
Development and Evaluation of a HS-SPME GC-MS Method for Determining the Retention of Volatile Phenols by Cyclodextrin in Model Wine



Abstract
:1. Introduction
2. Results and Discussion
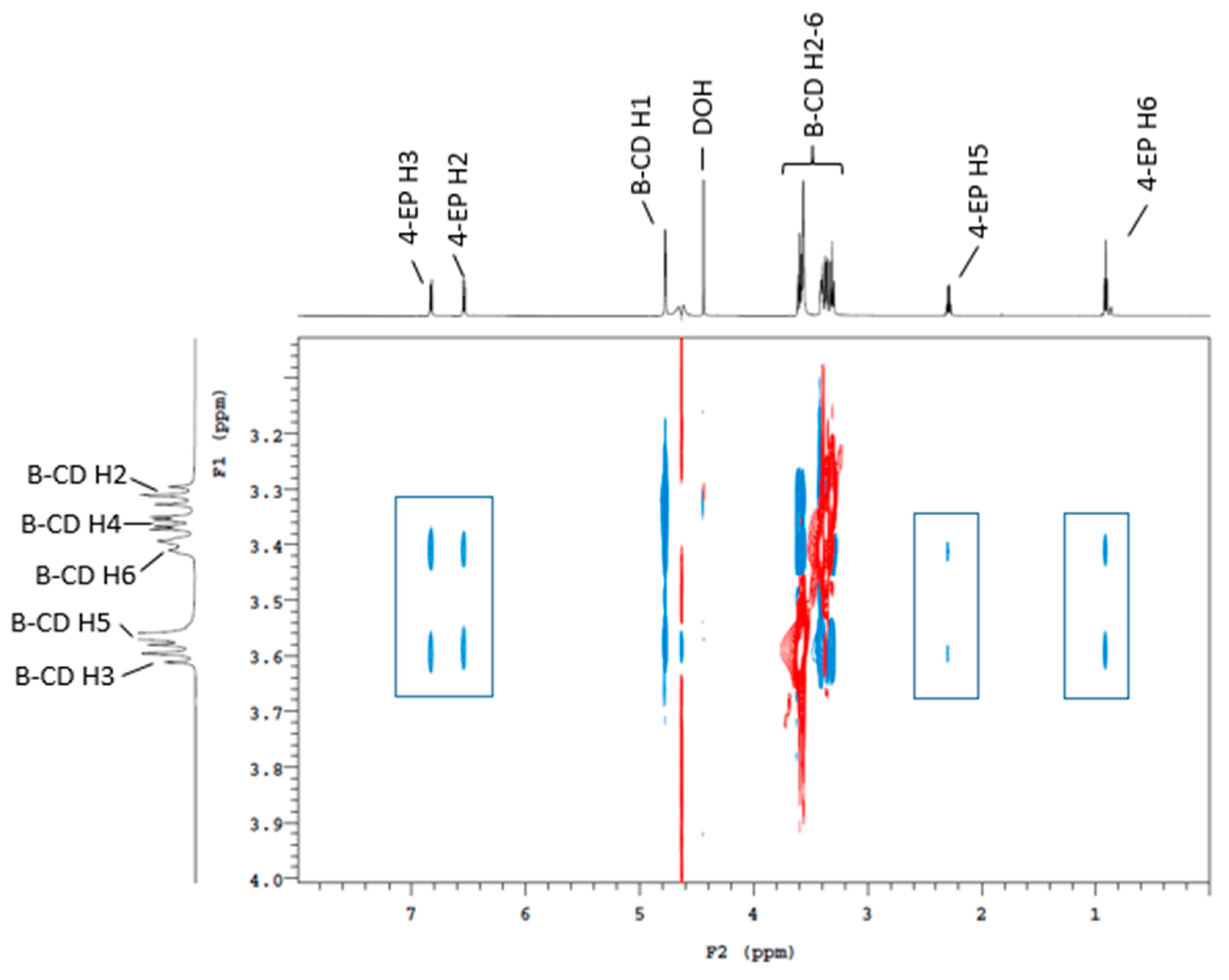
2.1. Traditional (Three-Phase) HS-SPME GC-MS, NMR, and Sensory Analysis
2.2. Development of A Four-Phase HS-SPME GC-MS Method
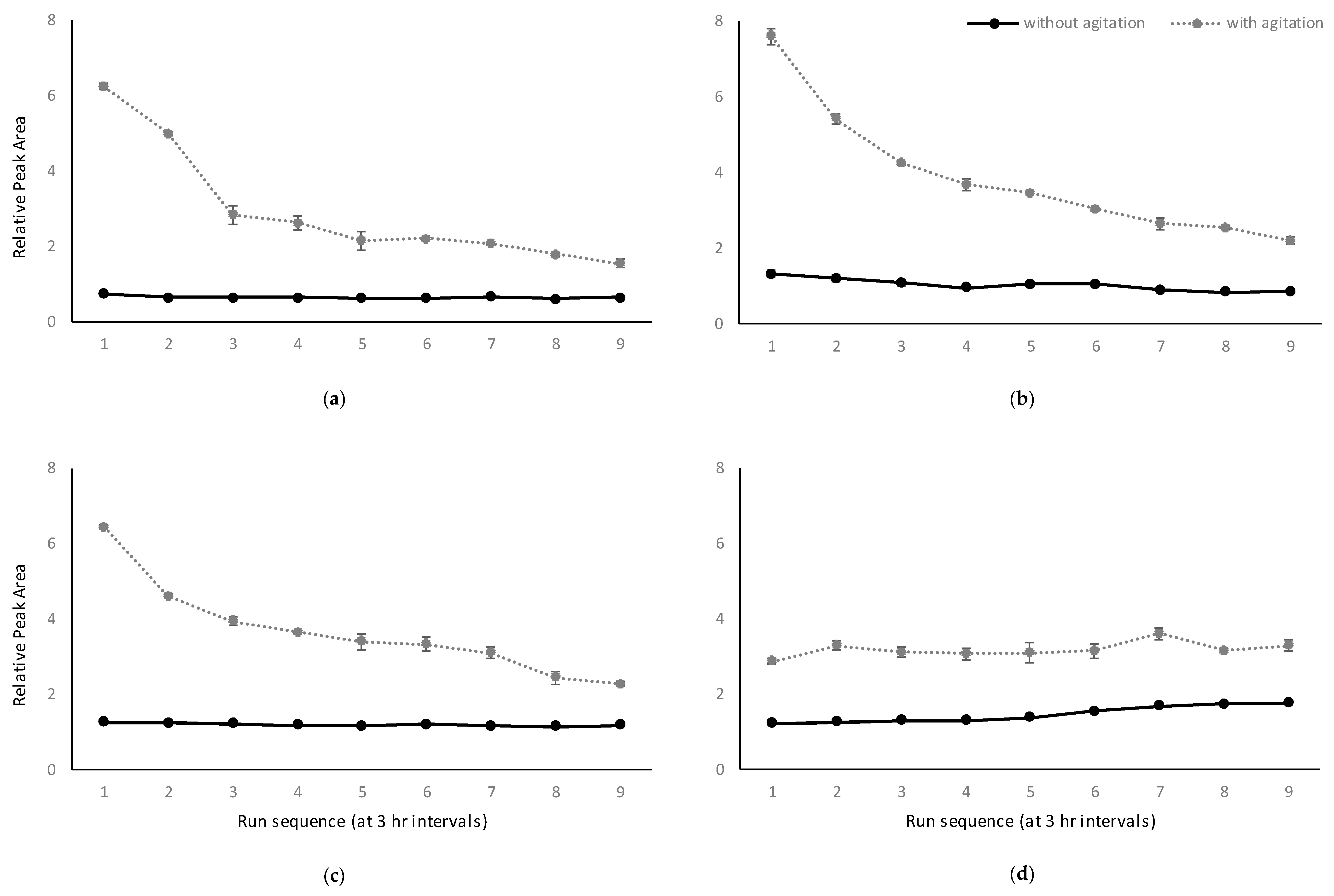
2.2.1. Influence of Agitation
2.2.2. Influence of Volume of Internal Standard Solution
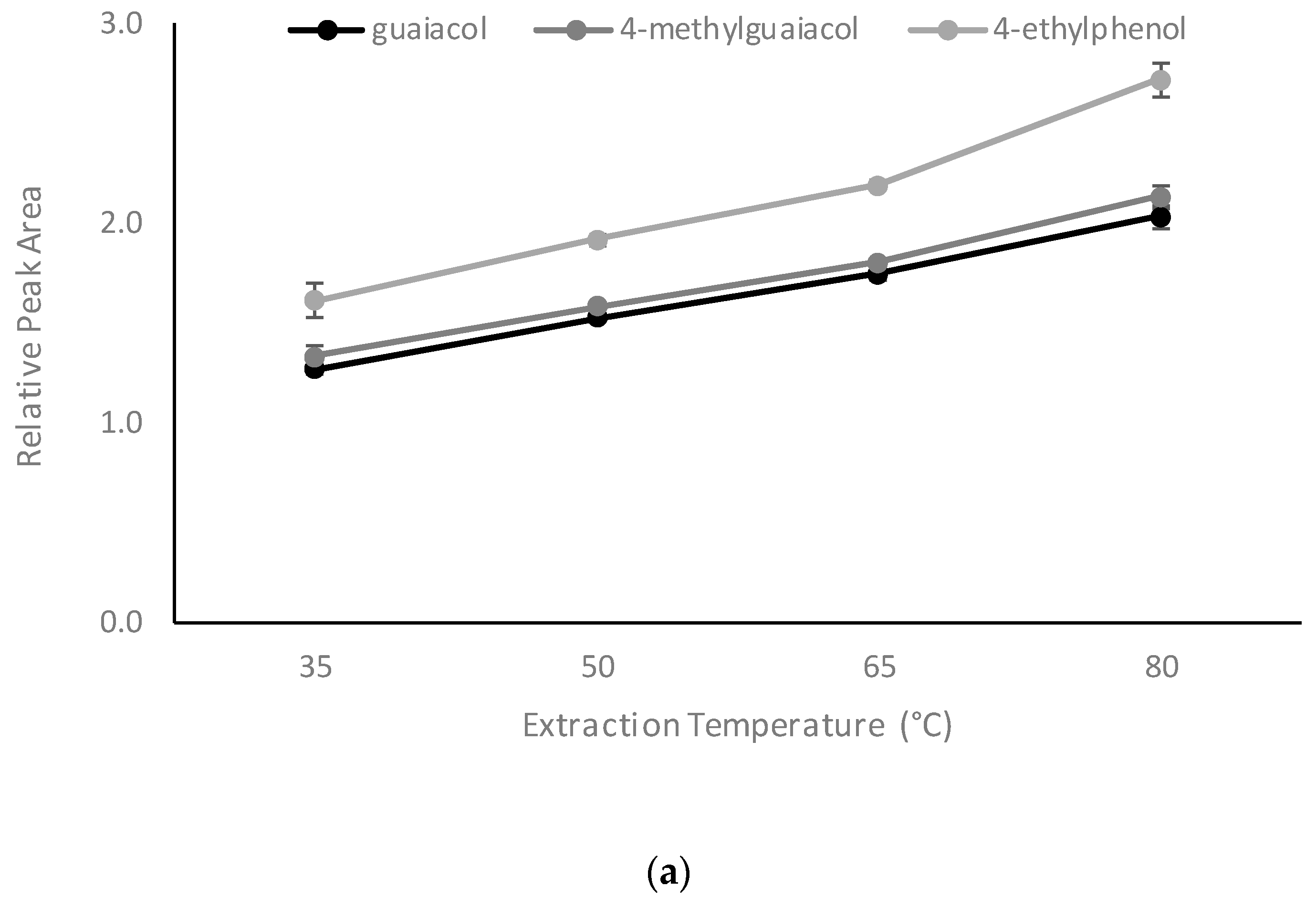
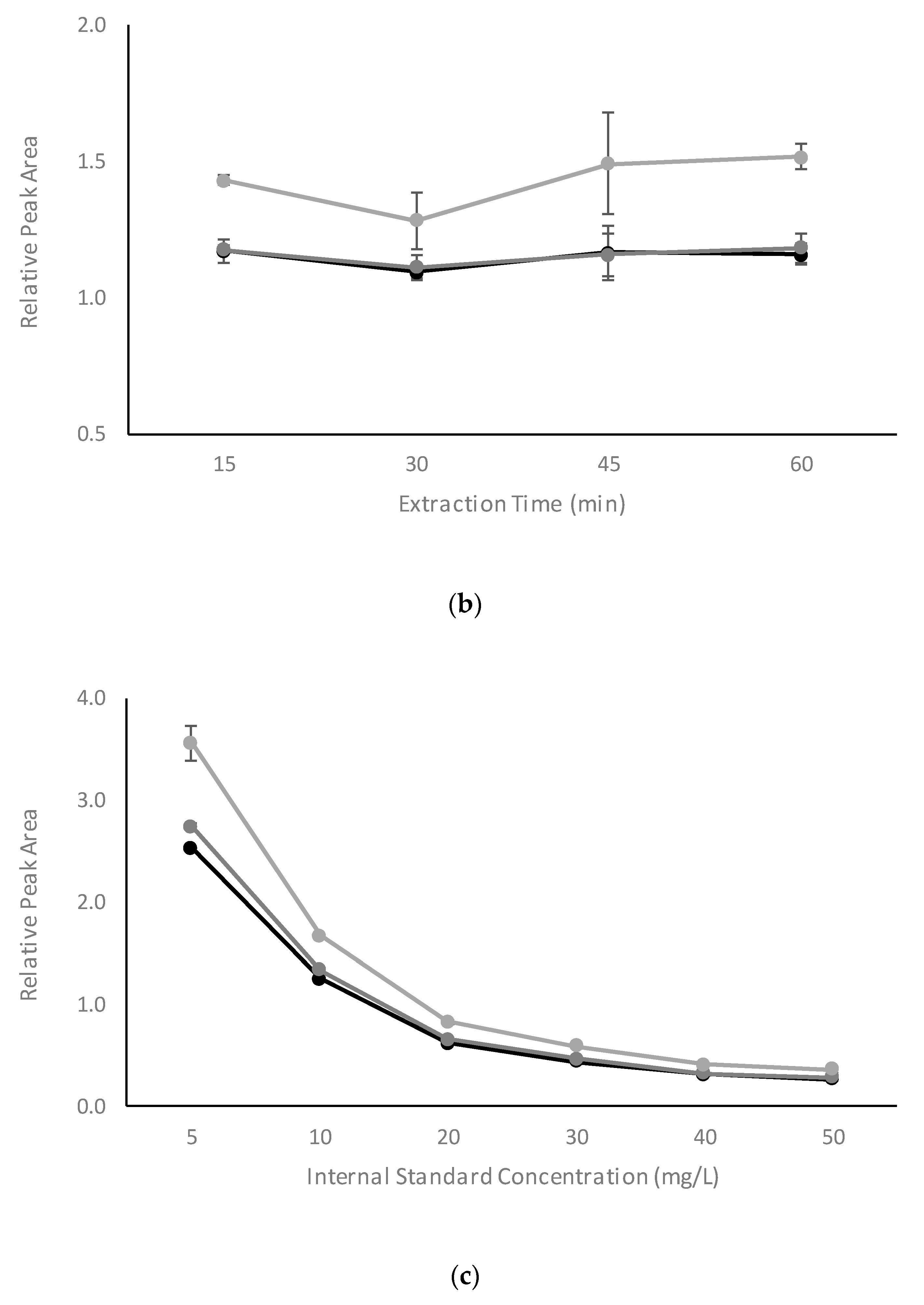
2.2.3. Influence of Extraction Temperature, Extraction Time, and Internal Standard Concentration
2.3. Experimental Conditions for the Four-Phase HS-SPME GC-MS Method
2.4. Retention of Volatile Phenols by α-CD, β-CD, and γ-CD in Model Wine
3. Materials and Methods
3.1. Chemicals
3.2. Nuclear Magnetic Resonance Analysis
3.3. Sensory Analysis
3.4. GC-MS Instrumental Analysis
3.5. HS-SPME GC-MS Analysis of Volatile Phenols in Model Wine Following CD Addition
3.6. Method Development for the Four-Phase HS-SPME GC-MS Method
3.6.1. Influence of Agitation, Internal Standard Volume, and Pre-Analysis Equilibration Time
3.6.2. Influence of Extraction Time, Extraction Temperature, and Internal Standard Concentration
3.7. Method Performance for the Four-Phase HS-SPME GC-MS Method
3.8. Retention of Volatile Phenols in Model Wine by Cyclodextrins
3.9. Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Maga, J.A. The contribution of wood to the flavour of alcoholic beverages. Food Rev. Int. 1989, 5, 39–99. [Google Scholar] [CrossRef]
