
Two Approaches for Evaluating the Effects of Galangin on the Activities and mRNA Expression of Seven CYP450

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
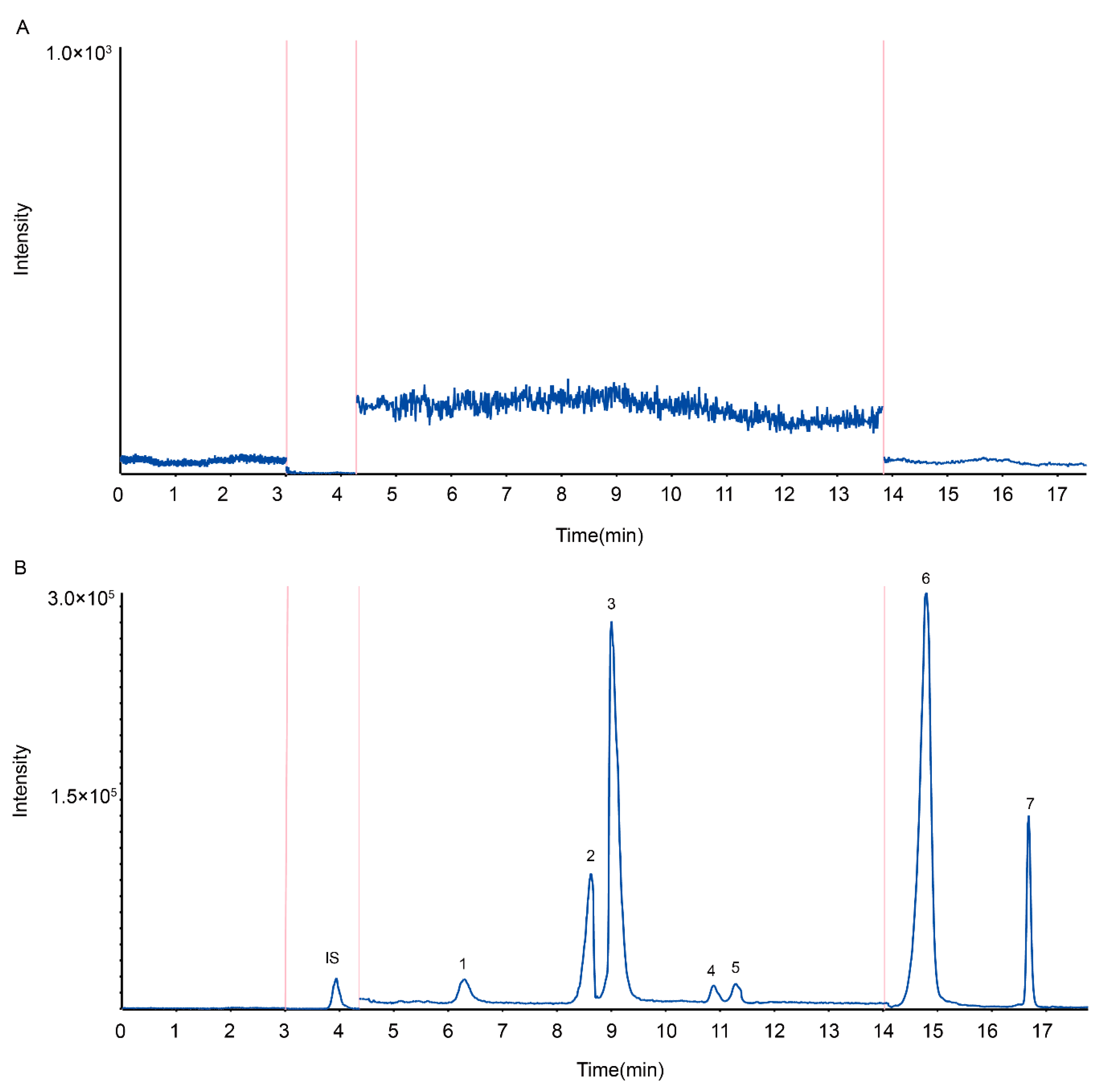
2.1. High-Performance Liquid Chromatography–Tandem Mass Spectrometry (HPLC-MS/MS) Method Development
2.2. Method Validation
2.3. Selection of CYP450 Isozymes
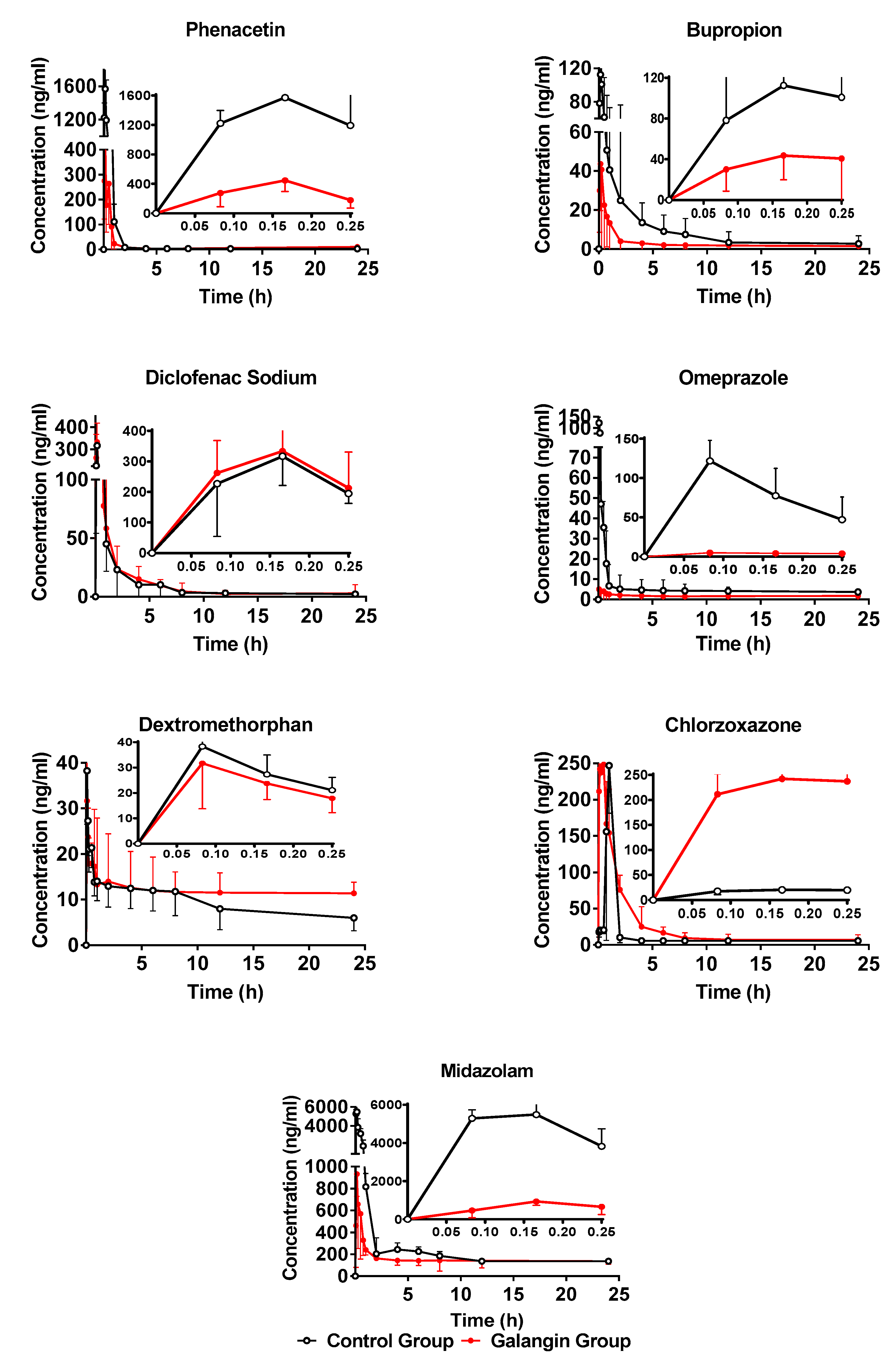
2.4. Effect of Galangin on the Activities of Rat Liver CYPs
2.4.1. Effect of Galangin on Rat Hepatic CYP1A2
2.4.2. Effect of Galangin on Rat Hepatic CYP2B3
2.4.3. Effect of Galangin on Rat Hepatic CYP2C13
2.4.4. Effect of Galangin on Rat Hepatic CYP3A1
2.4.5. Effect of Galangin on Rat Hepatic CYP2C11, CYP2D4, and CYP2E1
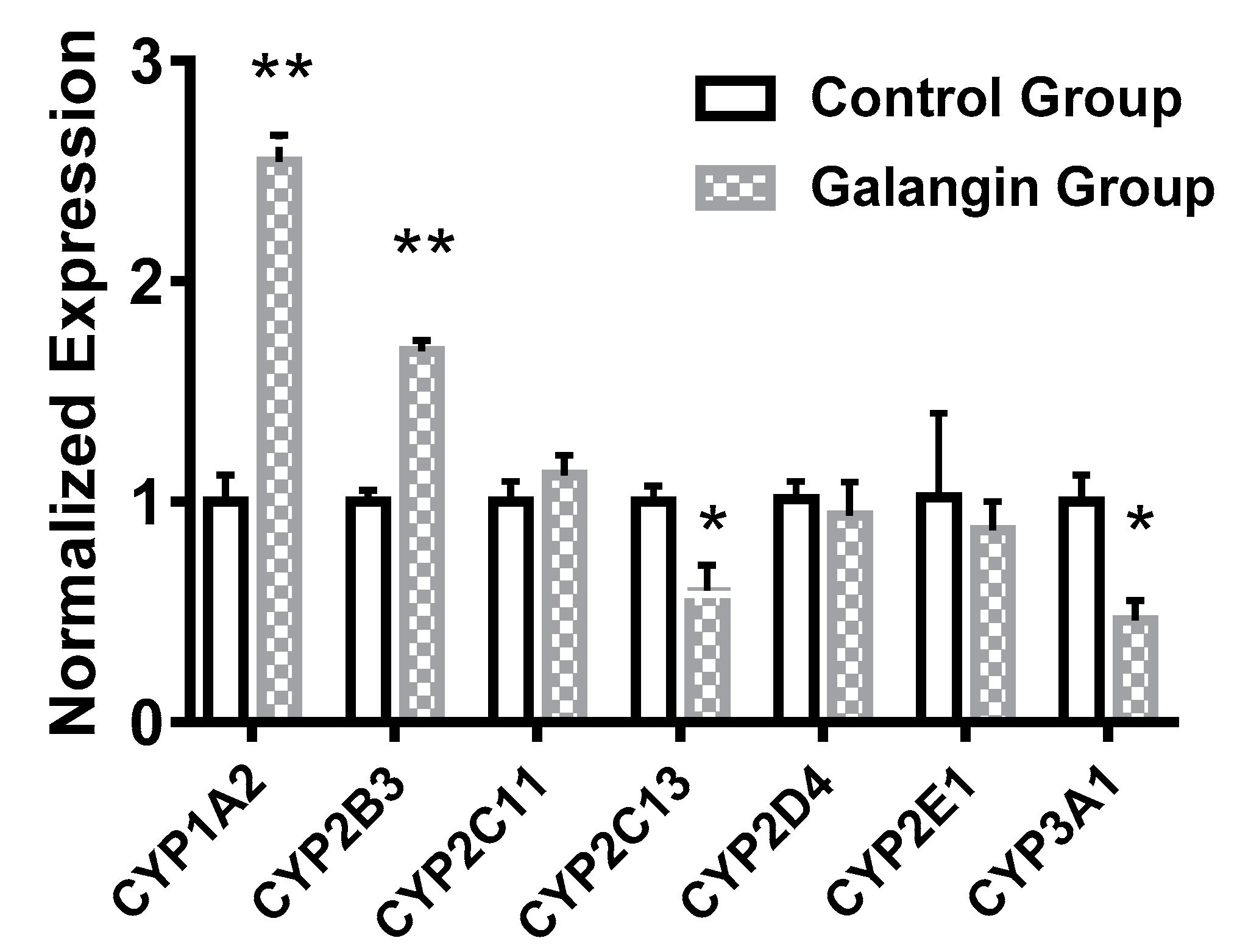
2.5. Effects of Galangin on Rat Liver CYP mRNA-Expression Levels
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Animals and Experimental Design
3.3. Pharmacokinetic Study
3.4. Sample Preparation
3.5. Preparation of Calibration Curves and Quality Control (QC) Samples
3.6. LC-MS Analytical Conditions
3.7. Method Validation
3.8. Effects of Galangin on mRNA Expression of CYP Enzymes in Rats
3.9. Statistical Processing Method
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Nebert, D.W.; Russell, D.W. Clinical importance of the cytochromes P450. Lancet 2002, 360, 1155–1162. [Google Scholar] [CrossRef]
- Lee, J.J.; Lee, J.H.; Yim, N.H.; Han, J.H.; Ma, J.Y. Application of galangin, an active component of Alpinia officinarum Hance (Zingiberaceae), for use in drug-eluting stents. Sci. Rep. 2017, 7, 8207. [Google Scholar] [CrossRef]
- Cushnie, T.P.; Hamilton, V.E.; Chapman, D.G.; Taylor, P.W.; Lamb, A.J. Aggregation of Staphylococcus aureus following treatment with the antibacterial flavonol galangin. J. Appl. Microbiol. 2007, 103, 1562–1567. [Google Scholar] [CrossRef] [PubMed]
