Insight into Improved Thermostability of Cold-Adapted Staphylococcal Lipase by Glycine to Cysteine Mutation

 ,
,
Abstract
:1. Introduction
2. Results
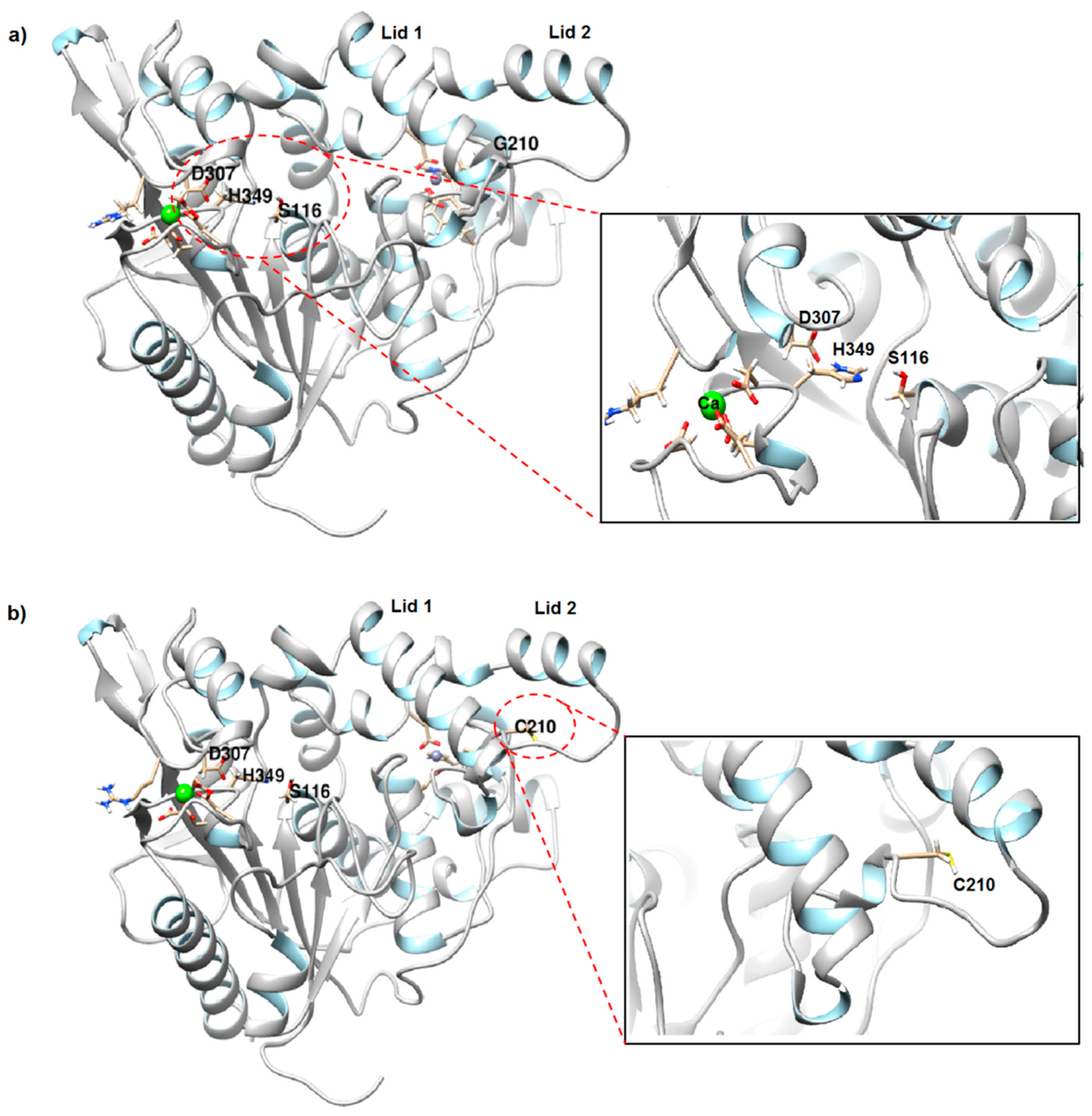
2.1. Structure Prediction and Model Validation of rT-M386 and G210C Lipases
2.2. Comparative Structural Analysis of rT-M386 and G210C Lipase Models
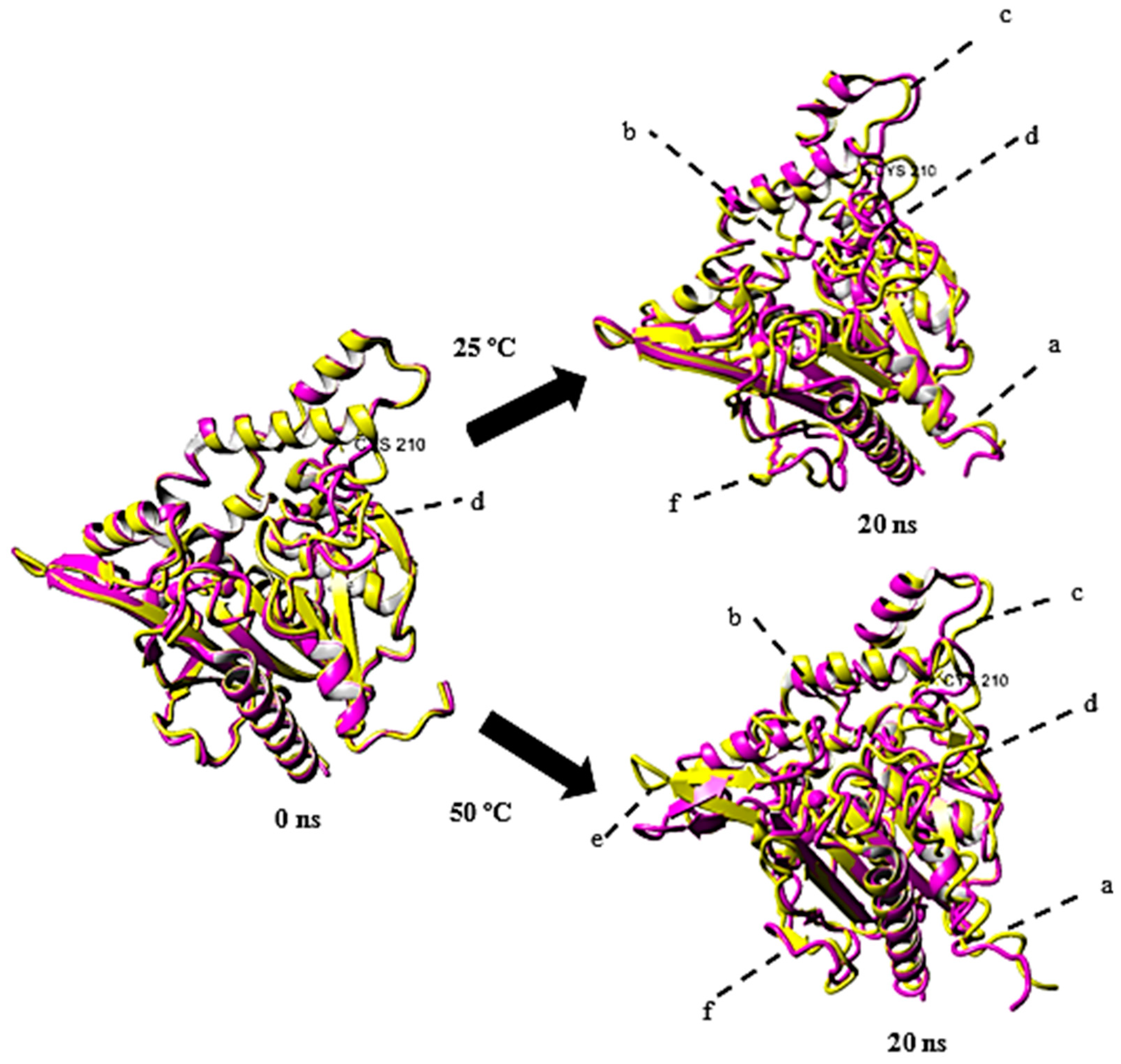
2.2.1. Superimposition of rT-M386 and G210C
2.2.2. Cysteine Substitution
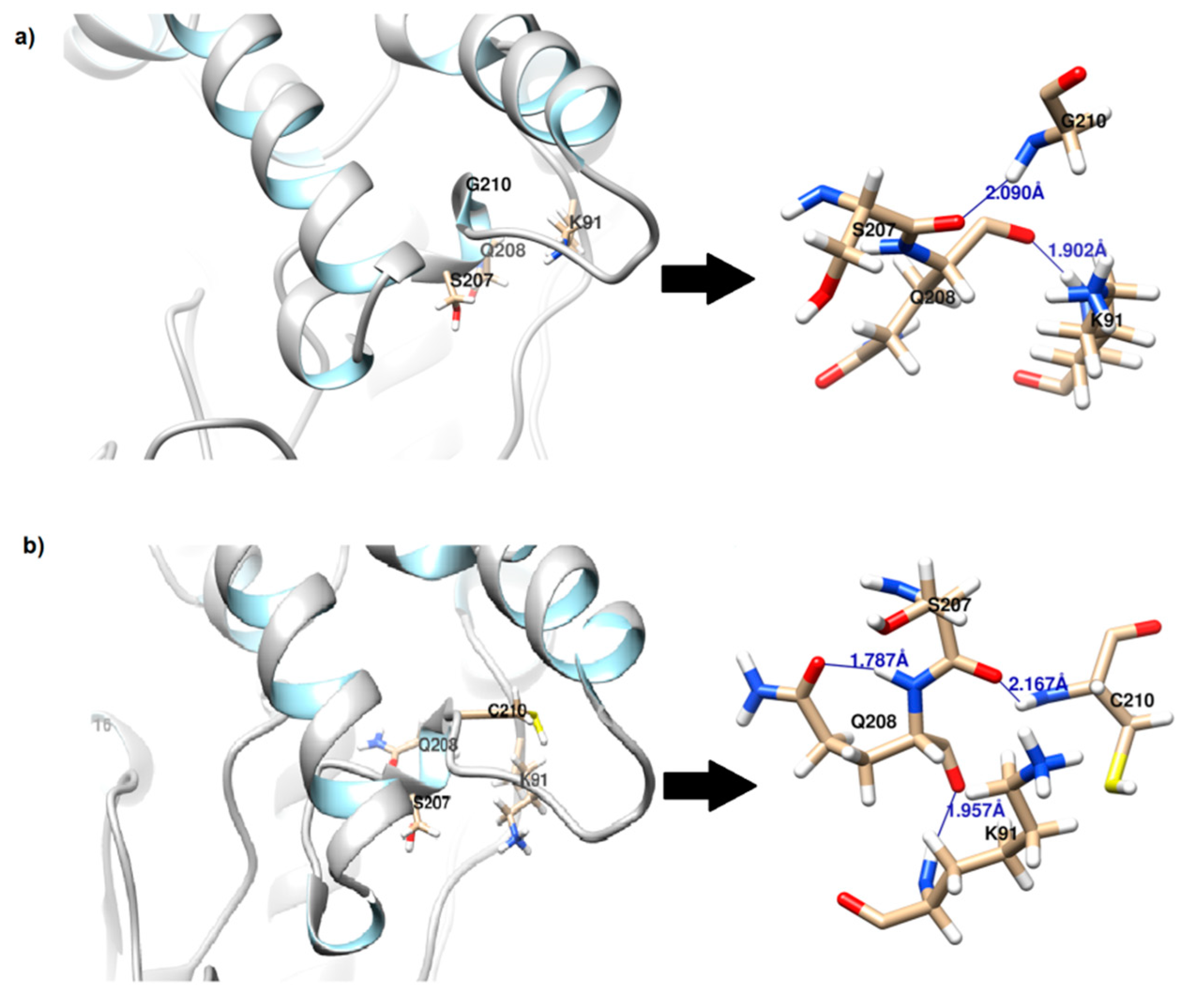
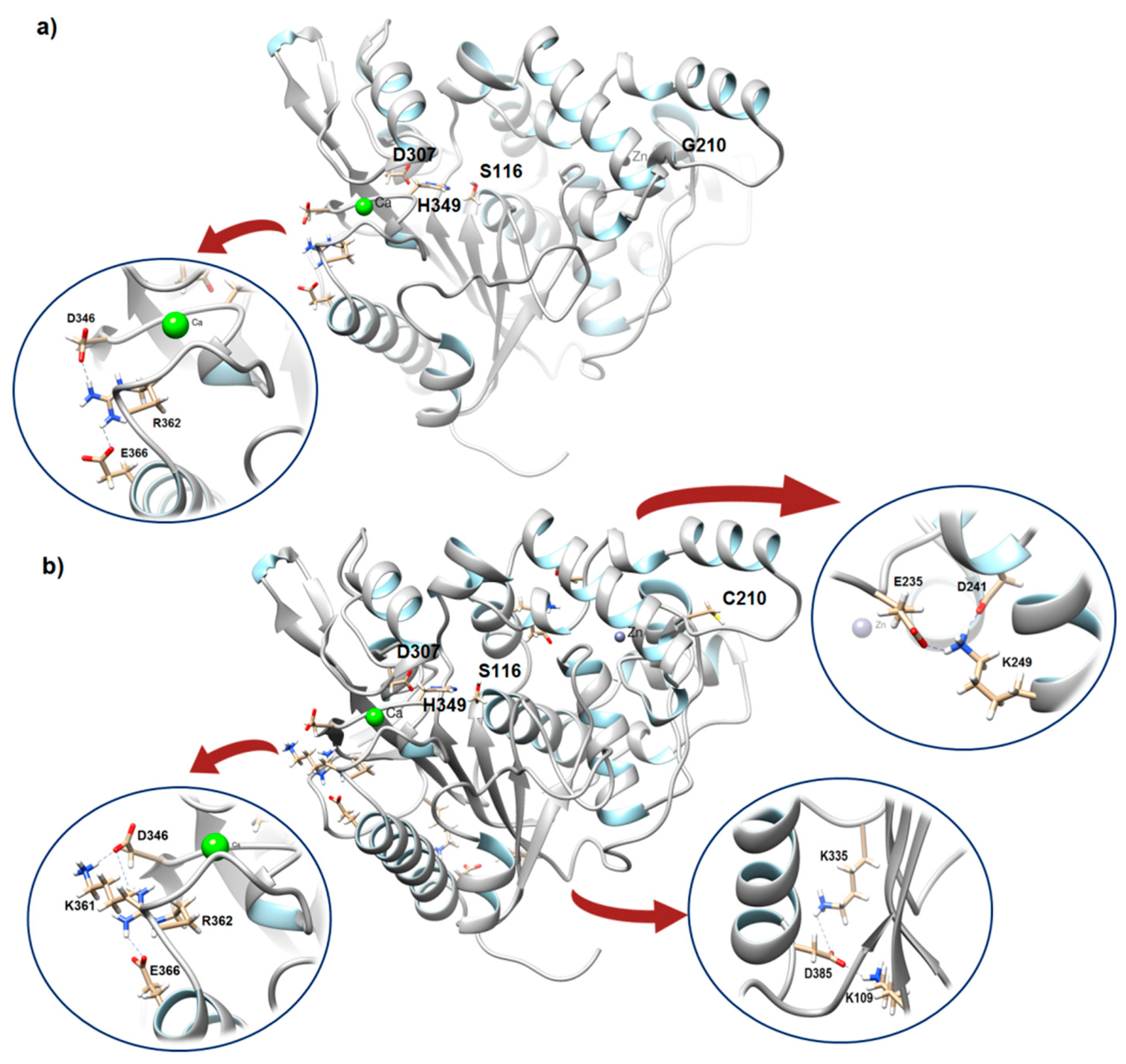
2.2.3. Hydrogen Bond and Salt Bridge Analysis
2.2.4. Cation-π Interactions
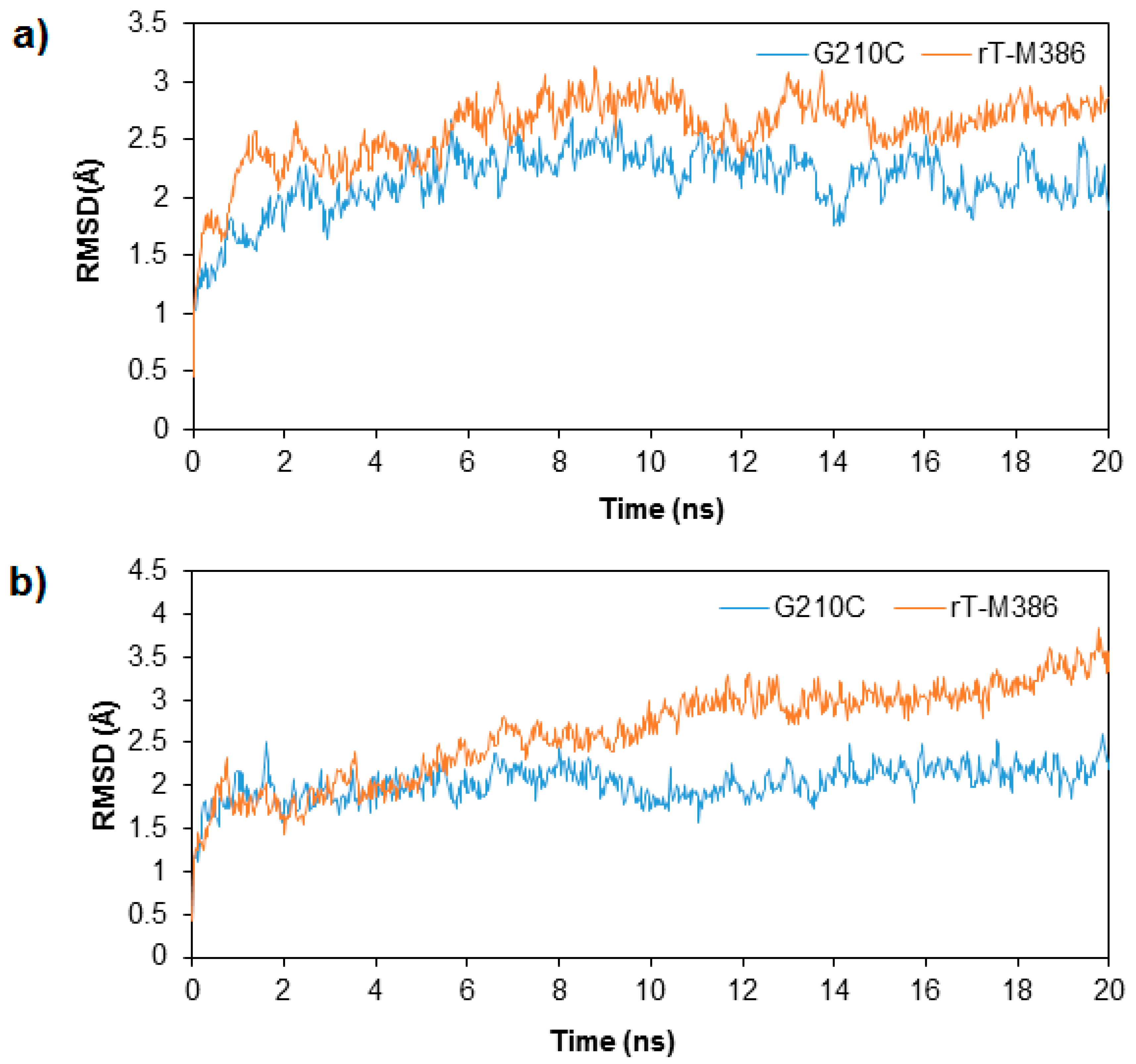
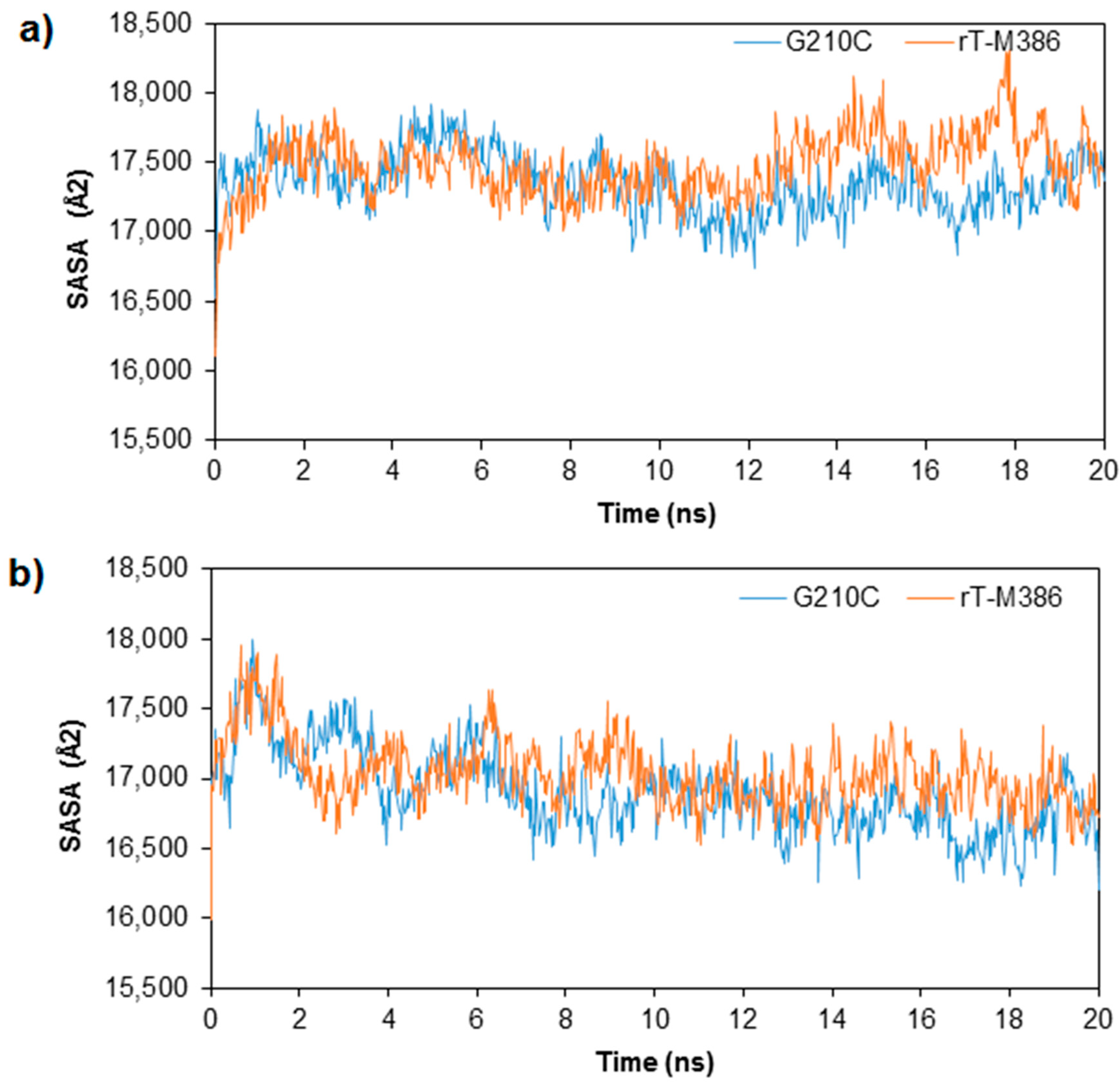
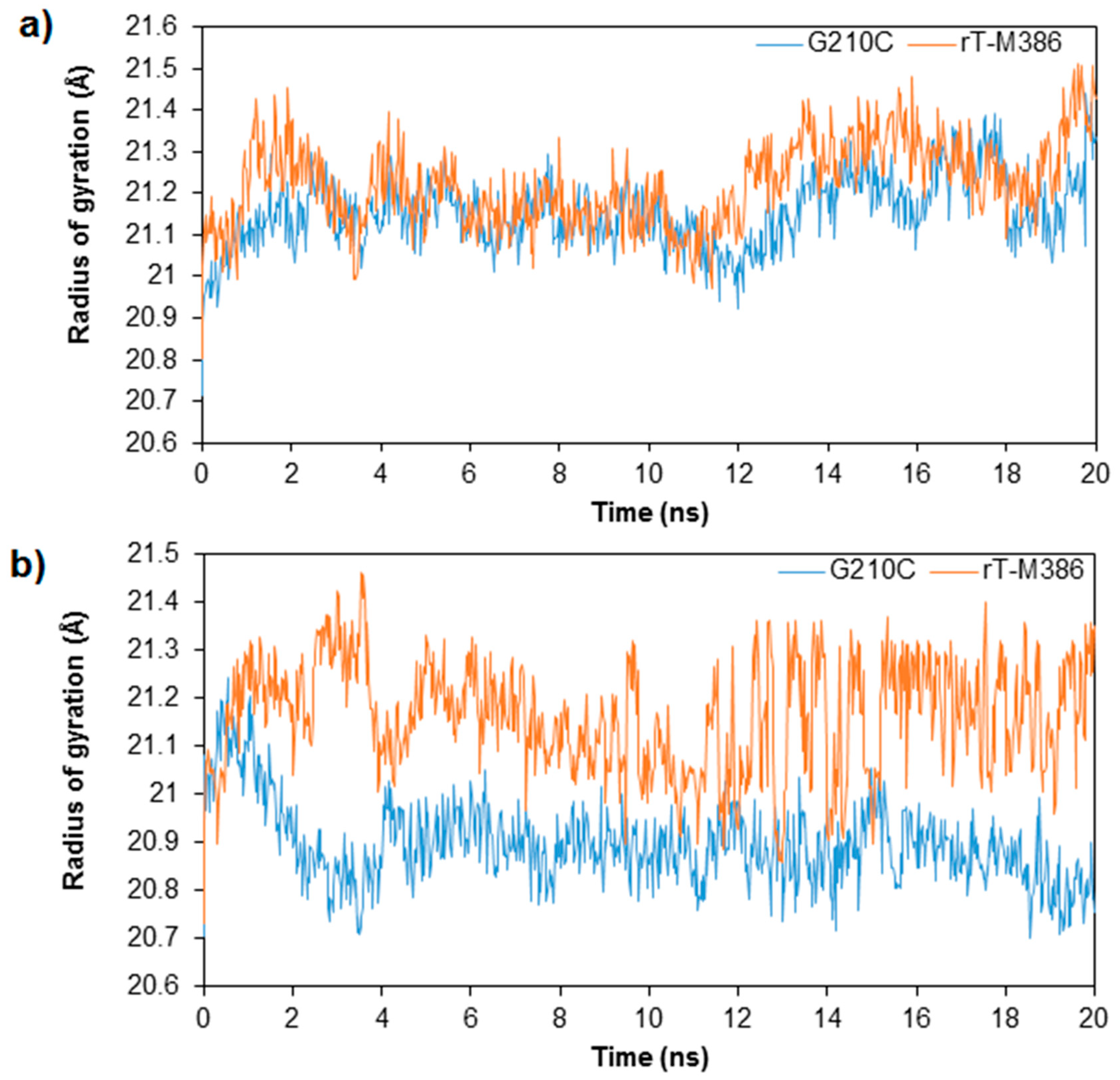
2.3. MD Simulation of rT-M386 and G210C Lipases
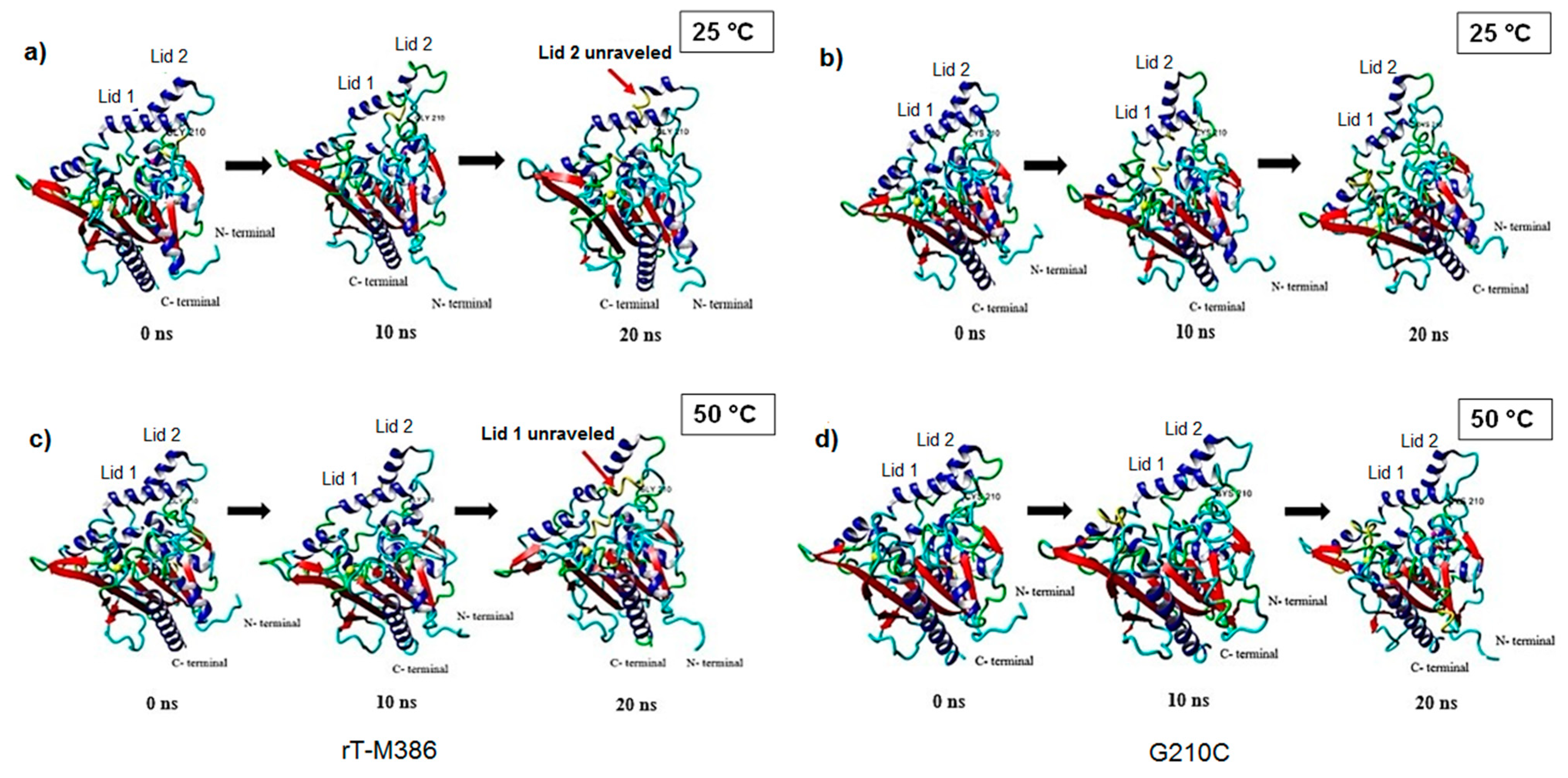
2.4. Structural Dynamics of Mutant G210C and Its Wild-Type
3. Discussion
4. Materials and Methods
4.1. Protein Sequence
4.2. Structure Prediction of rT-M386 and G210C Lipases
4.3. Covalent and Noncovalent Interactions Analysis
4.4. Solvent Accessibility Prediction
4.5. Molecular Dynamics Simulations of rT-M386 and G210C Lipases
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ó’Fágáin, C. Engineering Protein Stability. In Methods in Molecular Biology (Clifton, N.J.); Humana Press: New York, UK, USA, 2011; Volume 681, pp. 103–136. [Google Scholar]
