
Dicentrine Potentiates TNF-α-Induced Apoptosis and Suppresses Invasion of A549 Lung Adenocarcinoma Cells via Modulation of NF-κB and AP-1 Activation


 and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
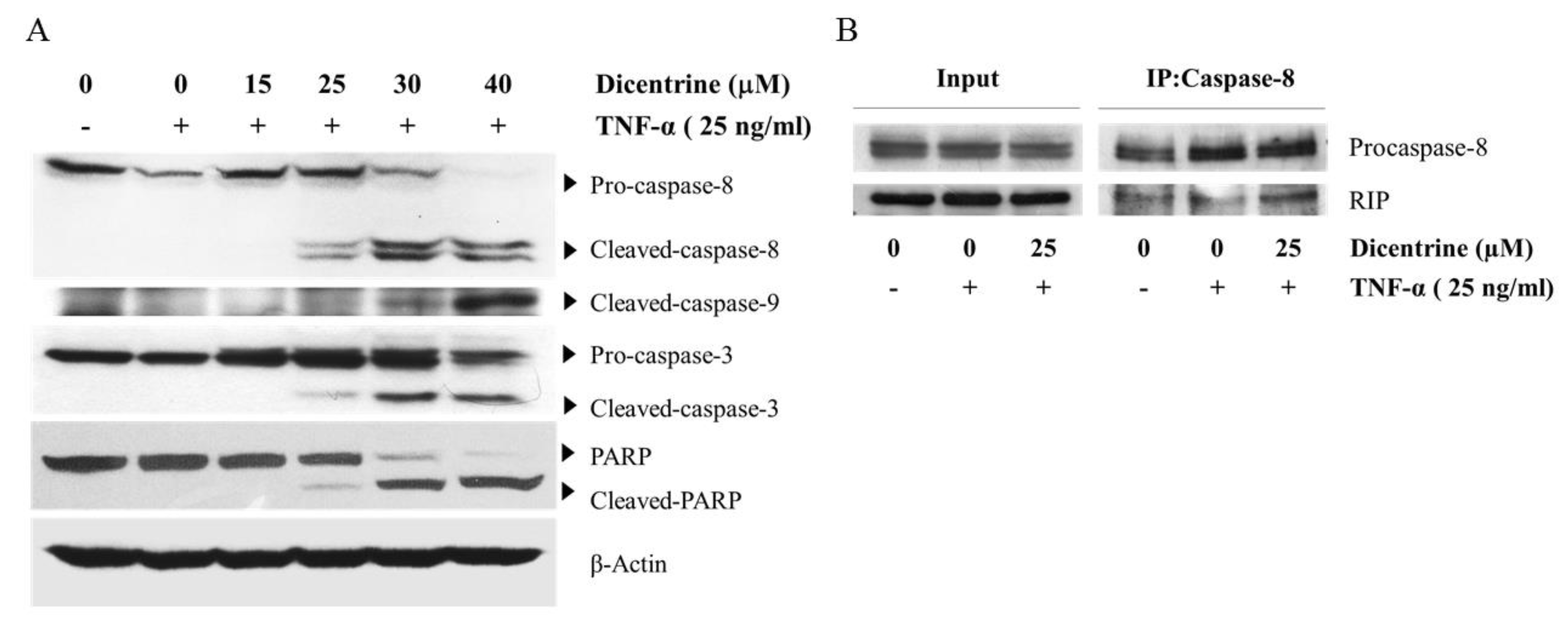
2.1. Dicentrine Potentiates TNF-α-Induced Apoptosis in A549 Lung Adenocarcinoma Cells
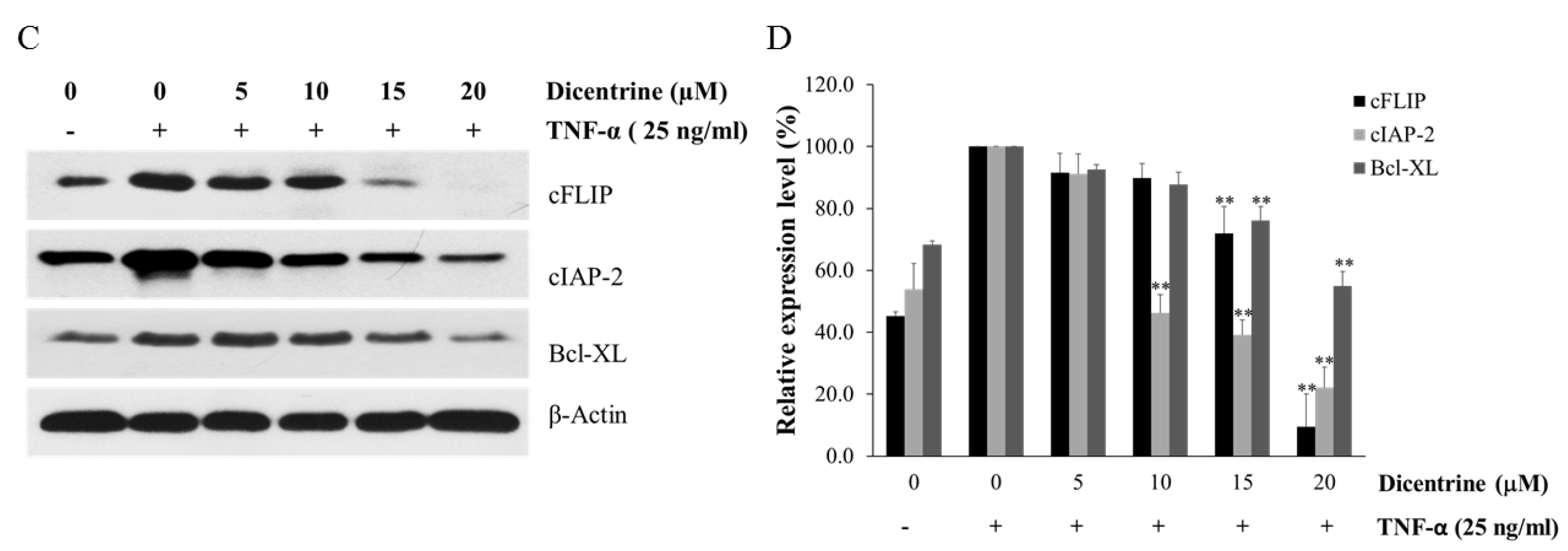
2.2. Dicentrine Enhances TNF-α-Induced Apoptosis in a Caspase-Dependent Manner and Inhibits the Expression of Antiapoptotic Proteins
2.3. Dicentrine Inhibits TNF-α-Induced A549 Cells Invasion and Migration
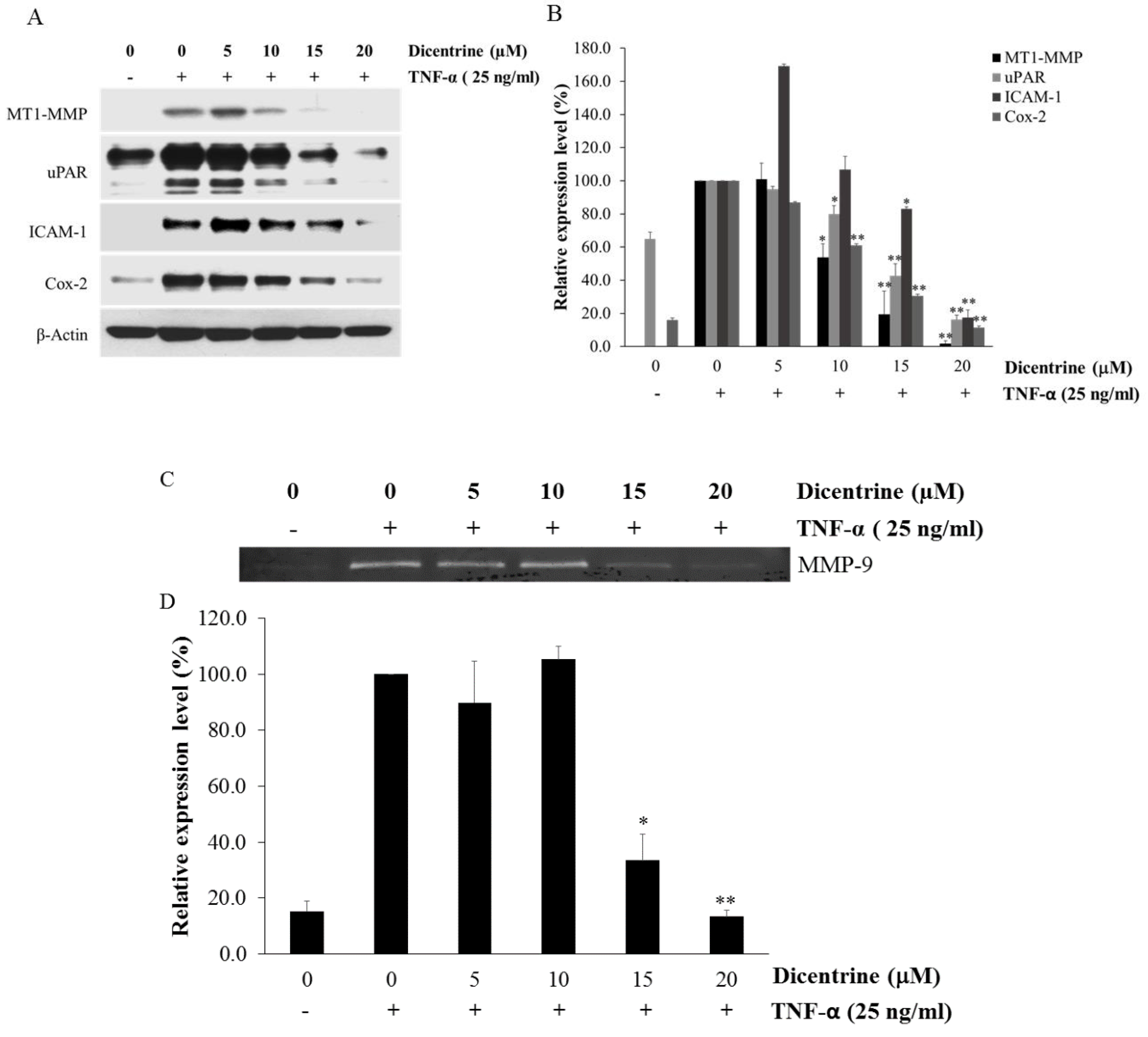
2.4. Dicentrine Inhibits TNF-α-Induced Expression of Metastasis-Associated Proteins
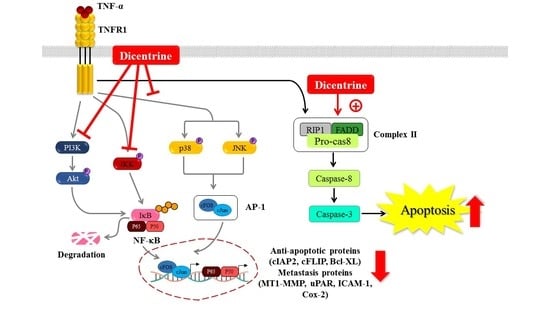
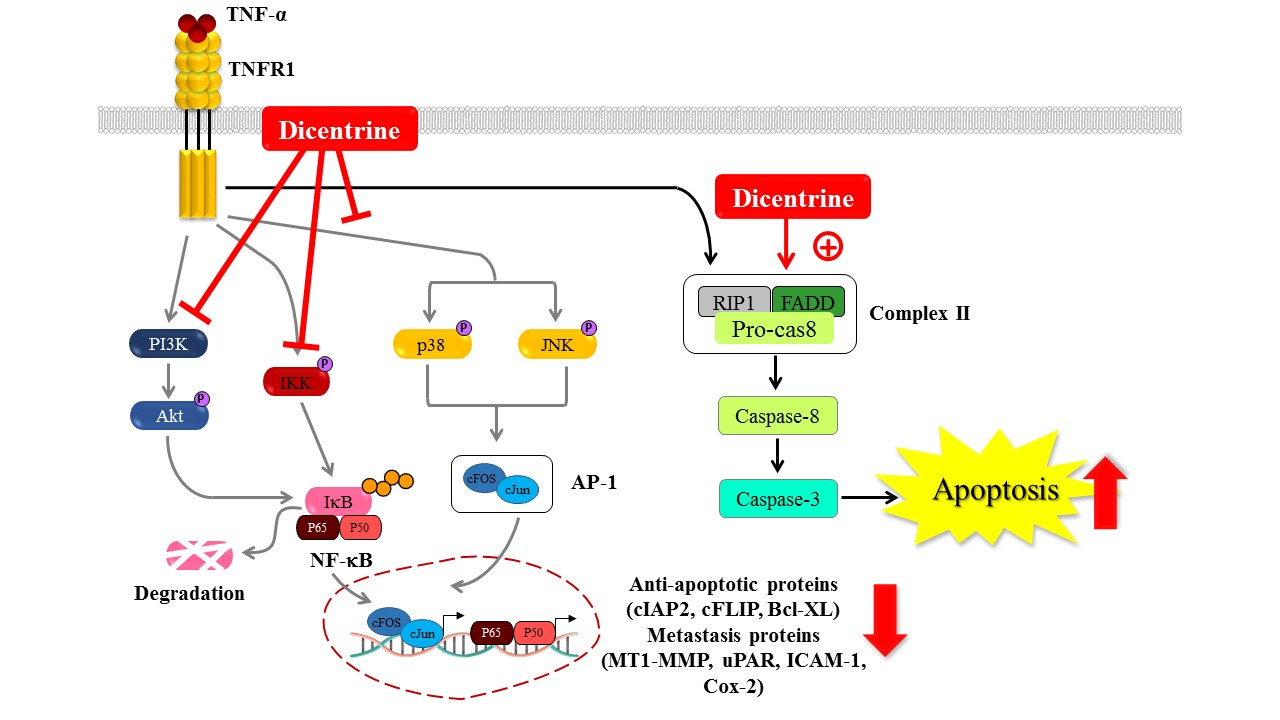
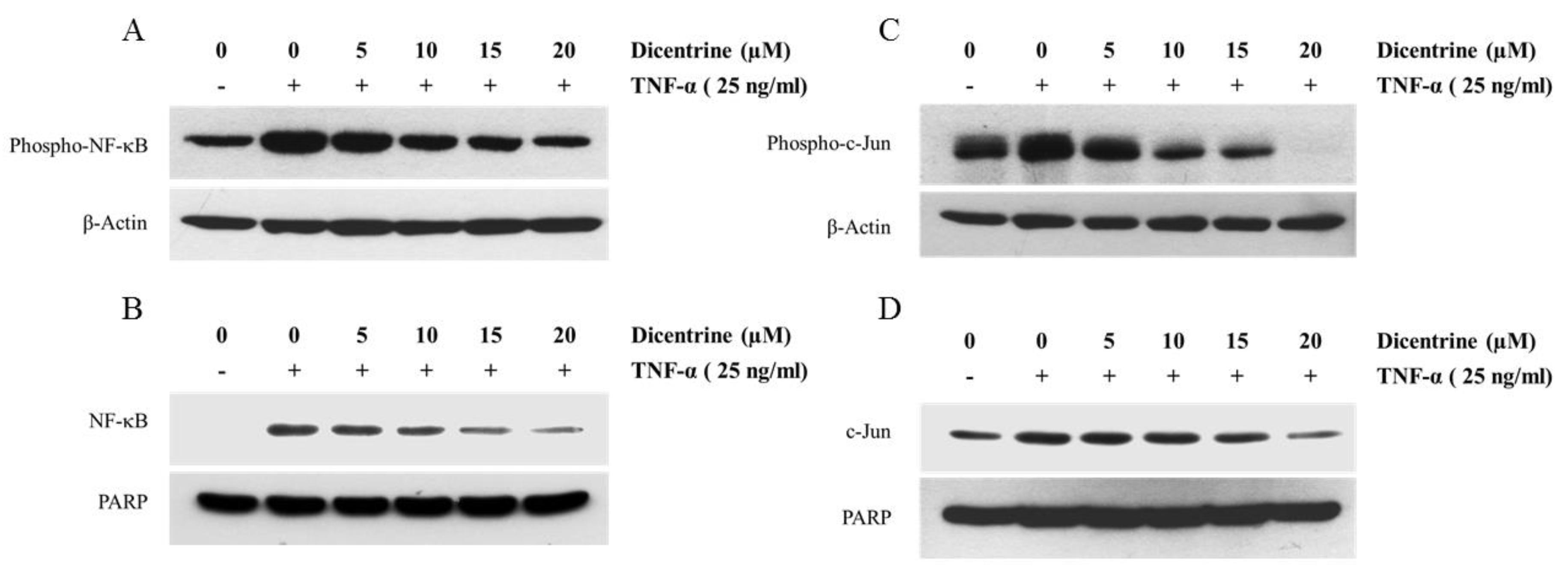
2.5. Dicentrine Inhibits TNF-α-Induced NF-κB and AP-1 Activation
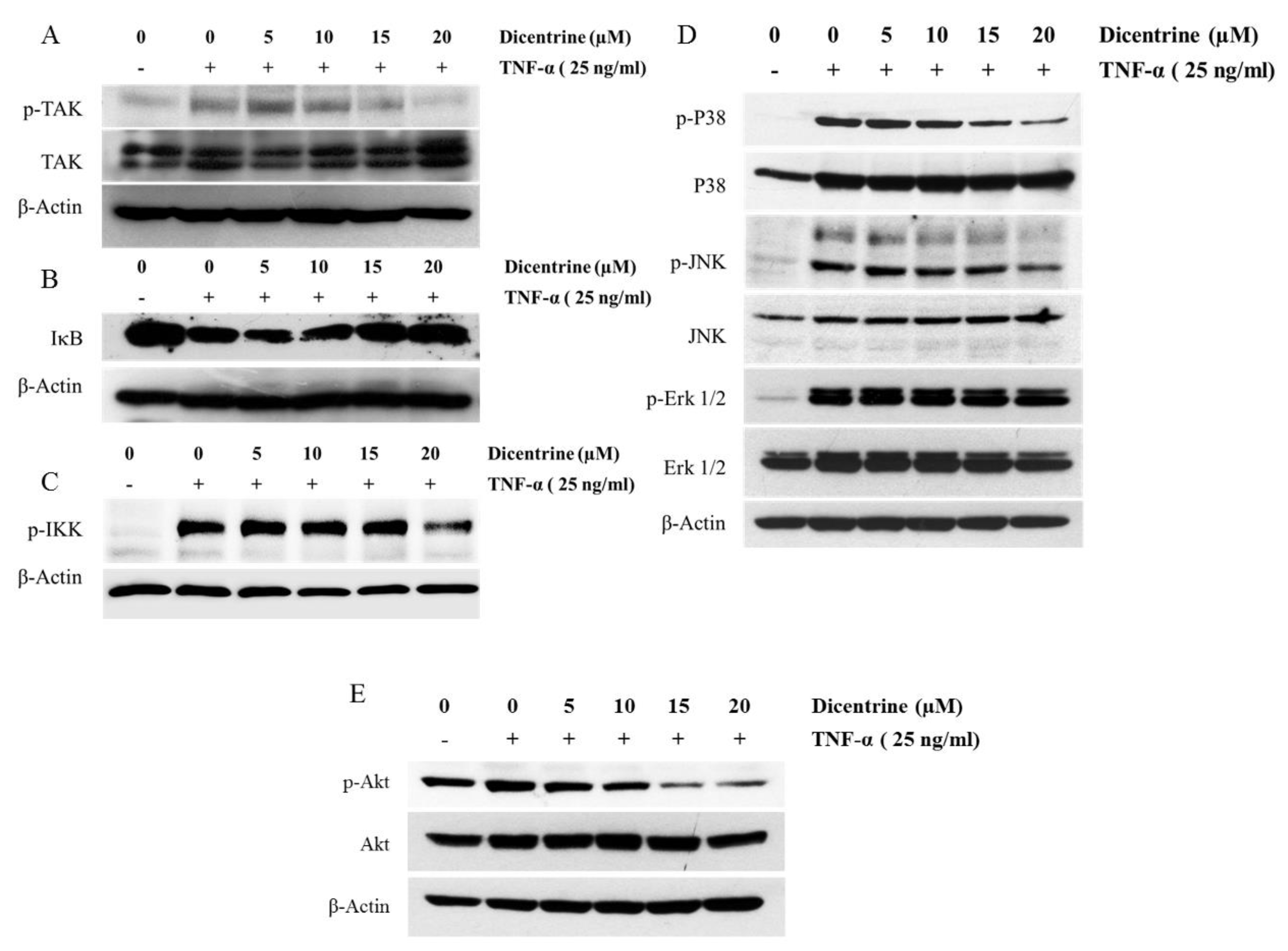
2.6. Effects of Dicentrine on TNF-α-Induced TAK-1, IκB-α, Akt, and MAPKs Signaling Pathways
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Cell Viability
4.4. Cell Cycle Arrest Assay
4.5. Cell Invasion and Migration Assay
4.6. Gelatin Zymography
4.7. Coimmunoprecipitation and Western Blot
4.8. Extraction of Nuclear and Whole Cell Lysate
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Malhotra, J.; Malvezzi, M.; Negri, E.; La Vecchia, C.; Boffetta, P. Risk factors for lung cancer worldwide. Eur. Respir. J. 2016, 48, 889–902. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Walser, T.C.; Dubinett, S.M. Chronic inflammation, chronic obstructive pulmonary disease, and lung cancer. Curr. Opin. Pulm. Med. 2009, 15, 303–307. [Google Scholar] [PubMed]
