
Identification of Potential Dipeptidyl Peptidase (DPP)-IV Inhibitors among Moringa oleifera Phytochemicals by Virtual Screening, Molecular Docking Analysis, ADME/T-Based Prediction, and In Vitro Analyses

 , and
, and
Abstract
:1. Introduction
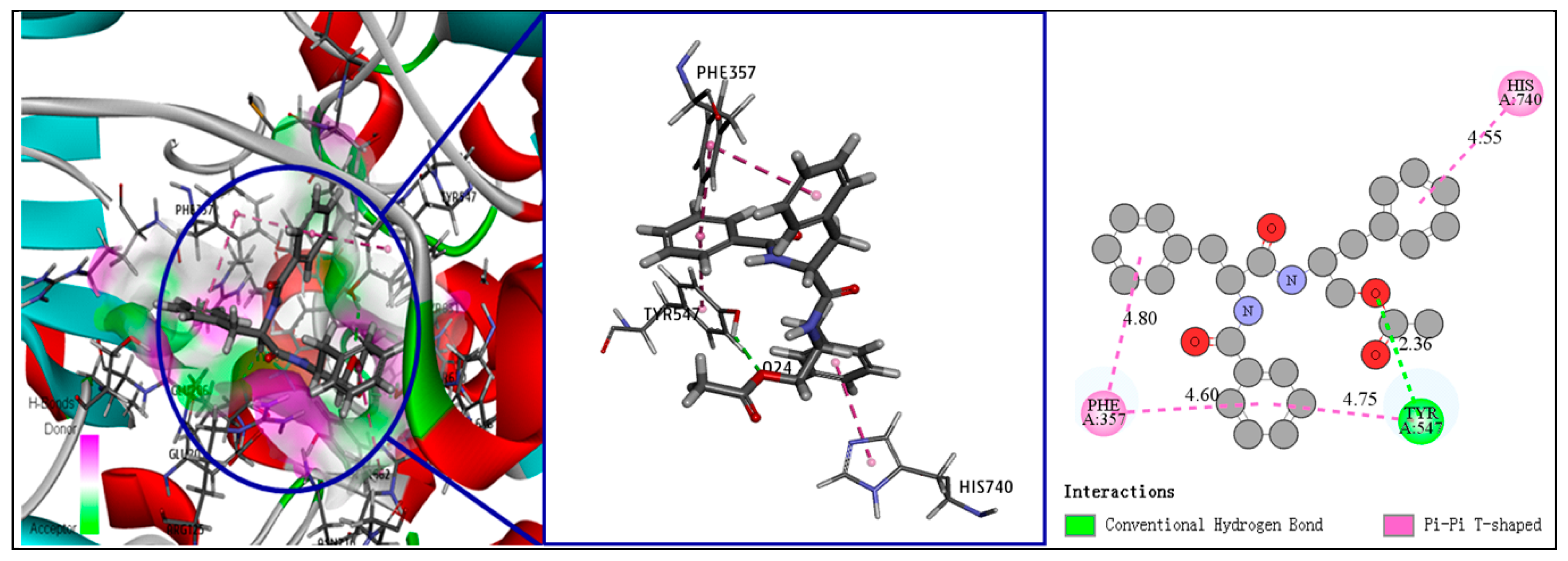
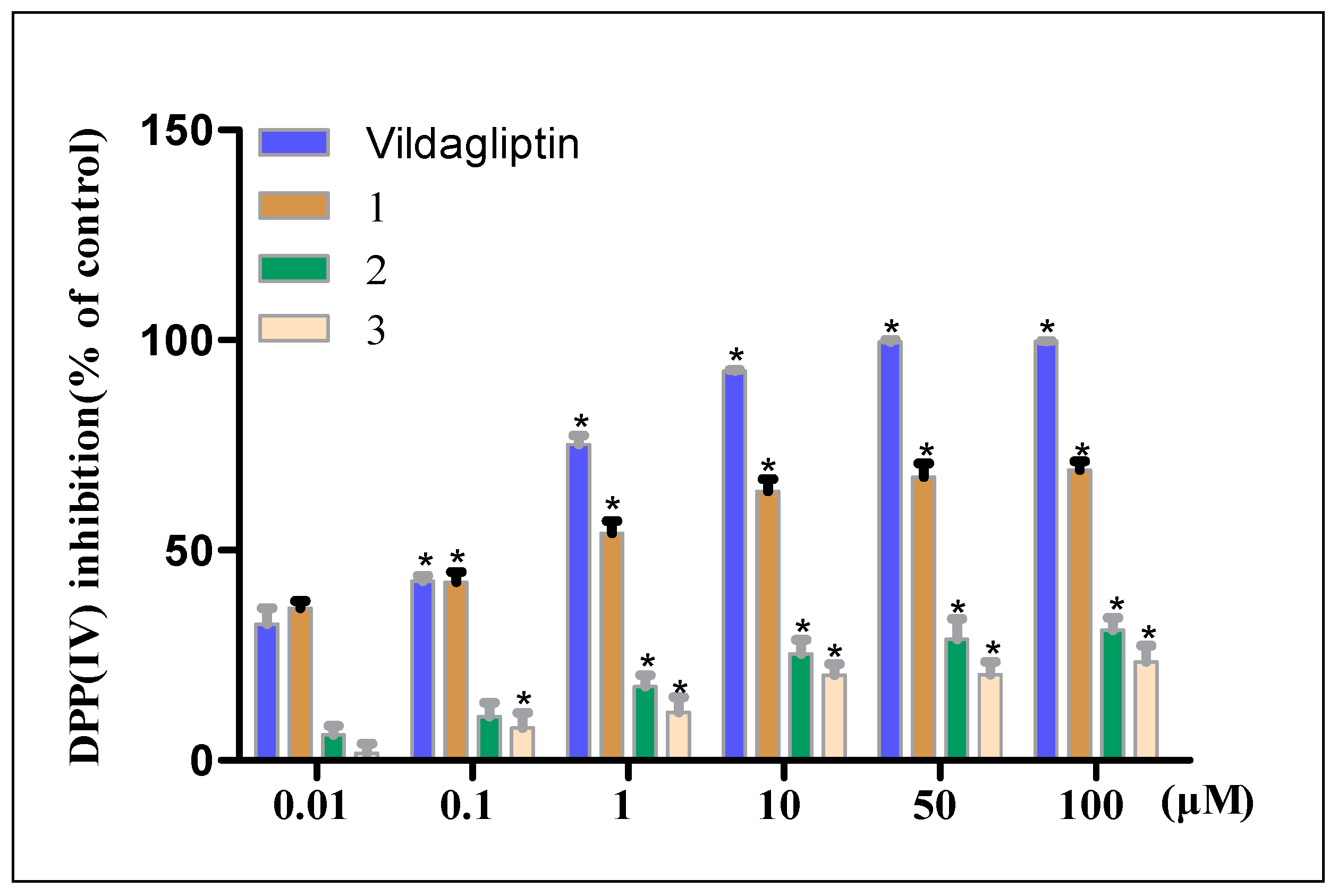
2. Results and Discussion
3. Materials and Methods
3.1. Literature Search and Establishment of Ligand Library
3.2. First-Round Screening Using Lipinski’s “Rule of Five”
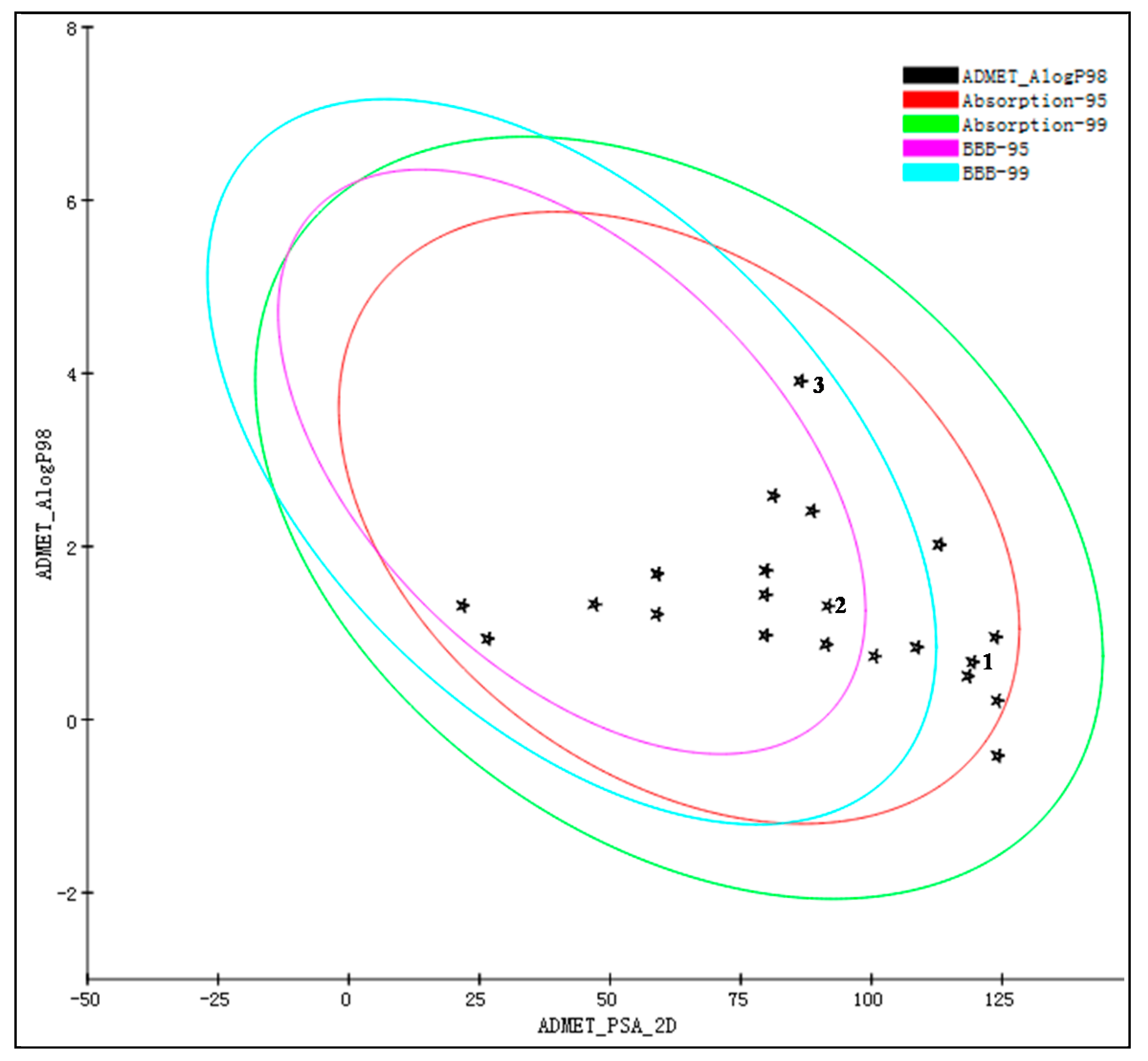
3.3. Second-Round Screening on the Basis of ADME/T Properties
3.4. Third-Round Screening Using LibDock
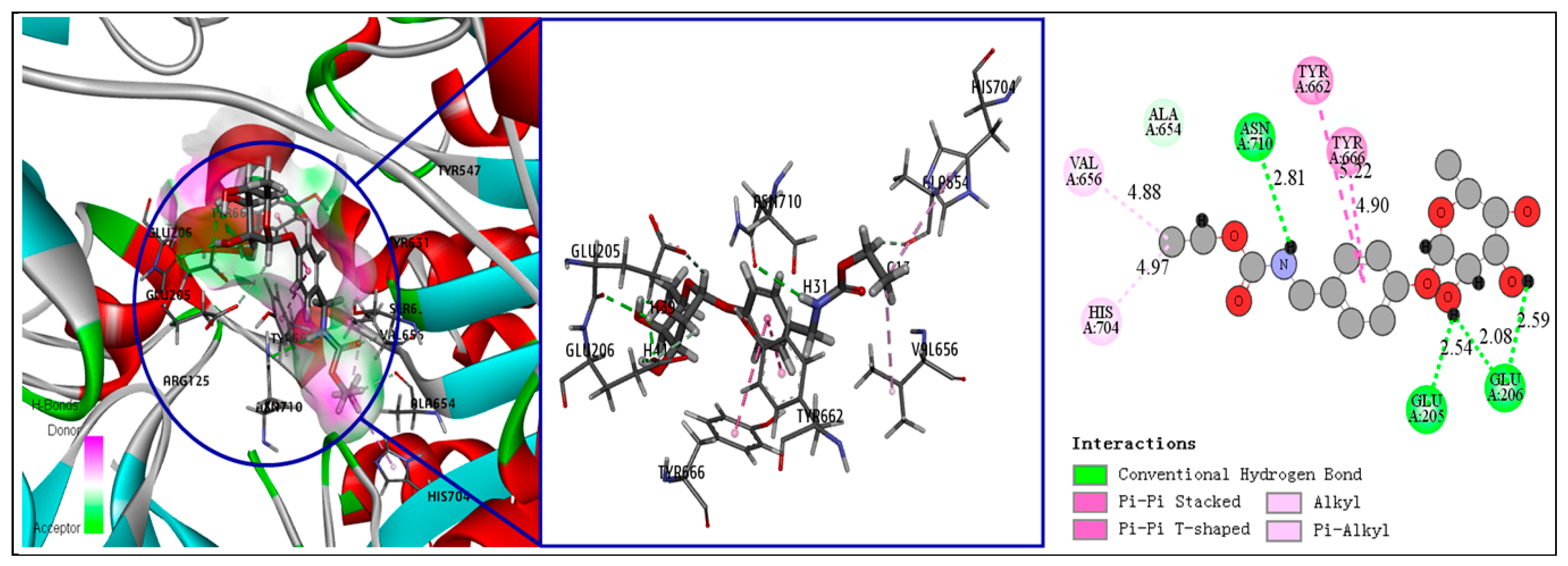
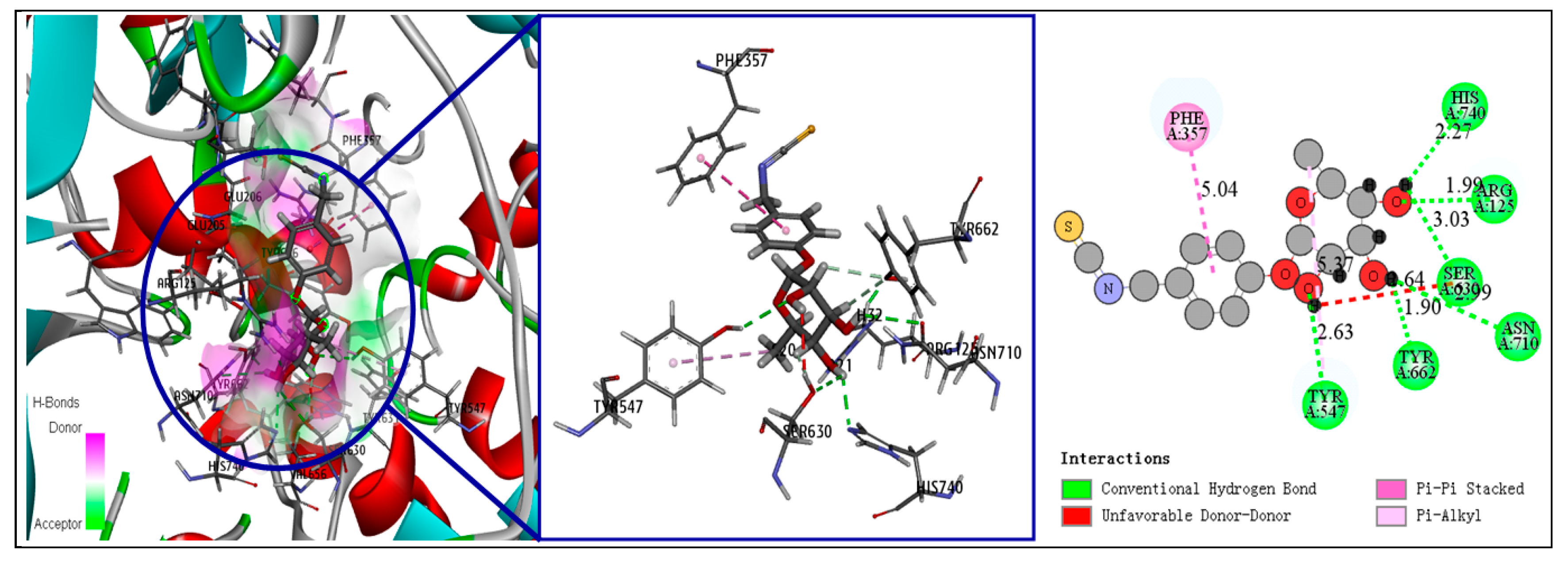
3.5. Fourth-Round Screening Using CDOCKER Molecular Docking Analysis and Docking Mode Analysis
3.6. In Vitro DPP-IV Inhibition Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Yki-Järvinen, H. Non-alcoholic fatty liver disease as a cause and a consequence of metabolic syndrome. Lancet Diabetes Endocrinol. 2014, 2, 901–910. [Google Scholar] [CrossRef]
- Biswas, T.; Islam, A.; Rawal, L.; Islam, S. Increasing prevalence of diabetes in Bangladesh: A scoping review. Public Health 2016, 138, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Liu, Z.; Chen, H.; Ye, N.; Cheng, X.; Zhou, J. Exchange proteins directly activated by cAMP (EPACs): Emerging therapeutic targets. Bioorg. Med. Chem. Lett. 2017, 27, 1633–1639. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.T.; Mandard, S.; Dray, C.; Deckert, V.; Valet, P.; Besnard, P.; Drucker, D.J.; Lagrost, L.; Grober, J. Lipopolysaccharides-mediated increase in glucose-stimulated insulin secretion: Involvement of the GLP-1 pathway. Diabetes 2014, 63, 471–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.Y. Glucagon-Like peptide-1 formulation—The present and future development in diabetes treatment. Basic Clin. Pharm. Toxicol. 2016, 118, 173–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juillerat-Jeanneret, L. Dipeptidyl peptidase IV and its inhibitors: Therapeutics for type 2 diabetes and what else? J. Med. Chem. 2013, 57, 2197–2212. [Google Scholar] [CrossRef] [PubMed]
- Makrilakis, K. The Role of DPP-4 Inhibitors in the Treatment Algorithm of Type 2 Diabetes Mellitus: When to Select, What to Expect. Int. J. Environ. Res. Public Health 2019, 16, 2720. [Google Scholar] [CrossRef] [Green Version]
- Guasch, L.; Sala, E.; Ojeda, M.J.; Valls, C.; Bladé, C.; Mulero, M.; Blay, M.; Ardévol, A.; Garcia-Vallvé, S.; Pujadas, G. Identification of novel human dipeptidyl peptidase-IV inhibitors of natural origin (Part II): In silico prediction in antidiabetic extracts. PLoS ONE 2012, 7, e44972. [Google Scholar] [CrossRef]
- Gao, Y.; Zhang, Y.; Zhu, J.; Li, B.; Li, Z.; Zhu, W.; Shi, J.; Jia, Q.; Li, Y. Recent progress in natural products as DPP-4 inhibitors. Future Med. Chem. 2015, 7, 1079–1089. [Google Scholar] [CrossRef]
- DiMasi, J.A.; Grabowski, H.G.; Hansen, R.W. Innovation in the pharmaceutical industry: New estimates of R&D costs. J. Health Econ. 2016, 47, 20–33. [Google Scholar]
