
Synthesis of Uracil-Iodonium(III) Salts for Practical Utilization as Nucleobase Synthetic Modules

 , ,
, ,
Abstract
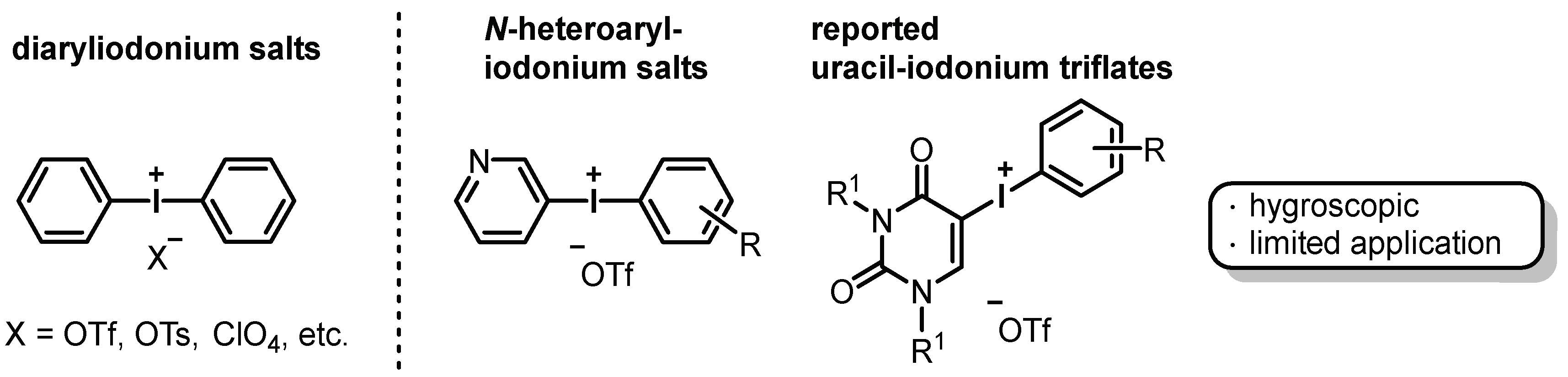
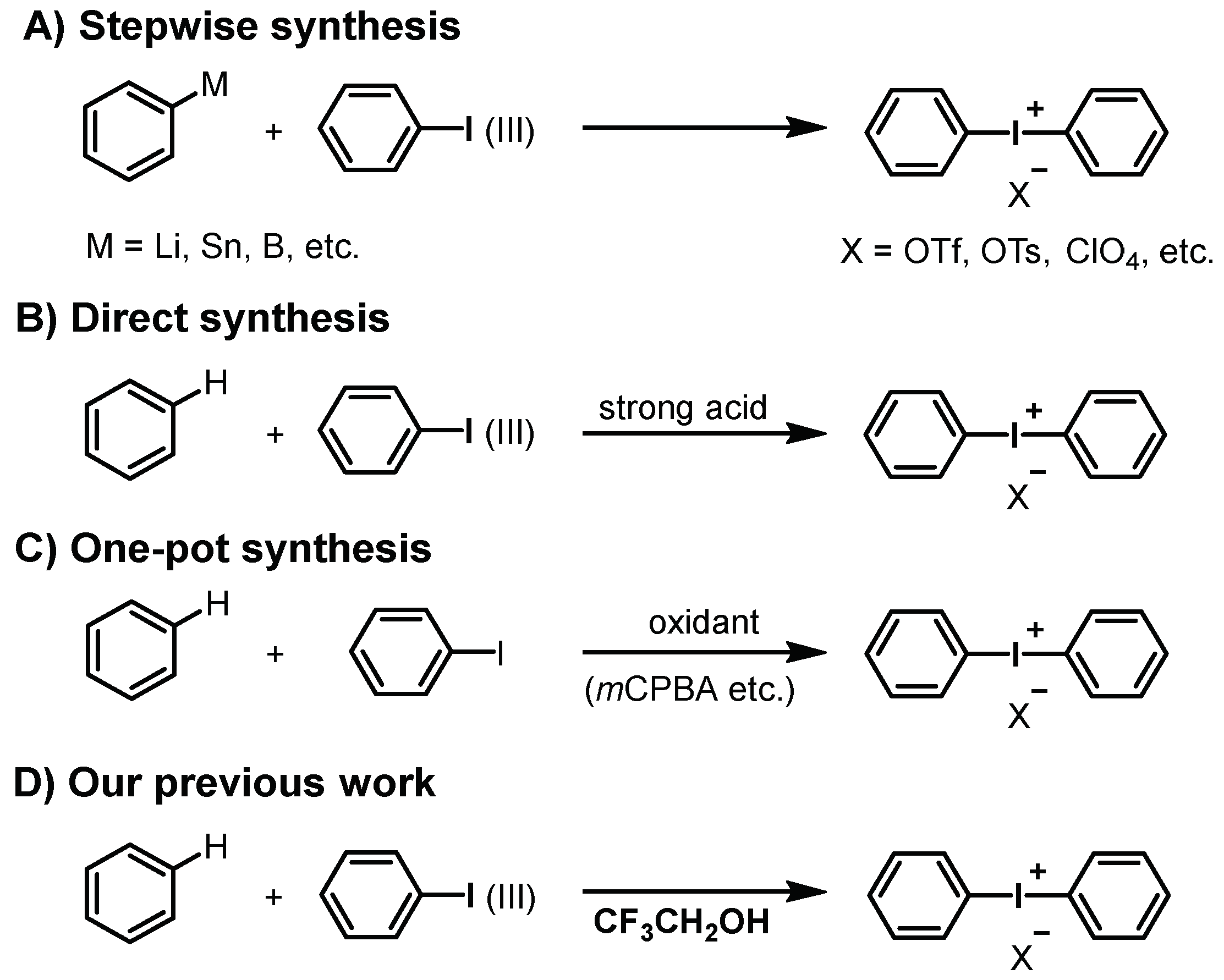
:1. Introduction
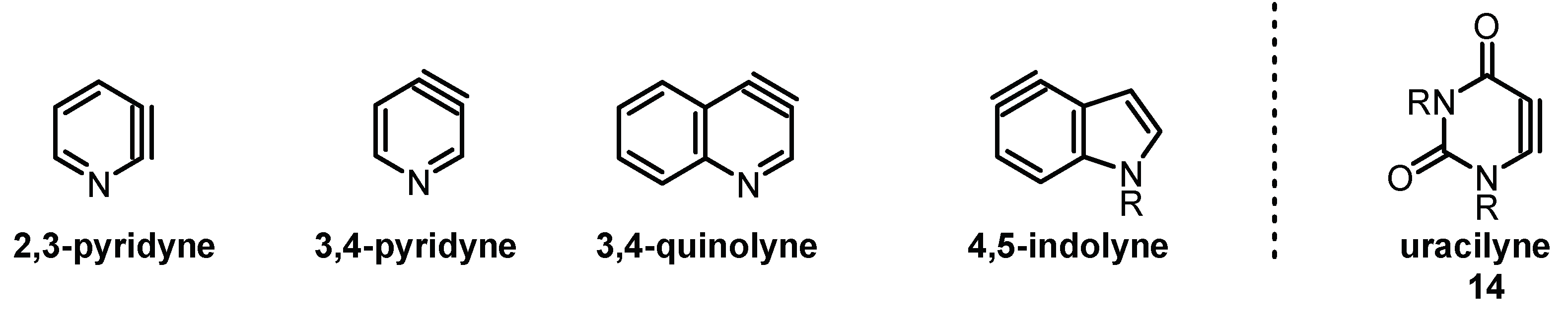
2. Results and Discussions
3. Conclusions
4. Experimental Section
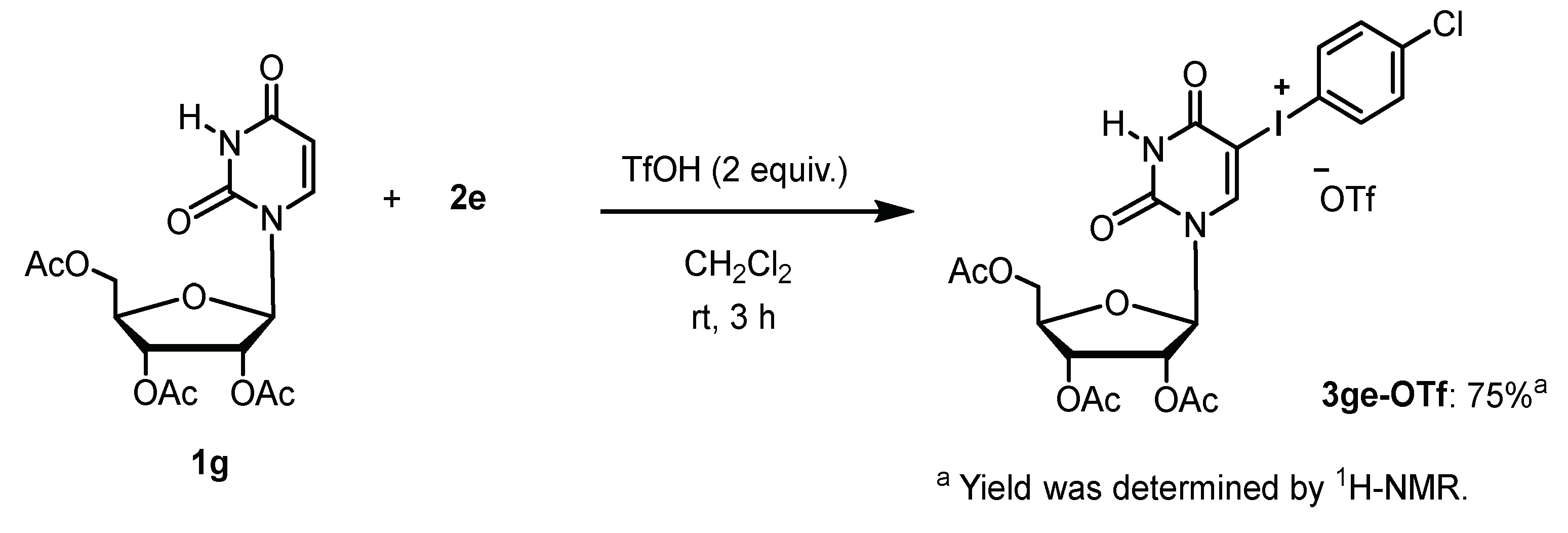
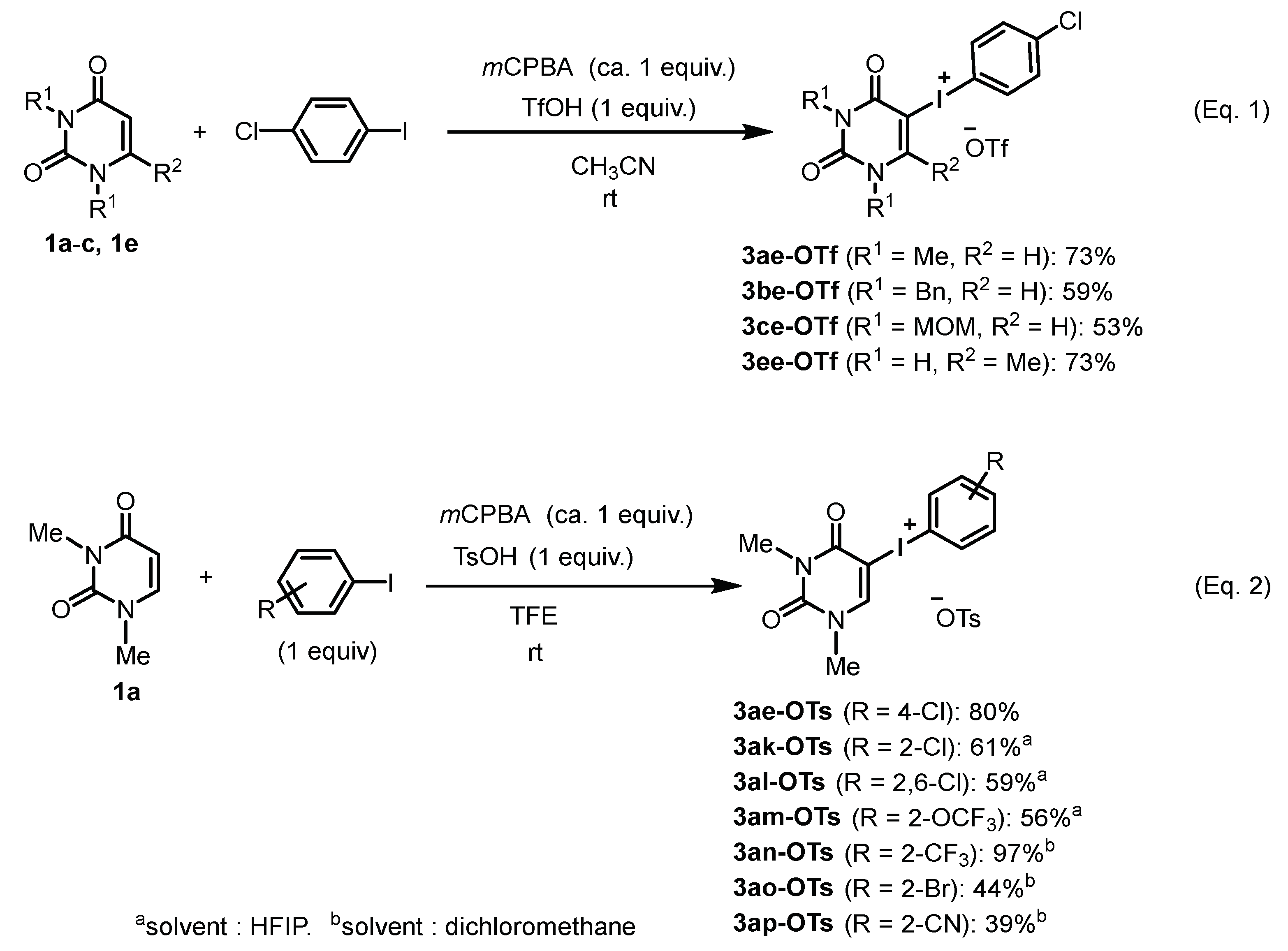
4.1. General Procedure for the Synthesis of Uracil-Iodonium(III) Triflates
4.2. General Procedure for the Synthesis of Uracil-Iodonium(III) Tosylates
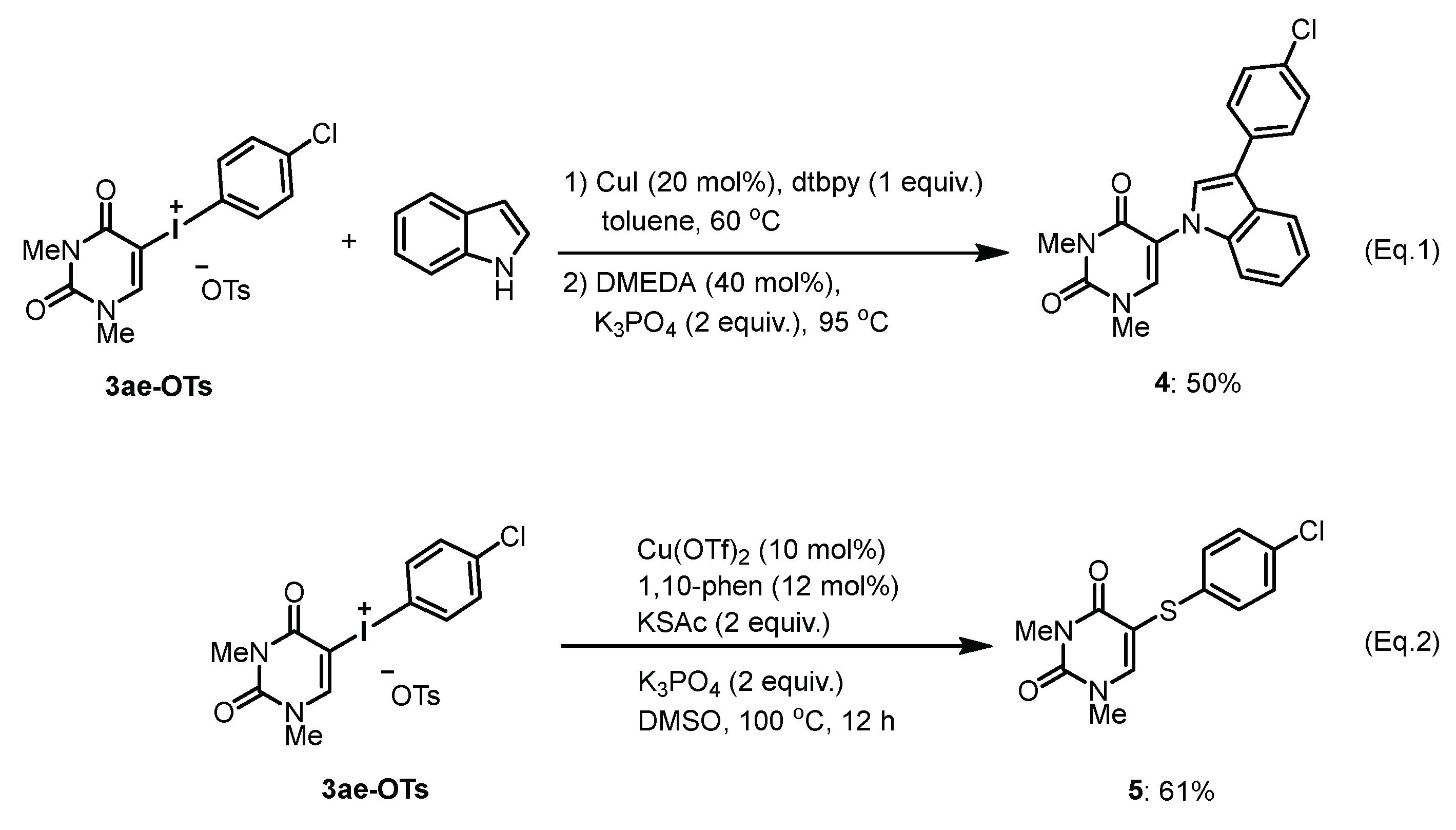
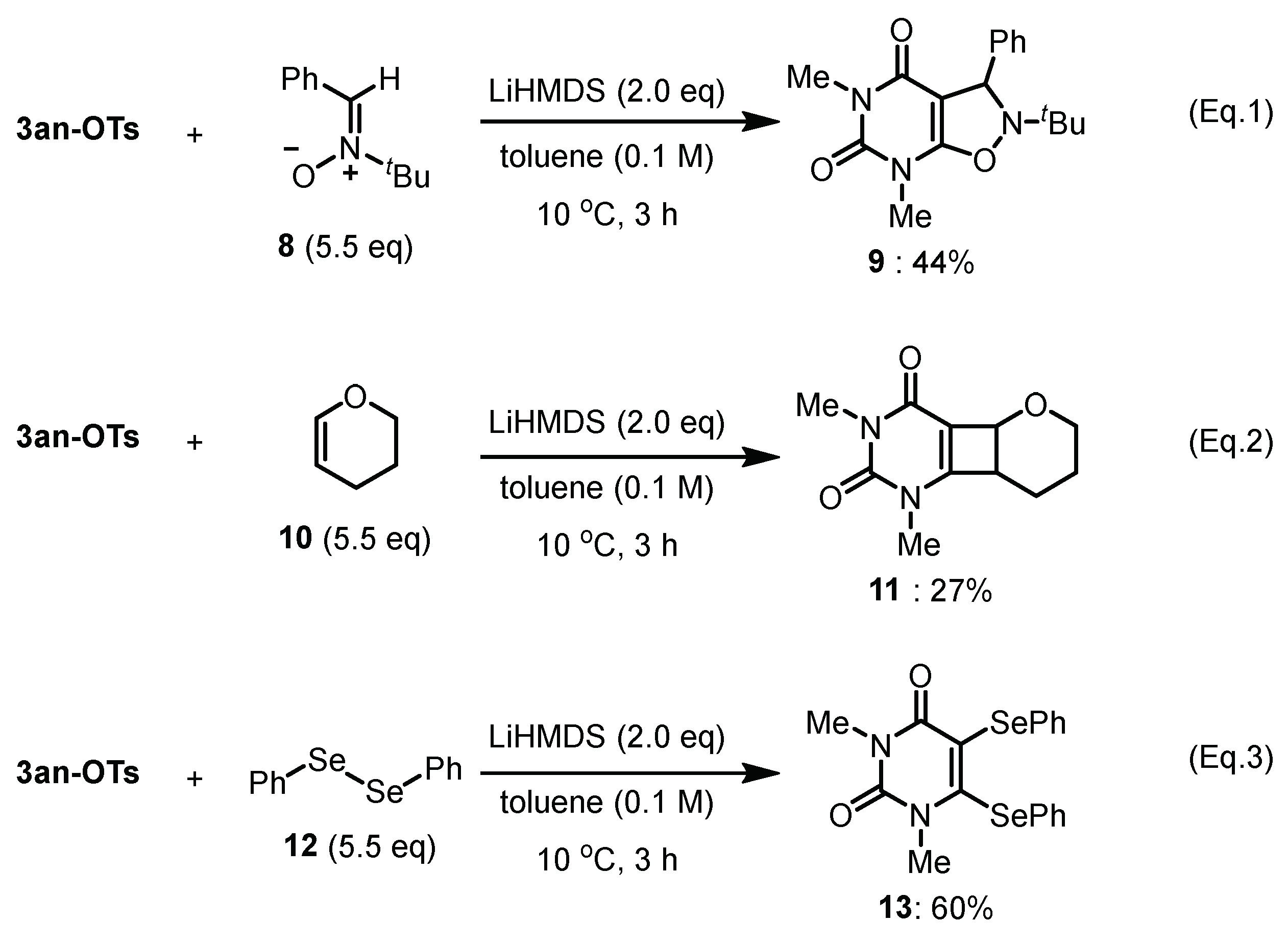
4.3. Reaction of Uracil-Iodonium(III) Tosylates with Arynophiles
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References and Notes
- Varvoglis, A. Hypervalent Iodine in Organic Synthesis; Academic Press: San Diego, CA, USA, 1997. [Google Scholar]
- Ochiai, M. Chemistry of Hypervalent Compounds; Akiba, K., Ed.; Wiley-VCH: New York, NY, USA, 1999; Volume 12. [Google Scholar]
- Zhdankin, V.V.; Stang, P.J. Recent developments in the chemistry of polyvalent iodine compounds. Chem. Rev. 2002, 102, 2523–2584. [Google Scholar] [CrossRef] [PubMed]
- Wirth, T. (Ed.) Hypervalent Iodine Chemistry; Springer-Verlag: Berlin, Germany, 2003. [Google Scholar]
- Merritt, E.A.; Olofsson, B. Diaryliodonium salts: A journey from obscurity to fame. Angew. Chem. Int. Ed. 2009, 48, 9052–9070. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, A.; Zhdankin, V.V. Advances in Synthetic Applications of Hypervalent Iodine Compounds. Chem. Rev. 2016, 116, 3328–3435. [Google Scholar] [CrossRef] [PubMed]