- Gomes, M.; Teixeira, A.L.; Coelho, A.; Araujo, A.; Medeiros, R. The role of inflammation in lung cancer. Adv. Exp. Med. Biol. 2014, 816, 1–23. [Google Scholar] [PubMed]
- Aggarwal, B.B.; Shishodia, S.; Sandur, S.K.; Pandey, M.K.; Sethi, G. Inflammation and cancer: How hot is the link? Biochem. Pharmacol. 2006, 72, 1605–1621. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef]
- Mantovani, A. Molecular pathways linking inflammation and cancer. Curr. Mol. Med. 2010, 10, 369–373. [Google Scholar] [CrossRef]
- Landskron, G.; De la Fuente, M.; Thuwajit, P.; Thuwajit, C.; Hermoso, M.A. Chronic inflammation and cytokines in the tumor microenvironment. J. Immunol. Res. 2014, 2014, 149185. [Google Scholar] [CrossRef]
- Shang, G.S.; Liu, L.; Qin, Y.W. IL-6 and TNF-alpha promote metastasis of lung cancer by inducing epithelial-mesenchymal transition. Oncol. Lett. 2017, 13, 4657–4660. [Google Scholar] [CrossRef]
- Derin, D.; Soydinc, H.O.; Guney, N.; Tas, F.; Camlica, H.; Duranyildiz, D.; Yasasever, V.; Topuz, E. Serum levels of apoptosis biomarkers, survivin and TNF-alpha in nonsmall cell lung cancer. Lung Cancer 2008, 59, 240–245. [Google Scholar] [CrossRef]
- Idriss, H.T.; Naismith, J.H. TNF alpha and the TNF receptor superfamily: Structure-function relationship(s). Microsc. Res. Tech. 2000, 50, 184–195. [Google Scholar]
- Chen, G.; Goeddel, D.V. TNF-R1 signaling: A beautiful pathway. Science 2002, 296, 1634–1635. [Google Scholar] [CrossRef]
