Derivatization of Natural Compound β-Pinene Enhances Its In Vitro Antifungal Activity against Plant Pathogens


Abstract
:1. Introduction
2. Results
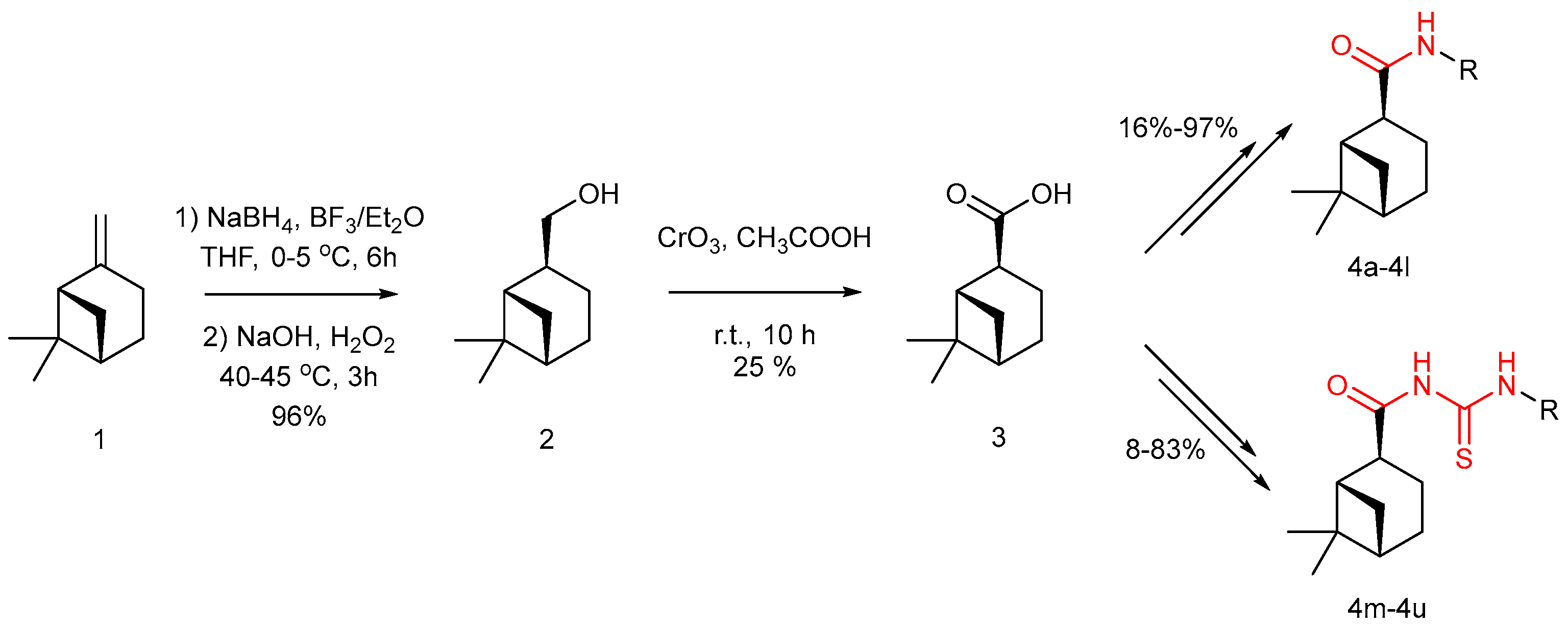
2.1. Chemisty
2.2. Biological Activity
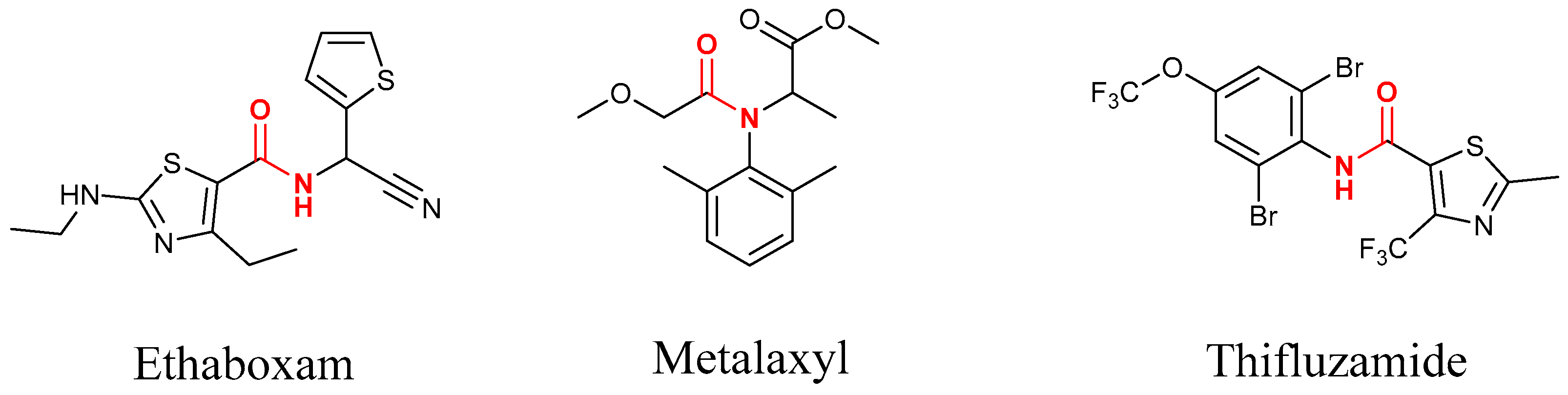
3. Discussion
4. Materials and Methods
4.1. General
4.2. Synthesis of Derivatives
4.2.1. Synthesis of Myrtanol (Compound 2)
4.2.2. Synthesis of Myrtanyl Acid (Compound 3)
4.2.3. Synthesis of Myrtanyl Acid Amide Derivatives (Compound 4a–4l)
4.2.4. Synthesis of Myrtanyl Acid Acylthiourea Derivative (Compounds 4m–4t)
4.3. Biological Activity Evaluation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Knogge, W. Fungal infection of plants. Plant Cell 1996, 8, 1711. [Google Scholar] [CrossRef] [PubMed]
- Mendgen, K.; Hahn, M. Plant infection and the establishment of fungal biotrophy. Trends Plant Sci. 2002, 7, 352–356. [Google Scholar] [CrossRef] [Green Version]
- Bebber, D.P.; Gurr, S.J. Crop-destroying fungal and oomycete pathogens challenge food security. Fungal Genet. Biol. 2015, 74, 62–64. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, F.P. Pesticides, environment, and food safety. Food Energy Secur. 2017, 6, 48–60. [Google Scholar] [CrossRef]
- Russell, P.E. A century of fungicide evolution. J. Agri. Sci. 2005, 143, 11–25. [Google Scholar] [CrossRef]
- Fisher, M.C.; Hawkins, N.J.; Sanglard, D.; Gurr, S.J. Worldwide emergence of resistance to antifungal drugs challenges human health and food security. Science 2018, 360, 739–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robbins, N.; Caplan, T.; Cowen, L.E. Molecular evolution of antifungal drug resistance. Ann. Rev. Microbiol. 2017, 71, 753–775. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.C.R.; Lopes, P.M.; Azevedo, M.M.B.; Costa, D.C.M.; Alviano, C.S.; Alviano, D.S. Biological activities of a-pinene and β-pinene enantiomers. Molecules 2012, 17, 6305–6316. [Google Scholar] [CrossRef]
- De Macêdo, A.; Cláudia, A.; Rosalen, P.L.; de Macêdo, A.; Cláudia, A.; Rosalen, P.L.; Freires, I.A.; Scotti, L.; Scotti, M.T.; Aquino, S.G.; et al. Antifungal activity, mode of action, docking prediction and anti-biofilm effects of (+)-β-pinene enantiomers against Candida spp. Curr. Top. Med. Chem. 2018, 18, 2481–2490. [Google Scholar] [CrossRef]
- Wilson, C.L.; Solar, J.M.; El Ghaouth, A.; Wisniewski, M.E. Rapid evaluation of plant extracts and essential oils for antifungal activity against Botrytis cinerea. Plant Dis. 1997, 81, 204–210. [Google Scholar] [CrossRef]
- Nikitina, L.E.; StartsevaI, V.A.; VakulenkoI, A.; Khismatulina, M.; Lisovskaya, S.A.; Glushko, N.P.; Fassakhov, R.S. Synthesis and antifungal activity of compounds of the pinene series. Pharm. Chem. J. 2009, 43, 251–254. [Google Scholar] [CrossRef]
- Gavrilov, V.V.; Startseva, V.A.; Nikitina, L.E.; Lodochnikova, O.A.; Gnezdilov, O.I.; Lisovskaya, S.A.; Glushko, N.I.; Klimovitskii, E.N. Synthesis and antifungal activity of sulfides, sulfoxides, and sulfones based on (1S)-(−)-β-pinene. Pharm. Chem. J. 2010, 44, 126–129. [Google Scholar] [CrossRef]