- Aradi, K.; Tóth, B.L.; Tolnai, G.L.; Novák, Z. Diaryliodonium Salts in Organic Syntheses: A Useful Compound: Class for Novel Arylation Strategies. Synlett 2016, 27, 1456–1485. [Google Scholar] [CrossRef]
- Stang, P.J. Polyvalent Iodine in Organic Chemistry. J. Org. Chem. 2003, 68, 2997–3008. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, V.K.; Olofsson, B. Enantioselective alpha-arylation of cyclohexanones with diaryl iodonium salts: Application to the synthesis of (-)-epibatidine. Angew. Chem. Int. Ed. 2005, 44, 5516–5519. [Google Scholar] [CrossRef] [PubMed]
- Stang, P.J.; Chen, K. Hybrid, Iodonium-Transition Metal, Cationic Tetranuclear Macrocyclic Squares. J. Am. Chem. Soc. 1995, 117, 1667–1668. [Google Scholar] [CrossRef]
- Beringer, F.M.; Nathan, R.A. Diaryliodonium salts from aryllithium reagents with trans-chlorovinyliodoso dichloride. J. Org. Chem. 1969, 34, 685–689. [Google Scholar] [CrossRef]
- Stang, P.J.; Olenyuk, B.; Chen, K. Preparation of Nitrogen-Containing Bis(heteroaryl)iodonium Salts. Synthesis 1995, 937–938. [Google Scholar] [CrossRef]
- Pirguliyev, N.S.; Brel, V.K.; Akhmedov, N.G.; Zefirov, N.S. An Efficient Ligand Exchange Reaction of (E)-[(β-(Trifluoromethanesulfonyloxy)ethenyl](phenyl)iodonium Triflates with Aryl-and Alkynyllithium Reagents Leading to Diaryl- and Alkynyliodonium Triflates. Synthesis 2000, 81–83. [Google Scholar] [CrossRef]