- Zelova, H.; Hosek, J. TNF-alpha signalling and inflammation: Interactions between old acquaintances. Inflamm. Res. 2013, 62, 641–651. [Google Scholar] [CrossRef]
- Wang, L.; Du, F.; Wang, X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell 2008, 133, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Devin, A.; Rodriguez, Y.; Liu, Z.G. Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev. 1999, 13, 2514–2526. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F. TNF-alpha in promotion and progression of cancer. Cancer Metast. Rev. 2006, 25, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Gaur, U.; Aggarwal, B.B. Regulation of proliferation, survival and apoptosis by members of the TNF superfamily. Biochem. Pharmacol. 2003, 66, 1403–1408. [Google Scholar] [CrossRef]
- Wajant, H.; Pfizenmaier, K.; Scheurich, P. Tumor necrosis factor signaling. Cell Death Differ. 2003, 10, 45–65. [Google Scholar]
- Teng, C.M.; Yu, S.M.; Ko, F.N.; Chen, C.C.; Huang, Y.L.; Huang, T.F. Dicentrine, a natural vascular alpha 1-adrenoceptor antagonist, isolated from Lindera megaphylla. Br. J. Pharmacol. 1991, 104, 651–656. [Google Scholar] [CrossRef]
- Lin, H.F.; Huang, H.L.; Liao, J.F.; Shen, C.C.; Huang, R.L. Dicentrine Analogue-Induced G2/M Arrest and Apoptosis through Inhibition of Topoisomerase II Activity in Human Cancer Cells. Planta Med. 2015, 81, 830–837. [Google Scholar] [CrossRef]
- Montrucchio, D.P.; Cordova, M.M.; Santos, A.R. Plant derived aporphinic alkaloid S-(+)-dicentrine induces antinociceptive effect in both acute and chronic inflammatory pain models: Evidence for a role of TRPA1 channels. PLoS ONE 2013, 8, e67730. [Google Scholar] [CrossRef]