- Aloud, A.A.; Veeramani, C.; Govindasamy, C.; Alsaif, M.A.; El Newehy, A.S.; Al-Numair, K.S. Galangin, a dietary flavonoid, improves antioxidant status and reduces hyperglycemia-mediated oxidative stress in streptozotocin-induced diabetic rats. Redox Rep. 2017, 22, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Alagawadi, K.R. Anti-obesity effects of galangin, a pancreatic lipase inhibitor in cafeteria diet fed female rats. Pharm. Biol. 2013, 51, 607–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heo, M.Y.; Sohn, S.J.; Au, W.W. Anti-genotoxicity of galangin as a cancer chemopreventive agent candidate. Mutat. Res. 2001, 488, 135–150. [Google Scholar] [CrossRef]
- Matsuda, H.; Ando, S.; Kato, T.; Morikawa, T.; Yoshikawa, M. Inhibitors from the rhizomes of Alpinia officinarum on production of nitric oxide in lipopolysaccharide-activated macrophages and the structural requirements of diarylheptanoids for the activity. Bioorgan. Med. Chem. 2006, 14, 138–142. [Google Scholar] [CrossRef]
- Sivakumar, A.S.; Anuradha, C.V. Effect of galangin supplementation on oxidative damage and inflammatory changes in fructose-fed rat liver. Chem. Biol. Interact. 2011, 193, 141–148. [Google Scholar] [CrossRef]
- Su, Y.; Chen, Y.; Liu, Y.; Yang, Y.; Deng, Y.; Gong, Z.; Chen, J.; Wu, T.; Lin, S.; Cui, L. Antiosteoporotic effects of Alpinia officinarum Hance through stimulation of osteoblasts associated with antioxidant effects. J. Orthop. Transl. 2016, 4, 75–91. [Google Scholar] [CrossRef]
- Morello, S.; Vellecco, V.; Alfieri, A.; Mascolo, N.; Cicala, C. Vasorelaxant effect of the flavonoid galangin on isolated rat thoracic aorta. Life. Sci. 2006, 78, 825–830. [Google Scholar] [CrossRef]
- Lu, Y.Y.; Du, Z.Y.; Li, Y.; Wang, J.L.; Zhao, M.B.; Jiang, Y.; Guo, X.Y.; Tu, P.F. Effects of Baoyuan decoction, a traditional Chinese medicine formula, on the activities and mRNA expression of seven CYP isozymes in rats. J. Ethnopharmacol. 2018, 225, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Kim, H.; Cha, I.J.; Park, J.S.; Shon, J.H.; Liu, K.H.; Shin, J.G. High-throughput screening of inhibitory potential of nine cytochrome P450 enzymes in vitro using liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2005, 19, 2651–2658. [Google Scholar] [CrossRef]
