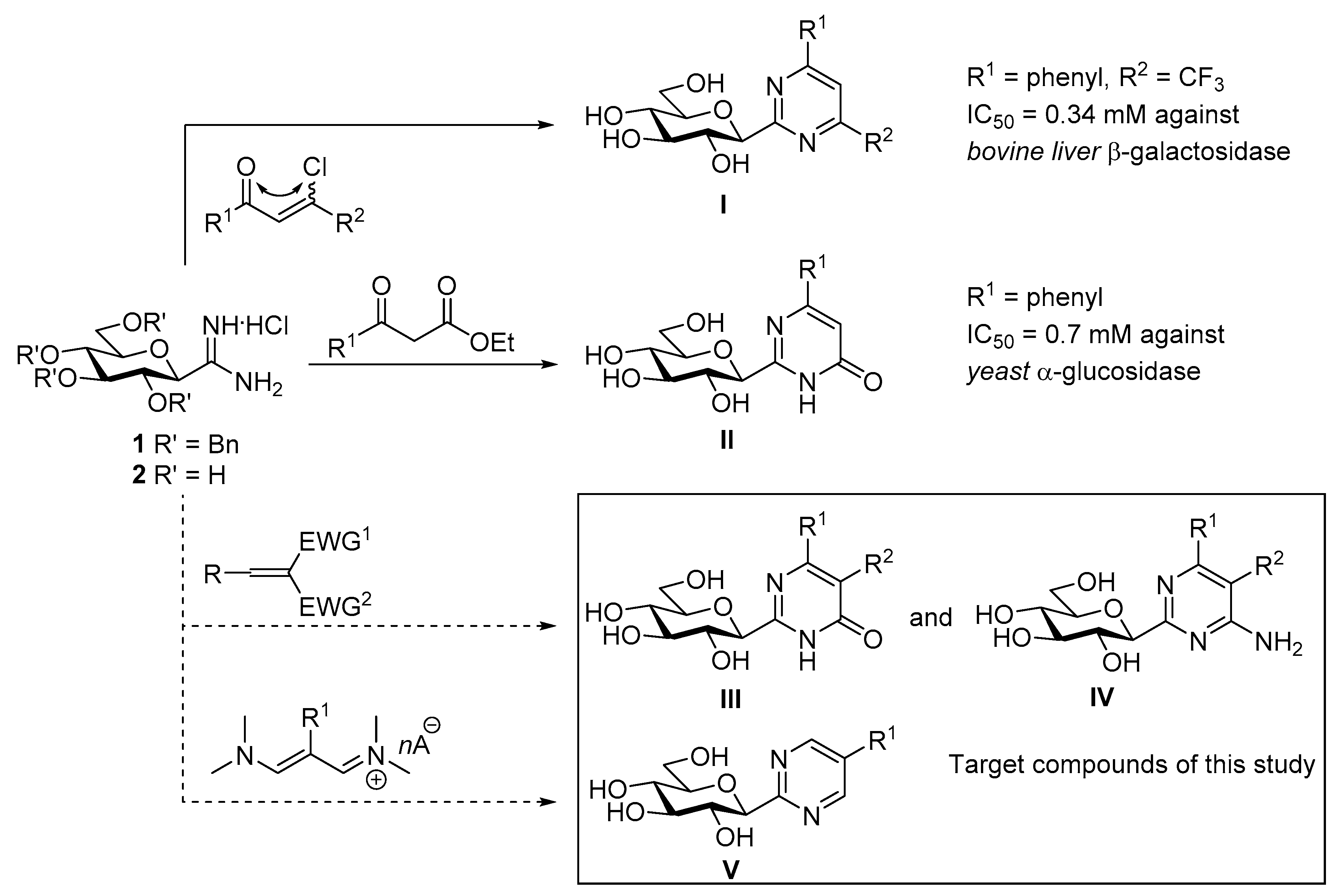
3.1.6. Synthesis and Characterization of the New Compounds
4-Amino-2-(2′,3′,4′,6′-tetra-O-benzyl-β-D-glucopyranosyl)-pyrimidine-5-carbonitrile (10a). Prepared from compound 1 (400 mg, 0.66 mmol) and 2-(ethoxymethylene)malononitrile 3 (162 mg, 1.33 mmol) according to genereal procedure 1. Reaction time: 30 min. The title compound precipitated from the reaction mixture as a pale yellow amorphous solid. Yield: 325 mg (76%). Rf = 0.55 (EtOAc-hexane = 1:1); [α]D = ‒1 (c 0.20, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ (ppm): 8.87 (2H, br s, NH2), 8.33 (1H, s, H-6), 7.40–6.99 (20H, m, aromatics), 4.93, 4.89 (2 × 1H, 2d, J = 11.0 Hz in each, PhCH2), 4.92, 4.69 (2 × 1H, 2d, J = 10.8 Hz in each, PhCH2), 4.82, 4.74 (2 × 1H, 2d, J = 12.2 Hz in each, PhCH2), 4.65, 4.22 (2 × 1H, 2d, J = 11.3 Hz in each, PhCH2), 4.38 (1H, d, J = 9.5 Hz, H-1′), 3.94 (1H, pt, J = 9.5, 9.2 Hz, H-3′ or H-4′), 3.85 (1H, pt, J = 9.4, 9.3 Hz, H-2′ or H-3′ or H-4′), 3.84 (1H, pt, J = 9.5, 9.3 Hz, H-2′ or H-3′ or H-4′), 3.79 (1H, dd, J = 11.9, 5.2 Hz, H-6′a), 3.64 (1H, dd, J = 11.9, 1.9 Hz, H-6′b), 3.52-3.49 (1H, m, H-5′); 13C NMR (100 MHz, CDCl3) δ (ppm): 168.8, 163.3 (C-2, C-4), 160.5 (C-6), 138.3, 138.1, 137.7, 136.9, 129.0–127.9 (aromatics), 114.8 (CN), 91.0 (C-5), 87.0, 83.2, 82.2, 79.0, 77.5 (C-1′–C-5′), 76.2, 75.5, 75.0, 73.8 (4 × PhCH2), 67.8 (C-6′). ESI-MS positive mode (m/z): Calcd for C39H39N4O5+ [M + H]+ 643.3. Found: 643.5.
Ethyl 4-amino-2-(2′,3′,4′,6′-tetra-O-benzyl-β-D-glucopyranosyl)-pyrimidine-5-carboxylate (10b) and 2-(2′,3′,4′,6′-tetra-O-benzyl-β-D-glucopyranosyl)-6-oxo-1,6-dihydropyrimidine-5-carbonitrile (10c). The title compounds were prepared from compound 1 (400 mg, 0.66 mmol) and ethyl 2-cyano-3-ethoxyacrylate 4 (224 mg, 1.33 mmol) according to general procedure 1. Reaction time: 1 h. Purification by column chromatography (EtOAc-hexane = 1:3) yielded 10b as the first and 10c as the second fraction. 10b: Yield: 167 mg (37%), colourless syrup. Rf = 0.25 (EtOAc-hexane = 1:2); [α]D = +54 (c 0.20, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ (ppm): 8.81 (1H, s, H-6), 7.84 (1H, br s, NH2), 7.31–6.97 (20H, m, aromatics), 6.38 (1H, br s, NH2), 4.93, 4.89 (2 × 1H, 2d, J = 11.2 Hz in each, PhCH2), 4.84, 4.57 (2 × 1H, 2d, J = 10.7 Hz in each, PhCH2), 4.60, 4.27 (2 × 1H, 2d, J = 11.4 Hz in each, PhCH2), 4.60, 4.27 (2 × 1H, 2d, J = 12.2 Hz in each, PhCH2), 4.36 (2H, q, i = 7.2 Hz, CH2CH3), 4.36 (1H, d, J = 9.6 Hz, H-1′), 4.03 (1H, pt, J = 9.6, 9.0 Hz, H-2′), 3.84 (1H, pt, J = 9.2, 9.0 Hz, H-3′), 3.76–3.3.71 (3H, m, H-4′, H-6′a, H-6′b), 3.65 (1H, ddd, J = 9.5, 4.5, 2.2 Hz, H-5′), 1.40 (3H, t, J = 7.2 Hz, CH2CH3); 13C NMR (100 MHz, CDCl3) δ (ppm): 168.9, 166.0, 162.8 (C-2, C-4, COOEt), 159.6 (C-6), 138.8, 138.2, 138.2, 138.1, 128.5–127.5 (aromatics), 104.3 (C-5), 87.1, 82.9, 81.3, 79.8, 77.3 (C-1′–C-5′), 75.7, 75.2, 74.8, 73.5 (4 × PhCH2), 69.1 (C-6′), 61.3 (CH2CH3), 14.4 (CH2CH3). ESI-MS positive mode (m/z): Calcd for C41H44N3O7+ [M + H]+ 690.3. Found: 690.5. 10c: Yield: 128 mg (30%), colourless syrup. Rf = 0.23 (EtOAc-hexane = 1:2); [α]D = ‒12 (c 0.22, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ (ppm): 12.52 (1H, br s, NH), 8.15 (1H, s, H-4), 7.33-7.09 (20H, m, aromatics), 4.91, 4.88 (2 × 1H, 2d, J = 11.3 Hz in each, PhCH2), 4.86, 4.60 (2 × 1H, 2d, J = 10.8 Hz in each, PhCH2), 4.71, 4.46 (2 × 1H, 2d, J = 11.5 Hz in each, PhCH2), 4.54, 4.48 (2 × 1H, 2d, J = 12.0 Hz in each, PhCH2), 4.37 (1H, d, J = 9.5 Hz, H-1′), 3.86-3.70 (6H, m, H-2′–H-6′a,b); 13C NMR (100 MHz, CDCl3) δ (ppm): 162.9, 160.0 (C-2, C-6), 161.1 (C-4), 138.1, 137.9, 137.6, 137.1, 128.7–127.9 (aromatics), 113.3 (CN), 103.2 (C-5), 85.8, 79.2, 78.9, 78.2, 77.7 (C-1′–C-5′), 75.6, 75.2, 74.6, 73.4 (4 × PhCH2), 69.0 (C-6′). ESI-MS positive mode (m/z): Calcd for C39H38N3O6+ [M + H]+ 644.3. Found: 644.5.
