
Synthetic Approaches to a Challenging and Unusual Structure—An Amino-Pyrrolidine Guanine Core

 ,
,  and
and
Abstract
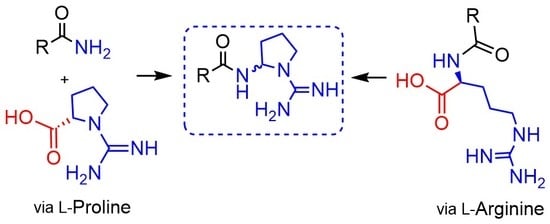
:
1. Introduction
2. Results and Discussion
2.1. Studies on the Oxidative Decarboxylation of l-arginine Derivatives (3)
2.2. Studies on the Oxidative Decarboxylation of Carbamimidoyl-l-proline (2)
3. Materials and Methods
3.1. General Procedure for the Synthesis of Arginine Derivatives
3.2. Synthesis of Carbamimidoyl-l-proline (2) [54]
3.3. General Procedure for the Preparation of Compounds 4 from Decarboxylation of Arginine Derivatives
3.4. General Procedure for the Preparation of Compounds 4 from Decarboxylation of Carbamimidoyl-l-proline (2)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nadin, A.; Hattotuwagama, C.; Churcher, I. Lead-oriented synthesis: A new opportunity for synthetic chemistry. Angew. Chem. Int. Edit. 2012, 51, 1114–1122. [Google Scholar] [CrossRef] [PubMed]
- Mitchison, T.J. Small-molecule screening and profiling by using automated microscopy. Chembiochem 2005, 6, 33–39. [Google Scholar] [CrossRef] [PubMed]
- de la Torre, B.G.; Albericio, F. The pharmaceutical industry in 2018. An analysis of fda drug approvals from the perspective of molecules. Molecules 2019, 24, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobson, C.M. Chemical space and biology. Nature 2004, 432, 824–828. [Google Scholar] [CrossRef]
- Erlanson, D.A.; Fesik, S.W.; Hubbard, R.E.; Jahnke, W.; Jhoti, H. Twenty years on: The impact of fragments on drug discovery. Nat. Rev. Drug Discov. 2016, 15, 605–619. [Google Scholar] [CrossRef]
- Lourenco, A.M.; Ferreira, L.M.; Branco, P.S. Molecules of natural origin, semi-synthesis and synthesis with anti-inflammatory and anticancer utilities. Curr. Pharm. Design 2012, 18, 3979–4046. [Google Scholar] [CrossRef]
- Karageorgis, G.; Waldmann, H. Guided by evolution: Biology-oriented synthesis of bioactive compound classes. Synthesis 2019, 51, 55–66. [Google Scholar] [CrossRef] [Green Version]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [Green Version]
- Berlinck, R.G.S.; Bertonha, A.F.; Takaki, M.; Rodriguez, J.P.G. The chemistry and biology of guanidine natural products. Nat. Prod. Rep. 2017, 34, 1264–1301. [Google Scholar] [CrossRef]