- Liao, S.; Shang, S.; Shen, M.; Rao, X.; Si, H.; Song, J.; Song, Z. One-pot synthesis and antimicrobial evaluation of novel 3-cyanopyridine derivatives of (−)-β-pinene. Bioorg. Med. Chem. Lett. 2016, 26, 1512–1515. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Tian, X.; Gao, Y.; Shang, S.; Feng, J.; Zhang, X. A value-added use of volatile turpentine: Antifungal activity and QSAR study of β-pinene derivatives against three agricultural fungi. RSC Adv. 2015, 5, 66947–66955. [Google Scholar] [CrossRef]
- Gao, Y.; Hao, J.; Li, J.; Song, Z.; Shang, S. Structural modification of turpentine with natural chiral preservation and low-risk application prospects in crop protection. ACS Omega 2019, 4, 6392–6398. [Google Scholar] [CrossRef]
- Gao, Y.; Wang, Y.; Li, J.; Shang, S.; Song, Z. Improved application of natural forest product terpene for discovery of potential botanical fungicide. Ind. Crop. Prod. 2018, 126, 103–112. [Google Scholar] [CrossRef]
- Gao, Y.; Li, J.; Li, J.; Song, Z.; Shang, S.; Rao, X. High add valued application of turpentine in crop production through structural modification and QSAR analysis. Molecules 2018, 23, 356. [Google Scholar] [CrossRef]
- Antypenko, L.; Meyer, F.; Kholodniak, O.; Sadykova, Z.; JiráskovákovirvTroianova, A.; Buhaiova, V.; Cao, S.; Kovalenko, S.; Garbe, L.A.; Steffens, K.G. Novel acyl thiourea derivatives: Synthesis, antifungal activity, gene toxicity, drug-like and molecular docking screening. ARCH Pham. 2019, 352, 1800275. [Google Scholar] [CrossRef]
- Qin, Y.; Liu, W.; Xing, R.; Liu, S.; Li, K.; Li, P. Cyclization reaction of acyl thiourea chitosan: Enhanced antifungal properties via structural optimization. Molecules 2018, 23, 594. [Google Scholar] [CrossRef]
- Liu, J.; He, L.; Zhou, G. Specific and rapid detection of Camellia oleifera anthracnose pathogen by nested-PCR. Afr. J. Biotechnol. 2009, 8, 1056–1061. [Google Scholar]
- Huang, S.; Wang, L.; Liu, L.; Tang, S.; Zhu, D.; Savary, S. Rice spikelet rot disease in China- 1. Characterization of fungi associated with the disease. Crop. Prot. 2011, 30, 1–9. [Google Scholar] [CrossRef]
- Masratul Hawa, M.; Salleh, B.; Latiffah, Z. Characterization and pathogenicity of Fusarium proliferatum causing stem rot of Hylocereus polyrhizus in Malaysia. Ann. Appl. Biol. 2013, 163, 269–280. [Google Scholar] [CrossRef]
- Chang, K.; Hwang, S.; Conner, R.; Ahmed, H.U.; Zhou, Q.; Turnbull, G.D.; Strelkov, S.E.; McLaren, D.L.; Gossen, B.D. First report of Fusarium proliferatum causing root rot in soybean (Glycine max L.) in Canada. Crop. Prot. 2015, 67, 52–58. [Google Scholar] [CrossRef]
- Baudry, A.; Morzieres, J.P.; Larue, P. First report of Japanese pear black spot caused by Alternaria kikuchiana in France. Plant Dis. 1993, 77, 428. [Google Scholar] [CrossRef]
- Li, L.; Pan, H.; Chen, M.; Zhang, S.; Zhong, C. Isolation and identification of pathogenic fungi causing postharvest fruit rot of kiwifruit (Actinidia chinensis) in China. J. Phytopathol. 2017, 165, 782–790. [Google Scholar] [CrossRef]
- Li, Q.; Chen, L.; Yu, Y.; Wang, Y.; Zhu, P.; Xu, L.; Nonomura, T.; Matsuda, Y.; Toyoda, H. Phomopsis vaccinii: The Main pathogen causing market diseases in Kiwifruit. Ann. Rept. Kansai Plant Prot. 2015, 57, 31–35. [Google Scholar] [CrossRef]
- Babadoost, M.; Pavon, C.; Islam, S.Z.; Tian, D. Phytophthora blight (Phytophthora capsici) of pepper and its management. Acta Hortic. 2015, 1105, 61–66. [Google Scholar] [CrossRef]
- Hausbeck, M.K.; Lamour, K.H. Phytophthora capsici on Vegetable crops: Research progress and management challenges. Plant Dis. 2014, 88, 1292–1303. [Google Scholar] [CrossRef]