- Huang, R.L.; Chen, C.C.; Huang, Y.L.; Ou, J.C.; Hu, C.P.; Chen, C.F.; Chang, C. Anti-tumor effects of d-dicentrine from the root of Lindera megaphylla. Planta Med. 1998, 64, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Yodkeeree, S.; Ooppachai, C.; Pompimon, W.; Limtrakul (Dejkriengkraikul), P. O-Methylbulbocapnine and Dicentrine Suppress LPS-Induced Inflammatory Response by Blocking NF-kappaB and AP-1 Activation through Inhibiting MAPKs and Akt Signaling in RAW264.7 Macrophages. Biol. Pharm. Bull. 2018, 41, 1219–1227. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Mayo, M.W.; Korneluk, R.G.; Goeddel, D.V.; Baldwin, A.S., Jr. NF-kappaB antiapoptosis: Induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science 1998, 281, 1680–1683. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, F.; Carturan, E.; Chimenti, C.; Pieroni, M.; Agostini, C.; Angelini, A.; Crosato, M.; Valente, M.; Boffa, G.M.; Frustaci, A.; et al. Overexpression of tumor necrosis factor (TNF)alpha and TNFalpha receptor I in human viral myocarditis: Clinicopathologic correlations. Mod. Pathol. 2004, 17, 1108–1118. [Google Scholar] [CrossRef] [PubMed]
- Edinger, A.L.; Thompson, C.B. Death by design: Apoptosis, necrosis and autophagy. Curr. Opin. Cell Biol. 2004, 16, 663–669. [Google Scholar] [PubMed]
- Munoz-Gamez, J.A.; Rodriguez-Vargas, J.M.; Quiles-Perez, R.; Aguilar-Quesada, R.; Martin-Oliva, D.; de Murcia, G.; de Murcia, J.M.; Almendros, A.; de Almodovar, M.R.; Oliver, F.J. PARP-1 is involved in autophagy induced by DNA damage. Autophagy 2009, 5, 61–74. [Google Scholar]