- Pink, R.; Hudson, A.; Mouriès, M.-A.; Bendig, M. Opportunities and challenges in antiparasitic drug discovery. Nat. Rev. Drug Discov. 2005, 4, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.-L.; Chan, D.S.-H.; Leung, C.-H. Drug repositioning by structure-based virtual screening. Chem. Soc. Rev. 2013, 42, 2130–2141. [Google Scholar] [CrossRef] [PubMed]
- Beg, A.; Khan, F.I.; Lobb, K.A.; Islam, A.; Ahmad, F.; Hassan, M.I. High throughput screening, docking, and molecular dynamics studies to identify potential inhibitors of human calcium/calmodulin-dependent protein kinase IV. J. Biomol. Struct. Dyn. 2019, 37, 2179–2192. [Google Scholar] [CrossRef] [PubMed]
- Katsuno, K.; Burrows, J.N.; Duncan, K.; Van Huijsduijnen, R.H.; Kaneko, T.; Kita, K.; Mowbray, C.E.; Schmatz, D.; Warner, P.; Slingsby, B. Hit and lead criteria in drug discovery for infectious diseases of the developing world. Nat. Rev. Drug Discov. 2015, 14, 751–758. [Google Scholar] [CrossRef]
- Meng, X.-Y.; Zhang, H.-X.; Mezei, M.; Cui, M. Molecular docking: A powerful approach for structure-based drug discovery. Curr. Comput. Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef]
- Karaman, B.; Alhalabi, Z.; Swyter, S.; Mihigo, S.; Andrae-Marobela, K.; Jung, M.; Sippl, W.; Ntie-Kang, F. Identification of bichalcones as sirtuin inhibitors by virtual screening and in vitro testing. Molecules 2018, 23, 416. [Google Scholar] [CrossRef] [Green Version]
- Shaikh, F.; Zhao, Y.; Alvarez, L.; Iliopoulou, M.; Lohans, C.; Schofield, C.J.; Padilla-Parra, S.; Siu, S.W.; Fry, E.E.; Ren, J. Structure-Based in silico screening identifies a potent ebolavirus inhibitor from a traditional chinese medicine library. J. Med. Chem. 2019, 62, 2928–2937. [Google Scholar] [CrossRef] [Green Version]
