
High Impact: The Role of Promiscuous Binding Sites in Polypharmacology

 , , and
, , and
Abstract
:1. Introduction
2. Results
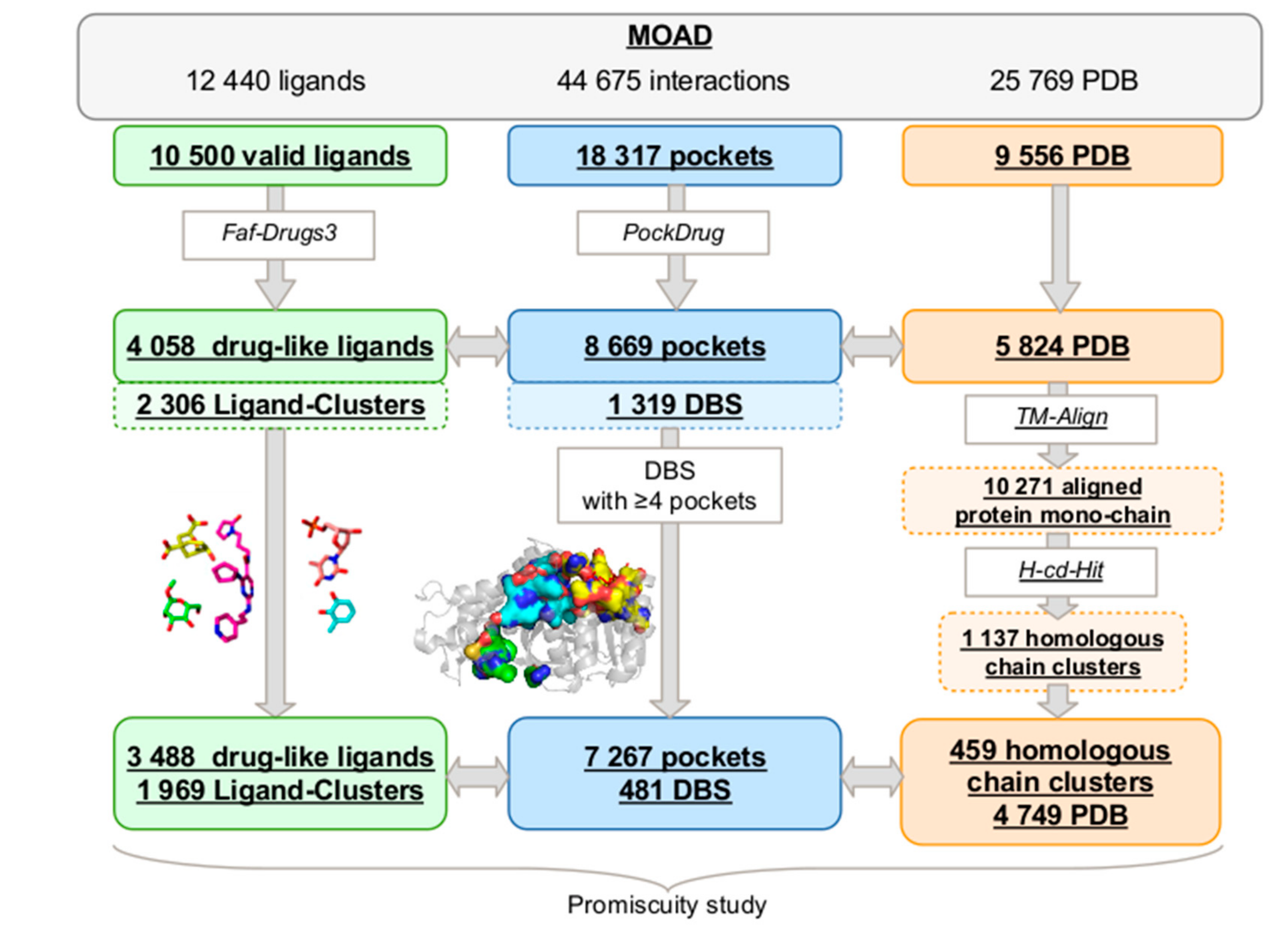
2.1. MOAD Druggable Binding Site Identification
2.1.1. MOAD Protein and Ligand Space
2.1.2. Druggable Binding Site Extraction
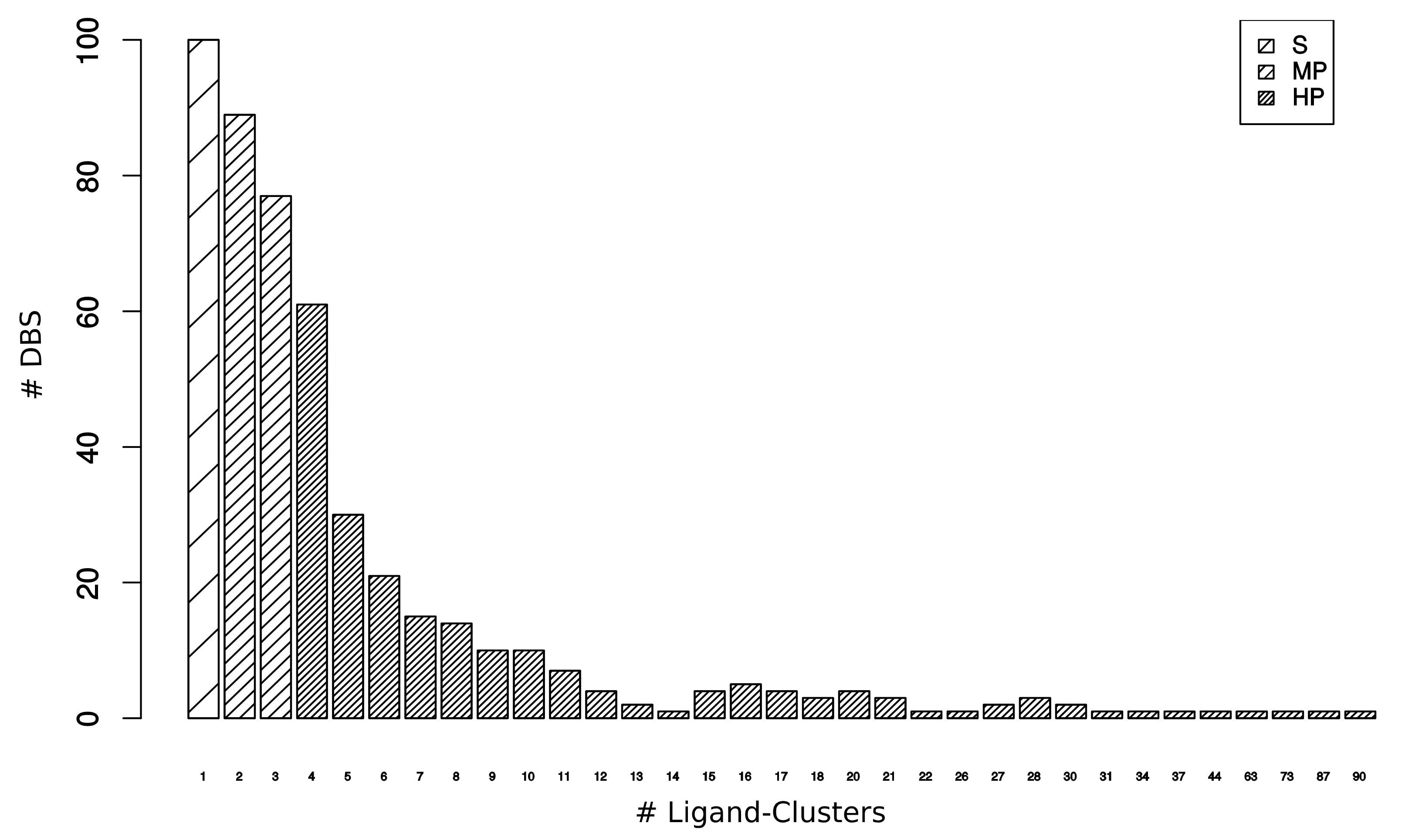
2.2. DBS Promiscuity Characterization
2.2.1. Druggable Binding Site Promiscuity Quantification