- D’Amico, S.; Claverie, P.; Collins, T.; Georlette, D.; Gratia, E.; Hoyoux, A.; Meuwis, M.A.; Feller, G.; Gerday, C. Molecular basis of cold adaptation. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2002, 357, 917–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerday, C.; Aittaleb, M.; Bentahir, M.; Chessa, J.P.; Claverie, P.; Collins, T.; D’Amico, S.; Dumont, J.; Garsoux, G.; Georlette, D.; et al. Cold-adapted enzymes: From fundamentals to biotechnology. Trends Biotechnol. 2000, 18, 103–107. [Google Scholar] [CrossRef]
- Smalås, A.O.; Leiros, H.K.; Os, V.; Willassen, N.P. Cold adapted enzymes. Biotechnol. Annu. Rev. 2000, 6, 1–57. [Google Scholar] [PubMed]
- Ganjalikhany, M.R.; Ranjbar, B.; Taghavi, A.H.; Tohidi Moghadam, T. Functional motions of candida antarctica lipase b: A survey through open-close conformations. PLoS ONE 2012, 7, 327. [Google Scholar] [CrossRef] [PubMed]
- Socha, R.D.; Tokuriki, N. Modulating protein stability—Directed evolution strategies for improved protein function. FEBS J. 2013, 280, 5582–5595. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, D.; Biswas, S.; Roy, S.; Dattagupta, J.K. Improving thermostability of papain through structure-based protein engineering. Protein Eng. Des. Sel. 2010, 23, 457–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pucci, F.; Bourgeas, R.; Rooman, M. Predicting protein thermal stability changes upon point mutations using statistical potentials: Introducing HoTMuSiC. Sci. Rep. 2016, 6, 23257. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui, K.S. Some like it hot, some like it cold: Temperature dependent biotechnological applications and improvements in extremophilic enzymes. Biotechnol. Adv. 2015, 33, 1912–1922. [Google Scholar] [CrossRef]
- Trejo, E.S.U. Protein adaptations in archael extremophiles. Archaea 2013, 2013, 373275. [Google Scholar]