Ethyl 2-(2′,3′,4′,6′-tetra-O-benzyl-β-D-glucopyranosyl)-6-oxo-1,6-dihydropyrimidine-5-carboxylate (10d). Prepared from compound 1 (400 mg, 0.66 mmol) and diethyl 2-(ethoxymethylene)malonate 5 (265 μL, 1.33 mmol) according to general procedure 1. Reaction time: 1 h. Purified by column chromatography (EtOAc-hexane 1:1) to give 367 mg (80%) colourless syrup. Rf = 0.21 (EtOAc-hexane = 1:1); [α]D = +9 (c 0.50, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ (ppm): 11.35 (1H, br s, NH), 8.55 (1H, s, H-4), 7.32–7.11 (20H, m, aromatics), 4.88, 4.84 (2 × 1H, 2d, J = 11.2 Hz in each, PhCH2), 4.82, 4.55 (2 × 1H, 2d, J = 10.9 Hz in each, PhCH2), 4.66, 4.45 (2 × 1H, 2d, J = 11.4 Hz in each, PhCH2), 4.55, 4.48 (2 × 1H, 2d, J = 12.1 Hz in each, PhCH2), 4.37 (2H, q, J = 7.2 Hz, CH2CH3), 4.37 (1H, d, J = 9.5 Hz, H-1′), 3.86–3.65 (6H, m, H-2′–H-6′a,b), 1.38 (3H, t, J = 7.2 Hz, CH2CH3); 13C NMR (90 MHz, CDCl3) δ (ppm): 163.2, 162.6, 160.0 (C-2, C-6, COOEt), 159.2 (C-4), 138.2, 138.0, 137.6, 137.2, 128.5–127.8 (aromatics), 116.7 (C-5), 86.1, 79.4, 78.9, 78.4, 77.4 (C-1′–C-5′), 75.5, 75.0, 74.6, 73.4 (4 × PhCH2), 68.8 (C-6′), 61.3 (CH2CH3), 14.4 (CH2CH3). ESI-MS positive mode (m/z): Calcd for C41H43N2O8+ [M + H]+ 691.3. Found: 691.4.
4-Amino-2-(2′,3′,4′,6′-tetra-O-benzyl-β-D-glucopyranosyl)-6-phenylpyrimidine-5-carbonitrile (10e). Prepared from compound 1 (400 mg, 0.66 mmol) and 2-benzylidenemalononitrile 6 (204 mg, 1.33 mmol) according to general procedure 1. Reaction time: 1 h. The title compound precipitated from the reaction mixture was a pale yellow amorphous solid. Yield: 373 mg (78%). Rf = 0.41 (EtOAc-hexane = 2:3); [α]D = ‒12 (c 0.27, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ (ppm): 9.00 (2H, br s, NH2), 7.96–6.97 (25H, m, aromatics), 4.96–4.69 (6H, m, PhCH2), 4.73, 4.27 (2 × 1H, 2d, J = 11.3 Hz in each, PhCH2), 4.50 (1H, d, J = 9.6 Hz, H-1′), 4.03 (1H, pt, J = 9.5, 9.2 Hz, H-4′), 3.98 (1H, pt, J = 9.6, 9.1 Hz, H-2′), 3.85 (1H, pt, J = 9.2, 9.1 Hz, H-3′), 3.82 (1H, dd, J = 11.8, 3.8 Hz, H-6′a), 3.66 (1H, dd, J = 11.8, 1.9 Hz, H-6′b), 3.52 (1H, ddd, J = 9.5, 3.8, 1.9 Hz, H-5′); 13C NMR (100 MHz, CDCl3) δ (ppm): 168.3, 167.9, 165.4 (C-2, C-4, C-6), 138.5, 138.2, 137.8, 137.1, 136.0, 131.4, 129.0–127.9 (aromatics), 116.0 (CN), 87.4 (C-5), 87.1, 83.5, 82.5, 79.0, 77.7 (C-1′–C-5′), 76.2, 75.6, 75.3, 73.9 (4 × PhCH2), 67.9 (C-6′). ESI-MS positive mode (m/z): Calcd for C45H43N4O5+ [M + H]+ 719.3. Found: 791.6.
2-(2′,3′,4′,6′-Tetra-O-benzyl-β-D-glucopyranosyl)-4-phenyl-6-oxo-1,6-dihydropyrimidine-5-carbonitrile (10f). Prepared from compound 1 (400 mg, 0.66 mmol) and ethyl 2-cyano-3-phenylacrylate 7 (267 mg, 1.33 mmol) according to general procedure 1. Reaction time: 1 h. Purified by column chromatography (EtOAc-hexane = 2:3) to give 334 mg (70%) colourless syrup. Rf = 0.51 (EtOAc-hexane = 1:1); [α]D = +11 (c 0.25, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ (ppm): 8.07–7.02 (25H, m, aromaics), 4.98, 4.93 (2 × 1H, 2d, J = 11.2 Hz in each, PhCH2), 4.93, 4.72 (2 × 1H, 2d, J = 10.7 Hz in each, PhCH2), 4.77, 4.47 (2 × 1H, 2d, J = 11.3 Hz in each, PhCH2), 4.61, 4.53 (2 × 1H, 2d, J = 12.3 Hz in each, PhCH2), 4.52 (1H, d, J = 9.5 Hz, H-1′), 3.97-3.84 (6H, m, H-2′–H-6′a,b); 13C NMR (100 MHz, CDCl3) δ (ppm): 168.7, 162.8, 160.6 (C-2, C-4, C-6), 138.2, 138.1, 137.8, 137.1, 134.6, 132.3, 129.3-127.6 (aromatics), 114.8 (CN), 97.7 (C-5), 86.1, 79.7, 79.2, 78.7, 78.1 (C-1′–C-5′), 75.6, 75.4, 74.7, 73.3 (4 × PhCH2), 69.4 (C-6′). ESI-MS positive mode (m/z): Calcd for C45H42N3O6+ [M + H]+ 720.3. Found: 720.6.