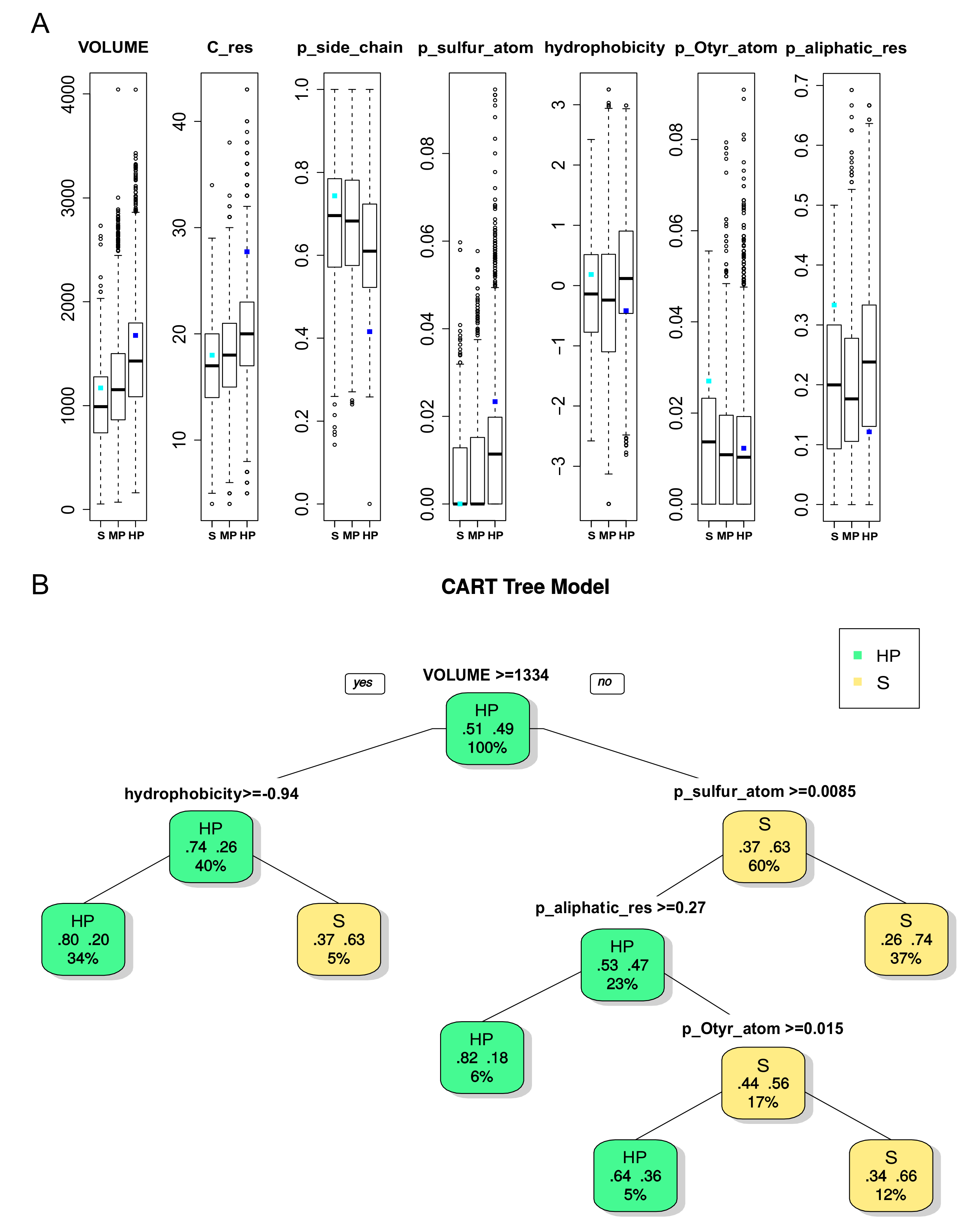
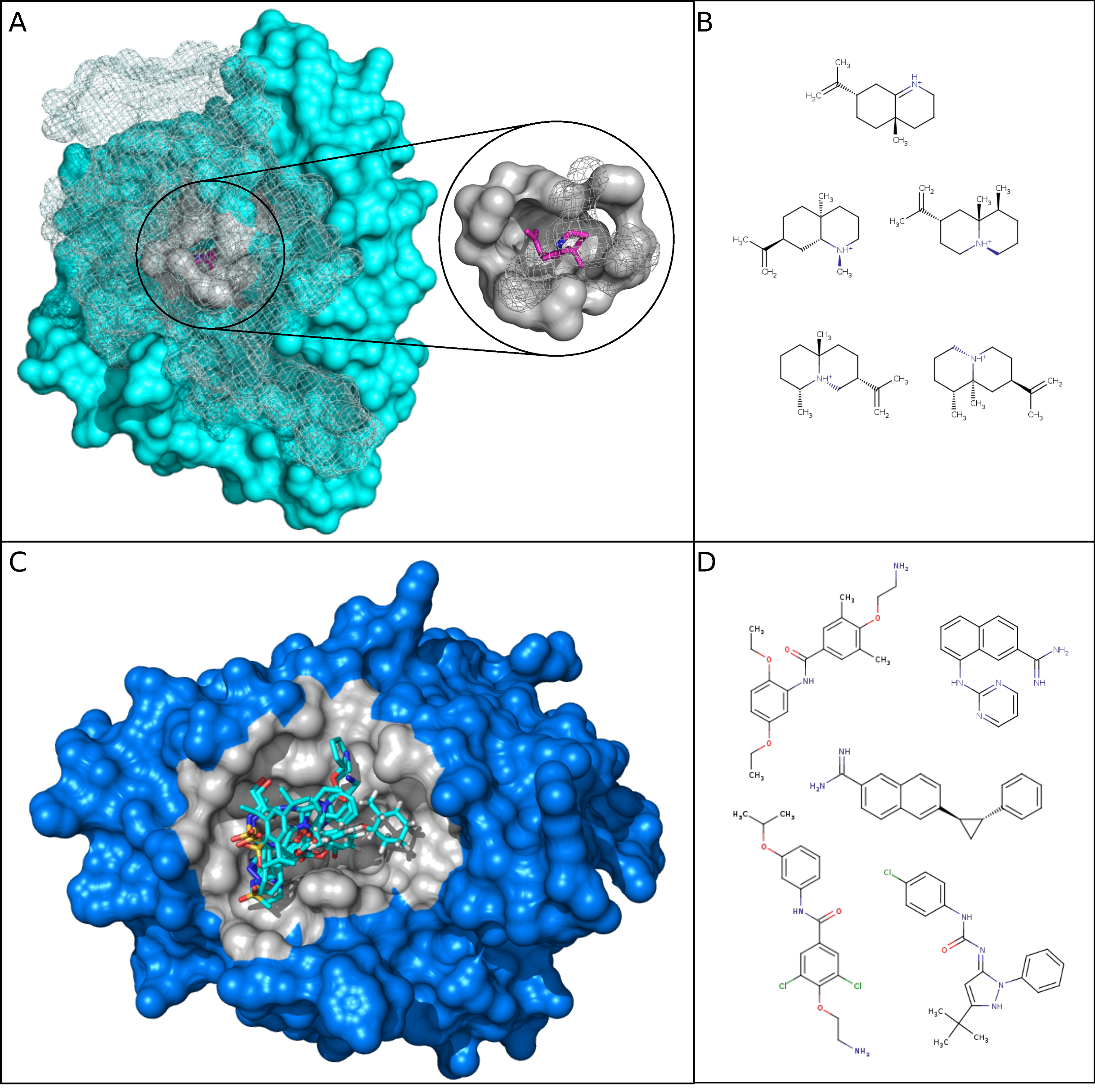
2.2.2. Binding Pocket Characteristics of DBS with Different Promiscuity Levels
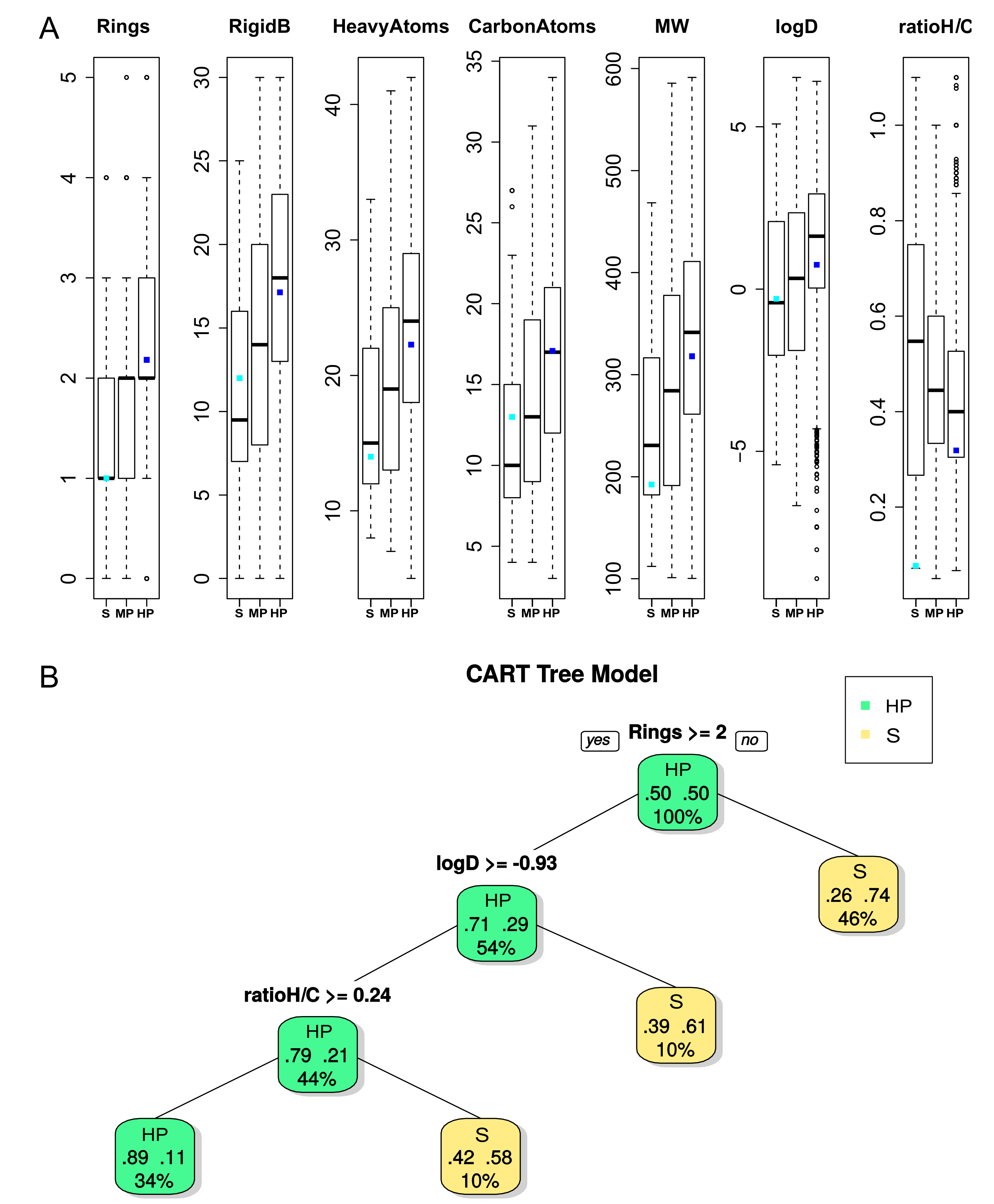
2.2.3. Ligand Characteristics interacting with DBS with Different Promiscuity Levels
2.2.4. Pocket and Ligand Property Correspondence
2.3. DBS Promiscuity Contribution to Multiple Interactions of Ligands with Different MOAD Protein Classes
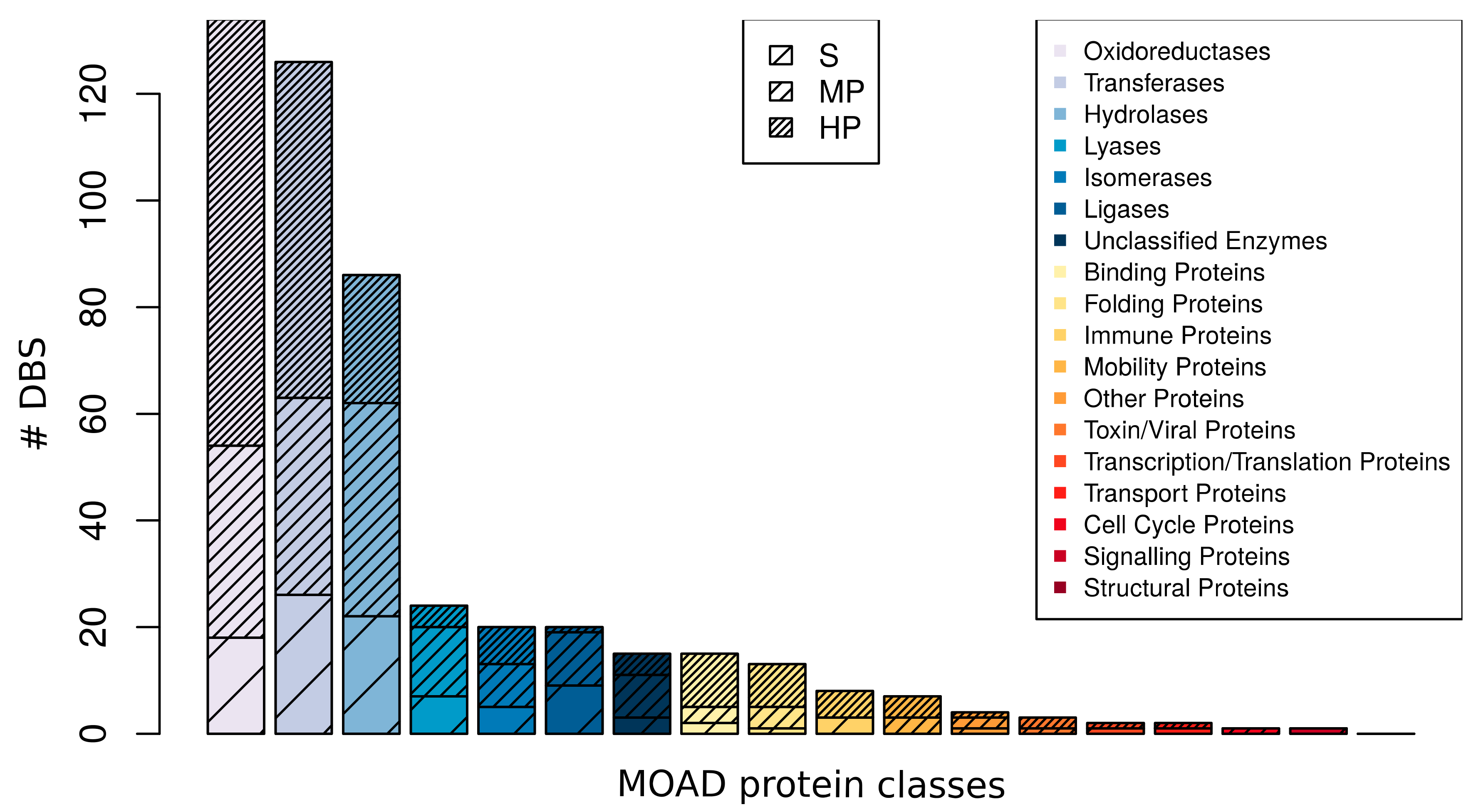