- Villeneuve, P.; Muderhwa, J.M.; Graille, J.; Haas, M.J. Customizing lipases for biocatalysis: A survey of chemical, physical and molecular biological approaches. J. Mol. Catal. B Enzym. 2000, 9, 113–148. [Google Scholar] [CrossRef]
- Veno, J.; Ahmad Kamarudin, N.; Mohamad Ali, M.; Masomian, M.; Raja Abd Rahman, R. Directed evolution of recombinant C-terminal truncated Staphylococcus epidermidis lipase AT2 for the enhancement of thermostability. Int. J. Mol. Sci. 2017, 18, 2202. [Google Scholar] [CrossRef] [PubMed]
- Carugo, O.; Pongor, S. Protein fold similarity estimated by a probabilistic approach based on C (alpha) -C (alpha) distance comparison. J. Mol. Biol. 2002, 315, 887–898. [Google Scholar] [CrossRef] [PubMed]
- Tyndall, J.D.A.; Sinchaikul, S.; Fothergill-Gilmore, L.A.; Taylor, P.; Walkinshaw, M.D. Crystal structure of a thermostable lipase from Bacillus stearothermophilus P1. J. Mol. Biol. 2002, 323, 859–869. [Google Scholar] [CrossRef]
- Abd Rahman, R.N.Z.R.; Shariff, F.M.; Basri, M.; Salleh, A.B. 3D structure Elucidation of thermostable l2 lipase from thermophilic Bacillus sp. L2. Int. J. Mol. Sci. 2012, 13, 9207–9217. [Google Scholar] [CrossRef] [PubMed]
- Tiesinga, J.J.W.; Van Pouderoyen, G.; Nardini, M.; Ransac, S.; Dijkstra, B.W. Structural Basis of Phospholipase Activity of Staphylococcus hyicus lipase. J. Mol. Biol. 2007, 371, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Fomenko, D.E.; Marino, S.M.; Gladyshev, V.N. Functional diversity of cysteine residues in proteins and unique features of catalytic redox-active cysteines in thiol oxidoreductases. Mol. Cells 2008, 26, 228–235. [Google Scholar] [PubMed]