- Carroll, M.A.; Nairne, J.; Woodcraft, J.L. Diaryliodonium salts: A solution to 3-[18F] fluoropyridine. J. Label. Compd. Radiopharm. 2007, 50, 452–454. [Google Scholar] [CrossRef]
- Bielawski, M.; Malmgren, J.; Pardo, L.M.; Wikmark, Y.; Olofsson, B. One-pot synthesis and applications of N-heteroaryl iodonium salts. ChemistryOpen 2014, 3, 19–22. [Google Scholar] [CrossRef] [PubMed]
- Huryn, D.M.; Okabe, M. AIDS-driven nucleoside chemistry. Chem. Rev. 1992, 92, 1745–1768. [Google Scholar] [CrossRef]
- Knapp, S. Synthesis of Complex Nucleoside Antibiotics. Chem. Rev. 1995, 95, 1859–1876. [Google Scholar] [CrossRef]
- Roh, K.R.; Kim, J.Y.; Kim, Y.H. Novel Synthesis of 5-Phenyliodonium Triflate Substituted Uracil Nucleosides. Chem. Lett. 1998, 27, 1095–1096. [Google Scholar] [CrossRef]
- Roh, K.R.; Kim, J.Y.; Kim, Y.H. Palladium Catalyzed Alkenylation or Alkynylation at C-5 of Uracil Nucleosides Using Novel Phenyliodonium Triflate. Tetrahedron Lett. 1999, 40, 1903–1906. [Google Scholar] [CrossRef]
- Toh, Q.Y.; McNally, A.; Vera, S.; Erdmann, N.; Gaunt, M.J. Organocatalytic C-H bond arylation of aldehydes to bis-heteroaryl ketones. J. Am. Chem. Soc. 2013, 135, 3772–3775. [Google Scholar] [CrossRef] [PubMed]
- Modha, S.G.; Greaney, M.F. Atom-economical transformation of diaryliodonium salts: tandem C-H and N-H arylation of indoles. J. Am. Chem. Soc. 2015, 137, 1416–1419. [Google Scholar] [CrossRef]