2.3.1. DBS Promiscuity Frequency Related to MOAD Protein Classes
2.3.2. Complementary Study of the Ligand–Cluster Promiscuity
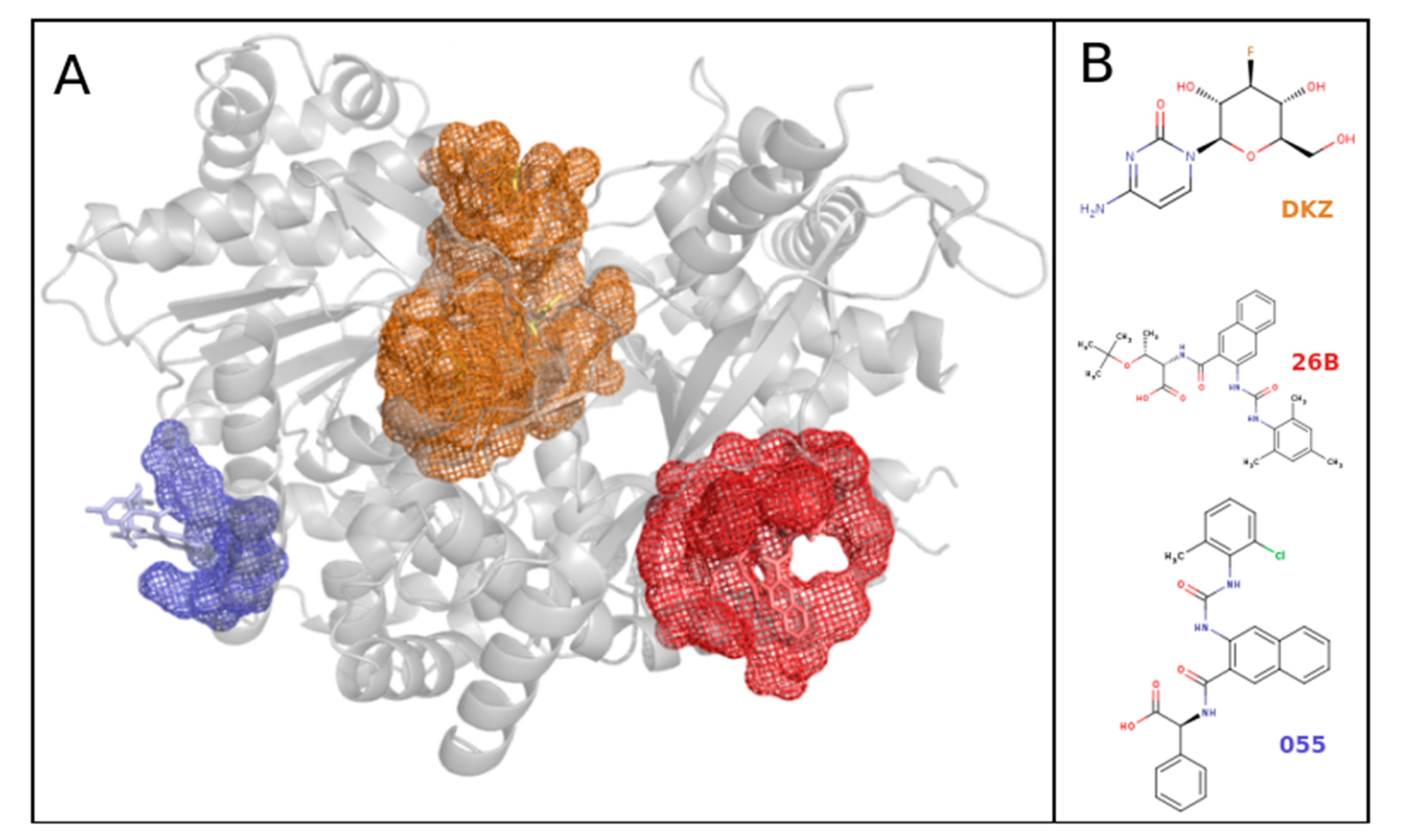
2.3.3. Ligand–Cluster-DBS Interaction Network Examples
3. Discussion
4. Materials and Methods
4.1. MOAD Mining
4.1.1. Drug-Like Ligand Space Analysis and Clustering
Ligand Selection
Drug-Like Ligand Clustering and Description
4.1.2. Protein Space Analysis and Clustering
4.2. MOAD Druggable Binding Sites Extraction Protocol
4.2.1. Ligand-Binding Pocket Estimation
4.2.2. Superimposition of Mono-Chains from Each Homologous Chain Cluster
4.2.3. Cluster of Pockets Associated with a Druggable Binding Site
- let n(i) be the number of atoms of pocket i,
- let g(i) be the barycenter of the n(i) atoms of the pocket i,
- let d(i,j) be the Euclidean distance between g(i) and atom j of pocket i and the maximum value among the n(i) distances between the n(i) atoms and the barycenter g(i) of pocket i,
- let D and be the average value and standard deviation respectively of the population of the p observed values of and
- the cutoff value based on D and from the p pockets from the homologous chain cluster used is (1):
- Two pockets and are fall in the same Pocket-Cluster when meaning that the distance between barycenter and barycenter is lower than the cutoff value.