- Nagano, N.; Ota, M.; Nishikawa, K. Strong hydrophobic nature of cysteine residues in proteins. FEBS Lett. 1999, 458, 69–71. [Google Scholar] [CrossRef]
- Hummer, G.; Garde, S.; Garca, A.E.; Paulaitis, M.E.; Pratt, L.R.; Wolynes, P.G. The pressure dependence of hydrophobic interactions is consistent with the observed pressure denaturation of proteins (protein foldingprotein folding kineticshydrophobic effectactivation volumesprotein unfolding). Biophysics 1998, 95, 1552–1555. [Google Scholar]
- Imai, K.; Mitaku, S. Mechanisms of secondary structure breakers in soluble proteins. Biophysics 2005, 1, 55–65. [Google Scholar] [CrossRef] [Green Version]
- Qiu, H.; Honey, D.M.; Kingsbury, J.S.; Park, A.; Boudanova, E.; Wei, R.R.; Pan, C.Q.; Edmunds, T. Impact of cysteine variants on the structure, activity, and stability of recombinant human α-galactosidase A. Protein Sci. 2015, 24, 1401–1411. [Google Scholar] [CrossRef]
- Klausen, M.S.; Jespersen, M.C.; Nielsen, H.; Jensen, K.K.; Jurtz, V.I.; Sønderby, C.K.; Sommer, M.O.A.; Winther, O.; Nielsen, M.; Petersen, B.; et al. NetSurfP-2.0: Improved prediction of protein structural features by integrated deep learning. Protein 2019, 87, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.B.; He, Y.; Pan, X.M. Predicting protein secondary structure and solvent accessibility with an improved multiple linear regression method. Proteins Struct. Funct. Bioinform. 