- Li, X.; Chen, X.; Li, Q.; Wang, L.; Zhong, D. Validated method for rapid inhibition screening of six cytochrome P450 enzymes by liquid chromatography-tandem mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2007, 852, 128–137. [Google Scholar] [CrossRef] [PubMed]
- De Bock, L.; Boussery, K.; Colin, P.; De Smet, J.; T’Jollyn, H.; Van Bocxlaer, J. Development and validation of a fast and sensitive UPLC-MS/MS method for the quantification of six probe metabolites for the in vitro determination of cytochrome P450 activity. Talanta 2012, 89, 209–216. [Google Scholar] [CrossRef]
- US Food and Drug Administration, Guidance for Industry: Bioanalytical Method Validation. 2001. Available online: http://www.fda.gov/downloads /Drugs/Guidance/ucm070107.pdf (accessed on 10 January 2017).
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.S.; Hu, X.C.; Chen, G.L.; Yuan, X.; Yang, R.N.; Liang, S.; Ren, J.; Sun, J.C.; Kong, G.Q.; Gao, S.G.; Feng, X.S. Effects of vitexin on the pharmacokinetics and mRNA expression of CYP isozymes in rats. Phytother. Res. 2015, 29, 366–372. [Google Scholar] [CrossRef]
- Dorne, J.L.; Walton, K.; Renwick, A.G. Uncertainty factors for chemical risk assessment. human variability in the pharmacokinetics of CYP1A2 probe substrates. Food Chem. Toxicol. 2001, 39, 681–696. [Google Scholar] [CrossRef]
- Mo, S.L.; Liu, Y.H.; Duan, W.; Wei, M.Q.; Kanwar, J.R.; Zhou, S.F. Substrate specificity, regulation, and polymorphism of human cytochrome P450 2B6. Curr. Drug Metab. 2009, 10, 730–753. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.F.; Xue, C.C.; Yu, X.Q.; Li, C.; Wang, G. Clinically important drug interactions potentially involving mechanism-based inhibition of cytochrome p450 3A4 and the role of therapeutic drug monitoring. Ther. Drug Monit. 2007, 29, 687–710. [Google Scholar] [CrossRef]
- Mohammadi-Bardbori, A.; Bengtsson, J.; Rannug, U.; Rannug, A.; Wincent, E. Quercetin, resveratrol, and curcumin are indirect activators of the aryl hydrocarbon receptor (AHR). Chem. Res. Toxicol. 2012, 25, 1878–1884. [Google Scholar] [CrossRef]