- Moyo, B.; Masika, P.J.; Hugo, A.; Muchenje, V. Nutritional characterization of Moringa (Moringa oleifera Lam.) leaves. Food Chem. 2011, 10, 12925–12933. [Google Scholar]
- Somova, L.; Shode, F.; Ramnanan, P.; Nadar, A. Antihypertensive, antiatherosclerotic and antioxidant activity of triterpenoids isolated from Olea europaea, subspecies africana leaves. J. Ethnopharmacol. 2003, 84, 299–305. [Google Scholar] [CrossRef]
- Sosa-Gutiérrez, J.; Valdéz-Solana, M.; Forbes-Hernández, T.; Avitia-Domínguez, C.; Garcia-Vargas, G.; Salas-Pacheco, J.; Flores-Herrera, O.; Téllez-Valencia, A.; Battino, M.; Sierra-Campos, E. Effects of Moringa oleifera leaves extract on high glucose-induced metabolic changes in HepG2 cells. Biology 2018, 7, 37. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, C.A. Lead-and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-J.; Zhang, J.; Wang, S.-Q.; Xu, W.-R.; Cheng, X.-C.; Wang, R.-L. Identification of novel multitargeted PPARα/γ/δ pan agonists by core hopping of rosiglitazone. Drug Des. Devel. Ther. 2014, 8, 2255–2262. [Google Scholar] [PubMed] [Green Version]
- Shekhawat, P.B.; Pokharkar, V.B. Understanding peroral absorption: Regulatory aspects and contemporary approaches to tackling solubility and permeability hurdles. Acta Pharm. Sin. B 2017, 7, 260–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, S.L.; Kirk, R.D.; DaSilva, N.A.; Ma, H.; Seeram, N.P.; Bertin, M.J. Polyphenol microbial metabolites exhibit gut and blood–brain barrier permeability and protect murine microglia against LPS-induced inflammation. Metabolites 2019, 9, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poulin, P.; Burczynski, F.J.; Haddad, S. The role of extracellular binding proteins in the cellular uptake of drugs: Impact on quantitative in vitro-to-in vivo extrapolations of toxicity and efficacy in physiologically based pharmacokinetic-pharmacodynamic research. J. Pharm. Sci. 2016, 105, 497–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, S.N.; Head, M.S.; Kulkarni, A.; LaLonde, J.M. Validation studies of the site-directed docking program LibDock. J. Chem. Inf. Model. 2007, 47, 2159–2171. [Google Scholar] [CrossRef] [Green Version]
- Ekins, S.; Freundlich, J.S.; Coffee, M. A common feature pharmacophore for FDA-approved drugs inhibiting the Ebola virus. F1000Research 2014, 3, 277. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Li, W.; Zhao, Y.; Zhong, S.; Wang, X.; Jiang, S.; Cheng, Y.; Xu, H.; Zhao, G. Computational study of novel natural inhibitors targeting O6-methylguanine-DNA methyltransferase. World Neurosurg. 2019, 130, e294–e306. [Google Scholar] [CrossRef]
- Meduru, H.; Wang, Y.-T.; Tsai, J.; Chen, Y.-C. Finding a potential dipeptidyl peptidase-4 (DPP-4) inhibitor for type-2 diabetes treatment based on molecular docking, pharmacophore generation, and molecular dynamics simulation. Int. J. Mol. Sci. 2016, 17, 920. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, M.A. Molecular mechanical study of halogen bonding in drug discovery. J. Comput. Chem. 2011, 32, 2564–2574. [Google Scholar] [CrossRef]