- Maienfisch, P.; Hall, R.G. The importance of fluorine in the life science industry. CHIMIA Int. J. Chem. 2004, 58, 93–99. [Google Scholar] [CrossRef]
- Exner, O.; Krygowski, T.M. The nitro group as substituent. Chem. Soc. Rev. 1996, 25, 71–75. [Google Scholar] [CrossRef]
- Boyd, D.B. Electronic structures of cephalosporins and penicillins. 15. Inductive effect of the 3-position side chain in cephalosporins. J. Med. Chem. 1984, 27, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Sahu, P.K. Design, structure activity relationship, cytotoxicity and evaluation of antioxidant activity of curcumin derivatives/analogues. Eur. J. Med. Chem. 2016, 121, 510–516. [Google Scholar] [CrossRef] [PubMed]
- Cherdtrakulkiat, R.; Boonpangrak, S.; Sinthupoom, N.; Prachayasittikul, S.; Ruchirawat, S.; Prachayasittikul, A. Derivatives (halogen, nitro and amino) of 8-hydroxyquinoline with highly potent antimicrobial and antioxidant activities. Biochem. Biophys. Rep. 2016, 6, 135–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillis, E.P.; Eastman, K.J.; Hill, M.D.; Donnelly, D.J.; Meanwell, N.A. Applications of fluorine in medicinal chemistry. J. Med. Chem. 2015, 58, 8315–8359. [Google Scholar] [CrossRef] [PubMed]
- Smart, B.E. Fluorine substituent effects (on bioactivity). J. Fluor. Chem. 2001, 109, 3–11. [Google Scholar] [CrossRef]
- Liang, T.; Neumann, C.N.; Ritter, T. Introduction of fluorine and fluorine-containing functional groups. Angew. Chem. Int. Ed. 2013, 52, 8214–8264. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.; Shang, S.; Si, H.; Shen, M.; Rao, X.; Song, Z. Synthesis and antibacterial activity of myristyl carboxylate. Chem. Ind. For. Prod. 2015, 35, 33–38. [Google Scholar]
- Liao, S.; Rao, X.; Shen, M.; Si, H.; Song, J.; Shang, S.; Song, Z. New hybrids derived from the natural compound (−)-β-pinene and amides or acylthioureas as antitumor agents. Lett. Drug Des. Discov. 2019. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |



{kind=link}
{kind=link}
| Compounds | Skeleton | R | Colletotrichum gloeosporioides | Fusarium proliferatum | Alternaria kikuchiana | Phomopsis sp. | Phytophthora capsici |
|---|---|---|---|---|---|---|---|
| 1 |  | - | >1000 | >1000 | >1000 | >1000 | >1000 |
| 2 |  | - | >1000 | >1000 | >1000 | >1000 | >1000 |
| 3 |  | - | >1000 | 417.54 | >1000 | >1000 | 224.10 |
| 4a |  |  | 364.60 | 147.38 | 134.62 | 98.89 | 626.25 |
| 4b |  | >1000 | 554.30 | 151.44 | 270.73 | 423.72 | |
| 4c |  | 755.05 | 397.69 | 64.17 | 68.35 | 499.01 | |
| 4d |  | >1000 | 200.61 | >1000 | 123.45 | 140.99 | |
| 4e |  | 161.40 | 39.21 | 38.80 | 40.98 | NT | |
| 4f |  | >1000 | >1000 | >1000 | >1000 | NT | |
| 4g |  | 165.61 | 72.84 | 240.96 | 335.23 | NT | |
| 4h |  | 77.06 | 41.98 | 68.17 | 20.43 | 350.63 | |
| 4i |  | 346.00 | 376.94 | 389.92 | 112.70 | 864.54 | |
| 4j |  | 179.64 | 549.59 | 320.34 | 184.46 | 375.65 | |
| 4k |  | >1000 | >1000 | >1000 | 490.82 | NT | |
| 4l |  | >1000 | NT | >1000 | 217.90 | >1000 | |
| 4m |  |  | >1000 | >1000 | 894.20 | 120.23 | >1000 |
| 4n |  | >1000 | >1000 | >1000 | >1000 | >1000 | |
| 4o |  | 498.34 | >1000 | >1000 | 136.54 | 0.18 | |
| 4p |  | >1000 | >1000 | >1000 | >1000 | >1000 | |
| 4q |  | >1000 | 143.84 | >1000 | 60.25 | 157.76 | |