- Okada, N.; Murakami, A.; Urushizaki, S.; Matsuda, M.; Kawazoe, K.; Ishizawa, K. Extracts of immature orange (Aurantii fructus immaturus) and citrus unshiu peel (Citri unshiu pericarpium) induce P-glycoprotein and cytochrome P450 3A4 expression via upregulation of pregnane X receptor. Front. Pharmacol. 2017, 8, 84. [Google Scholar] [CrossRef]
- Carazo Fernandez, A.; Smutny, T.; Hyrsova, L.; Berka, K.; Pavek, P. Chrysin, baicalein and galangin are indirect activators of the human constitutive androstane receptor (CAR). Toxicol. Lett. 2015, 233, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Li, L.A. Flavonoids exhibit diverse effects on CYP11B1 expression and cortisol synthesis. Toxicol. Appl. Pharmacol. 2012, 258, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Baeuerle, P.A.; Henkel, T. Function and Activation of NF-κB in the Immune System. Annu. Rev. Immunol. 1994, 12, 141–179. [Google Scholar] [CrossRef] [PubMed]
- Aloud, A.A.; Chinnadurai, V.; Chandramohan, G.; Alsaif, M.A.; Al-Numair, K.S. Galangin controls streptozotocin-caused glucose homeostasis and reverses glycolytic and gluconeogenic enzyme changes in rats. Arch. Physiol. Biochem. 2018, 1–6. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of compounds used in this study are available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
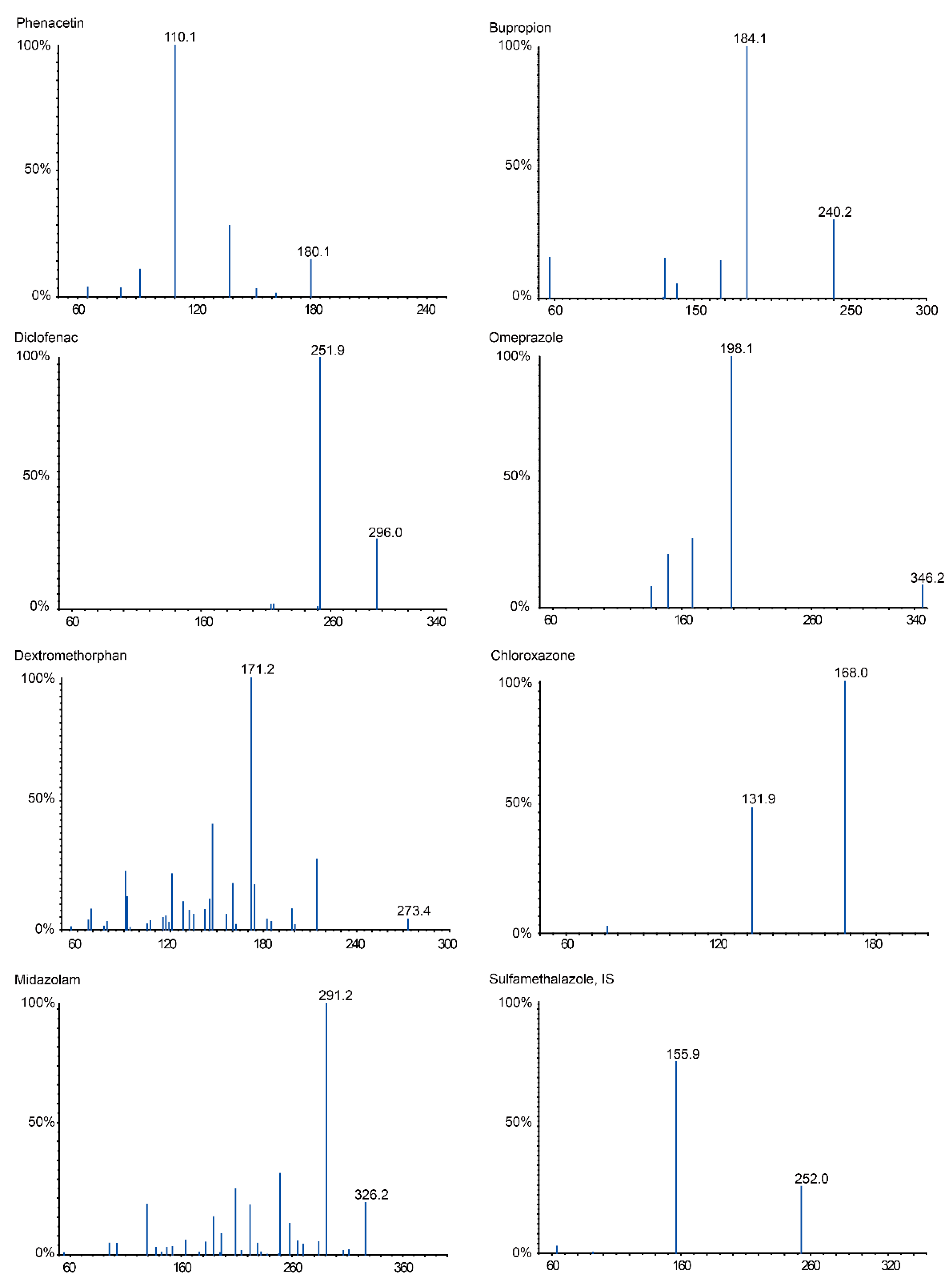
| Enzyme Isoform | Probe Drug | Retention Time (min) | C (Probe Drug μmol/L) | MRM Condition | |||
|---|---|---|---|---|---|---|---|
| Precursor-Ion (m/z) | Daughter-Ion (m/z) | Fragment Energy (V) | Collision Energy (eV) | ||||
| CYP1A2 | Phenacetin | 9.01 | 5 | 180.1 | 110.1 | 57.56 | 27.71 |
| CYP2B3 | Bupropion | 6.29 | 10 | 240.2 | 184.1 | 64.64 | 17.15 |
| CYP2C11 | Diclofenac | 16.68 | 5 | 296.0 | 251.9 | −24.50 | −16.50 |
| CYP2C13 | Omeprazole | 11.31 | 10 | 346.2 | 198.1 | 31.38 | 15.29 |
| CYP2D4 | Dextromethor-phan | 10.87 | 5 | 273.4 | 172.1 | 77.95 | 50.72 |
| CYP2E1 | Chloroxazone | 14.79 | 10 | 168.0 | 131.9 | −65.05 | −27.00 |
| CYP3A1 | Midazolam | 8.63 | 5 | 326.2 | 291.2 | 71.86 | 36.50 |
| Internal standard | Sulfamethalaz-ole | 3.99 | - | 252.0 | 155.9 | −30.00 | −20.00 |
| Compounds | Concentration (ng/mL) | Precision RSD (%) | Accuracy (%) | ||