2005, 61, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Pletneva, E.V.; Laederach, A.T.; Bruce Fulton, D.; Kostić, N.M. The Role of Cation−π Interactions in Biomolecular Association. Design of Peptides Favoring Interactions between Cationic and Aromatic Amino Acid Side Chains. J. Am. Chem. Soc. 2001, 123, 6232–6245. [Google Scholar] [CrossRef] [PubMed]
- Prajapati, R.S.; Sirajuddin, M.; Durani, V.; Sreeramulu, S.; Varadarajan, R. Contribution of Cation−π Interactions to Protein Stability. Biochemistry 2006, 45, 15000–15010. [Google Scholar] [CrossRef] [PubMed]
- Gallivan, J.P.; Dougherty, D.A. Cation-pi interactions in structural biology. Proc. Natl. Acad. Sci. USA 1999, 96, 9459–9464. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.; Woo, S.M.; Siu, T.; Cortopassi, W.A.; Duarte, F.; Paton, R.S. Cation–π interactions in protein–ligand binding: Theory and data-mining reveal different roles for lysine and arginine. Chem. Sci. 2018, 9, 2655–2665. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.I.; Lan, D.; Durrani, R.; Huan, W.; Zhao, Z.; Wang, Y. The Lid Domain in Lipases: Structural and Functional Determinant of Enzymatic Properties. Front. Bioeng. Biotechnol. 2017, 5, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Secundo, F.; Carrea, G.; Tarabiono, C.; Gatti-Lafranconi, P.; Brocca, S.; Lotti, M.; Jaeger, K.E.; Puls, M.; Eggert, T. The lid is a structural and functional determinant of lipase activity and selectivity. J. Mol. Catal. B Enzym. 2006, 39, 166–170. [Google Scholar] [CrossRef]
- Kumar, R.; Singh, R.; Kaur, J. Characterization and molecular modelling of an engineered organic solvent tolerant, thermostable lipase with enhanced enzyme activity. J. Mol. Catal. B Enzym. 2013, 97, 243–251. [Google Scholar] [CrossRef]