- Boidron, J.N.; Chatonnet, P.; Pons, M. Influence du bois sur certaines substances odorantes des vins. Conn. Vigne Vin 1988, 22, 275–294. [Google Scholar] [CrossRef]
- Prida, A.; Chatonnet, P. Impact of oak-derived compounds on the olfactory perception of barrel-aged wines. Am. J. Enol. Vitic. 2010, 61, 408–413. [Google Scholar]
- Chatonnet, P.; Dubourdieu, D.; Boidron, J.N. The influence of Brettanomyces/Dekkera sp. yeasts and lactic acid bacteria on the ethylphenol content of red wines. Am. J. Enol. Vitic. 1995, 46, 463–468. [Google Scholar]
- Kennison, K.R.; Wilkinson, K.L.; Williams, H.G.; Smith, J.H.; Gibberd, M.R. Smoke-derived taint in wine: Effect of postharvest smoke exposure of grapes on the chemical composition and sensory characteristics of wine. J. Agric. Food Chem. 2007, 55, 10897–10901. [Google Scholar] [CrossRef]
- Kennison, K.R.; Gibberd, M.R.; Pollnitz, A.P.; Wilkinson, K.L. Smoke-derived taint in wine: The release of smoke-derived volatile phenols during fermentation of Merlot juice following grapevine exposure to smoke. J. Agric. Food Chem. 2008, 56, 7379–7383. [Google Scholar] [CrossRef] [PubMed]
- Kennison, K.R.; Wilkinson, K.L.; Pollnitz, A.P.; Williams, H.G.; Gibberd, M.R. Effect of timing and duration of grapevine exposure to smoke on the composition and sensory properties of wine. Aust. J. Grape Wine Res. 2009, 15, 228–237. [Google Scholar] [CrossRef]
- Chassagne, D.; Guilloux-Benatier, M.; Alexandre, H.; Voilley, A. Sorption of wine volatile phenols by yeast lees. Food Chem. 2005, 91, 39–44. [Google Scholar] [CrossRef]