|---|---|---|---|---|---|
| Intra-Day | Inter-Day | Intra-Day | Inter-Day | ||
| Phenacetin | 1.006 | 8.21 | 7.06 | 4.12 | 6.23 |
| 2.012 | 5.96 | 4.51 | −2.34 | −4.04 | |
| 201.20 | 7.35 | 6.30 | 8.41 | 11.79 | |
| 1609.6 | 4.79 | 4.66 | 3.23 | 5.34 | |
| Bupropion | 0.801 | 2.91 | 6.43 | 3.52 | 9.23 |
| 2.034 | 7.34 | 5.25 | 6.14 | 9.35 | |
| 20.34 | 6.16 | 2.71 | 5.65 | 7.11 | |
| 203.4 | 2.44 | 3.55 | −2.47 | −3.76 | |
| Diclofenac | 1.01 | 2.80 | 8.34 | 4.98 | 5.92 |
| 2.02 | 1.21 | 5.26 | 6.90 | 9.45 | |
| 20.20 | 4.37 | 4.91 | 7.04 | 8.61 | |
| 202.00 | 3.55 | 4.27 | 4.93 | 5.85 | |
| Omeprazole | 1.015 | 2.89 | 3.55 | 11.71 | 12.13 |
| 2.03 | 4.10 | 2.78 | 7.26 | 6.41 | |
| 20.30 | 3.62 | 4.40 | 5.85 | 8.28 | |
| 406 | 1.23 | 6.05 | 2.92 | 8.66 | |
| Dextromethorphan | 0.99 | 6.41 | 7.03 | −4.16 | −6.84 |
| 1.98 | 5.12 | 6.31 | 6.97 | 12.02 | |
| 9.9 | 4.75 | 2.26 | 8.77 | 9.52 | |
| 99 | 3.69 | 1.85 | 8.55 | 10.61 | |
| Chloroxazone | 1.287 | 5.81 | 4.53 | 6.83 | 7.13 |
| 2.574 | 2.33 | 3.75 | 6.32 | 9.06 | |
| 12.87 | 2.69 | 5.97 | 2.91 | 4.75 | |
| 257.4 | 5.13 | 4.32 | 5.45 | 8.27 | |
| Midazolam | 2.005 | 2.01 | 7.08 | 5.86 | 7.85 |
| 4.01 | 1.89 | 4.93 | −3.7.83 | −8.33 | |
| 401 | 1.13 | 6.12 | 3.39 | 4.27 | |
| 4010 | 0.71 | 0.63 | 4.11 | 8.31 | |
| Compounds | Spiked Conc. (ng/mL) | Extraction Recovery (%) | Matrix Effect (%) | ||
|---|---|---|---|---|---|
| Mean ± SD | RSD (%) | Intra-Day | Inter-Day | ||
| Phenacetin | 2.01 | 81.48 ± 5.97 | 7.33 | 102.33 ± 7.45 | 7.22 |
| 1609.60 | 90.01 ± 3.26 | 3.62 | 92.25 ± 4.27 | 4.64 | |
| Bupropion | 2.03 | 88.95 ± 6.84 | 7.61 | 93.51 ± 6.30 | 6.56 |
| 203.40 | 94.92 ± 3.40 | 3.22 | 111.13 ± 4.16 | 3.77 | |
| Diclofenac | 2.02 | 92.74 ± 4.25 | 3.67 | 85.63 ± 4.65 | 4.84 |
| 404 | 90.20 ± 4.58 | 8.98 | 89.02 ± 8.03 | 3.99 | |
| Omeprazole | 2.03 | 91.66 ± 5.25 | 4.51 | 85.64 ± 10.13 | 6.89 |
| 406 | 93.49 ± 5.75 | 8.76 | 91.21 ± 4.63 | 4.56 | |
| Dextromethorphan | 1.98 | 93.54 ± 3.68 | 12.58 | 95.23 ± 54.0 | 5.34 |
| 99 | 94.61 ± 4.33 | 10.31 | 97.04 ± 6.21 | 6.30 | |
| Chloroxazone | 2.574 | 93.52 ± 5.46 | 3.82 | 88.01 ± 7.82 | 5.46 |