- Lee, E.W.; Seo, J.; Jeong, M.; Lee, S.; Song, J. The roles of FADD in extrinsic apoptosis and necroptosis. BMB Rep. 2012, 45, 496–508. [Google Scholar] [CrossRef]
- Ofengeim, D.; Yuan, J. Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nat. Rev. Mol. Cell Biol. 2013, 14, 727–736. [Google Scholar]
- Brenner, D.; Blaser, H.; Mak, T.W. Regulation of tumour necrosis factor signalling: Live or let die. Nat. Rev. Immunol. 2015, 15, 362–374. [Google Scholar] [CrossRef]
- Grethe, S.; Ares, M.P.; Andersson, T.; Porn-Ares, M.I. p38 MAPK mediates TNF-induced apoptosis in endothelial cells via phosphorylation and downregulation of Bcl-x(L). Exp. Cell Res. 2004, 298, 632–642. [Google Scholar] [CrossRef]
- Tamatani, M.; Che, Y.H.; Matsuzaki, H.; Ogawa, S.; Okado, H.; Miyake, S.; Mizuno, T.; Tohyama, M. Tumor necrosis factor induces Bcl-2 and Bcl-x expression through NFkappaB activation in primary hippocampal neurons. J. Biol. Chem. 1999, 274, 8531–8538. [Google Scholar] [PubMed]
- Herrmann, J.L.; Beham, A.W.; Sarkiss, M.; Chiao, P.J.; Rands, M.T.; Bruckheimer, E.M.; Brisbay, S.; McDonnell, T.J. Bcl-2 suppresses apoptosis resulting from disruption of the NF-kappa B survival pathway. Exp. Cell Res. 1997, 237, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, D.J.; Cheung, H.H.; Mrad, R.L.; Plenchette, S.; Simard, C.; Enwere, E.; Arora, V.; Mak, T.W.; Lacasse, E.C.; Waring, J.; et al. Both cIAP1 and cIAP2 regulate TNFalpha-mediated NF-kappaB activation. Proc. Natl. Acad. Sci. USA 2008, 105, 11778–11783. [Google Scholar]