- Pradelles, R.; Alexandre, H.; Ortiz-Julien, A.; Chassagne, D. Effects of yeast cell-wall characteristics on 4-ethylphenol sorption capacity in model wine. J. Agric. Food Chem. 2008, 56, 11854–11861. [Google Scholar] [CrossRef]
- Fudge, A.L.; Schiettecatte, M.; Ristic, R.; Hayasaka, Y.; Wilkinson, K.L. Amelioration of smoke taint in wine by treatment with commercial fining agents. Aust. J. Grape Wine Res. 2012, 18, 302–307. [Google Scholar] [CrossRef]
- Lisanti, M.T.; Gambuti, A.; Genovese, A.; Piombino, P.; Moio, L. Treatment by fining agents of red wine affected by phenolic off-odour. Eur. Food Res. Technol. 2017, 243, 501–510. [Google Scholar] [CrossRef]
- Ugarte, P.; Agosin, E.; Bordeu, E.; Villalobos, J.I. Reduction of 4-ethylphenol and 4-ethylguaiacol concentration in red wines using reverse osmosis and adsorption. Am. J. Enol. Vitic. 2005, 56, 30–36. [Google Scholar]
- Fudge, A.L.; Ristic, R.; Wollan, D.; Wilkinson, K.L. Amelioration of smoke taint in wine by reverse osmosis and solid phase adsorption. Aust. J. Grape Wine Res. 2011, 17, S41–S48. [Google Scholar] [CrossRef]
- Larcher, R.; Puecher, C.; Rohregger, S.; Malacarne, M.; Nicolini, G. 4-Ethylphenol and 4-ethylguaiacol depletion in wine using esterified cellulose. Food Chem. 2012, 132, 2126–2130. [Google Scholar] [CrossRef]
- Carrasco-Sánchez, V.; John, A.; Marican, A.; Santos, L.S.; Laurie, V.F. Removal of 4-ethylphenol and 4-ethylguaiacol with polyaniline-based compounds in wine-like model solutions and red wine. Molecules 2015, 20, 14312–14325. [Google Scholar] [CrossRef] [PubMed]
- Garde-Cerdán, T.; Zalacain, A.; Lorenzo, C.; Alonso, J.L.; Salinas, M.R. Molecularly imprinted polymer-assisted simple clean-up of 2, 4, 6-trichloroanisole and ethylphenols from aged red wines. Am. J. Enol. Vitic. 2008, 59, 396–400. [Google Scholar]