- Liu, J.; Li, X.W.; Guo, Y.W. Recent advances in the isolation, synthesis and biological activity of marine guanidine alkaloids. Mar. Drugs 2017, 15, 324. [Google Scholar] [CrossRef] [Green Version]
- McTaggart, F.; Buckett, L.; Davidson, R.; Holdgate, G.; McCormick, A.; Schneck, D.; Smith, G.; Warwick, M. Preclinical and clinical pharmacology of rosuvastatin, a new 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitor. Am. J. Cardiol. 2001, 87, 28–32. [Google Scholar] [CrossRef]
- Capuzzi, D.M.; Cevallos, W.H. Inhibition of hepatic cholesterol and triglyceride synthesis by guanabenz acetate. J. Cardiovasc. Pharmacol. 1984, 6, S847–S852. [Google Scholar] [CrossRef]
- Fabbro, D.; Parkinson, D.; Matter, A. Protein tyrosine kinase inhibitors: New treatment modalities? Curr. Opin. Pharmacol. 2002, 2, 374–381. [Google Scholar] [CrossRef]
- Carter, D.C.; Logan, R.; Forrest, J.A.H.; Ansell, I.; Heading, R.C.; Shearman, D.J.C. Effect of histamine h2 receptor antagonist, cimetidine, on insulin-induced gastric-secretion in man. Br. J. Surg. 1975, 62, 664–665. [Google Scholar]
- Saczewski, F.; Balewski, L. Biological activities of guanidine compounds, 2008-2012 update. Expert Opin. Ther. Patents 2013, 23, 965–995. [Google Scholar] [CrossRef]
- Coles, M.P. Bicyclic-guanidines, -guanidinates and -guanidinium salts: Wide ranging applications from a simple family of molecules. Chem. Commun. 2009, 3659–3676. [Google Scholar] [CrossRef]
- Lopes, L.C.; Roman, B.; Medeiros, M.A.; Mukhopadhyay, A.; Utrilla, P.; Galvez, J.; Maurino, S.G.; Moltiva, V.; Lourenco, A.; Feliciano, A.S. Cernumidine and isocernumidine, new type of cyclic guanidine alkaloids from solanum cernuum. Tetrahedron Lett. 2011, 52, 6392–6395. [Google Scholar] [CrossRef]
- Kundu, J.K.; Surh, Y.J. Breaking the relay in deregulated cellular signal transduction as a rationale for chemoprevention with anti-inflammatory phytochemicals. Mutat. Res.-Fundam. Mol. Mech. Mutagen. 2005, 591, 123–146. [Google Scholar] [CrossRef]
- Kundu, J.K.; Surh, Y.J. Emerging avenues linking inflammation and cancer. Free Radic. Biol. Med. 2012, 52, 2013–2037. [Google Scholar] [CrossRef]