- Altomonte, S.; Telu, S.; Lu, S.; Pike, V.W. Pd(0)-Mediated 11C-Carbonylation of Aryl(mesityl)iodonium Salts as a Route to [11C]Arylcarboxylic Acids and Derivatives. J. Org. Chem. 2017, 82, 11925–11932. [Google Scholar] [CrossRef] [PubMed]
- Takenaga, N.; Ueda, S.; Hayashi, T.; Dohi, T.; Kitagaki, S. Facile Synthesis of Stable Uracil-Iodonium(III) Salts with Various Counterions. Heterocycles 2018, 97, 1248–1256. [Google Scholar] [CrossRef]
- Beringer, F.M.; Dehn, J.W., Jr.; Winicov, M. Diaryliodonium Salts. XIV. Reactions of Organometallic Compounds with Iodosobenzene Dichlorides and with Iodonium Salts. J. Am. Chem. Soc. 1960, 82, 2948–2952. [Google Scholar] [CrossRef]
- Koser, G.F.; Wettach, R.H.; Smith, C.S. New methodology in iodonium salt synthesis. Reactions of [hydroxy (tosyloxy) iodo] arenes with aryltrimethylsilanes. J. Org. Chem. 1980, 45, 1543–1544. [Google Scholar] [CrossRef]
- Pike, V.W.; Butt, F.; Shah, A.; Widdowson, D.A. Facile synthesis of substituted diaryliodonium tosylates by treatment of aryltributylstannanes with Koser’s reagent. J. Chem. Soc. Perkin Trans. 1 1999, 245–248. [Google Scholar] [CrossRef]
- Carroll, M.A.; Pike, V.W.; Widdowson, D.A. New synthesis of diaryliodonium sulfonates from arylboronic acids. Tetrahedron Lett. 2000, 41, 5393–5396. [Google Scholar] [CrossRef]
- Kitamura, T.; Matsuyuki, J.; Taniguchi, H. Improved Preparation of Diaryliodonium Triflates. Synthesis 1994, 147–148. [Google Scholar] [CrossRef]