- Teixeira, R.; Dopico-García, S.; Andrade, P.B.; Valentão, P.; López-Vilariño, J.M.; González-Rodríguez, V.; Cela-Pérez, C.; Silva, L.R. Volatile phenols depletion in red wine using molecular imprinted polymers. J. Food Sci. Technol. 2015, 52, 7735–7746. [Google Scholar] [CrossRef] [Green Version]
- Szejtli, J. Introduction and general overview of cyclodextrin chemistry. Chem. Rev. 1998, 98, 1743–1754. [Google Scholar] [CrossRef]
- Kant, A.; Linforth, R.S.T.; Hort, J.; Taylor, A.J. Effect of β-cyclodextrin on aroma release and flavor perception. J. Agric. Food Chem. 2004, 52, 2028–2035. [Google Scholar] [CrossRef]
- Buschmann, H.-J.; Schollmeyer, E. Applications of cyclodextrins in cosmetic products: A review. J. Cosmetic Sci. 2002, 53, 185–191. [Google Scholar]
- Challa, R.; Ahuja, A.; Ali, J.; Khar, R.K. Cyclodextrins in drug delivery: An updated review. AAPS Pharm. Sci. Tech. 2005, 6, E329–E357. [Google Scholar] [CrossRef] [PubMed]
- Astray, G.; Gonzalez-Barreiro, C.; Mejuto, J.C.; Rial-Otero, R.; Simal-Gándara, J. A review on the use of cyclodextrins in foods. Food Hydrocoll. 2009, 23, 1631–1640. [Google Scholar] [CrossRef]
- Marques, H.M.C. A review on cyclodextrin encapsulation of essential oils and volatiles. Flavour Fragr. J. 2010, 25, 313–326. [Google Scholar] [CrossRef]
- Botelho, G.; Valiau, C.; Moreira da Silva, A. Effect of cyclodextrins on off-odours removal of red wine: An innovative approach. Ciência e Técnica Vitivinícola 2011, 26, 63–68. [Google Scholar]