| 257.4 | 101.24 ± 5.17 | 9.82 | 86.84 ± 6.74 | 6.47 | |
| Midazolam | 4.01 | 93.33 ± 5.06 | 4.63 | 84.34 ± 4.96 | 2.66 |
| 4010.00 | 92.43 ± 2.36 | 6.36 | 89.16 ± 4.03 | 4.40 | |
| Compounds | Spiked conc. (ng/mL) | Blood Sample Stored at RT for 2 h | Blood Sample Stored at | Blood Sample for Freeze-Thawing 3 Cycles | Post−Preparative Sample Stored at 4 °C for 24 h | Post−Preparative Sample Stored at RT for 4 h | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| −80 °C for 15 Days | |||||||||||
| Calc. conc | Accuracy | Calc. conc | Accuracy | Calc. conc | Accuracy | Calc. conc | Accuracy | Calc. conc | Accuracy | ||
| (ng/mL) | (%) | (ng/mL) | (%) | (ng/mL) | (%) | (ng/mL) | (%) | (ng/mL) | (%) | ||
| Phenacetin | 10.08 | 9.79 ± 0.20 | −2.9 | 9.85 ± 1.51 | −2.25 | 9.73 ± 0.24 | −3.5 | 9.97 ± 0.28 | 0.28 | 9.77 ± 0.50 | −2.26 |
| 201.2 | 203.6 ± 6.66 | 1.22 | 205.00 ± 12.53 | 1.89 | 192.30 ± 5.20 | −4.42 | 196.96 ± 5.13 | −2.12 | 195.72 ± 17.41 | −2.73 | |
| 1609.6 | 1630.19 ± 61.37 | 1.28 | 1560.30 ± 12.44 | −3.06 | 1564.93 ± 32.09 | −2.78 | 1576.26 ± 38.13 | −2.07 | 1561.74 ± 35.53 | −2.97 | |
| Bupropion | 2.3 | 2.42 ± 0.28 | 4.91 | 2.38 ± 0.23 | 3.33 | 2.22 ± 0.39 | −3.33 | 2.35 ± 0.26 | 2.17 | 2.25 ± 0.35 | −2.32 |
| 20.34 | 20.47 ± 1.92 | 0.62 | 20.55 ± 1.92 | 1.02 | 20.98 ± 2.57 | 3.13 | 20.54 ± 0.18 | 0.97 | 19.93 ± 1.45 | −2.02 | |
| 203.4 | 198.67 ± 12.84 | −2.32 | 212.26 ± 22.29 | 4.36 | 196.30 ± 17.49 | −3.49 | 206.79 ± 30.07 | 1.67 | 201.45 ± 29.70 | −0.96 | |
| Diclofenac | 2.02 | 1.95 ± 0.19 | −3.63 | 1.99 ± 0.43 | −1.65 | 2.05 ± 0.27 | 1.32 | 2.07 ± 0.12 | 2.64 | 2.06 ± 0.20 | 2.15 |
| 20.2 | 19.75 ± 2.76 | −2.22 | 20.52 ± 1.06 | 1.6 | 20.58 ± 1.42 | 1.88 | 20.45 ± 3.53 | 1.25 | 19.75 ± 2.76 | −2.22 | |
| 404 | 401.17 ± 19.48 | −0.7 | 404.99 ± 50.97 | 0.24 | 409.20 ± 29.18 | 1.29 | 406.63 ± 39.90 | 0.65 | 395.24 ± 14.96 | −2.17 | |
| Omeprazole | 2.03 | 2.09 ± 0.31 | 2.79 | 2.05 ± 0.34 | 0.99 | 2.00 ± 0.33 | −1.48 | 2.07 ± 0.10 | 1.81 | 1.99 ± 0.24 | −1.97 |
| 203 | 205.96 ± 35.04 | 1.46 | 202.63 ± 30.88 | −0.18 | 209.30 ± 29.66 | 3.1 | 202.63 ± 30.88 | −0.18 | 200.91 ± 35.05 | −1.02 | |