- Bielawski, M.; Olofsson, B. High-yielding one-pot synthesis of diaryliodonium triflates from arenes and iodine or aryl iodides. Chem. Commun. 2007, 2521–2523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dohi, T.; Ito, M.; Morimoto, K.; Minamitsuji, Y.; Takenaga, N.; Kita, Y. Versatile direct dehydrative approach for diaryliodonium(III) salts in fluoroalcohol media. Chem. Commun. 2007, 4152–4154. [Google Scholar] [CrossRef] [PubMed]
- Jalalian, N.; Petersen, T.B.; Olofsson, B. Metal-free arylation of oxygen nucleophiles with diaryliodonium salts. Chem. Eur. J. 2012, 18, 14140–14149. [Google Scholar] [CrossRef]
- Wang, M.; Wei, J.; Fan, Q.; Jiang, X. Cu(II)-catalyzed sulfide construction: both aryl groups utilization of intermolecular and intramolecular diaryliodonium salt. Chem. Commun. 2017, 2918–2922. [Google Scholar] [CrossRef]
- Takenaga, N.; Ueda, S.; Hayashi, T.; Dohi, T.; Kitagaki, S. Vicinal Functionalization of Uracil Heterocycles with Base Activation of Iodonium(III) Salts. Heterocycles 2019, 99, 865–874. [Google Scholar] [CrossRef]
- Matsumoto, T.; Sohma, T.; Hatazaki, S.; Suzuki, K. On the Regiochemistry of Cycloaddition of Unsymmetrical Aryne with Nitrone Remarkable Effect of Trialkylsilyl Substituent. Synlett 1993, 11, 843–846. [Google Scholar] [CrossRef]
- Yedulla, V.R.; Pradhan, P.; Yang, L.; Lakshman, M.K. Cycloaddition of Arynes and Cyclic Enol Ethers as a Platform for Access to Stereochemically Defined 1,2-Disubstituted Benzocyclobutenes. Eur. J. Org. Chem. 2015, 2015, 750–764. [Google Scholar] [CrossRef] [PubMed]