- Ratnasooriya, C.C.; Rupasinghe, H.P.V. Extraction of phenolic compounds from grapes and their pomace using β-cyclodextrin. Food Chem. 2012, 134, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Whiton, R.S.; Zoecklein, B.W. Optimization of headspace solid-phase microextraction for analysis of wine aroma compounds. Am. J. Enol. Vitic. 2000, 51, 379–382. [Google Scholar]
- Rocha, S.; Ramalheira, V.; Barros, A.; Delgadillo, I.; Coimbra, M.A. Headspace solid phase microextraction (SPME) analysis of flavor compounds in wines. Effect of the matrix volatile composition in the relative response factors in a wine model. J. Agric. Food Chem. 2001, 49, 5142–5151. [Google Scholar] [CrossRef]
- Castro-Mejías, R.; Natera-Marín, R.; Valme García-Moreno, M.; Barroso, C.G. Optimisation of headspace solid-phase microextraction for the analysis of volatile phenols in wine. J. Chromatogr. A 2003, 995, 11–20. [Google Scholar] [CrossRef]
- Tsoutsi, C.; Konstantinou, I.; Hela, D.; Albanis, T. Screening method for organophosphorus insecticides and their metabolites in olive oil samples based on headspace solid-phase microextraction coupled with gas chromatography. Anal. Chim. Acta 2006, 573, 216–222. [Google Scholar] [CrossRef]
- Pawliszyn, J.; Yang, M.J.; Orton, M.L. Quantitative determination of caffeine in beverages using a combined SPME-GC/MS method. J. Chem. Edu. 1997, 74, 1130. [Google Scholar] [CrossRef]
- Pawliszyn, J. Theory of solid-phase microextraction. J. Chromatogr. Sci. 2000, 38, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Sander, R. Compilation of Henry’s law constants (version 4.0) for water as solvent. Atmos. Chem. Phys. 2015, 15, 4399–4981. [Google Scholar] [CrossRef]