| 406 | 392.14 ± 32.82 | −3.41 | 397.14 ± 35.26 | −2.18 | 400.47 ± 38.99 | −1.36 | 403.81 ± 23.84 | −0.54 | 401.22 ± 31.33 | −1.52 | |
| Dextromethorp−han | 1.98 | 1.96 ± 0.16 | −1.04 | 1.99 ± 0.12 | 0.67 | 1.93 ± 0.13 | −2.53 | 2.01 ± 0.14 | 1.51 | 1.95 ± 0.33 | −1.52 |
| 9.9 | 9.88 ± 0.22 | −0.24 | 10.03 ± 0.23 | 1.31 | 9.99 ± 2.02 | 0.88 | 9.76 ± 0.45 | −1.45 | 9.65 ± 0.22 | −2.49 | |
| 99 | 99.92 ± 2.14 | 0.93 | 97.2 ± 2.56 | −1.82 | 97.87 ± 5.15 | −1.14 | 97.55 ± 4.77 | −1.99 | 100.38 ± 12.69 | 1.39 | |
| Chloroxazone | 2.574 | 2.57 ± 0.37 | −1.18 | 2.56 ± 0.13 | 0.43 | 2.53 ± 0.20 | −0.54 | 2.59 ± 0.22 | 1.83 | 2.45 ± 0.31 | −3.93 |
| 12.87 | 12.99 ± 0.44 | −1.63 | 13.12 ± 0.71 | 1.97 | 12.79 ± 0.44 | −0.6 | 12.79 ± 1.24 | −0.6 | 12.53 ± 1.43 | −2.67 | |
| 257.4 | 250.20 ± 2.88 | 0.96 | 252.73 ± 17.27 | −1.82 | 251.16 ± 10.37 | −2.42 | 254.5 ± 16.71 | −1.13 | 253.83 ± 17.43 | −1.39 | |
| Midazolam | 4.01 | 3.98 ± 0.29 | −0.75 | 3.96 ± 0.27 | −1.33 | 3.94 ± 0.35 | −1.75 | 399.03 ± 11.57 | 2.16 | 4.08 ± 0.17 | 1.75 |
| 401 | 395.70 ± 22.26 | −1.32 | 405.7 ± 26.34 | 1.17 | 405.70 ± 25.59 | 1.17 | 399.03 ± 11.57 | −0.49 | 406.67 ± 15.79 | 1.41 | |
| 4010 | 3969.06 ± 233.65 | −1.02 | 4062.30 ± 108.90 | 1.3 | 4095.64 ± 87.80 | 2.14 | 3962.30 ± 66.21 | −1.18 | 3995.64 ± 112.78 | −0.36 | |
| Analytes | Group | AUC(0–t) (μg·h/L) | AUC(0–∞) (μg·h/L) | Cmax(ug/L) | Tmax(h) | T1/2 (h) |
|---|---|---|---|---|---|---|
| Phenacetin | Blank | 1194.97 ±620.95 | 1276.56 ±617.86 | 1442.54 ±250.87 | 0.14 ±0.04 | 2.46 ±0.94 |
| Treat | 327.63 ** ±228.18 | 353.22 ** ±224.12 | 433.42 ** ±147.95 | 0.16 ±0.06 | 1.38 * ±1.45 | |
| Bupropion | Blank | 165.25 ±2.94 | 201.94 ±24.20 | 101.5 ±14.49 | 0.22 ±0.04 | 3.25 ±1.18 |
| Treat | 51.13 ** ±14.68 | 64.91 ** ±17.01 | 57.97 ** ±18.09 | 0.19 ±0.05 | 4.05 ±1.42 | |
| Diclofenac | Blank | 386.52 ±132.62 | 400.40 ±127.67 | 425.78 ±124.74 | 0.12 ±0.04 | 1.38 ±0.46 |
| Treat | 397.96 ±18.43 | 430.27 ±238.07 | 354.53 ±84.72 | 0.15 ±0.03 | 2.01 ±0.630 | |
| Omeprazole | Blank | 75.30 ±12.22 | 129.70 ±23.10 | 95.76 ±35.93 | 0.083 ±0.01 | 10.86 ±4.08 |