- Toledo, F.T.; Marques, H.; Comasseto, J.V.; Raminelli, C. The diorgano dichalcogenides addition to benzyne under mild conditions. Tetrahedron Lett. 2007, 48, 8125–8127. [Google Scholar] [CrossRef]
- Okuyama, T.; Takino, T.; Sueda, T.; Ochiai, M. Solvolysis of Cyclohexenyliodonium Salt, a New Precursor for the Vinyl Cation: Remarkable Nucleofugality of the Phenyliodonio Group and Evidence for Internal Return from an Intimate Ion—Molecule Pair. J. Am. Chem. Soc. 1995, 117, 3360–3367. [Google Scholar] [CrossRef]
- Cheong, P.H.; Paton, R.S.; Bronner, S.M.; Im, G.Y.; Garg, N.K.; Houk, K.N. Indolyne and aryne distortions and nucleophilic regioselectivites. J. Am. Chem. Soc. 2010, 132, 1267–1269. [Google Scholar] [CrossRef]
- Bronner, S.M.; Mackey, J.L.; Houk, K.N.; Garg, N.K. Steric effects compete with aryne distortion to control regioselectivities of nucleophilic additions to 3-silylarynes. J. Am. Chem. Soc. 2012, 134, 13966–13969. [Google Scholar] [CrossRef]
- Medina, J.M.; Mackey, J.L.; Garg, N.K.; Houk, K.N. The role of aryne distortions, steric effects, and charges in regioselectivities of aryne reactions. J. Am. Chem. Soc. 2014, 136, 15798–15805. [Google Scholar] [CrossRef]
- Picazo, E.; Houk, K.N.; Garg, N.K. Computational predictions of substituted benzyne and indolyne regioselectivities. Tetrahedron Lett. 2015, 56, 3511–3514. [Google Scholar] [CrossRef] [Green Version]
- Heaney, H. The Benzyne and Related Intermediates. Chem. Rev. 1962, 62, 81–97. [Google Scholar] [CrossRef]