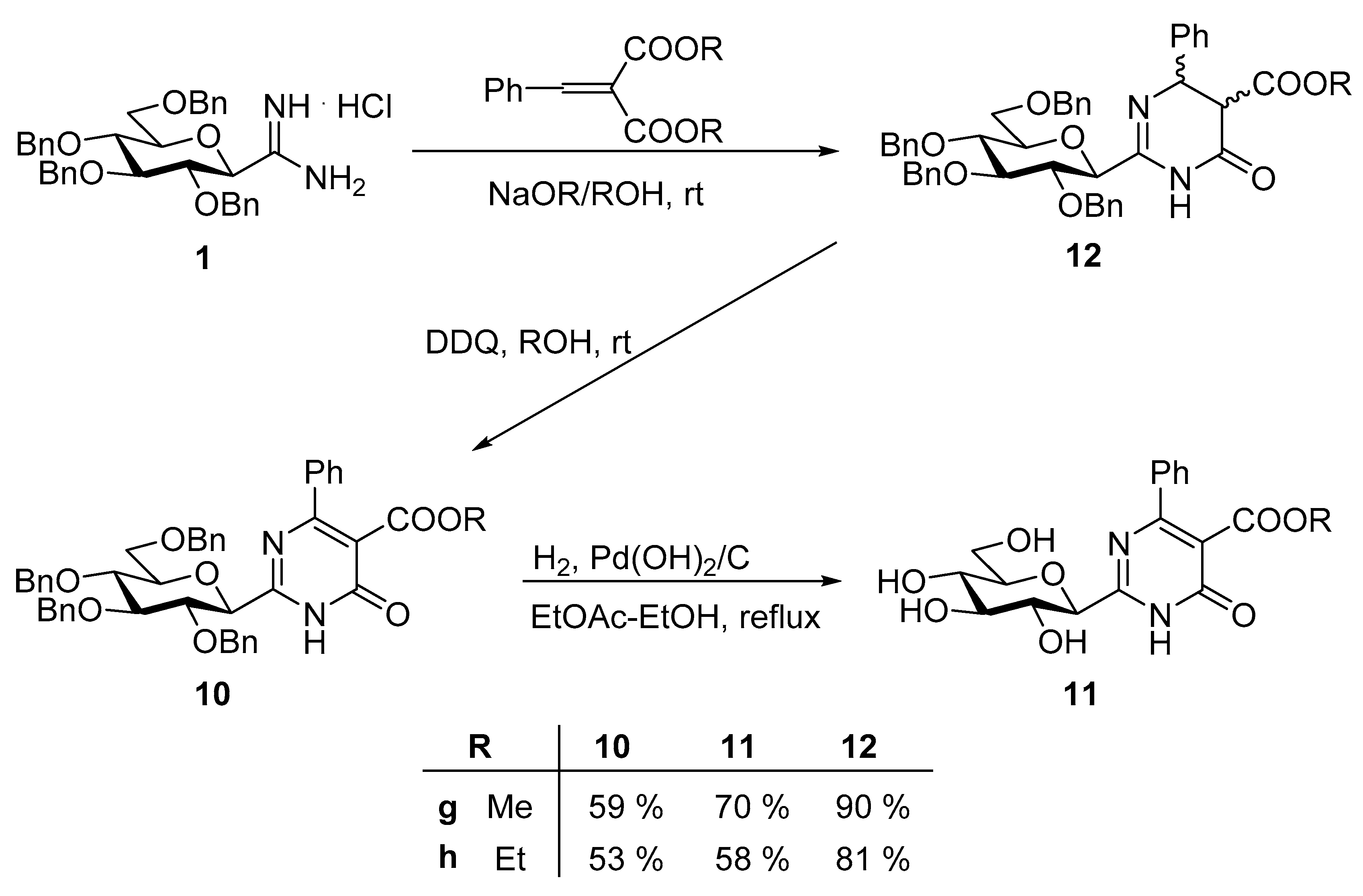
Methyl 2-(2′,3′,4′,6′-tetra-O-benzyl-β-D-glucopyranosyl)-4-phenyl-6-oxo-1,6-dihydropyrimidine-5-carboxylate (10g). Prepared from compound 12g (300 mg, 0.40 mmol) and DDQ (90 mg, 0.40 mmol) according to general procedure 3. Purified by column chromatography (EtOAc-hexane = 2:3) to give 177 mg (59%) pale yellow syrup. Rf = 0.48 (EtOAc-hexane = 1:1); [α]D = +10 (c 0.40, CH2Cl2); 1H NMR (360 MHz, CDCl3) δ (ppm): 12.40 (1H, br s, NH), 7.65–7.01 (25H, m, aromatics), 4.93, 4.90 (2 × 1H, 2d, J = 11.5 Hz, PhCH2), 4.88, 4.64 (2 × 1H, 2d, J = 10.8 Hz, PhCH2), 4.73, 4.47 (2 × 1H, 2d, J = 11.0 Hz, PhCH2), 4.58, 4.54 (2 × 1H, 2d, J = 12.1 Hz, PhCH2), 4.42 (1H, d, J = 9.4 Hz, H-1′), 3.94 (1H, pt, J = 9.2, 9.0 Hz, H-2′or H-3′ or H-4′), 3.86–3.72 (5H, m, H-2′ and/or H-3′ and/or H-4′, H-5′–H-6′), 3.64 (3H, s, OCH3); 13C NMR (90 MHz, CDCl3) δ (ppm): 166.0, 161.3 (2), 157.9 (C-2, C-4, C-6, COOMe), 138.3, 138.0, 137.9, 137.2, 136.8, 130.5, 128.5-127.8 (aromatics), 118.4 (C-5), 86.3, 79.2, 79.2, 78.8, 77.8 (C-1′–C-5′), 75.7, 75.3, 74.7, 73.4 (4 × PhCH2), 69.0 (C-6′), 52.6 (OCH3). ESI-MS positive mode (m/z): Calcd for C46H45N2O8+ [M + H]+ 753.3. Found: 753.6.
Ethyl 2-(2′,3′,4′,6′-tetra-O-benzyl-β-D-glucopyranosyl)-4-phenyl-6-oxo-1,6-dihydropyrimidine-5-carboxylate (10h). Prepared from compound 12h (300 mg, 0.40 mmol) and DDQ (90 mg, 0.40 mmol) according to general procedure 3. Purified by column chromatography (EtOAc-hexane = 2:3) to give 159 mg (53%) pale yellow syrup. Rf = 0.50 (EtOAc-hexane = 1 : 1); [α]D = +62 (c 0.23, CH2Cl2); 1H NMR (360 MHz, CDCl3) δ (ppm): 12.67 (1H, br s, NH), 7.66–7.02 (25H, m, aromatics), 4.94, 4.91 (2 × 1H, 2d, J = 11.2 Hz in each, PhCH2), 4.89, 4.64 (2 × 1H, 2d, J = 10.8 Hz in each, PhCH2), 4.74, 4.49 (2 × 1H, 2d, J = 11.2 Hz in each, PhCH2), 4.58, 4.52 (2 × 1H, 2d, J = 12.2 Hz in each, PhCH2), 4.43 (1H, d, J = 9.5 Hz, H-1′), 4.14 (2H, q, J = 7.1 Hz, CH2CH3), 3.97 (1H, pt, J = 9.2, 9.0 Hz, H-2′ or H-3′ or H-4′), 3.87–3.70 (5H, m, H-2′ and/or H-3′ and/or H-4′, H-5′–H-6′a,b), 1.00 (3H, t, J = 7.1 Hz, CH2CH3); 13C NMR (90 MHz, CDCl3) δ (ppm): 165.3, 161.4, 161.3, 157.9 (C-2, C-4, C-6, COOEt), 138.4, 138.1, 137.8, 137.2, 136.9, 130.3, 128.5-127.7 (aromatics), 118.7 (C-5), 86.4, 79.2 (2), 78.9, 77.8 (C-1′–C-5′), 75.7, 75.2, 74.7, 73.4 (4 × PhCH2), 69.0 (C-6′), 61.6 (CH2CH3), 13.8 (CH2CH3). ESI-MS positive mode (m/z): Calcd for C47H47N2O8+ [M + H]+ 767.3. Found: 767.6.
4-Amino-2-(β-D-glucopyranosyl)-pyrimidine-5-carbonitrile (11a). Prepared from compound 2 (100 mg, 0.41 mmol) and 2-(ethoxymethylene)malononitrile 3 (101 mg, 0.82 mmol) according to general procedure 1. Reaction time: 30 min. Purified by column chromatography (CHCl3-MeOH = 5:1) to give 85 mg (73%) pale yellow syrup. Rf = 0.31 (CHCl3-MeOH = 3:1); [α]D = +42 (c 0.16, MeOH); 1H NMR (400 MHz, CD3OD) δ (ppm): 8.56 (1H, s, H-6), 4.18 (1H, d, J = 9.5 Hz, H-1′), 3.85 (1H, dd, J = 12.2, 1.9 Hz, H-6′a), 3.70 (1H, dd, J = 12.2, 4.9 Hz, H-6′b), 3.67 (1H, pt, J = 9.5, 9.0 Hz, H-2′), 3.50 (1H, pt, J = 9.1, 9.0 Hz, H-3′), 3.44 (1H, pt, J = 9.4, 9.1 Hz, H-4′), 3.39 (1H, ddd, J = 9.4, 4.9, 1.9 Hz, H-5′); 13C NMR (100 MHz, CD3OD) δ (ppm): 170.2, 164.4 (C-2, C-4), 161.8 (C-6), 115.4 (CN), 90.8 (C-5), 83.5, 82.3, 79.2, 74.4, 71.1 (C-1′–C-5′), 62.7 (C-6′). ESI-HRMS positive mode (m/z): calcd for C11H15N4O5+ [M + H]+ 283.1037; C11H14N4NaO5+ [M + Na]+ 305.0856. Found: [M + H]+ 283.1034; [M + Na]+ 305.0852.