- Bertrand, M.J.M.; Milutinovic, S.; Dickson, K.M.; Ho, W.C.; Boudreault, A.; Durkin, J.; Gillared, J.W.; Jaquith, J.B.; Morris, S.J.; Barker, P.A. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol. Cell 2008, 30, 689–700. [Google Scholar] [PubMed]
- Safa, A.R. c-FLIP, a master anti-apoptotic regulator. Exp. Oncol. 2012, 34, 176–184. [Google Scholar]
- Zhang, X.; Jin, T.; Yang, H.; DeWolf, W.C.; Khosravi, R.; Olumi, A.F. Persistent c-FLIP(L) Expression Is Necessary and Sufficient to Maintain Resistance to Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand–Mediated Apoptosis in Prostate Cancer. Cancer Res. 2004, 64, 7086–7091. [Google Scholar]
- Ham, B.; Fernandez, M.C.; D’Costa, Z.; Brodt, P. The diverse roles of the TNF axis in cancer progression and metastasis. Trends Cancer Res. 2016, 11, 1–27. [Google Scholar]
- Rao, J.S.; Gondi, C.; Chetty, C.; Chittivelu, S.; Joseph, P.A.; Lakka, S.S. Inhibition of invasion, angiogenesis, tumor growth, and metastasis by adenovirus-mediated transfer of antisense uPAR and MMP-9 in non-small cell lung cancer cells. Mol. Cancer Ther. 2005, 4, 1399–1408. [Google Scholar]
- Lin, Y.C.; Shun, C.T.; Wu, M.S.; Chen, C.C. A novel anticancer effect of thalidomide: Inhibition of intercellular adhesion molecule-1-mediated cell invasion and metastasis through suppression of nuclear factor-kappaB. Clin. Cancer Res. 2006, 12, 7165–7173. [Google Scholar]
- Castelao, J.E.; Bart, R.D., 3rd; DiPerna, C.A.; Sievers, E.M.; Bremner, R.M. Lung cancer and cyclooxygenase-2. Ann. Thorac. Surg. 2003, 76, 1327–1335. [Google Scholar]