| 4r |  | 21.64 | 492.13 | 341.24 | 109.93 | 176.39 | |
| 4s |  | >1000 | >1000 | >1000 | >1000 | >1000 | |
| 4t |  | >1000 | 555.32 | >1000 | 240.79 | 557.82 | |
| carbendazim |  | - | 0.534 | 0.426 | 0.431 | 0.217 | 0.386 |
| Compound | Characterization Data |
|---|---|
| 2 | Colorless liquid; yield 96.8%; purity 94.5%; FT-IR v (cm−1): 3313 (O–H), 1041 (C–O); 1H NMR (300 MHz, CDCl3) δ: 3.55 (dd, J = 7.6, 5.2 Hz, 2H), 2.44–2.31 (m, 1H), 2.31–2.14 (m, 1H), 2.05–1.97 (m, 2H), 1.98–1.78 (m, 4H), 1.54–1.35 (m, 1H), 1.19 (s, 3H), 0.97 (s, 3H), 0.93 (d, J = 9.6 Hz, 1H). 13C NMR (75 MHz, CDCl3) δ: 66.95, 43.77, 42.37, 40.93, 38.07, 32.63, 27.44, 25.48, 22.79, 18.29. GC-MS m/z = 154.1 [M]+. |
| Compound | Characterization Data |
|---|---|
| 3 | White solid; m.p. 92–94 °C; yield 24.5%; purity 98.7%; FT-IR v (cm−1): 3660, 3638, 3061 (O–H), 2990, 2950, 2920, 2903, 2869 (C–H), 1675 (C=O), 1478, 1458 (C–H), 1414 (O–H), 1386, 1364, 1338, 1320 (C–H), 1250 (C–O), 940 (O–H). 1H NMR (500 MHz, CDCl3) δ: 11.91 (s, 1H), 3.02 (dt, J = 10.3, 3.5 Hz, 1H), 2.54 (dd, J = 9.1, 5.5 Hz, 1H), 2.42–2.29 (m, 2H), 2.07–1.84 (m, 4H), 1.26 (s, 3H), 1.23 (d, J = 10.0 Hz, 1H), 0.91 (s, 3H). 13C NMR (126 MHz, CDCl3) δ: 183.04, 43.76, 42.98, 40.34, 38.74, 29.03, 26.88, 24.60, 21.51, 15.09. ESI-MS: m/z 191.1 [M + Na]+; 167.1 [M − H]−. |
| Compound | Characterization Data |
|---|---|
| 4a | Light yellow solid; m.p. 75–77 °C; yield 97.4%; purity 98.3%; FT-IR v (cm−1): 3292, 3267 (N–H), 3195, 3134 (C–H), 2992, 2912, 2866 (C–H), 1674, 1657 (C=O, N–H), 1595, 1532, 1491, 1465 (C=C), 1438, 1367 (C–H), 1332, 1300 (C–N), 753,694; 1H NMR (300 MHz, CDCl3) δ: 7.49 (d, J = 7.9 Hz, 2H), 7.31 (t, J = 7.9 Hz, 2H), 7.09 (t, J = 7.4 Hz, 2H), 3.00 (dd, J = 6.4, 2.7 Hz, 1H), 2.46 (ddd, J = 17.9, 10.5, 5.8Hz, 3H), 2.10–1.84 (m, 4H), 1.25 (s, 3H), 1.22 (s, 1H), 0.94 (s, 3H). 13C NMR (75 MHz, CDCl3) δ: 173.92, 138.18, 128.92, 123.97, 119.95, 46.03, 44.06, 40.63, 38.83, 30.10, 27.43,24.90, 21.97, 15.24. ESI-MS: m/z 266.1 [M + Na]+; 242.1 [M − H]−. |
| 4b | Light yellow solid; m.p. 53–55 °C; yield 90.5%; purity 96.7%; FT-IR v (cm−1): 3406, 3310 (N–H), 3066 (C–H), 2949, 2927, 2902, 2869 (C–H), 1655 (C=O), 1621, 1588, 1502, 1472 (C=C), 1436, 1383 (C–H), 1293 (C–N), 753, 579 (C–Br). 1H NMR (300 MHz, CDCl3) δ: 8.40 (dd, J = 8.3, 1.5 Hz, 1H), 7.84 (s, 1H), 7.53 (dd, J = 8.0, 1.4 Hz, 1H), 7.35–7.28 (m, 1H), 6.96 (td, J = 7.9, 1.6 Hz, 1H), 3.14–3.02 (m, 1H), 2.58–2.38 (m, 3H), 2.10–1.87 (m, 4H), 1.32 (s, 1H), 1.29 (s, 3H), 0.90 (s, 3H). 13C NMR (75 MHz, CDCl3) δ: 173.58, 135.37, 131.62, 127.93, 124.22, 121.13, 112.73, 45.65, 43.17, 40.00, 38.44, 29.03, 26.71, 24.26, 21.39, 14.53. ESI-MS: m/z 322.1 [M + H]+; 344.1 [M + Na]+. |
| 4c | Light yellow solid; m.p. 50–52 °C; yield 91.2%; purity 95.4%; FT-IR v (cm−1): 3435, 3321 (N–H), 3189, 3120 (C–H), 2984, 2950, 2915, 2868 (C–H), 1670 (C=O), 1592,1524, 1476 (C=C), 1417, 1368 (C–H), 1331, 1302 (C–N), 774, 681, 569 (C–Br). 1H NMR (300 MHz, CDCl3) δ: 7.77 (d, J = 1.9 Hz, 1H), 7.39 (d, J =7.7 Hz, 1H), 7.20 (ddd, J = 18.8, 11.1, 4.7 Hz, 3H), 2.99 (dd, J = 6.2, 3.3 Hz, 1H), 2.54–2.34 (m, 3H), 2.07–2.01 (m, 1H), 2.00–1.86 (m, 3H), 1.25 (s, 3H), 1.21 (d, J = 4.3Hz, 1H), 0.91 (s, 3H).13C NMR (75 MHz, CDCl3) δ: 173.82, 138.93, 129.70, 126.46, 122.42, 122.07, 117.96, 45.54, 43.43, 40.04, 38.33, 29.57, 26.90, 24.35, 21.54, 14.70. ESI-MS: m/z 322.1 [M + H]+; 344.1 [M + Na]+; 320.1 [M − H]−. |