- Brew, R.; Erikson, J.S.; West, D.C.; Kinsella, A.R.; Slavin, J.; Christmas, S.E. Interleukin-8 as an autocrine growth factor for human colon carcinoma cells in vitro. Cytokine 2000, 12, 78–85. [Google Scholar] [CrossRef]
- Miranda, M.A.; Mondal, A.; Sachdeva, M.; Cabral, H.; Neto, Y.; Khan, I.; Groppo, M.; McChesney, J.D.; Bastos, J.K. Chemosensitizing effect of cernumidine extracted from solanum cernuum on bladder cancer cells in vitro. Chem. Biodivers. 2019, 16, e1900334. [Google Scholar] [CrossRef]
- Damasceno, J.L.; de Oliveira, P.F.; Miranda, M.A.; Lima, M.; Bastos, J.K.; Tavares, D.C. Antigenotoxic and antioxidant properties of solanum cernuum and its alkaloid, cernumidine. Biol. Pharm. Bull. 2016, 39, 920–926. [Google Scholar] [CrossRef] [Green Version]
- Ramos, A.C.; de Oliveira, R.R. A new alkaloid and flavonoids isolated from solanum cernuum leaves by high-performance countercurrent chromatography. Nat. Prod. Res. 2017, 31, 2405–2412. [Google Scholar] [CrossRef] [PubMed]
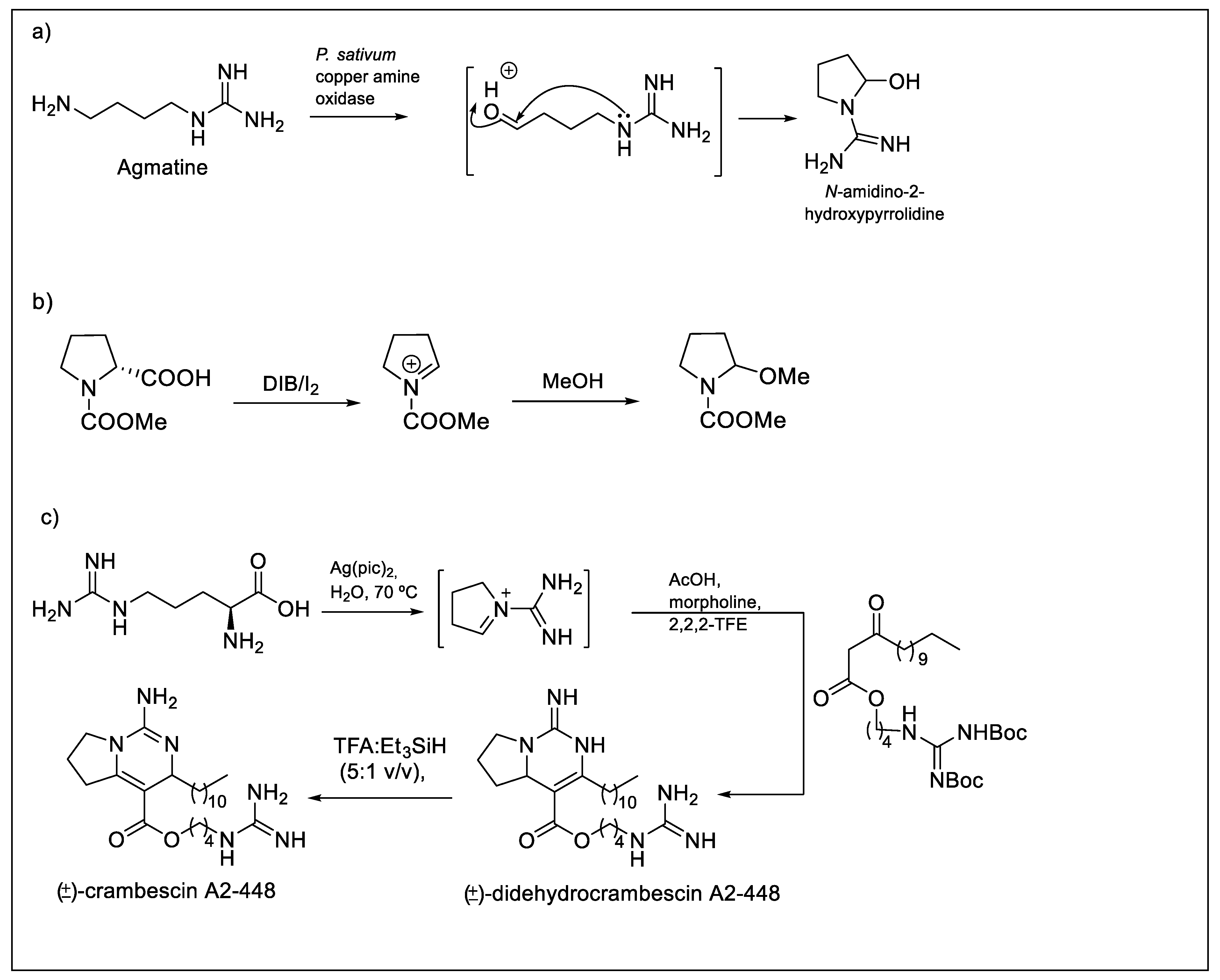
- Ascenzi, P.; Fasano, M.; Marino, M.; Venturini, G.; Federico, R. Agmatine oxidation by copper amine oxidase—Biosynthesis and biochemical characterization of N-amidino-2-hydroxypyrrolidine. Eur. J. Biochem. 2002, 269, 884–892. [Google Scholar] [CrossRef] [PubMed]
- Boto, A.; Hernandez, R.; Suarez, E. Oxidative decarboxylation of alpha-amino acids: A mild and efficient method for the generation of N-acyliminium ions. Tetrahedron Lett. 1999, 40, 5945–5948. [Google Scholar] [CrossRef]
- Boto, A.; Hernandez, R.; Suarez, E. Synthesis of alkaloids from amino acids via N-acyliminium ions generated by one-pot radical decarboxylation-oxidation. Tetrahedron Lett. 2000, 41, 2899–2902. [Google Scholar] [CrossRef]
- Boto, A.; Hernandez, R.; Suarez, E. Tandem radical decarboxylation-oxidation of amino acids: A mild and efficient method for the generation of N-acyliminium ions and their nucleophilic trapping. J. Org. Chem. 2000, 65, 4930–4937. [Google Scholar] [CrossRef]
- Boto, A.; Hernandez, R.; Suarez, E. Tandem oxidative radical decarboxylation-beta-iodination of amino acids. Application to the synthesis of chiral 2,3-disubstituted pyrrolidines. Tetrahedron Lett. 2000, 41, 2495–2498. [Google Scholar] [CrossRef]