- Gilani, A.H.; Aftab, K.; Suria, A.; Siddiqui, S.; Salem, R.; Siddiqui, B.S.; Faizi, S. Pharmacological studies on hypotensive and spasmolytic activities of pure compounds from Moringa oleifera. Phytother. Res. 1994, 8, 87–91. [Google Scholar] [CrossRef]
- Waterman, C.; Rojas-Silva, P.; Tumer, T.B.; Kuhn, P.; Richard, A.J.; Wicks, S.; Stephens, J.M.; Wang, Z.; Mynatt, R.; Cefalu, W. Isothiocyanate-rich Moringa oleifera extract reduces weight gain, insulin resistance, and hepatic gluconeogenesis in mice. Mol. Nutr. Food Res. 2015, 59, 1013–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isshiki, K.; Asai, Y.; Tanaka, S.; Nishio, M.; Uchida, T.; Okuda, T.; Komatsubara, S.; Sakurai, N. Aurantiamide acetate, a selective cathepsin inhibitor, produced by Aspergillus penicilloides. Biosci. Biotechnol. Biochem. 2001, 65, 1195–1197. [Google Scholar] [CrossRef] [PubMed]
- Yoon, C.-S.; Kim, D.-C.; Lee, D.-S.; Kim, K.-S.; Ko, W.; Sohn, J.H.; Yim, J.H.; Kim, Y.-C.; Oh, H. Anti-neuroinflammatory effect of aurantiamide acetate from the marine fungus Aspergillus sp. SF-5921: Inhibition of NF-κB and MAPK pathways in lipopolysaccharide-induced mouse BV2 microglial cells. Int. Immunopharmacol. 2014, 23, 568–574. [Google Scholar] [CrossRef]
- Yang, H.; Sun, L.; Li, W.; Liu, G.; Tang, Y. In silico prediction of chemical toxicity for drug design using machine learning methods and structural alerts. Front Chem. 2018, 6, 30. [Google Scholar] [CrossRef]
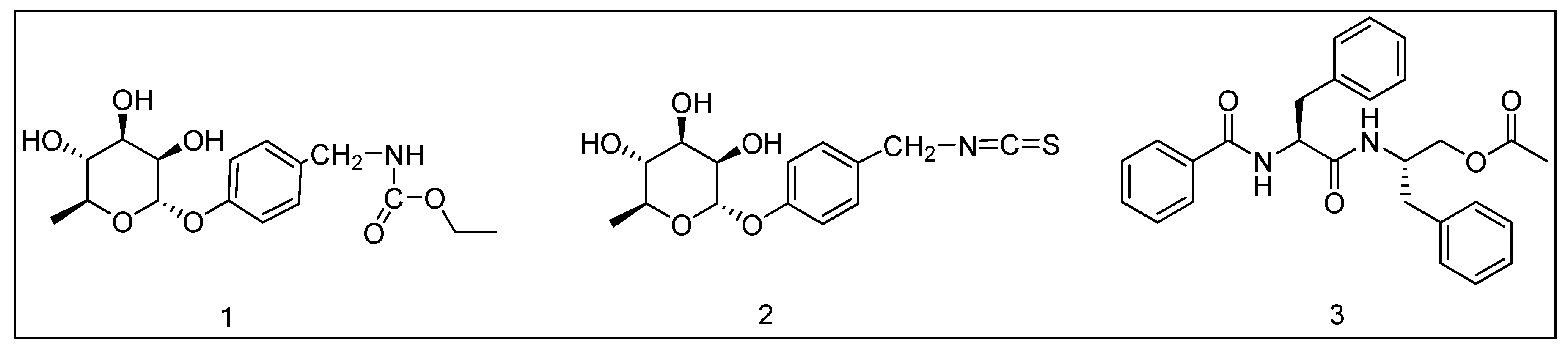
Sample Availability: Samples of the compounds 1, 2 and 3 are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Number | Absorption Level a | BBB Level b | CYP2D6 Inhibition | Plasma Protein Binding | Solubility Level c |