- Reinecke, M.G. Heteroarynes. Tetrahedron 1982, 38, 427–498. [Google Scholar] [CrossRef]
- Kauffmann, T.; Wirthwein, R. Progress in the Hetaryne Field. Angew. Chem. Int. Ed. 1971, 10, 20–33. [Google Scholar] [CrossRef]
- Bronner, S.M.; Bahnck, K.B.; Garg, N.K. Indolynes as electrophilic indole surrogates: fundamental reactivity and synthetic applications. Org. Lett. 2009, 11, 1007–1010. [Google Scholar] [CrossRef] [PubMed]
- Im, G.Y.; Bronner, S.M.; Goetz, A.E.; Paton, R.S.; Cheong, P.H.; Houk, K.N.; Garg, N.K. Indolyne experimental and computational studies: synthetic applications and origins of selectivities of nucleophilic additions. J. Am. Chem. Soc. 2010, 132, 17933–17944. [Google Scholar] [CrossRef] [PubMed]
- Goetz, A.E.; Garg, N.K. Regioselective reactions of 3,4-pyridynes enabled by the aryne distortion model. Nat. Chem. 2013, 5, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Saito, N.; Nakamura, K.; Shibano, S.; Ide, S.; Minami, M.; Sato, Y. Addition of cyclic ureas and 1-methyl-2-oxazolidone to pyridynes: A new approach to pyridodiazepines, pyridodiazocines, and pyridooxazepines. Org. Lett. 2013, 15, 386–389. [Google Scholar] [CrossRef]
- Fang, Y.; Larock, R.C. Nucleophilic addition to 2, 3-pyridyne and synthesis of benzonaphthyridinones. Tetrahedron 2012, 68, 2819–2826. [Google Scholar] [CrossRef] [PubMed]
- The reaction of 5-iodouracil, instead of iodonium(III) salt 3, with furan 6a under our optimized conditions did not proceed, and cycloadduct 7a was not obtained at all.