Ethyl 4-amino-2-(β-D-glucopyranosyl)-pirimidine-5-carboxylate (11b) and 2-(β-D-glucopyranosyl)-6-oxo-1,6-dihydropyrimidine-5-carbonitrile (11c). Method A: The Pd-catalyst (35 mg, 20% Pd(OH)2/C) was suspended in an anhydrous EtOAc-EtOH solvent mixture (10 mL in 1:5 ratio) under Ar and the suspension was saturatated with H2 (3×). To this heterogenous mixture, a solution of compound 10b (70 mg, 0.10 mmol) in EtOAc (1 mL) and one drop of ccHCl were added. The reaction mixture was stirred under H2 at rt for two days, and the transformation was monitored by TLC (EtOAc-hexane 1:1 and CHCl3-MeOH 7:3). After complete conversion of the starting material, the mixture was neutralized with NaHCO3. The catalyst and insoluble inorganic salts were filtered off through a pad of Celite and washed three times with MeOH (3 × 3 mL). The resulting combined solution was concentrated under diminished pressure and the residue was purified by column chromatography (CHCl3-MeOH = 5:1). Yield of compound 11b: 17 mg (51%) colourless syrup. Rf = 0.55 (CHCl3-MeOH = 7:3); [α]D = ‒62 (c 0.15, MeOH); 1H NMR (400 MHz, D2O) δ (ppm): 8.82 (1H, s, H-6), 4.37 (2H, q, J = 7.1 Hz, CH2CH3), 4.28 (1H, d, J = 9.4 Hz, H-1′), 3.93 (1H, dd, J = 12.2, 1.9 Hz, H-6′a), 3.81 (1H, dd, J = 12.2, 4.5 Hz, H-6′b), 3.73 (1H, pt, J = 9.4, 9.0 Hz, H-2′), 3.69–3.54 (3H, m, H-3′, H-4′, H-5′), 1.38 (3H, t, J = 7.1 Hz, CH2CH3); 13C NMR (90 MHz, CD3OD) δ (ppm): 170.4, 166.9, 164.0, 159.7 (C-2, C-4, C-6, COOEt), 105.2 (C-5), 83.0, 82.3, 79.3, 74.5, 71.2 (C-1′–C-5′), 62.9 (C-6′), 62.4 (CH2CH3), 14.5 (CH2CH3). ESI-HRMS positive mode (m/z): calcd for C13H20N3O7+ [M + H]+ 330.1296; C13H19N3NaO7+ [M + Na]+ 352.1115. Found: [M + H]+ 330.1294; [M + Na]+ 352.1114. Method B: The title compounds 11b and 11c were prepared from compound 2 (200 mg, 0.82 mmol) and ethyl 2-cyano-3-ethoxyacrylate 4 (279 mg, 1.65 mmol) according to general procedure 1. Reaction time: 1 h. Purification by column chromatography (CHCl3-MeOH = 5:1 → 3:1) yielded 11b (55 mg, 20 %) as the first and 11c 105 mg (45%) as the second fraction. Compound 11c: colourless syrup. Rf = 0.39 (CHCl3-MeOH 1:1); [α]D = ‒66 (c 0.16, MeOH); 1H NMR (400 MHz, CD3OD) δ (ppm): 8.33 (1H, s, H-4), 4.11 (1H, d, J = 9.4 Hz, H-1′), 3.88 (1H, dd, J = 12.0, 1.8 Hz, H-6′a), 3.71 (1H, dd, J = 12.0, 4.8 Hz, H-6′b), 3.58 (1H, pt, J = 9.3, 9.2 Hz, H-2′ or H-3′ or H-4′), 3.53–3.41 (3H, m, H-2′ and/or H-3′ and/or H-4′, H-5′); 13C NMR (100 MHz, CD3OD) δ (ppm): 173.1, 171.1 (C-2, C-6), 161.4 (C-4), 118.1 (CN), 97.6 (C-5), 81.8, 81.4, 79.1, 74.6, 71.0 (C-1′–C-5′), 62.5 (C-6′). ESI-HRMS positive mode (m/z): Calcd. for C11H13NaN3O6+ [M + Na]+ 306.0697. Found: 306.0696.
Ethyl 2-(β-D-glucopyranosyl)-6-oxo-1,6-dihydropyrimidine-5-carboxylate (11d). Method A: The Pd-catalyst (150 mg, 20% Pd(OH)2/C) was suspended in an anhydrous EtOAc-EtOH solvent mixture (30 mL in 1:5 ratio) under Ar. This degased suspension was saturatated with H2 (3×). To this heterogenous mixture, a solution of compound 10d (335 mg, 0.48 mmol) in EtOAc (3 mL) and three drops of ccHCl were added. The reaction mixture was stirred under H2 at rt for two days, and the transformation was monitored by TLC (EtOAc-hexane 1:1 and CHCl3-MeOH 7:3). After the complete conversion of the starting material, the mixture was neutralized with NaHCO3. The catalyst and the insoluble inorganic salts were filtered off through a pad of Celite and washed three times with MeOH (3 × 10 mL). The resulting solution was concentrated under reduced pressure and the residue was purified by column chromatography (CHCl3-MeOH = 3:1). Yield: 107 mg (67%), colourless syrup. Method B: Prepared from compound 2 (100 mg, 0.41 mmol) and diethyl 2-(ethoxymethylene)malonate 5 (165 μL, 0.82 mmol) according to general procedure 1. Reaction time: 1 h. Purified by column chromatography (CHCl3-MeOH = 3:1) to give 70 mg (51%) colourless syrup. Rf = 0.38 (CHCl3-MeOH = 1:1); [α]D = +75 (c 0.15, MeOH); 1H NMR (360 MHz, D2O) δ (ppm): 8.70 (1H, s, H-4), 4.39–4.32 (3H, m, H-1′, CH2CH3), 3.94 (1H, dd, J = 12.2, 2.4 Hz, H-6′a), 3.82 (1H, dd, J = 12.2, 4.0 Hz, H-6′b), 3.72–3.60 (4H, m, H-2′–H-5′), 1.36 (3H, t, J = 6.9 Hz, CH2CH3); 13C NMR (90 MHz, D2O) δ (ppm): 167.2, 166.7, 164.9, 157.3 (C-2, C-4, C-6, COOEt), 114.2 (C-5), 80.4, 79.7, 77.3, 73.0, 69.7 (C-1′–C-5′), 62.8 (C-6′), 61.2 (CH2CH3), 14.0 (CH2CH3). ESI-HRMS positive mode (m/z): calcd for C13H19N2O8+ [M + H]+ 331.1136; C13H18N2NaO8+ [M + Na]+ 353.0955. Found: [M + H]+ 331.1140; [M + Na]+ 353.0953.
4-Amino-2-(β-D-glucopyranosyl)-6-phenyl-pirimidine-5-carbonitrile (11e). Prepared from compound 2 (50 mg, 0.21 mmol) and 2-benzylidenemalononitrile 6 (64 mg, 0.41 mmol) according to general procedure 1. Reaction time: 1 h. Purified by column chromatography (CHCl3-MeOH = 3:1) to give 63 mg (85%) pale yellow syrup. Rf = 0.49 (CHCl3-MeOH 7:3); [α]D = +34 (c 0.17, MeOH); 1H NMR (400 MHz, CD3OD) δ (ppm): 7.89-7.87 (2H, d, J = 7.9 Hz, Ph), 7.57–7.50 (3H, m, Ph ), 4.29 (1H, d, J = 9.6 Hz, H-1′), 3.86 (1H, dd, J = 12.1, 2.1 Hz, H-6′a), 3.81 (1H, pt, J = 9.5, 9.3 Hz, H-2′), 3.77 (1H, dd, J = 12.1, 4.7 Hz, H-6′b), 3.59–3.52 (2H, m, H-3′, H-4′), 3.45–3.42 (1H, m, H-5′); 13C NMR (100 MHz, CD3OD) δ (ppm): 170.4, 169.5, 166.2 (C-2, C-4, C-6), 137.5, 132.2, 129.9, 129.8, 129.6 (2) (Ph), 116.4 (CN), 90.8 (C-5), 87.8, 83.8, 82.2, 79.0, 70.9 (C-1′–C-5′), 62.4 (C-6′). ESI-HRMS positive mode (m/z): Calcd for C17H19N4O5+ [M + H]+ 359.1350; C13H18N2NaO8+ [M + Na]+ 381.1169. Found: [M + H]+ 359.1350; [M + Na]+ 381.1169.
2-(β-D-Glucopyranosyl)-4-phenyl-6-oxo-1,6-dihydropyrimidine-5-carbonitrile (11f). Prepared from compound 2 (50 mg, 0.21 mmol) and ethyl 2-cyano-3-phenylacrylate 7 (83 mg, 0.41 mmol) according to general procedure 1. Reaction time: 1 h. Purified by column chromarography (CHCl3-MeOH = 7:3) to give 30 mg (41%) colourless syrup. Rf = 0.21 (CHCl3-MeOH 7:3); [α]D = +23 (c 0.16, MeOH); 1H NMR (360 MHz, D2O) δ (ppm): 7.81 (2H, d, J = 6.9 Hz, Ph), 7.64–7.58 (3H, m, Ph), 4.31 (1H, d, J = 9.5 Hz, H-1′), 3.91–3.78 (3H, m, H-2′, H-6′a, H-6′b), 3.69–3.60 (3H, m, H-3′–H-5′); 13C NMR (90 MHz, D2O) δ (ppm): 173.2, 171.1, 167.5 (C-2, C-4, C-6), 136.3, 131.7, 129.3 (2), 129.0 (2) (Ph), 118.7 (CN), 95.1 (C-5), 81.9, 80.4, 77.5, 73.3, 69.7 (C-1′–C-5′), 61.1 (C-6′). ESI-HRMS positive mode (m/z): Calcd for C17H18N3O6+ [M + H]+ 360.1190; C17H17N3NaO6+ [M + Na]+ 382.1010. Found: [M + H]+ 360.1190; [M + Na]+ 382.1009.