|---|---|---|---|---|---|
| 1 | 0 | 4 | No | Weak | 4 |
| 2 | 0 | 3 | No | Weak | 4 |
| 3 | 0 | 2 | No | Weak | 2 |
| 4 | 0 | 4 | No | Weak | 4 |
| 5 | 0 | 4 | No | Weak | 4 |
| 6 | 0 | 3 | No | Weak | 3 |
| 7 | 0 | 3 | No | Weak | 4 |
| 8 | 0 | 4 | No | Weak | 4 |
| 9 | 0 | 2 | No | Weak | 4 |
| 10 | 0 | 2 | No | Weak | 4 |
| 11 | 0 | 3 | No | Weak | 3 |
| 12 | 0 | 4 | No | Weak | 3 |
| 13 | 0 | 3 | No | Weak | 3 |
| 14 | 0 | 3 | No | Weak | 4 |
| 15 | 0 | 3 | No | No | 4 |
| 16 | 0 | 3 | No | No | 4 |
| 17 | 0 | 3 | No | No | 4 |
| 18 | 0 | 3 | No | No | 4 |
| 19 | 0 | 3 | No | No | 4 |
| 20 | 1 | 4 | No | No | 4 |
| 21 | 0 | 2 | No | No | 4 |
| 22 | 0 | 3 | No | No | 3 |
| 23 | 0 | 3 | No | No | 4 |
| Compound Number | LibDock Score |
|---|---|
| 3 | data |
| 5 | 120.126 |
| 1 | 110.991 |
| 6 | 109.801 |
| 4 | 103.673 |
| 7 | 102.232 |
| 2 | 99.719 |
| Vildagliptin | 93.424 |
| Compound Number | CDOCKER Interaction Energy (kcal/mol) | Binding Energy (kcal/mol) | Number of Hydrogen Bonds | Number of Hydrophilic Bonds |
|---|---|---|---|---|
| 1 | 44.9575 | −84.9987 | 4 | 4 |
| 2 | 39.3594 | −81.1002 | 6 | 1 |
| 3 | 35.7187 | −47.3644 | 1 | 4 |
| Vildagliptin | 35.6244 | −42.0109 | 4 | 3 |
| Compound | 1 | 2 | 3 | Vildagliptin |
|---|---|---|---|---|
| Molecular weight | 341.36 | 311.35 | 377.39 | 303.40 |
| H-bond acceptor | 8 | 6 | 8 | 5 |
| H-bond donor | 4 | 3 | 4 | 2 |
| No. of ionization states | 1 | 1 | 1 | 3 |
| No. of tautomers | 1 | 1 | 1 | 1 |
| Aerobic biodegradability | Degradable | Nondegradable | Degradable | Degradable |
| Ames mutagenicity | Nonmutagen | Nonmutagen | Nonmutagen | Nonmutagen |