| Treat | 126.86 * ±29.32 | 165.33 * ±29.54 | 159.49 * ±58.26 | 0.081 ±0.01 | 3.76 * ±1.72 | |
| Dextrometho-rphan | Blank | 298.32 ±8.06 | 463.11 ±88.17 | 32.76 ±19.27 | 0.14 ±0.04 | 22.57 ±8.07 |
| Treat | 399 ±46.39 | 758.12 ±599.98 | 61.47 ±62.32 | 0.32 ±0.38 | 23.39 ±7.91 | |
| Chloroxazone | Blank | 218 ±274.51 | 306.99 ±399.66 | 380.06 ±499.13. | 0.50 ±0.52 | 2.38 ±0.78 |
| Treat | 302 ±124.51 | 496.20 ±192.58 | 232.33 ±100.08 | 0.23 ±0.21 | 2.84 ±3.19 | |
| Midazolam | Blank | 6454.1 ±1345.7 | 11194.82 ±6581.06 | 5252.44 ±654.62 | 0.11 ±0.04 | 3.61 ±1.62 |
| Treat | 1558.15 * ±732.44 | 4712.50 * ±1748.06 | 903.98 ** ±255.58 | 0.15 ±0.03 | 17.74 ** ±6.22 |
| CYPs | Forward Primer Sequence | Reverse Primer Sequence |
|---|---|---|
| 1A2 | GTCACCTCAGGGAATGCTGTG | GTTGACAATCTTCTCCTGAGG |
| 2B3 | AGGACCCCGTCCCTTACC | CCGGCCAGAGAAAGCCTC |
| 2C11 | CTGCTGCTGCTGAAACACG | TTTCATGCAGGGGCTCCG |
| 2C13 | TGGTCCACGAGGTTCAGAGATACA | GGTTGGGAAACTCCTTGCTGTCAT |
| 2D4 | TGCGAGAGGCACTGGTGA | CGTGGTCCAAAGCCCGAC |
| 2E1 | GACCTTTCCCTCTTCCCATCCTTG | GTAGCACCTCCTTGACAGCCTTG |
| 3A1 | GGCAAACCTGTCCCTGTGAAAGA | CTGGCGTGAGGAATGGAAAGAGT |
| GAPDH | TGCTGAGTATGTCGTGGAG | GTCTTCTGAGTGGCAGTGAT |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, Y.-L.; Zhao, F.; Yin, J.-T.; Liang, C.-J.; Niu, X.-L.; Qiu, Z.-H.; Zhang, L.-T. Two Approaches for Evaluating the Effects of Galangin on the Activities and mRNA Expression of Seven CYP450. Molecules 2019, 24, 1171. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24061171
Ma Y-L, Zhao F, Yin J-T, Liang C-J, Niu X-L, Qiu Z-H, Zhang L-T. Two Approaches for Evaluating the Effects of Galangin on the Activities and mRNA Expression of Seven CYP450. Molecules. 2019; 24(6):1171. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24061171
Chicago/Turabian StyleMa, Yin-Ling, Feng Zhao, Jin-Tuo Yin, Cai-Juan Liang, Xiao-Li Niu, Zhi-Hong Qiu, and Lan-Tong Zhang. 2019. "Two Approaches for Evaluating the Effects of Galangin on the Activities and mRNA Expression of Seven CYP450" Molecules 24, no. 6: 1171. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24061171