Methyl 2-(β-D-glucopyranosyl)-4-phenyl-6-oxo-1,6-dihydropyrimidine-5-carboxylate (11g). The Pd-catalyst (50 mg, 20% Pd(OH)2/C) was suspended in anhydrous EtOH (10 mL) under Ar. This degased suspension was saturatated with H2 (3×). To this heterogenous mixture, a solution of compound 10g (200 mg, 0.27 mmol) in anhydrous EtOAc (2 mL) was added. The reaction mixture was heated at reflux temperature under H2 atmosphere until the TLC indicated (EtOAc-hexane 1:1 and CHCl3-MeOH 4:1) the complete conversion of the starting material (6 h). After the completion of the reaction, the catalyst was filtered off through a pad of Celite, and washed with MeOH (3 × 5 mL). The combined organic solution was concentrated under reduced pressure and the crude product was purified by column chromatography (CHCl3-MeOH = 8:1). Yield: 73 mg (70%), colourless syrup. Rf = 0.43 (CHCl3-MeOH = 7:1); [α]D = +37 (c 0.18, MeOH); 1H NMR (400 MHz, CD3OD) δ (ppm): 7.63–7.43 (5H, m, Ph), 4.28 (1H, d, J = 9.5 Hz, H-1′), 3.90 (1H, dd, J = 12.0, 2.0 Hz, H-6′a), 3.79 (1H, dd, J = 12.0, 4.3 Hz, H-6′b), 3.68 (3H, s, OCH3), 3.65 (1H, pt, J = 9.5, 9.2 Hz, H-2′), 3.55–3.43 (3H, m, H-3′, H-4′, H-5′); 13C NMR (100 MHz, CD3OD) δ (ppm): 167.7, 162.8, 162.2, 161.2 (C-2, C-4, C-6, COOMe), 137.9, 131.6, 129.6 (2), 129.2 (2) (Ph), 119.4 (C-5), 82.1, 80.0, 78.8, 74.0, 70.5 (C-1′–C-5′), 62.2 (C-6′), 53.0 (OCH3). ESI-HRMS positive mode (m/z): Calcd for C18H21N2O8+ [M + H]+ 393.1292; C18H20N2NaO8+ [M + Na]+ 415.1112. Found: [M + H]+ 393.1292; [M + Na]+ 415.1111.
Ethyl 2-(β-D-glucopyranosyl)-4-phenyl-6-oxo-1,6-dihydropyrimidine-5-carboxylate (11h). The Pd-catalyst (50 mg, 20% Pd(OH)2/C) was suspended in anhydrous EtOH (10 mL) under Ar, and the suspension was saturatated with H2 (3×). To this heterogenous mixture, a solution of compound 11h (200 mg, 0.26 mmol) in anhydrous EtOAc (2 mL) was added. The reaction mixture was heated at reflux temperature under H2 atmosphere until the TLC indicated (EtOAc-hexane 1:1 and CHCl3-MeOH 4:1) the complete conversion of the starting material (6 h). After completion of the reaction, the catalyst was filtered off through a pad of Celite, and washed with MeOH (3 × 5 mL). The combined organic solution was concentrated under reduced pressure and the crude product was purified by column chromatography (CHCl3-MeOH = 8:1). Yield: 61 mg (58%), colourless syrup. Rf = 0.43 (CHCl3-MeOH = 7:1); [α]D = +62 (c 0.11, MeOH); 1H NMR (400 MHz, CD3OD) δ (ppm): 7.63–7.43 (5H, m, Ph), 4.28 (1H, d, J = 9.5 Hz, H-1′), 4.16 (2H, q, J = 7.1 Hz, CH2CH3), 3.90 (1H, dd, J = 11.9, 2.0 Hz, H-6′a), 3.79 (1H, dd, J = 11.9, 4.3 Hz, H-6′b), 3.64 (1H, pt, J = 9.5, 9.1 Hz, H-2′), 3.54-3.46 (3H, m, H-3′, H-4′, H-5′), 1.08 (3H, t, J = 7.1 Hz, CH2CH3); 13C NMR (90 MHz, CD3OD) δ (ppm): 167.2, 162.9, 162.2, 161.2 (C-2, C-4, C-6, COOEt), 138.1, 131.5, 129.5 (2), 129.3 (2) (Ph), 119.7 (C-5), 82.1, 80.1, 78.8, 74.0, 70.5 (C-1′–C-5′), 62.8 (C-6′), 62.2 (CH2CH3), 14.0 (CH2CH3). ESI-HRMS positive mode (m/z): calcd for C19H23N2O8+ [M + H]+ 407.1449; C19H22N2NaO8+ [M + Na]+ 429.1268. Found: [M + H]+ 407.1445; [M + Na]+ 429.1262.
Methyl 2-(2′,3′,4′,6′-tetra-O-benzyl-β-D-glucopyranosyl)-4-phenyl-6-oxo-1,4,5,6-tetrahydropyrimi-dine-5-carboxylate (12g). Prepared from compound 1 (400 mg, 0.66 mmol) and dimethyl benzylidenemalonate 8 (292 mg, 1.33 mmol) according to general procedure 2. Purified by column chromatography (EtOAc-hexane = 2:3) to give 451 mg (90%) colourless syrup. Rf = 0.46 (EtOAc-hexane = 2:3). 1H NMR (400 MHz, CDCl3) δ (ppm): 8.74, 8.67 (br s, 2 × NH), 7.33–7.10 (m, aromatics), 5.00 (d, J = 13.5 Hz, H-4 or H-5), 4.94 (d, J = 11.6 Hz, H-4 or H-5), 4.87–4.48 (m, PhCH2), 4.10, 4.05 (2d, J = 9.1 Hz in each, 2 × H-1′), 3.79–3.55 (m, 2 × [H-2′–H-6′a,b]), 3.61, 3.55 (2s, 2 × OMe), 3.49 (d, J = 11.5 Hz, H-4 or H-5), 3.22 (d, J = 13.8 Hz, H-4 or H-5); 13C NMR (100 MHz, CDCl3) δ (ppm): 168.3, 168.1, 166.2 (2) (2 × [C-6, COOMe]), 150.7, 150.5 (2 × C-2), 139.9 (2), 138.4, 138.2, 138.0, 137.9, 137.9, 137.8, 137.7 (2), 128.8-127.2 (aromatics), 86.3 (2), 79.2, 79.1, 79.1, 78.9, 78.7 (2), 77.4 (2) (2 × [C-1′–C-5′]), 75.7 (2), 75.2 (2), 74.8 (2), 73.6, 73.6 (8 × PhCH2), 68.8, 68.6 (2 × C-6′), 61.5, 61.4, 53.8, 53.7, 52.7, 52.7 (2 × [C-4, C-5, OCH3]). ESI-HRMS positive mode (m/z): Calcd for C46H47N2O8+ [M + H]+ = 755.3. Found: 755.5.
Ethyl 2-(2′,3′,4′,6′-tetra-O-benzyl-β-D-glucopyranosyl)-4-phenyl-6-oxo-1,4,5,6-tetrahydropyrimidine-5-carboxylate (12h). Prepared from compound 2 (400 mg, 0.66 mmol) and diethyl benzylidinemalonate 9 (329 mg, 1.33 mmol) according to general procedure 2. Purified by column chromatography (EtOAc-hexane = 2:3) to give 413 mg (81%) colourless syrup. Rf = 0.46 (EtOAc-hexane = 2:3). 1H NMR (400 MHz, CDCl3) δ (ppm): 8.68, 8.60 (br s, 2 × NH), 7.36–7.11 (m, aromatics), 4.99 (d, J = 13.3 Hz, H-4 or H-5), 4.92 (d, J = 11.8 Hz, H-4 or H-5), 4.90-4.49 (m, PhCH2), 4.08, 4.07 (2q, J = 7.1 Hz in each, 2 × CH2CH3), 4.05, 4.01 (2d, J = 9.1 Hz in each, 2 × H-1′), 3.79–3.53 (m, 2 × [H-2′–H-6′]), 3.47 (d, J = 11.8 Hz, H-4 or H-5), 3.20 (d, J = 13.4 Hz, H-4 or H-5), 1.08, 1.06 (2t, J = 7.1 Hz in each, 2 × CH2CH3); 13C NMR (100 MHz, CDCl3) δ (ppm): 167.8, 167.6, 166.3, 166.2 (2 × C-6, 2 × COOEt), 150.7, 150.5 (2 × C-2), 139.9, 139.9, 138.4, 138.2, 138.0, 137.9, 137.9, 137.8, 137.7, 137.7, 128.7–127.3 (aromatics), 86.3 (2), 79.2, 79.1, 79.1, 78.9, 78.7, 78.7, 77.5, 77.4 (2 × [C-1′–C-5′]), 75.7 (2), 75.2 (2), 74.8 (2), 73.6, 73.6, (8 × PhCH2), 68.8, 68.6 (2 × C-6′), 61.8, 61.7 (2 × CH2CH3), 61.6, 61.5, 53.8 (2) (2 × C-4, 2 × C-5), 14.0, 14.0 (2 × CH2CH3). ESI-MS positive mode (m/z): Calcd for C47H49N2O8+ [M + H]+ 769.4. Found: 769.6.