4.3. Promiscuity Characterization of Druggable Binding Site
4.3.1. Determination of DBS Promiscuity
4.3.2. Analysis of DBS Promiscuity in Terms of Pocket and Ligand Properties
4.4. Ligand–Cluster–DBS Interaction Network Illustration
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Lavecchia, A.; Cerchia, C. In silico methods to address polypharmacology: Current status, applications and future perspectives. Drug Discov. Today 2016, 21, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Haupt, V.J.; Daminelli, S.; Schroeder, M. Drug Promiscuity in PDB: Protein Binding Site Similarity Is Key. PLoS ONE 2013, 8, e65894. [Google Scholar] [CrossRef]
- Zhou, H.; Gao, M.; Skolnick, J.; Gao, M.; Skolnick, J.; Skolnick, J.; Gao, M.; von Eichborn, J.; Paolini, G.; Shapland, R.; et al. Comprehensive prediction of drug-protein interactions and side effects for the human proteome. Sci. Rep. 2015, 5, 11090. [Google Scholar] [CrossRef] [PubMed]
- Mei, Y.; Yang, B. Rational application of drug promiscuity in medicinal chemistry. Future Med. Chem. 2018, 10, 1835–1851. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Bajorath, J. What is the likelihood of an active compound to be promiscuous? Systematic assessment of compound promiscuity on the basis of PubChem confirmatory bioassay data. AAPS J. 2013, 15, 808–815. [Google Scholar] [CrossRef] [PubMed]
- Jalencas, X.; Mestres, J. Identification of Similar Binding Sites to Detect Distant Polypharmacology. Mol. Inform. 2013, 32, 976–990. [Google Scholar] [CrossRef] [PubMed]
- Paolini, G.V.; Shapland, R.H.B.; Van Hoorn, W.P.; Mason, J.S.; Hopkins, A.L. Global mapping of pharmacological space. Nat. Biotechnol. 2006, 24, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Govindaraj, R.G.; Brylinski, M. Comparative assessment of strategies to identify similar ligand-binding pockets in proteins. BMC Bioinform. 2018, 19, 1–17. [Google Scholar] [CrossRef]
- Mestres, J.; Gregori-Puigjané, E.; Valverde, S.; Solé, R.V. The topology of drug-target interaction networks: Implicit dependence on drug properties and target families. Mol. Biosyst. 2009, 5, 1051–1057. [Google Scholar] [CrossRef]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; et al. The Protein Data Bank. Acta Crystallogr. Sect. D Biol. Crystallogr. 2002, 58, 899–907. [Google Scholar] [CrossRef]
- Naderi, M.; Lemoine, J.M.; Govindaraj, R.G.; Kana, O.Z.; Feinstein, W.P.; Brylinski, M. Binding site matching in rational drug design: Algorithms and applications. Brief. Bioinform. 2018, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Barnash, K.D.; James, L.I.; Frye, S.V. Target class drug discovery. Nat. Chem. Biol. 2017, 13, 1053–1056. [Google Scholar] [CrossRef] [PubMed]
- Duran-Frigola, M.; Siragusa, L.; Ruppin, E.; Barril, X.; Cruciani, G.; Aloy, P. Detecting similar binding pockets to enable systems polypharmacology. PLoS Comput. Biol. 2017, 13, e1005522. [Google Scholar] [CrossRef] [PubMed]
- Meyers, J.; Chessum, N.E.A.; Ali, S.; Mok, N.Y.; Wilding, B.; Pasqua, A.E.; Rowlands, M.; Tucker, M.J.; Evans, L.E.; Rye, C.S.; et al. Privileged Structures and Polypharmacology within and between Protein Families. ACS Med. Chem. Lett. 2018, 9, 1199–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Bajorath, J. How Promiscuous Are Pharmaceutically Relevant Compounds? A Data-Driven Assessment. AAPS J. 2013, 15, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Kufareva, I.; Ilatovskiy, A.V.; Abagyan, R. Pocketome: An encyclopedia of small-molecule binding sites in 4D. Nucleic Acids Res. 2012, 40, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Skolnick, J.; Gao, M.; Roy, A.; Srinivasan, B.; Zhou, H. Implications of the small number of distinct ligand binding pockets in proteins for drug discovery, evolution and biochemical function. Bioorgan. Med. Chem. Lett. 2015, 25, 1163–1170. [Google Scholar] [CrossRef] [Green Version]
- Ember, S.W.J.; Zhu, J.-Y.; Olesen, S.H.; Martin, M.P.; Becker, A.; Berndt, N.; Georg, G.I.; Schönbrunn, E. Acetyl-lysine Binding Site of Bromodomain-Containing Protein 4 (BRD4) Interacts with Diverse Kinase Inhibitors. ACS Chem. Biol. 2014, 9, 1160–1171. [Google Scholar] [CrossRef]
- Antolín, A.A.; Jalencas, X.; Yélamos, J.; Mestres, J. Identification of Pim Kinases as Novel Targets for PJ34 with Confounding Effects in PARP Biology. ACS Chem. Biol. 2012, 7, 1962–1967. [Google Scholar] [CrossRef]
- Barelier, S.; Sterling, T.; O’Meara, M.J.; Shoichet, B.K. The Recognition of Identical Ligands by Unrelated Proteins. ACS Chem. Biol. 2015, 10, 2772–2784. [Google Scholar] [CrossRef] [Green Version]
- Feixas, F.; Lindert, S.; Sinko, W.; McCammon, J.A. Exploring the Role of Receptor Flexibility in Structure-Based Drug Discovery. Biophys. Chem. 2014, 186, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Pabon, N.A.; Camacho, C.J. Probing protein flexibility reveals a mechanism for selective promiscuity. eLife 2017, 6, e22889. [Google Scholar] [CrossRef]
