment of solid tumors (2005). It was withdrawn in phase 1 in the treatment of brain and central nervous system tumors, and malignant lymphomas (2008). It was withdrawn in phase 2 in the treatment of non-small-cell lung carcinoma (NSCLC) and leukemia (2011) | Neurotoxic effect such as fatigue and hypoxia. At higher and cumulative doses, it may produce fatal leukoencephalopathy |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raimondi, M.V.; Randazzo, O.; La Franca, M.; Barone, G.; Vignoni, E.; Rossi, D.; Collina, S. DHFR Inhibitors: Reading the Past for Discovering Novel Anticancer Agents. Molecules 2019, 24, 1140. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24061140
Raimondi MV, Randazzo O, La Franca M, Barone G, Vignoni E, Rossi D, Collina S. DHFR Inhibitors: Reading the Past for Discovering Novel Anticancer Agents. Molecules. 2019; 24(6):1140. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24061140
Chicago/Turabian StyleRaimondi, Maria Valeria, Ornella Randazzo, Mery La Franca, Giampaolo Barone, Elisa Vignoni, Daniela Rossi, and Simona Collina. 2019. "DHFR Inhibitors: Reading the Past for Discovering Novel Anticancer Agents" Molecules 24, no. 6: 1140. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24061140