2-Bromo-1,3-bis(dimethylamino)trimethinium perchlorate (15). 1,3-Bis(dimethylamino)trimethinium perchlorate 13 (5 g, 22.06 mmol) and NBS (3.93 g, 22.06 mmol) were stirred in dry CH2Cl2 at rt for 5 h. The solvent was then removed under diminished pressure and the residue was triturated with cold EtOH (15 mL) and the precipitate was filtered off. The obtained pale yellow solid (yield: 6.67 g, 99%) was used in the next step without further purification. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 7.95 (2H, s), 3.43 (6H, s), 3.23 (6H, s); 13C NMR (100 MHz, DMSO-d6) δ (ppm): 161.9, 75.9, 49.4, 39.8.
2-(2′,3′,4′,6′-Tetra-O-benzyl-β-D-glucopyranosyl)-pyrimidine(17a). Prepared from amidine 1 (200 mg, 0.33 mmol) and 1,3-bis(dimethylamino)trimethinium perchlorate 13 (82 mg, 0.36 mmol) according to general procedure 4. Reaction time: 16 h. Purified by column chromatography (EtOAc-hexane = 1:2) to give 120 mg (60 %) white solid. Mp: 87–89 °C; Rf = 0.45 (EtOAc-hexane = 1:1); [α]D = +66 (c 0.27, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ (ppm): 8.72 (2H, d, J = 4.9 Hz, H-4, H-6), 7.34–6.85 (21H, m, aromatics, H-5), 4.94, 4.91 (2 × 1H, 2d, J = 11.2 Hz, PhCH2), 4.85, 4.57 (2 × 1H, 2d, J = 10.8 Hz, PhCH2), 4.59, 4.16 (2 × 1H, 2d, J = 11.3 Hz, PhCH2), 4.57 (1H, d, J = 9.6 Hz, H-1′), 4.55, 4.50 (2 × 1H, 2d, J = 12.2 Hz, PhCH2), 4.15 (1H, pt, J = 9.6, 9.2 Hz, H-2′), 3.90 (1H, pt, J = 9.2, 9.1 Hz, H-3′), 3.77–3.70 (4H, m, H-4′–H-6′a,b); 13C NMR (100 MHz, CDCl3) δ (ppm): 166.5 (C-2), 157.3 (C-4, C-6), 138.8, 138.2, 138.1 (2), 128.5-127.5 (aromatics), 120.6 (C-5), 87.2, 83.2, 81.4, 79.9, 78.4 (C-1′–C-5′), 75.7, 75.2, 74.7, 73.5 (4 × PhCH2), 69.3 (C-6′). ESI-MS positive mode (m/z): Calcd for C38H39N2O5+ [M + H]+ 603.3; Found: [M + H]+ 603.5.
2-(2′,3′,4′,6′-Tetra-O-benzyl-β-D-glucopyranosyl)-5-chloropyrimidine (17b). Prepared from amidine 1 (200 mg, 0.33 mmol) and 2-chloro-1,3-bis(dimethylamino)trimethinium hexafluorophosphate 14 (111 mg, 0.36 mmol) according to general procedure 4. Reaction time: 6 h. Purified by column chromatography (EtOAc-hexane = 1:3) to give 205 mg (97%) white solid. Mp: 68–70 °C; Rf = 0.40 (EtOAc-hexane = 1 : 3); [α]D = +4 (c 0.25, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ (ppm): 8.54 (2H, s, H-4, H-6), 7.36–6.86 (20H, m, aromatics), 4.94 (2H, s, PhCH2), 4.85, 4.56 (2 × 1H, 2d, J = 10.7 Hz, PhCH2), 4.64, 4.26 (2 × 1H, 2d, J = 11.6 Hz, PhCH2), 4.52 (1H, d, J = 9.7 Hz, H-1′), 4.54, 4.49 (2 × 1H, 2d, J = 12.4 Hz, PhCH2), 4.06 (1H, pt, J = 9.7, 9.3 Hz, H-2′), 3.89 (1H, pt, J = 9.3, 9.2 Hz, H-3′), 3.75–3.67 (4H, m, H-4′–H-6′a,b); 13C NMR (100 MHz, CDCl3) δ (ppm): 164.1 (C-2), 155.6 (C-4, C-6), 138.7, 138.2, 138.1, 138.0, 128.6–127.5 (aromatics), 130.8 (C-5), 87.3, 82.3, 80.9, 79.9, 78.4 (C-1′–C-5′), 75.8, 75.2, 74.7, 73.6 (4 × PhCH2), 69.2 (C-6′). ESI-MS positive mode (m/z): Calcd for C38H38ClN2O5+ [M + H]+ 637.2464; C38H37ClN2NaO5+ [M + Na]+ 659.2283. Found: [M + H]+ 637.2464; [M + Na]+ 659.2284.
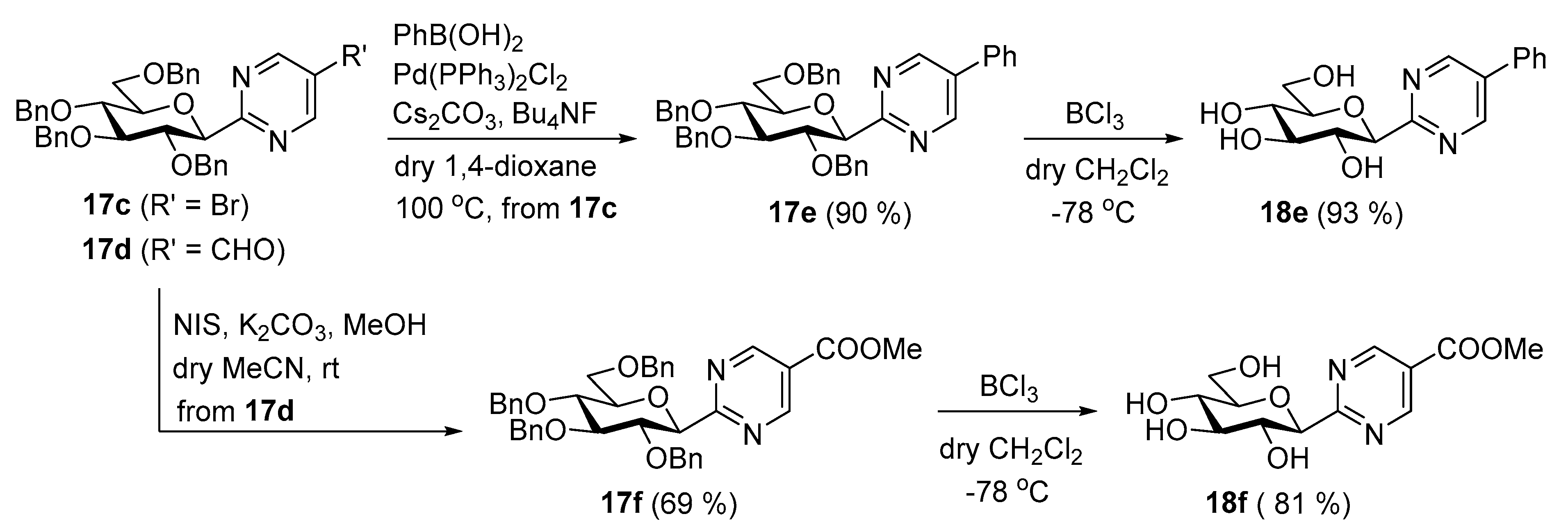
2-(2′,3′,4′,6′-Tetra-O-benzyl-β-D-glucopyranosyl)-5-bromopyrimidine (17c). Prepared from amidine 1 (200 mg, 0.33 mmol) and 2-bromo-1,3-bis(dimethylamino)trimethinium perchlorate 15 (111 mg, 0.36 mmol) according to general procedure 4. Reaction time: 6 h. Purified by column chromatography (EtOAc-hexane = 1:3) to give 203 mg (90%) white amorphous solid. Rf = 0.48 (EtOAc-hexane = 1:2); [α]D = +69 (c 0.17, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ (ppm): 8.62 (2H, s, H-4, H-6), 7.36–6.86 (20H, m, aromatics), 4.94 (2H, s, PhCH2), 4.85, 4.57 (2 × 1H, 2d, J = 10.8 Hz, PhCH2), 4.64, 4.28 (2 × 1H, 2d, J = 11.6 Hz, PhCH2), 4.54, 4.48 (2 × 1H, 2d, J = 12.2 Hz, PhCH2), 4.50 (1H, d, J = 9.6 Hz, H-1′), 4.06 (1H, pt, J = 9.6, 9.2 Hz, H-2′), 3.89 (1H, pt, J = 9.2, 9.1 Hz, H-3′), 3.75–3.67 (4H, m, H-4′–H-6′a,b); 13C NMR (100 MHz, CDCl3) δ (ppm): 164.3 (C-2), 157.7 (C-4, C-6), 138.6, 138.1, 138.0, 137.9, 128.5–127.5 (aromatics), 119.9 (C-5), 87.3, 82.3, 80.8, 79.9, 78.3 (C-1′–C-5′), 75.7, 75.2, 74.6, 73.5 (4 × PhCH2), 69.1 (C-6′). ESI-MS positive mode (m/z): Calcd for C38H38BrN2O5+ [M + H]+ 681.1959; C38H37BrN2NaO5+ [M + Na]+ 703.1778. Found: [M + H]+ 681.1965; [M + Na]+ 703.1782.