- Bu, X.; Zhao, C.; Dai, X. Involvement of COX-2/PGE2 pathway in the upregulation of MMP-9 expression in pancreatic cancer. Gastroent. Res. Pract. 2011, 2011. [Google Scholar] [CrossRef]
- Dempsey, P.W.; Doyle, S.E.; He, J.Q.; Cheng, G. The signaling adaptors and pathways activated by TNF superfamily. Cytokine Growth Factor Rev. 2003, 14, 193–209. [Google Scholar] [CrossRef]
- Jackson-Bernitsas, D.G.; Ichikawa, H.; Takada, Y.; Myers, J.N.; Lin, X.L.; Darnay, B.G.; Chaturvedi, M.M.; Aggarwal, B.B. Evidence that TNF-TNFR1-TRADD-TRAF2-RIP-TAK1-IKK pathway mediates constitutive NF-kappaB activation and proliferation in human head and neck squamous cell carcinoma. Oncogene 2007, 26, 1385–1397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Choi, J.H.; Kim, J.B.; Nam, S.J.; Yang, J.H.; Kim, J.H.; Lee, J.E. Berberine suppresses TNF-alpha-induced MMP-9 and cell invasion through inhibition of AP-1 activity in MDA-MB-231 human breast cancer cells. Molecules 2008, 13, 2975–2985. [Google Scholar] [CrossRef]
- Jung, Y.S.; Lee, S.O. Apomorphine suppresses TNF-alpha-induced MMP-9 expression and cell invasion through inhibition of ERK/AP-1 signaling pathway in MCF-7 cells. Biochem. Biophys. Res. Commun. 2017, 487, 903–909. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, W.Z.; Liu, T.; Feng, X.; Yang, N.; Zhou, H.F. Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J. Recept. Signal Transduct. Res. 2015, 35, 600–604. [Google Scholar]