- Qian, H.; Zhang, C.; Lu, Z.; Xia, B.; Bie, X.; Zhao, H.; Lu, F. Consensus design for improved thermostability of lipoxygenase from Anabaena sp. PCC 7120. BMC Biotechnol. 2018, 18, 1–7. [Google Scholar] [CrossRef]
- Gromiha, M.M.; Thomas, S.; Santhosh, C. Role of cation-π interactions to the stability of thermophilic proteins. Prep. Biochem. Biotechnol. 2002, 32, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Vogt, G.; Woell, S.; Argos, P. Protein thermal stability, hydrogen bonds, and ion pairs. J. Mol. Biol. 1997, 269, 631–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newberry, R.W.; Raines, R.T. A prevalent intraresidue hydrogen bond stabilizes proteins. Nat. Chem. Biol. 2016, 12, 1084–1088. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.H.; Huang, S.W.; Lai, Y.L.; Lin, C.P.; Shih, C.H.; Huang, C.C.; Hsu, W.L.; Hwang, J.K. On the relationship between the protein structure and protein dynamics. Proteins 2008, 72, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Biundo, A.; Steinkellner, G.; Gruber, K.; Spreitzhofer, T.; Ribitsch, D.; Guebitz, G.M. Engineering of the zinc-binding domain of an esterase from Clostridium botulinum towards increased activity on polyesters. Catal. Sci. Technol. 2017, 7, 1440–1447. [Google Scholar] [CrossRef]
- Karshikoff, A.; Nilsson, L.; Ladenstein, R. Rigidity versus flexibility: The dilemma of understanding protein thermal stability. FEBS J. 2015, 282, 3899–3917. [Google Scholar] [CrossRef]