2-(2′,3′,4′,6′-Tetra-O-benzyl-β-D-glucopyranosyl)-pyrimidine-5-carbaldehyde (17d). Prepared from amidine 1 (200 mg, 0.33 mmol) and 2-dimethylaminomethylene-1,3-bis(dimethylimonio)propane diperchlorate 16 (139 mg, 0.36 mmol) according to general procedure 4. Reaction time: 4 h. Purified by column chromatography (EtOAc-hexane = 1:2) to give 180 mg (86%) white solid. Mp: 80–82 °C; Rf = 0.62 (EtOAc-hexane = 1:1); [α]D = +80 (c 0.21, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ (ppm): 10.06 (1H, s, CHO), 9.01 (2H, s, H-4, H-6), 7.36–6.84 (20H, m, aromatics), 4.94 (2H, s, PhCH2), 4.86, 4.58 (2 × 1H, 2d, J = 10.7 Hz, PhCH2), 4.65, 4.25 (2 × 1H, 2d, J = 11.6 Hz, PhCH2), 4.63 (1H, d, J = 9.5 Hz, H-1′), 4.54, 4.49 (2 × 1H, 2d, J = 12.2 Hz, PhCH2), 4.12 (1H, pt, J = 9.5, 9.3 Hz, H-2′), 3.92 (1H, pt, J = 9.2, 9.1 Hz, H-3′), 3.77-3.69 (4H, m, H-4′–H-6′); 13C NMR (100 MHz, CDCl3) δ (ppm): 188.8 (CHO), 170.3 (C-2), 158.2 (C-4, C-6), 138.6, 138.1, 138.0, 137.9, 128.6–127.5 (aromatics), 127.7 (C-5), 87.3, 82.7, 81.0, 80.0, 78.3 (C-1′–C-5′), 75.8, 75.3, 74.7, 73.6 (4 × PhCH2), 69.2 (C-6′). ESI-MS positive mode (m/z): Calcd for C39H39N2O6+ [M + H]+ 631.2803; C39H38N2NaO6+ [M + Na]+ 653.2622. Found: [M + H]+ 631.2806; [M + Na]+ 653.2626.
2-(2′,3′,4′,6′-Tetra-O-benzyl-β-D-glucopyranosyl)-5-phenylpyrimidine (17e). Compound 17c (380 mg, 0.56 mmol), phenylboronic acid (136 mg, 1.12 mmol, 2 equiv.), Pd(PPh3)2Cl2 (79 mg, 0.11 mmol, 0.2 equiv.), Cs2CO3 (363 mg, 1.12 mmol, 2 equiv.), and Bu4NF (1.12 mL, 1.12 mmol, 2 equiv., 1M solution in dry THF) were heated at 100 °C in dry 1,4-dioxane (10 mL). After 16 h, the solvent was removed under diminished pressure and the residue was purified by column chromatography (EtOAc-hexane = 1:2). Yield: 340 mg (90%), white amorphous solid. Rf = 0.29 (EtOAc-hexane = 1:2); [α]D = +55 (c 0.27, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ (ppm): 8.86 (2H, s, H-4, H-6), 7.56–6.87 (25H, m, aromatics), 4.97, 4.94 (2 × 1H, 2d, J = 11.1 Hz, PhCH2), 4.87, 4.59 (2 × 1H, 2d, J = 10.8 Hz, PhCH2), 4.65, 4.28 (2 × 1H, 2d, J = 11.4 Hz, PhCH2), 4.63 (1H, d, J = 9.6 Hz, H-1′), 4.56, 4.50 (2 × 1H, 2d, J = 12.2 Hz, PhCH2), 4.20 (1H, pt, J = 9.6, 9.3 Hz, H-2′), 3.93 (1H, pt, J = 9.3, 9.1 Hz, H-3′), 3.80–3.72 (4H, m, H-4′–H-6′a,b); 13C NMR (100 MHz, CDCl3) δ (ppm): 165.0 (C-2), 155.1 (C-4, C-6), 138.7, 138.1 (3), 134.2, 133.2, 129.5–127.1 (aromatics, C-5), 87.3, 82.9, 81.2, 79.8, 78.4 (C-1′–C-5′), 75.7, 75.2, 74.7, 73.5 (4 × PhCH2), 69.2 (C-6′). ESI-MS positive mode (m/z): Calcd for C44H43N2O5+ [M + H]+ 679.3. Found: [M + H]+ 679.6.
Methyl 2-(2′,3′,4′,6′-tetra-O-benzyl-β-D-glucopyranosyl)-pyrimidine-5-carboxylate (17f). To a solution of compound 17d (100 mg, 0.16 mmol) in dry CH3CN (2 mL) NIS (107 mg, 0.48 mmol, 3 equiv.), K2CO3 (67 mg, 0.48 mmol, 3 equiv.) and MeOH (32 μL, 0.79 mmol, 5 equiv.) were added. The reaction mixture was stirred at rt until the TLC (EtOAc-hexane = 2:3) showed complete transformation of the starting material (5 h). The reaction was then quenched with 10% aq. solution of Na2S2O3 (10 mL) and the mixture was extracted with EtOAc (3 × 10 mL). The combined organic phase was washed with brine (10 mL), dried over MgSO4, filtered, and the solvent was removed under diminished pressure. Column chromatographic purification of the residue (EtOAc-hexane = 1:2) gave 72 mg (69%) white amorphous solid. Rf = 0.33 (EtOAc-hexane = 1:2); [α]D = +53 (c 0.20, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ (ppm): 9.14 (2H, s, H-4, H-6), 7.36–6.84 (20H, m, aromatics), 4.94 (2H, s, PhCH2), 4.86, 4.58 (2 × 1H, 2d, J = 10.8 Hz, PhCH2), 4.61, 4.21 (2 × 1H, 2d, J = 11.5 Hz, PhCH2), 4.61 (1H, d, J = 9.6 Hz, H-1′), 4.55, 4.49 (2 × 1H, 2d, J = 12.2 Hz, PhCH2), 4.11 (1H, pt, J = 9.6, 9.3 Hz, H-2′), 3.99 (3H, s, OCH3), 3.91 (1H, pt, J = 9.3, 9.1 Hz, H-3′), 3.77–3.69 (4H, m, H-4′–H-6′a,b); 13C NMR (100 MHz, CDCl3) δ (ppm): 169.4, 164.1 (COOMe, C-2), 158.2 (C-4, C-6), 138.6, 138.1, 138.0, 137.8, 128.5–127.5 (aromatics), 123.1 (C-5), 87.2, 82.7, 8, 1.0, 79.9, 78.3 (C-1′–C-5′), 75.8, 75.2, 74.7, 73.5 (4 × PhCH2), 69.1 (C-6′), 52.8 (OCH3). ESI-MS positive mode (m/z): Calcd for C40H41N2O7+ [M + H]+ 661.3. Found: [M + H]+ 661.6.