- Stank, A.; Kokh, D.B.; Fuller, J.C.; Wade, R.C. Protein Binding Pocket Dynamics. Acc. Chem. Res. 2016, 49, 809–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, M.; Skolnick, J. A Comprehensive Survey of Small-Molecule Binding Pockets in Proteins. PLoS Comput. Biol. 2013, 9, e1003302. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Bajorath, J. Systematic Assessment of Molecular Selectivity at the Level of Targets, Bioactive Compounds, and Structural Analogues. ChemMedChem 2016, 11, 1362–1370. [Google Scholar] [CrossRef]
- Ahmed, A.; Smith, R.D.; Clark, J.J.; Dunbar, J.B., Jr.; Carlson, H.A. Recent improvements to Binding MOAD: A resource for protein–ligand binding affinities and structure. Nucleic Acids Res. 2015, 43, D465–D469. [Google Scholar] [CrossRef] [PubMed]
- Abi Hussein, H.; Geneix, C.; Petitjean, M.; Borrel, A.; Flatters, D.; Camproux, A.-C. Global vision of druggability issues: Applications and perspectives. Drug Discov. Today 2017, 22, 404–415. [Google Scholar] [CrossRef]
- Zhang, Y.; Skolnick, J. TM-align: A protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005, 33, 2302–2309. [Google Scholar] [CrossRef]
- Huang, Y.; Niu, B.; Gao, Y.; Fu, L.; Li, W. CD-HIT Suite: A web server for clustering and comparing biological sequences. Bioinformatics 2010, 26, 680–682. [Google Scholar] [CrossRef]
- Mentese, A.; Erkut, N.; Demir, S.; Yaman, S.O.; Sumer, A.; Erdem, M.; Alver, A.; Sönmez, M.G. Serum carbonic anhydrase I and II autoantibodies in patients with chronic lymphocytic leukaemia. Cent. J. Immunol. 2018, 43, 276–280. [Google Scholar] [CrossRef]
- Chohan, T.A.; Chen, J.-Z.J.-J.; Qian, H.-Y.; Pan, Y.-L.; Chen, J.-Z.J.-J. Molecular modeling studies to characterize N-phenylpyrimidin-2-amine selectivity for CDK2 and CDK4 through 3D-QSAR and molecular dynamics simulations. Mol. BioSyst. 2016, 12, 1250–1268. [Google Scholar] [CrossRef] [PubMed]
- Van Bergen, L.A.H.; Alonso, M.; Palló, A.; Nilsson, L.; De Proft, F.; Messens, J. Revisiting sulfur H-bonds in proteins: The example of peroxiredoxin AhpE. Sci. Rep. 2016, 6, 30369. [Google Scholar] [CrossRef] [PubMed]
- Hussein, H.A.; Borrel, A.; Geneix, C.; Petitjean, M.; Regad, L.; Camproux, A.-C.C. PockDrug-Server: A new web server for predicting pocket druggability on holo and apo proteins. Nucleic Acids Res. 2015, 43, W436–W442. [Google Scholar] [CrossRef] [PubMed]
- Borrel, A.; Regad, L.; Xhaard, H.; Petitjean, M.; Camproux, A.-C.C. PockDrug: A model for predicting pocket druggability that overcomes pocket estimation uncertainties. J. Chem. Inf. Model. 2015, 55, 882–895. [Google Scholar] [CrossRef] [PubMed]
- Degryse, B. The urokinase receptor system as strategic therapeutic target: Challenges for the 21st century. Curr. Pharm. Des. 2011, 17, 1872–1873. [Google Scholar] [CrossRef] [PubMed]
- Cerisier, N.; Regad, L.; Triki, D.; Petitjean, M.; Flatters, D.; Camproux, A.-C.A.C. Statistical Profiling of One Promiscuous Protein Binding Site: Illustrated by Urokinase Catalytic Domain. Mol. Inform. 2017, 36, 1700040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrödinger, LLC. The PyMOL Molecular Graphics System; Version 2.0; Schrödinger, LLC.: New York, NY, USA, 2015. [Google Scholar]
- Cooney, C.M. Triclosan comes under scrutiny. Environ. Health Perspect. 2010, 118, A242. [Google Scholar] [CrossRef]
- Gao, M.; Skolnick, J. APoc: Large-scale identification of similar protein pockets. Bioinformatics 2013, 29, 597–604. [Google Scholar] [CrossRef]
- Liargkova, T.; Eleftheriadis, N.; Dekker, F.; Voulgari, E.; Avgoustakis, C.; Sagnou, M.; Mavroidi, B.; Pelecanou, M.; Hadjipavlou-Litina, D. Small Multitarget Molecules Incorporating the Enone Moiety. Molecules 2019, 24, 199. [Google Scholar] [CrossRef]
- Bajorath, J. Analyzing Promiscuity at the Level of Active Compounds and Targets. Mol. Inform. 2016, 35, 583–587. [Google Scholar] [CrossRef] [PubMed]
- Rifaioglu, A.S.; Atas, H.; Martin, M.J.; Cetin-Atalay, R.; Atalay, V.; Doğan, T. Recent applications of deep learning and machine intelligence on in silico drug discovery: Methods, tools and databases. Brief. Bioinform. 2018, 1–35. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, R.; Tan, Z.; Huang, B.; Zhang, S. Computational polypharmacology: A new paradigm for drug discovery. Expert Opin. Drug Discov. 2017, 12, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Benson, M.L.; Smith, R.D.; Lerner, M.G.; Carlson, H.A. Binding MOAD (Mother Of All Databases). Proteins Struct. Funct. Bioinform. 2005, 60, 333–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Ritchie, D.W.; Nancy, I.; Botanique, J.; Peérez-Nueno, V.I.; Ritchie, D.W. Using Consensus-Shape Clustering To Identify Promiscuous Ligands and Protein Targets and To Choose the Right Query for Shape-Based Virtual Screening. J. Chem. Inf. Model. 2011, 51, 1233–1248. [Google Scholar]
- Miteva, M.A.; Violas, S.; Montes, M.; Gomez, D.; Tuffery, P.; Villoutreix, B.O. FAF-Drugs: Free ADME/tox filtering of compound collections. Nucleic Acids Res. 2006, 34, W738–W744. [Google Scholar] [CrossRef]