| Mouse NTP classification a | Noncarcinogen | Noncarcinogen | Noncarcinogen | Noncarcinogen |
| Rat NTP classification a | Noncarcinogen | Noncarcinogen | Noncarcinogen | Noncarcinogen |
| WOE prediction | Noncarcinogen | Noncarcinogen | Noncarcinogen | Noncarcinogen |
| Hepatotoxicity | Yes | Yes | No | No |
| Skin sensitization | Mild | Mild | Mild | Mild |
| TD50 (mg/kg) | 32.81 | 19.78 | 5.14 | 1.48 |
| LC50 (g/L) | 0.58 | 0.15 | 0.05 | 0.26 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Y.; Shi, C.-Y.; Xie, J.; Dai, J.-H.; He, S.-L.; Tian, Y. Identification of Potential Dipeptidyl Peptidase (DPP)-IV Inhibitors among Moringa oleifera Phytochemicals by Virtual Screening, Molecular Docking Analysis, ADME/T-Based Prediction, and In Vitro Analyses. Molecules 2020, 25, 189. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25010189
Yang Y, Shi C-Y, Xie J, Dai J-H, He S-L, Tian Y. Identification of Potential Dipeptidyl Peptidase (DPP)-IV Inhibitors among Moringa oleifera Phytochemicals by Virtual Screening, Molecular Docking Analysis, ADME/T-Based Prediction, and In Vitro Analyses. Molecules. 2020; 25(1):189. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25010189
Chicago/Turabian StyleYang, Yang, Chong-Yin Shi, Jing Xie, Jia-He Dai, Shui-Lian He, and Yang Tian. 2020. "Identification of Potential Dipeptidyl Peptidase (DPP)-IV Inhibitors among Moringa oleifera Phytochemicals by Virtual Screening, Molecular Docking Analysis, ADME/T-Based Prediction, and In Vitro Analyses" Molecules 25, no. 1: 189. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25010189