- Adamczak, R.; Porollo, A.; Meller, J. Accurate prediction of solvent accessibility using neural networks-based regression. Proteins Struct. Funct. Bioinform. 2004, 56, 753–767. [Google Scholar] [CrossRef] [PubMed]
- Gianese, G.; Argos, P.; Pascarella, S. Structural adaptation of enzymes to low temperatures. Protein Eng. Des. Sel. 2001, 14, 141–148. [Google Scholar] [CrossRef] [Green Version]
- Momen-Roknabadi, A.; Sadeghi, M.; Pezeshk, H.; Marashi, S.A. Impact of residue accessible surface area on the prediction of protein secondary structures. BMC Bioinform. 2008, 9, 357. [Google Scholar] [CrossRef]
- Okumura, H. Temperature and pressure denaturation of chignolin: Folding and unfolding simulation by multibaric-multithermal molecular dynamics method. Proteins Struct. Funct. Bioinform. 2012, 80, 2397–2416. [Google Scholar] [CrossRef]
- Abd Rahman, R.N.Z.R.; Ahmad Kamarudin, N.H.; Yunus, J.; Salleh, A.B.; Basri, M. Expression of an organic solvent stable lipase from Staphylococcus epidermidis AT2. Int. J. Mol. Sci. 2010, 11, 3195–3208. [Google Scholar] [CrossRef] [PubMed]
- Kamarudin, N.H.A.; Rahman, R.N.Z.R.A.; Ali, M.S.M.; Leow, T.C.; Basri, M.; Salleh, A.B. Unscrambling the effect of C-terminal tail deletion on the stability of a cold-adapted, organic solvent stable lipase from Staphylococcus epidermidis AT2. Mol. Biotechnol. 2014, 56, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Joo, K.; Lee, J.; Lee, J.; Raman, S.; Thompson, J.; Tyka, M.; Baker, D.; Karplus, K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins Struct. Funct. Bioinform. 