- Ma, S.; Turino, G.M.; Hayashi, T.; Yanuma, H.; Usuki, T.; Lin, Y.Y. Stable deuterium internal standard for the isotope-dilution LC–MS/MS analysis of elastin degradation. Anal. Biochem. 2013, 440, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Wieland, F.; Neff, A.; Gloess, A.N.; Poisson, L.; Atlan, S.; Larrain, D.; Prêtre, D.; Blank, I.; Yeretzian, C. Temperature dependence of Henry’s law constants: An automated, high-throughput gas stripping cell design coupled to PTR-ToF-MS. Int. J. Mass Spectrom. 2015, 387, 69–77. [Google Scholar] [CrossRef]
- Liang, H.R.; Foltz, R.L.; Meng, M.; Bennett, P. Ionization enhancement in atmospheric pressure chemical ionization and suppression in electrospray ionization between target drugs and stable-isotope-labeled internal standards in quantitative liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2003, 17, 2815–2821. [Google Scholar] [CrossRef] [PubMed]
- Astray, G.; Mejuto, J.C.; Morales, J.; Rial-Otero, R.; Simal-Gándara, J. Factors controlling flavors binding constants to cyclodextrins and their applications in foods. Food Res. Int. 2010, 43, 1212–1218. [Google Scholar] [CrossRef]
- Meilgaard, M.C.; Carr, B.T.; Civille, G.V. Sensory Evaluation Techniques, 4th ed.; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
Sample Availability: Samples of the compounds are not available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Guaiacol | 4-Methylguaiacol | 4-Ethylphenol | |
|---|---|---|---|
| Control | 0.99 ± 0.02 | 1.00 ± 0.01 | 1.22 ± 0.02 |
| α-CD | 0.99 ± 0.02 (100%) | 1.00 ± 0.02 (100%) | 1.23 ± 0.02 (100.8%) |