| 4d | Yellow solid; m.p. 84–86 °C; yield 96.1%; purity 98.7%; FT-IR v (cm−1): 3288, 3253 (N–H), 3184, 3114, 3037 (C–H), 2987, 2962, 2914, 2865 (C–H), 1656 (C=O), 1595, 1514, 1462 (C=C), 1410, 1382, 1367 (C–H), 1329, 1298 (C–N), 826. 1H NMR (300 MHz, CDCl3) δ: 7.40 (d, J = 8.4 Hz, 2H), 7.27 (t, J = 9.6 Hz,1H), 7.13 (d, J = 8.3 Hz, 2H), 3.04–2.90 (m, 1H), 2.61 (q, J = 7.6 Hz, 2H), 2.46 (ddd, J = 23.4, 11.7, 5.9 Hz, 3H), 2.08–1.81 (m, 4H), 1.27–1.18 (m, 7H), 0.94 (s, 3H). 13C NMR (75 MHz, CDCl3) δ: 173.84, 140.00, 135.83, 128.19, 120.13, 45.92, 44.08, 40.64, 38.82, 30.10, 28.25, 27.44, 24.92, 21.97, 15.63, 15.25. ESI-MS: m/z 294.1 [M + Na]+; 270.1 [M − H]−. |
| 4e | Yellow solid; m.p. 107–109 °C; yield 94.4%; purity 97.3%; FT-IR v (cm −1): 3301 (N–H), 3198, 3128 (C–H), 2984, 2953, 2912, 2866 (C–H), 1670 (C=O), 1601, 1524, 1464 (C=C), 1407, 1384, 1367 (C–H), 1320 (C–N), 837. 1H NMR (300 MHz, CDCl3) δ: 7.62 (d, J = 8.7 Hz, 2H), 7.55 (d, J = 8.7 Hz, 2H), 7.32 (s, 1H), 3.01 (dd, J = 6.3, 2.6 Hz, 1H), 2.55–2.37 (m, 3H), 2.08–1.88 (m, 4H), 1.25 (s, 3H), 1.21 (d, J = 2.1 Hz, 1H), 0.92 (s, 3H). 13C NMR (75 MHz, CDCl3) δ: 175.16, 142.15, 127.18, 120.29, 47.16, 44.98, 41.53, 39.82, 31.00, 28.35, 25.77, 22.90, 16.13. ESI-MS: m/z 334.1 [M + Na]+; 310.1 [M − H]−. |
| 4f | Yellow solid; m.p. 107–109 °C; yield 88.2%; purity 95.4%; FT-IR v (cm−1): 3328, 3310 (N–H), 2987, 2948, 2917, 2867 (C–H), 1677, 1659 (C=O), 1622, 1597, 1510, 1465 (C=C), 1385, 1366 (C–H), 1288 (C–N), 1006 (C–F), 776, 702. 1H NMR (300 MHz, CDCl3) δ: 7.14 (s, 1H), 6.96–6.79 (m, 3H), 3.05 (s, 1H), 2.42 (d, J = 20.8 Hz, 3H), 1.95 (dd, J = 45.4, 21.0 Hz, 4H), 1.24 (s, 4H), 0.94 (s, 3H). 13C NMR (75 MHz, CDCl3) δ: 174.16, 172.21, 158.88, 156.89, 127.13, 114.41, 111.52, 111.37, 45.27, 44.03, 42.91, 40.52, 40.30, 38.81, 29.94, 29.11, 27.26, 26.87, 24.83, 24.49, 21.71, 15.05. ESI-MS: m/z 280.1 [M + H]+; 302.1 [M + Na]+; 278.1 [M − H]−. |
| 4g | Yellow solid; m.p. 78–80 °C; yield 89.5%; purity 97.7%; FT-IR v (cm−1): 3286, 3256 (N–H), 3209, 3145, 3067 (C–H), 2986, 2929, 2866 (C–H), 1669 (C=O), 1641, 1610, 1506, 1462 (C=C), 1406, 1383, 1366 (C–H), 1296 (C–N), 1012, 993 (C–F), 832. 1H NMR (300 MHz, CDCl3) δ: 7.48–7.38 (m, 2H), 7.11 (s, 1H), 7.05–6.94 (m, 2H), 3.04–2.93 (m, 1H), 2.55–2.35 (m, 3H), 2.08–1.85 (m, 4H), 1.25 (s, 3H), 1.21 (d, J = 3.8 Hz, 1H), 0.93 (s, 3H). 13C NMR (75 MHz, CDCl3) δ: 173.95, 160.21, 158.28, 134.10, 121.83, 115.57, 115.39, 45.87, 44.00, 40.60, 38.81, 30.04, 27.39, 24.84, 21.95, 15.23. ESI-MS: m/z 284.1 [M + Na]+; 260.1 [M − H]−. |
| 4h | Yellow solid; m.p. 103–105 °C; yield 84.4%; purity 96.2%; FT-IR v (cm−1): 3359 (N–H), 3117, 3084 (C–H), 2987, 2916, 2868 (C–H), 1703 (C=O), 1609, 1594, 1540, 1495, 1463 (C=C), 1405, 1384, 1368 (C–H), 1327 (N=O), 1296, 1249 (C–N), 854. 1H NMR (300 MHz, CDCl3) δ: 8.17 (d, J = 9.1 Hz, 2H), 7.69 (d, J = 9.2 Hz, 2H), 7.66 (d, J = 3.2 Hz, 1H), 3.03 (dd, J = 5.9, 2.6 Hz, 1H), 2.43 (dt, J = 12.4, 6.3 Hz, 3H), 2.07–1.87 (m, 4H), 1.23 (s, 3H), 1.20 (d, J = 4.0 Hz, 1H), 0.95–0.82 (m, 3H). 13C NMR (75 MHz, CDCl3) δ: 175.50, 145.16, 144.19, 125.99, 120.01, 47.42, 46.32, 44.93, 43.90, 41.38, 39.79, 31.13, 30.06, 28.33, 27.85, 25.75, 25.46, 22.86, 16.05. ESI-MS: m/z 311.1 [M + Na]+; 287.1 [M − H]−. |