2009, 77, 114–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Prot. Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costantini, S.; Colonna, G.; Facchiano, A.M. ESBRI: A web server for evaluating salt bridges in proteins. Bioinformation 2008, 3, 137–138. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Koraimann, G.; Vriend, G. Increasing the precision of comparative models with YASARA NOVA-a self-parameterizing force field. Proteins Struct. Funct. Bioinform. 2002, 47, 393–402. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lipases | No. of Hydrogen Bonds | Total Hydrogen Bond Energy [kJ/mol] |
|---|---|---|
| rT-M386 | 334 | 6848.55 |
| G210C | 349 | 7084.35 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Veno, J.; Rahman, R.N.Z.R.A.; Masomian, M.; Ali, M.S.M.; Kamarudin, N.H.A. Insight into Improved Thermostability of Cold-Adapted Staphylococcal Lipase by Glycine to Cysteine Mutation. Molecules 2019, 24, 3169. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24173169
Veno J, Rahman RNZRA, Masomian M, Ali MSM, Kamarudin NHA. Insight into Improved Thermostability of Cold-Adapted Staphylococcal Lipase by Glycine to Cysteine Mutation. Molecules. 2019; 24(17):3169. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24173169
Chicago/Turabian StyleVeno, Jiivittha, Raja Noor Zaliha Raja Abd Rahman, Malihe Masomian, Mohd Shukuri Mohamad Ali, and Nor Hafizah Ahmad Kamarudin. 2019. "Insight into Improved Thermostability of Cold-Adapted Staphylococcal Lipase by Glycine to Cysteine Mutation" Molecules 24, no. 17: 3169. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24173169