- Liu, L.; Zhu, H.; Wu, W.; Shen, Y.; Lin, X.; Wu, Y.; Liu, L.; Tang, J.; Zhou, Y.; Sun, F. Neoantimycin F, a Streptomyces-Derived Natural Product Induces Mitochondria-Related Apoptotic Death in Human Non-Small Cell Lung Cancer Cells. Front. Pharmacol. 2019, 10, 1042. [Google Scholar] [CrossRef]
- Wen, J.; Fu, J.H.; Zhang, W.; Guo, M. Lung carcinoma signaling pathways activated by smoking. Chin. J. Cancer 2011, 30, 551–558. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.K.; Choi, E.J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. ACTA 2010, 1802, 396–405. [Google Scholar]
Sample Availability: Samples of the compounds are not available from the authors. |







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ooppachai, C.; Limtrakul, P.; Yodkeeree, S. Dicentrine Potentiates TNF-α-Induced Apoptosis and Suppresses Invasion of A549 Lung Adenocarcinoma Cells via Modulation of NF-κB and AP-1 Activation. Molecules 2019, 24, 4100. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24224100
Ooppachai C, Limtrakul P, Yodkeeree S. Dicentrine Potentiates TNF-α-Induced Apoptosis and Suppresses Invasion of A549 Lung Adenocarcinoma Cells via Modulation of NF-κB and AP-1 Activation. Molecules. 2019; 24(22):4100. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24224100
Chicago/Turabian StyleOoppachai, Chanatip, Pornngarm Limtrakul (Dejkriengkraikul), and Supachai Yodkeeree. 2019. "Dicentrine Potentiates TNF-α-Induced Apoptosis and Suppresses Invasion of A549 Lung Adenocarcinoma Cells via Modulation of NF-κB and AP-1 Activation" Molecules 24, no. 22: 4100. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules24224100