| 4i | Brown solid; m.p. 41–43 °C; yield 85.6%; purity 96.1%; FT-IR v (cm−1): 2990, 2948, 2914, 2867 (C–H), 1695 (C=O), 1638, 1577, 1508, 1463 (C=C), 1429 (C=N), 1383,1367 (C–H), 1295 (C–N), 871, 776. 1H NMR (300 MHz, CDCl3) δ: 8.57 (s, 1H), 8.34–8.18 (m, 1H), 7.73 (s, 1H), 7.04 (s, 1H), 3.08 (s, 1H), 2.50 (dd, J = 51.2, 27.8 Hz, 3H), 1.97 (s, 4H), 1.27 (s, 4H), 0.95 (s, 3H). 13C NMR (75 MHz, CDCl3) δ: 181.14, 179.43, 174.63, 172.17, 151.77, 149.37, 146.99, 138.65, 124.36, 119.26, 114.18, 46.92, 46.32, 45.30, 43.98, 43.73, 43.19, 42.90, 40.73, 38.72, 38.39, 31.52, 30.20, 29.35, 29.11, 27.30, 26.94, 25.85, 25.48, 24.51, 22.02, 16.84, 15.31, 15.04. ESI-MS: m/z 267.1 [M + Na]+; 243.1 [M − H]−. |
| 4j | Light yellow solid; m.p. 41–43 °C; yield 87.4%; purity 96.5%; FT-IR v (cm−1): 3321, 3281 (N–H), 3090, 3063, 3025 (C–H), 2995, 2973, 2917, 2875 (C–H), 1641 (C=O), 1606, 1537, 1496, 1465 (C=C), 1453, 1384, 1363 (C–H), 1259, 1234 (C–N), 722, 694. 1H NMR (300 MHz, CDCl3) δ: 7.31 (dd, J = 13.6, 7.8 Hz, 5H), 5.76 (s, 1H), 4.52–4.37 (m, 2H), 2.86 (dd, J = 5.8, 3.3 Hz, 1H), 2.50–2.26 (m, 3H), 2.04–1.83 (m, 4H), 1.21 (d, J = 7.7 Hz, 3H), 1.16 (d, J = 9.3 Hz, 1H), 0.87 (s, 3H). 13C NMR (75 MHz, CDCl3) δ: 175.44, 138.70, 128.60, 127.97, 127.35, 45.09, 43.78, 40.65, 38.71, 30.12, 27.38, 24.96, 22.03, 15.44. ESI-MS: m/z 280.1 [M + Na]+; 256.1 [M − H]−. |
| 4k | Scarlet solid; m.p. 53–55 °C; yield 18.7%; purity 95.5%; FT-IR v (cm−1): 3432 (N–H), 3192, 3117, 3056 (C–H), 2919, 2869 (C–H), 1687, 1654 (C=O), 1597, 1539, 1491, 1465 (C=C), 1410, 1385, 1368 (C–H), 1321, 1270 (C–N), 1174, 1156, 1061 (C–S–C, C–F), 839. 1H NMR (300 MHz, CDCl3) δ: 11.91 (s, 1H), 7.74 (dd, J = 8.8, 5.2 Hz, 1H), 7.64–7.30 (m, 2H), 7.14 (t, J = 8.7 Hz, 1H), 7.02 (s, 1H), 3.24–3.12 (m, 1H), 2.54 (s, 1H), 2.48–2.36 (m, 1H), 2.31 (s, 1H), 2.10–1.83 (m, 4H), 1.27 (s, 1H), 1.23 (s, 3H), 0.85 (s, 3H). 13C NMR (75 MHz, CDCl3) δ: 173.66, 163.81, 160.52, 158.91, 148.09, 130.05, 127.36, 115.40, 115.12, 106.80, 44.60, 42.94, 39.90, 38.09, 31.44, 30.98, 29.69, 29.24, 28.78, 26.58, 24.08, 22.21, 21.27, 13.78. ESI-MS: m/z 345.1 [M + H]+; 343.1 [M − H]−. |
| 4l | Yellow solid; m.p. 64–66 °C; yield 20.9%; purity 96.6%; FT-IR v (cm−1): 3434 (N–H), 3117, 3045 (C–H), 2945, 2916, 2870, 2837 (C–H), 1687 (C=O), 1612, 1540, 1492, 1463 (C=C), 1440, 1419, 1385, 1367 (C–H), 1326, 1285, 1249 (C–N), 1173, 1110, 1062 (C–S–C), 834. 1H NMR (300 MHz, CDCl3) δ: 11.04 (s, 1H), 7.72 (dq, J = 4.5, 1.8 Hz, 2H), 7.03–6.89 (m, 3H), 3.85 (s, 3H), 3.19–3.11 (m, 1H), 2.60–2.53 (m, 1H), 2.49–2.38 (m, 2H), 2.09–1.89 (m, 4H), 1.25 (s, 3H), 1.23 (s, 1H), 0.85 (s, 3H). 13C NMR (75 MHz, CDCl3) δ: 174.68, 165.59, 164.21, 131.99, 126.84, 113.68 (s), 112.44, 105.36, 54.86, 44.63, 42.88, 39.95, 29.24, 26.60, 25.82, 24.13, 21.29, 14.15. ESI-MS: m/z 357.1 [M + H]+; 355.1 [M − H]−. |
| Compound | Characterization Data |
|---|---|
| 4m | Light yellow solid; m.p. 85–87 °C; yield 82.4%; purity 96.4%; FT-IR v (cm−1): 3635 (N–H), 3164, 3028 (C–H), 2987, 2915, 2868 (C–H), 1686 (C=O), 1563 (N–H), 1598, 1516, 1498, 1469 (C=C), 1447, 1384, 1356 (C–H), 1327, 1310, 1296 (C–N), 1238 (C–O), 1143, 1102 (C=S), 756, 685. 1H NMR (300 MHz, CDCl3) δ: 12.47 (s, 1H), 8.55 (s, 1H), 7.72 (d, J = 8.0 Hz, 2H), 7.44 (t, J = 7.8 Hz, 2H), 7.35–7.29 (m, 1H), 3.10–3.00 (m, 1H), 2.53–2.44 (m, 2H), 2.41 (ddd, J = 13.8, 6.9, 3.6 Hz, 1H), 2.13–1.93 (m, 4H), 1.32 (s, 3H), 1.25 (d, J = 9.7 Hz, 1H), 0.97 (s, 3H). 13C NMR (126 MHz, CDCl3) δ: 178.33, 176.81, 137.66, 129.25, 128.78, 126.69, 123.95, 46.22, 43.27, 40.36, 38.75, 30.00, 27.15, 24.61, 22.03, 14.81. ESI-MS: m/z 325.1 [M + Na]+; 301.2 [M − H]−. |