- MDL Information Systems, Inc. MACCS Drug Data Report, Release 2000.2; MDL Information Systems, Inc.: San Leandro, CA, USA, 2000. [Google Scholar]
- Durant, J.L.; Leland, B.A.; Henry, D.R.; Nourse, J.G. Reoptimization of MDL Keys for Use in Drug Discovery. J. Chem. Inf. Comput. Sci. 2002, 42, 1273–1280. [Google Scholar] [CrossRef] [Green Version]
- Butina, D. Unsupervised Data Base Clustering Based on Daylight’s Fingerprint and Tanimoto Similarity: A Fast and Automated Way To Cluster Small and Large Data Sets. J. Chem. Inf. Comput. Sci. 1999, 39, 747–750. [Google Scholar] [CrossRef]
- Lagorce, D.; Sperandio, O.; Baell, J.B.; Miteva, M.A.; Villoutreix, B.O. FAF-Drugs3: A web server for compound property calculation and chemical library design. Nucleic Acids Res. 2015, 43, W200–W207. [Google Scholar] [CrossRef]
- Hopkins, A.L.; Groom, C.R. The druggable genome. Nat. Rev. Drug Discov. 2002, 1, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Uddin, R.; Jamil, F. Prioritization of potential drug targets against P. aeruginosa by core proteomic analysis using computational subtractive genomics and Protein-Protein interaction network. Comput. Biol. Chem. 2018, 74, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Schalon, C.; Surgand, J.-S.; Kellenberger, E.; Rognan, D. A simple and fuzzy method to align and compare druggable ligand-binding sites. Proteins Struct. Funct. Bioinform. 2008, 71, 1755–1778. [Google Scholar] [CrossRef] [PubMed]
- Weill, N.; Rognan, D. Alignment-Free Ultra-High-Throughput Comparison of Druggable Protein−Ligand Binding Sites. J. Chem. Inf. Model. 2010, 50, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Yeturu, K.; Chandra, N. PocketMatch: A new algorithm to compare binding sites in protein structures. BMC Bioinform. 2008, 9, 543. [Google Scholar] [CrossRef] [PubMed]
- Feldman, H.J.; Labute, P. Pocket similarity: Are alpha carbons enough? J. Chem. Inf. Model. 2010, 50, 1466–1475. [Google Scholar] [CrossRef] [PubMed]
- Le Guilloux, V.; Schmidtke, P.; Tuffery, P. Fpocket: An open source platform for ligand pocket detection. BMC Bioinform. 2009, 10, 168. [Google Scholar] [CrossRef]
- Schneider, P.; Schneider, G. A Computational Method for Unveiling the Target Promiscuity of Pharmacologically Active Compounds. Angew. Chem. Int. Ed. 2017, 56, 11520–11524. [Google Scholar] [CrossRef]
- Breiman, L.; Friedman, J.; Stone, C.J.; Olshen, R.A. Classification and Regression Trees, 1st ed.; Chapman and Hall/CRC: Boca Raton, FL, USA, 1984; Volume 1, ISBN 978-0412048418. [Google Scholar]
- Zhang, P. Model Selection via Multifold Cross Validation. Ann. Stat. 1993, 21, 299–313. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2014. [Google Scholar]
- Therneau, T.M.; Atkinson, E.J. An Introduction to Recursive Partitioning Using the RPART Routine; Mayo Clinic, Division Of Biomedical Statistics And Informatics: Rochester, MN, USA, 1997. [Google Scholar]
- Csardi, G.; Nepusz, T. The igraph software package for complex network research. InterJournal Complex Syst. 2006, 1695, 1–9. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DBS | ||||
|---|---|---|---|---|
| S | 100 | (20.8%) | 791 | (10.9%) |
| MP | 166 | (34.5%) | 1447 | (19.9%) |
| HP | 215 | (44.7%) | 5029 | (69.2%) |
| Total | 481 | (100.0%) | 7267 | (100.0%) |
| Ligand–Cluster | Occurrence (Frequency) | Tanimoto coefficient average (std. dev.) | Corresponding Ligands: Occurrence (Frequency) | |||
|---|---|---|---|---|---|---|
| Dedicated to | S DBS | 29 | (1.5%) | 0.24 (0.11) | 53 | (1.5%) |
| MP DBS | 182 | (9.2%) | 0.29 (0.12) | 257 | (7.4%) | |
| HP DBS | 1631 | (82.8%) | 0.33 (0.13) | 2621 | (75.1%) | |
| Mixed | 127 | (6.4%) | 0.28 (0.13) | 557 | (16.0%) | |
| ALL | 1969 | (100.0%) | 0.33 (0.13) | 3488 | (100.0%) | |
| Ligand–Cluster | Total | Selective | Promiscuous | Promiscuous | Promiscuous |
|---|---|---|---|---|---|
| Occurrence | Occurrence | Occurrence | Number of DBS: Average (sd) | Number of Protein Class: Average (sd) | |
| Selective DBS | 68 | 29 | 39 | 6.7 (5.37) | 2.9 (1.45) |
| MP DBS | 301 | 168 | 133 | 4.2 (3.68) | 2.1 (1.32) |
| HP DBS | 1753 | 1421 | 332 | 3.2 (2.60) | 1.8 (1.02) |
| All DBS1 | 1969 | 1618 | 351 | 3.1 (2.54) | 1.8 (1.02) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cerisier, N.; Petitjean, M.; Regad, L.; Bayard, Q.; Réau, M.; Badel, A.; Camproux, A.-C. High Impact: The Role of Promiscuous Binding Sites in Polypharmacology. Molecules 2019, 24, 2529. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24142529
Cerisier N, Petitjean M, Regad L, Bayard Q, Réau M, Badel A, Camproux A-C. High Impact: The Role of Promiscuous Binding Sites in Polypharmacology. Molecules. 2019; 24(14):2529. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24142529
Chicago/Turabian StyleCerisier, Natacha, Michel Petitjean, Leslie Regad, Quentin Bayard, Manon Réau, Anne Badel, and Anne-Claude Camproux. 2019. "High Impact: The Role of Promiscuous Binding Sites in Polypharmacology" Molecules 24, no. 14: 2529. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24142529