2-(β-D-Glucopyranosyl)-pyrimidine (18a). Method A: Prepared from amidine 2 (100 mg, 0.41 mmol) and 1,3-bis(dimethylamino)trimethinium perchlorate 13 (103 mg, 0.45 mmol) according to general procedure 4. Reaction time: 16 h. Purified by column chromatography (CHCl3-MeOH = 5:1) to give 81 mg (81%) colourless syrup. Method B: Compound 17a (200 mg, 0.33 mmol) was dissolved in anhydrous CH2Cl2 (10 mL). The stirred reaction mixture was cooled to ‒78 °C and ~1M solution of BCl3 in CH2Cl2 (1.7 mL, 1.7 mmol, 5 eqiuv.) was added. The stirring was continued at this temperature and the reaction was monitored by TLC (EtOAc-hexane = 1:2 and CHCl3-MeOH = 3:1). After the complete disappearance of the starting material (3 h), MeOH (15 mL) was added to the reaction mixture and was left to warm to rt. The solvents were removed under diminished pressure and the residue was purified by column chromatography (CHCl3-MeOH = 5:1) to give 68 mg (85%) colourless syrup. Rf = 0.28 (CH3Cl-MeOH = 7:3); [α]D = +44 (c 0.24, H2O); 1H NMR (360 MHz, CD3OD) δ (ppm): 8.84 (2H, d, J = 5.0 Hz, H-4, H-6), 7.48 (1H, t, J = 5.0 Hz, H-5), 4.44 (1H, d, J = 9.6 Hz, H-1′), 3.88 (1H, dd, J = 12.2, 1.7 Hz, H-6′a), 3.76 (1H, pt, J = 9.5, 9.1 Hz, H-2′), 3.72 (1H, dd, J = 12.2, 4.7 Hz, H-6′b), 3.58 (1H, pt, J = 9.1, 9.0 Hz, H-3′ or H-4′), 3.52 (1H, pt, J = 9.3, 9.1 Hz, H-3′ or H-4′), 3.49–3.46 (1H, m, H-5′); 13C NMR (90 MHz, CD3OD) δ (ppm): 167.9 (C-2), 158.7 (2) (C-4, C-6), 122.2 (C-5), 83.6, 82.3, 79.3, 74.8, 71.2 (C-1′–C-5′), 62.7 (C-6′). ESI-MS positive mode (m/z): C10H14N2NaO5+ [M + Na]+ 265.0795. Found: 265.0795.
5-Chloro-2-(β-D-glucopyranosyl)-pyrimidine (18b). Prepared from amidine 2 (100 mg, 0.41 mmol) and 2-chloro-1,3-bis(dimethylamino)trimethinium hexafluorophosphate 14 (139 mg, 0.45 mmol) according to general procedure 4. Reaction time: 2 h. Purified by column chromatography (CHCl3-MeOH = 9:1) to give 100 mg (88%) white solid. Mp: 200–202 °C; Rf = 0.25 (CH3Cl-MeOH = 5:1); [α]D = ‒11 (c 0.22, H2O); 1H NMR (360 MHz, CD3OD) δ (ppm): 8.87 (2H, s, H-4, H-6), 4.43 (1H, d, J = 9.6 Hz, H-1′), 3.87 (1H, dd, J = 12.3, 1.7 Hz, H-6′a), 3.76 (1H, pt, J = 9.5, 9.1 Hz, H-2′), 3.70 (1H, dd, J = 12.3, 4.8 Hz, H-6′b), 3.54 (1H, pt, J = 9.2, 9.0 Hz, H-3′ or H-4′), 3.50–3.43 (2H, m, H-3′ or H-4′, H-5′); 13C NMR (90 MHz, CD3OD) δ (ppm): 165.9 (C-2), 157.1 (2) (C-4, C-6), 132.1 (C-5), 83.6, 82.6, 79.3, 74.7, 71.4 (C-1′–C-5′), 62.8 (C-6′). ESI-MS positive mode (m/z): C10H13ClN2NaO5+ [M + Na]+ 299.0405. Found: 299.0407.
5-Bromo-2-(β-D-glucopyranosyl)-pyrimidine (18c). Prepared from amidine 2 (100 mg, 0.41 mmol) and 2-bromo-1,3-bis(dimethylamino)trimethinium perchlorate 15 (138 mg, 0.45 mmol) according to general procedure 4. Reaction time: 2 h. Purified by column chromatography (CHCl3-MeOH = 9:1) to give 112 mg (85%) white solid. Mp: 224–226 °C; Rf = 0.25 (CH3Cl-MeOH = 5:1); [α]D = +19 (c 0.22, H2O); 1H NMR (360 MHz, CD3OD) δ (ppm): 8.96 (2H, s, H-4, H-6), 4.41 (1H, d, J = 9.6 Hz, H-1′), 3.87 (1H, dd, J = 12.2, 1.5 Hz, H-6′a), 3.75 (1H, pt, J = 9.6, 9.1 Hz, H-2′), 3.70 (1H, dd, J = 12.2, 4.6 Hz, H-6′b), 3.54 (1H, pt, J = 9.3, 9.1 Hz, H-3′ or H-4′), 3.50–3.43 (2H, m, H-3′ or H-4′, H-5′); 13C NMR (90 MHz, CD3OD) δ (ppm): 166.2 (C-2), 159.4 (2) (C-4, C-6), 120.9 (C-5), 83.7, 82.6, 79.3, 74.7, 71.4 (C-1′–C-5′), 62.8 (C-6′). ESI-MS positive mode (m/z): C10H13BrN2NaO5+ [M + Na]+ 342.9900. Found: 342.9901.
2-(β-D-Glucopyranosyl)-5-phenylpyrimidine (18e). Compound 17e (200 mg, 0.29 mmol) was dissolved in anhydrous CH2Cl2 (10 mL). The stirred reaction mixture was cooled to ‒78 °C and a ~1M solution of BCl3 in CH2Cl2 (1.5 mL, 1.5 mmol, 5 equiv.) was added. The stirring was continued at this temperature and the reaction was monitored by TLC (EtOAc-hexane = 1:2 and CHCl3-MeOH = 3:1). After the complete disappearance of the starting material (2 h), MeOH (10 mL) was added to the reaction mixture and was left to warm to rt. The solvents were removed under diminished pressure and the residue was purified by column chromatography (CHCl3-MeOH = 9:1) to give 87 mg (93%) colourless syrup. Rf = 0.50 (CH3Cl-MeOH = 3:1); [α]D = –31 (c 0.22, H2O); 1H NMR (360 MHz, CD3OD) δ (ppm): 9.07 (2H, s, H-4, H-6), 7.72 (2H, d, J = 7.1 Hz, Ph), 7.56–7.46 (3H, m, Ph), 4.49 (1H, d, J = 9.5 Hz, H-1′), 3.90 (1H, dd, J = 12.1, 1.7 Hz, H-6′a), 3.81 (1H, pt, J = 9.5, 9.1 Hz, H-2′), 3.74 (1H, dd, J = 12.1, 4.8 Hz, H-6′b), 3.59 (1H, pt, J = 9.1, 9.0 Hz, H-3′ or H-4′), 3.53 (1H, pt, J = 9.2, 9.0 Hz, H-3′ or H-4′), 3.53–3.51 (1H, m, H-5′); 13C NMR (90 MHz, CD3OD) δ (ppm): 166.5 (C-2), 156.3 (2) (C-4, C-6), 135.1, 134.9 (Ph, C-5), 130.6 (2), 130.3, 128.1 (2) (Ph), 83.6, 82.5, 79.4, 74.9, 71.3 (C-1′–C-5′), 62.9 (C-6′). ESI-MS positive mode (m/z): C16H18N2NaO5+ [M + Na]+ 341.1108. Found: 341.1108.
Methyl 2-(β-D-glucopyranosyl)-pyrimidine-5-carboxylate (18f). Compound 17f (200 mg, 0.30 mmol) was dissolved in anhydrous CH2Cl2 (10 mL). The stirred reaction mixture was cooled to ‒78 °C and a ~1M solution of BCl3 in CH2Cl2 (1.51 mL, 1.51 mmol, 5 equiv.) was added. The stirring was continued at this temperature and the reaction was monitored by TLC (EtOAc-hexane = 1:2 and CHCl3-MeOH = 3:1). After complete disappearance of the starting material (2 h), MeOH (15 mL) was added to the reaction mixture and was left to warm to rt. The solvents were removed under diminished pressure and the residue was purified by column chromatography (CHCl3-MeOH = 9:1) to give 74 mg (81%) colourless syrup. Rf = 0.31 (CH3Cl-MeOH = 5:1); [α]D = +51 (c 0.22, H2O); 1H NMR (360 MHz, CD3OD) δ (ppm): 9.29 (2H, s, H-4, H-6), 4.52 (1H, d, J = 9.6 Hz, H-1′), 3.99 (3H, s, OCH3), 3.88 (1H, dd, J = 12.2, 1.7 Hz, H-6′a), 3.78 (1H, pt, J = 9.5, 9.1 Hz, H-2′), 3.72 (1H, dd, J = 12.2, 4.6 Hz, H-6′b), 3.62–3.47 (3H, m, H-3′, H-4′, H-5′); 13C NMR (90 MHz, CD3OD) δ (ppm): 171.0 (C=O), 165.1 (C-2), 159.3 (2) (C-4, C-6), 124.8 (C-5), 83.8, 82.5, 79.3, 74.7, 71.2 (C-1′–C-5′), 62.8 (C-6′), 53.3 (OCH3). ESI-MS positive mode (m/z): C12H16N2NaO7+ [M + Na]+ 323.0850. Found: 323.0851.







{kind=link}
{kind=link}
{kind=link}






