| 4n | Light yellow solid; m.p. 116–118 °C; yield 80.7%; purity 96.9%; FT-IR v (cm−1): 3300 (N–H), 3141, 3002 (C–H), 2943, 2918, 2865 (C–H), 1681 (C=O), 1516 (N–H), 1576, 1467 (C=C), 1442, 1382, 1366 (C–H), 1332, 1309, 1285 (C–N), 1238 (C–O), 1159, 1122 (C=S), 744. 1H NMR (300 MHz, CDCl3) δ: 12.45 (s, 1H), 8.68 (s, 1H), 8.22 (d, J = 8.1 Hz, 1H), 7.68 (d, J = 8.0 Hz, 1H), 7.41 (t, J = 7.7 Hz, 1H), 7.19 (t, J = 7.7 Hz, 1H), 3.07 (d, J = 7.6 Hz, 1H), 2.52–2.41 (m, 3H), 2.13–1.92 (m, 4H), 1.32 (s, 3H), 1.27 (d, J = 7.7 Hz, 1H), 0.98 (s, 3H). 13C NMR (126 MHz, CDCl3) δ: 179.20, 176.60, 136.52, 132.86, 128.13, 127.37, 118.66, 46.11, 43.37, 40.38, 38.84, 29.84, 27.18, 24.56, 21.93, 14.65. ESI-MS: m/z 403.0 [M + Na]+; 379.1 [M − H]−. |
| 4o | Light yellow solid; m.p. 89–91 °C; yield 81.3%; purity 97.2%; FT-IR v (cm−1): 3295 (N–H), 3161, 3004 (C–H), 2965, 2922, 2862 (C–H), 1687, 1657 (C=O), 1518 (N–H), 1588, 1462 (C=C), 1412, 1384 (C–H), 1327 (C–N), 1249 (C–O), 1155, 1136 (C=S), 835. 1H NMR (300 MHz, CDCl3) δ: 12.37 (s, 1H), 8.47 (s, 1H), 7.61 (d, J = 8.1 Hz, 2H), 7.31 (s, 1H), 7.26 (s, 1H), 3.05 (s, 1H), 2.70 (q, J = 7.5 Hz, 2H), 2.48 (dd, J = 14.1, 6.9 Hz, 2H), 2.39 (d, J = 9.8 Hz, 1H), 2.02 (dd, J = 33.3, 21.1 Hz, 5H), 1.32 (s, 3H), 1.30 (d, J = 7.5 Hz, 3H), 1.28 (s, 1H), 0.97 (s, 3H). 13C NMR (126 MHz, CDCl3) δ: 178.26, 176.70, 142.94, 135.27, 128.19, 123.99, 46.21, 43.24, 40.37, 38.77, 29.96, 28.41, 27.14, 24.61, 22.02, 15.33, 14.81. ESI-MS: m/z 331.2 [M + H] +; 353.2 [M + Na]+; 329.1 [M − H]−. |
| 4p | Light yellow solid; m.p. 110–112 °C; yield 83.3%; purity 95.4%; FT-IR v (cm−1): 3247, 3200 (N–H), 3017 (C–H), 2999, 2921, 2870 (C–H), 1700 (C=O), 1524 (N–H), 1612, 1596, 1466 (C=C), 1409, 1387, 1369 (C–H), 1319 (C–N), 1256 (C–O), 1160, 1122, 1103 (C=S), 1063, 1015 (C–F), 841. 1H NMR (300 MHz, CDCl3) δ: 12.73 (s, 1H), 8.71 (s, 1H), 7.92 (d, J = 8.2 Hz, 2H), 7.69 (d, J = 8.3 Hz, 2H), 3.07 (dd, J = 6.0, 3.1 Hz, 1H), 2.55–2.43 (m, 2H), 2.39 (t, J = 10.2 Hz, 1H), 2.12–1.93 (m, 4H), 1.32 (s, 3H), 1.25 (d, J = 9.9 Hz, 1H), 0.97 (s, 3H). 13C NMR (126 MHz, CDCl3) δ: 178.50, 177.19, 140.71, 128.43, 128.17, 125.97, 124.92, 123.56, 122.75, 46.29, 43.52, 43.33, 40.34, 38.74, 30.11, 27.16, 24.60, 22.07, 14.83. ESI-MS: m/z 371.1 [M + H]+; 369.1 [M − H]−. |
| 4q | Light yellow solid; m.p. 108–110 °C; yield 82.1%; purity 95.8%; FT-IR v (cm−1): 3406 (N–H), 3152, 3001 (C–H), 2981, 2921, 2862 (C–H), 1685 (C=O), 1519 (N–H), 1628, 1595, 1469 (C=C), 1376 (C–H), 1345, 1304 (C–N), 1253, 1238 (C–O), 1164, 1138 (C=S), 995 (C–F), 740. 1H NMR (300 MHz, CDCl3) δ: 11.68 (s, 1H), 8.78 (s, 1H), 7.41–7.33 (m, 1H), 7.04 (t, J = 8.5 Hz, 2H), 3.07 (d, J = 10.4 Hz, 1H), 2.48 (dt, J = 11.7, 8.0 Hz, 2H), 2.44–2.39 (m, 1H), 2.12–1.94 (m, 4H), 1.32 (s, 3H), 1.26 (d, J = 9.6 Hz, 1H), 0.98 (s, 3H). 13C NMR (126 MHz, CDCl3) δ: 181.73, 176.87, 159.24, 157.22, 129.32, 115.22, 111.88, 111.77, 111.59, 46.19, 43.30, 40.38, 38.81, 29.94, 27.14, 24.59, 21.92, 14.76. ESI-MS: m/z 361.1 [M + Na]+; 337.1 [M − H]−. |