| β-CD | 0.99 ± 0.01 (100%) | 1.00 ± 0.01 (100%) | 1.29 ± 0.02 (105.7%) |
| γ-CD | 0.99 ± 0.02 (100%) | 1.00 ± 0.02 (100%) | 1.24 ± 0.03 (101.6%) |
| Guaiacol | 4-Methylguaiacol | 4-Ethylphenol | ||||
|---|---|---|---|---|---|---|
| RPA | CV (%) | RPA | CV (%) | RPA | CV (%) | |
| 0.25 mg/L | 0.35 | 2.7 | 0.38 | 2.5 | 0.48 | 1.7 |
| 0.5 mg/L | 0.63 (0.65) | 3.1 (4.5) | 0.68 (0.69) | 2.2 (5.8) | 0.86 (0.90) | 5.4 (8.9) |
| 1.0 mg/L | 1.12 | 7.5 | 1.16 | 8.9 | 1.50 | 9.3 |
| 1.25 mg/L | 1.61 (1.53) | 0.6 (5.4) | 1.74 (1.58) | 0.8 (5.8) | 2.27 (1.99) | 6.5 (7.0) |
| 1.75 mg/L | 2.12 (2.10) | 0.1 (2.2) | 2.27 (2.17) | 0.2 (3.1) | 3.01 (2.84) | 6.6 (6.1) |
| 2.0 mg/L | 2.31 | 0.7 | 2.36 | 0.8 | 2.97 | 2.4 |
| Guaiacol | 4-Methylguaiacol | 4-Ethylphenol | 4-Ethylguaiacol | o-Cresol | m-Cresol | p-Cresol | Eugenol | ||
|---|---|---|---|---|---|---|---|---|---|
| Control | 1.26 a ± 0.05 | 1.36 a ± 0.06 | 1.85 a ± 0.10 | 0.94 a ± 0.01 | 1.64 a ± 0.11 | 0.95 a ± 0.05 | 1.86 a ± 0.06 | 0.66 a ± 0.03 | |
| α-CD | 5 g/L | 1.14 abc ± 0.02 | 1.19 ab ± 0.04 | 1.51 ab ± 0.10 | 0.82 b ± 0.01 | 1.39 abc ± 0.06 | 0.81 b ± 0.02 | 0.92 b ± 0.03 | 0.59 ab ± 0.03 |
| (90.3%) | (87.9%) | (81.6%) | (87.3%) | (85.0%) | (85.1%) | (84.8%) | (71.1%) | ||
| 25 g/L | 1.17 ab ± 0.05 | 1.20 ab ± 0.04 | 1.42 b ± 0.11 | 0.76 b ± 0.02 | 1.41 ab ± 0.07 | 0.75 bc ± 0.03 | 0.85 bc ± 0.04 | 0.47 c ± 0.01 | |
| (92.9%) | (88.2%) | (76.9%) | (80.4%) | (86.2%) | (79.3%) | (78.3%) | (71.1%) | ||
| β-CD | 5 g/L | 1.01 cde ± 0.03 | 1.04 bcd ± 0.04 | 0.80 c ± 0.03 | 0.68 c ± 0.02 | 1.14 cd ± 0.03 | 0.61 d ± 0.01 | 0.61 d ± 0.01 | 0.46 c ± 0.01 |
| (80.2%) | (76.7%) | (43.1%) | (72.6%) | (70.0%) | (63.9%) | (56.5%) | (70.0%) | ||