- Medina, J.M.; Jackl, M.K.; Susick, R.B.; Garg, N.K. Synthetic studies pertaining to the 2,3-pyridyne and 4,5-pyrimidyne. Tetrahedron 2016, 72, 3629–3634. [Google Scholar] [CrossRef] [Green Version]
- Kazmieczak, P.; Skulski, L.; Karaszkiewicz, L. Syntheses of (Diacetoxyiodo) arenes or Iodylarenes from Iodoarenes, with Sodium Periodate as the Oxidant. Molecules 2001, 6, 881–891. [Google Scholar] [CrossRef]
- Miralles, N.; Romero, R.M.; Fernandez, E.; Muniz, K. A mild carbon-boron bond formation from diaryliodonium salts. Chem. Commun. 2015, 51, 14068–14071. [Google Scholar] [CrossRef]
- Iinuma, M.; Moriyama, K.; Togo, H. Simple and Practical Method for Preparation of [(Diacetoxy) iodo] arenes with Iodoarenes and m-Chloroperoxybenzoic Acid. Synlett 2012, 23, 2663–2666. [Google Scholar] [CrossRef]
- Merritt, E.A.; Carneiro, V.M.T.; Silva, L.F., Jr.; Olofsson, B. Facile synthesis of Koser’s reagent and derivatives from iodine or aryl iodides. J. Org. Chem. 2010, 75, 7416–7419. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.M.B.; Fujita, M.A.; Brocksom, T.J.; Oliveira, K.T. Synthesis and photophysical evaluations of β-fused uracil-porphyrin derivatives. Tetrahedron 2013, 69, 9986–9993. [Google Scholar] [CrossRef]
- Su, T.-L.; Huang, J.-T.; Burchenal, J.H.; Watanabe, K.A.; Fox, J.J. Synthesis and biological activities of 5-deaza analogs of aminopterin and folic acid. J. Med. Chem. 1986, 29, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Cernova, M.; Cerna, I.; Pohl, R.; Hocek, M. Regioselective direct C-H arylations of protected uracils. Synthesis of 5- and 6-aryluracil bases. J. Org. Chem. 2011, 76, 5309–5319. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the products are available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | R | I (III) | TfOH | Product | Yield (%) |
|---|---|---|---|---|---|
| 1 | H | I(OAc)2 (2a) | 2.0 equiv. | 3aa-OTf | hygroscopic |
| 2 | Me | I(OAc)2 (2b) | 2.0 equiv. | 3ab-OTf | decomp. |
| 3 | NO2 | I(OAc)2 (2c) | 2.0 equiv. | 3ac-OTf | 56 |
| 4 | CF3 | I(OAc)2 (2d) | 2.0 equiv. | 3ad-OTf | 75 |
| 5 | Cl | I(OAc)2 (2e) | 2.0 equiv. | 3ae-OTf | 78 |
| 6 | Cl | I(OAc)2 (2e) | 1.0 equiv. | 3ae-OTf | 19 |

| Entry | R1 | R | I (III) | Additive | X | Product | Yield (%) |
|---|---|---|---|---|---|---|---|
| 1 | Me | 4-Cl | I(OH)OTs (2f) | none | OTs | 3af-OTs | 98 |
| 2 a | Me | 4-Cl | I(OH)OTs (2f) | none | OTs | 3af-OTs | 89 |
| 3 | Me | 4-Cl | I(OH)OMs (2g) | none | OMs b | 3ag-OMs | 74 |
| 4 | Me | 4-Cl | I(OAc)2 (2e) | (+)-10-CSA c | (+)-10-OCs d | 3ae-OCs | 74 |
| 5 | Me | 4-Cl | I(OAc)2 (2e) | CF3CO2H | OCOCF3 | 3ae-OCOCF3 | 72 |
| 6 | Me | 4-Cl | I(OAc)2 (2e) | HClO4 | ClO4 | 3ae-ClO4 | 68 |
| 7 | Me | H | I(OH)OTs (2h) | none | OTs | 3ah-OTs | 78 |
| 8 | Me | 4-NO2 | I(OH)OTs (2i) | none | OTs | 3ai-OTs | 20 |
| 9 | Me | 4-CF3 | I(OH)OTs (2j) | none | OTs | 3aj-OTs | 16 |
| 10 | Me | 4-Cl | I(OAc)2 (2e) | TfOH | OTf | 3ae-OTf | 98 |
| 11 | Bn | 4-Cl | I(OAc)2 (2e) | TfOH | OTf | 3be-OTf | 75 |
| 12 | MEM | 4-Cl | I(OAc)2 (2e) | TfOH | OTf | 3fe-OTf | 55 |

| Entry | R1 | 3 | Yield (%) b |
|---|---|---|---|
| 1 | 4-Cl | 3ae-OTs | 31 |
| 2 | 4-CF3 | 3aj-OTs | 20 |
| 3 | 2-Cl | 3ak-OTs | 37 |
| 4 | 2,6-Cl | 3al-OTs | 24 |
| 5 | 2-F | 3aq-OTs | 22 |
| 6 | 2-OCF3 | 3am-OTs | 28 |
| 7 | 2-CF3 | 3an-OTs | 40 c |

| Entry | 6 | X | Y | Product 7 | Yield (%) |
|---|---|---|---|---|---|
 | |||||
| 1 | 6a | H | O | 7a | 40 |
| 2 | 6b | Me | O | 7b | 42 |
| 3 | 6c | H | NPh | 7c | 38 |
| 4 | 6d | H | NBoc | 7d | 45 |
| 5 | 6e | H | N-4-CF3C6H4 | 7e | 42 |
| 6 b |  6f | Ph | O | 7f | 55 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takenaga, N.; Hayashi, T.; Ueda, S.; Satake, H.; Yamada, Y.; Kodama, T.; Dohi, T. Synthesis of Uracil-Iodonium(III) Salts for Practical Utilization as Nucleobase Synthetic Modules. Molecules 2019, 24, 3034. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24173034
Takenaga N, Hayashi T, Ueda S, Satake H, Yamada Y, Kodama T, Dohi T. Synthesis of Uracil-Iodonium(III) Salts for Practical Utilization as Nucleobase Synthetic Modules. Molecules. 2019; 24(17):3034. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24173034
Chicago/Turabian StyleTakenaga, Naoko, Takumi Hayashi, Shohei Ueda, Hiroyuki Satake, Yoichi Yamada, Tetsuya Kodama, and Toshifumi Dohi. 2019. "Synthesis of Uracil-Iodonium(III) Salts for Practical Utilization as Nucleobase Synthetic Modules" Molecules 24, no. 17: 3034. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24173034