| 4r | Light yellow solid; m.p. 92–94 °C; yield 81.9%; purity 96.0%; FT-IR v (cm−1): 3638 (N–H), 3132, 3028 (C–H), 2986, 2920, 2866 (C–H), 1687 (C=O), 1524 (N–H), 1607, 1505, 1465 (C=C), 1411, 1385, 1367 C–H), 1331 (C–N), 1253 (C–O), 1215, 1151 (C=S), 1010 (C–F), 837. 1H NMR (300 MHz, CDCl3) δ: 12.39 (s, 1H), 8.57 (s, 1H), 7.66 (dd, J = 8.7, 4.7 Hz, 2H), 7.12 (t, J = 8.5 Hz, 2H), 3.05 (dd, J = 6.3, 3.4 Hz, 1H), 2.48 (dd, J = 16.8, 7.2 Hz, 2H), 2.43–2.36 (m, 1H), 2.11–1.97 (m, 4H), 1.32 (s, 3H), 1.25 (d, J = 9.9 Hz, 1H), 0.97 (s, 3H). 13C NMR (126 MHz, CDCl3) δ: 179.16, 178.87, 176.92, 161.87, 159.91, 133.67, 126.05, 115.74, 115.56, 46.24, 43.92, 43.28, 40.35, 39.92, 38.76, 30.02, 27.15, 26.27, 24.61, 23.91, 23.56, 22.05, 20.17, 16.16, 14.83. ESI-MS: m/z 343.1 [M + Na]+; 319.1 [M − H]−. |
| 4s | Light yellow solid; m.p. 172–174 °C; yield 9.7%; purity 90.9%; FT-IR v (cm−1): 3603 (N–H), 3245, 3187, 3007 (C–H), 2942, 2917, 2870 (C–H), 1697 (C=O), 1531 (N–H), 1546, 1484 (C=C), 1449, 1412, 1386 (C–H), 1309, 1293 (C–N), 1277 (C–O), 1226, 1209, 1158, 1126 (C=S), 1062 (C–S–C), 1012 (C–F), 837. 1H NMR (300 MHz, CDCl3) δ: 13.75 (s, 1H), 8.49 (s, 1H), 7.87 (dd, J = 8.5, 5.4 Hz, 2H), 7.11 (dd, J = 14.3, 5.6 Hz, 3H), 3.05 (dd, J = 6.6, 3.6 Hz, 1H), 2.44 (ddd, J = 20.7, 15.9, 9.3 Hz, 3H), 2.10–1.92 (m, 4H), 1.29 (s, 3H), 1.25 (d, J = 9.8 Hz, 1H), 0.93 (s, 3H). 13C NMR (126 MHz, CDCl3) δ: 176.57, 174.90, 158.50, 149.69, 127.86, 115.66, 115.49, 107.60, 46.24, 43.25, 40.33, 38.86, 29.86, 27.13, 24.55, 22.02, 14.73. ESI-MS: m/z 404.1 [M + H]+; 426.1 [M + Na]+; 402.1 [M − H]−. |
| 4t | Light yellow solid; m.p. 221–223 °C; yield 8.4%; purity 93.2%; FT-IR v (cm−1): 3418 (N–H), 3252, 3197 (C–H), 2942, 2921, 2870 (C–H), 1699 (C=O), 1527 (N–H), 1600, 1545, 1515, 1477 (C=C), 1446, 1412, 1385 (C–H), 1342 (N=O), 1316, 1300 (C–N), 1283 (C–O), 1209, 1166, 1127, 1107 (C=S), 1061 (C–S–C), 856,730. 1H NMR (300 MHz, CDC3) δ: 13.81 (s, 1H), 8.57 (s, 1H), 8.27 (d, J = 8.7 Hz, 2H), 8.04 (d, J = 8.7 Hz, 2H), 7.38 (s, 1H), 3.12–3.03 (m, 1H), 2.52–2.37 (m, 3H), 2.02 (ddd, J = 27.5, 21.0, 14.2 Hz, 4H), 1.29 (s, 3H), 1.25 (d, J = 9.8 Hz, 1H), 0.94 (s, 3H). 13C NMR (126 MHz, CDCl3) δ: 176.81, 175.24, 159.03, 148.21, 147.28, 139.96, 126.62, 124.11, 111.28, 46.27, 43.24, 40.29, 38.83, 29.89, 27.12, 24.53, 22.03, 14.73. ESI-MS: m/z 453.2 [M + Na]+; 429.1 [M − H]−. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, Y.; Si, H.; Wang, P.; Chen, S.; Shang, S.; Song, Z.; Wang, Z.; Liao, S. Derivatization of Natural Compound β-Pinene Enhances Its In Vitro Antifungal Activity against Plant Pathogens. Molecules 2019, 24, 3144. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24173144
Shi Y, Si H, Wang P, Chen S, Shang S, Song Z, Wang Z, Liao S. Derivatization of Natural Compound β-Pinene Enhances Its In Vitro Antifungal Activity against Plant Pathogens. Molecules. 2019; 24(17):3144. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24173144
Chicago/Turabian StyleShi, Yunfei, Hongyan Si, Peng Wang, Shangxing Chen, Shibin Shang, Zhanqian Song, Zongde Wang, and Shengliang Liao. 2019. "Derivatization of Natural Compound β-Pinene Enhances Its In Vitro Antifungal Activity against Plant Pathogens" Molecules 24, no. 17: 3144. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24173144