| 25 g/L | 0.98 de ± 0.00 | 0.95 cd ± 0.02 | 0.43 d ± 0.02 | 0.47 e ± 0.01 | 0.92 d ± 0.01 | 0.41 e ± 0.01 | 0.34 e ± 0.01 | 0.24 e ± 0.01 | |
| (77.5%) | (69.8%) | (23.1%) | (50.0%) | (56.0%) | (42.6%) | (31.1%) | (36.9%) | ||
| γ-CD | 5 g/L | 1.07 bcd ± 0.01 | 1.10 bc ± 0.01 | 1.29 b ± 0.01 | 0.79 b ± 0.00 | 1.24 bc ± 0.01 | 0.67 cd ± 0.01 | 0.76 c ± 0.01 | 0.56 b ± 0.01 |
| (84.8%) | (81.1%) | (69.8%) | (84.2%) | (75.7%) | (70.1%) | (70.0%) | (84.7%) | ||
| 25 g/L | 0.89 e ± 0.02 | 0.87 d ± 0.03 | 0.77 cd ± 0.03 | 0.57 d ± 0.01 | 0.93 d ± 0.01 | 0.49 e ± 0.01 | 0.56 d ± 0.01 | 0.35 d ± 0.01 | |
| (70.6%) | (64.1%) | (41.5%) | (60.0%) | (57.2%) | (51.1%) | (51.5%) | (53.0%) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dang, C.; Wilkinson, K.L.; Jiranek, V.; Taylor, D.K. Development and Evaluation of a HS-SPME GC-MS Method for Determining the Retention of Volatile Phenols by Cyclodextrin in Model Wine. Molecules 2019, 24, 3432. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24193432
Dang C, Wilkinson KL, Jiranek V, Taylor DK. Development and Evaluation of a HS-SPME GC-MS Method for Determining the Retention of Volatile Phenols by Cyclodextrin in Model Wine. Molecules. 2019; 24(19):3432. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24193432
Chicago/Turabian StyleDang, Chao, Kerry L. Wilkinson, Vladimir Jiranek, and Dennis K. Taylor. 2019. "Development and Evaluation of a HS-SPME GC-MS Method for Determining the Retention of Volatile Phenols by Cyclodextrin in Model Wine" Molecules 24, no. 19: 3432. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24193432