- Silva, S.B.L.; Oberhansli, F.; Tribalat, M.A.; Genta-Jouve, G.; Teyssie, J.L.; Dechraoui-Bottein, M.Y.; Gallard, J.F.; Evanno, L.; Poupon, E.; Thomas, O.P. Insights into the biosynthesis of cyclic guanidine alkaloids from crambeidae marine sponges. Angew. Chem. Int. Edit. 2019, 58, 520–525. [Google Scholar] [CrossRef]
- Fan, R.H.; Li, W.X.; Wang, B. A one-pot oxidative decarboxylation-friedel-crafts reaction of acyclic alpha-amino acid derivatives activated by the combination of iodobenzene diacetate/iodine and iron dust. Org. Biomol. Chem. 2008, 6, 4615–4621. [Google Scholar] [CrossRef]
- Cowden, C.J. Use of N-protected amino acids in the minisci radical alkylation. Org. Lett. 2003, 5, 4497–4499. [Google Scholar] [CrossRef]
- Bellale, E.V.; Huddar, S.N.; Mahajan, U.S.; Akamanchi, K.G. Oxidative decarboxylation of alpha-amino acids to nitriles using o-iodoxybenzoic acid in aqueous ammonia. Pure Appl. Chem. 2011, 83, 607–612. [Google Scholar] [CrossRef]
- Matthessen, R.; Claes, L.; Fransaer, J.; Binnemans, K.; De Vos, D.E. Decarboxylation of a wide range of amino acids with electrogenerated hypobromite. Eur. J. Org. Chem. 2014, 6649–6652. [Google Scholar] [CrossRef]
- Laval, G.; Golding, B.T. One-pot sequence for the decarboxylation of alpha-amino acids. Synlett 2003, 542–546. [Google Scholar]
- Bacon, R.G.R.; Hanna, W.J.W.; Stewart, D. Oxidation by persulphate. Part V. Silver-catalysed oxidation of secondary aliphatic amines and α-amino-acids. J. Chem. Soc. C 1966, 1388–1389. [Google Scholar] [CrossRef]
- Clarke, T.G.; Hampson, N.A.; Lee, J.B.; Morley, J.R.; Scanlon, B. Oxidations involving silver. II. Oxidation of alcohols and aldehydes with silver(II) picolinate. Can. J. Chem. 1969, 47, 1649–1654. [Google Scholar] [CrossRef]
- Zelechonok, Y.; Silverman, R.B. Silver(I)/peroxydisulfate-induced oxidative decarboxylation of amino-acids—A chemical-model for a possible intermediate in the monoamine oxidase-catalyzed oxidation of amines. J. Org. Chem. 1992, 57, 5787–5790. [Google Scholar] [CrossRef]
- Anderson, J.M.; Kochi, J.K. Silver(I)-catalyzed oxidative decarboxylation of acids by peroxydisulfate—Role of silver(II). J. Am. Chem. Soc. 1970, 92, 1651–1659. [Google Scholar] [CrossRef]
- Fristad, W.E.; Fry, M.A.; Klang, J.A. Persulfate silver ion decarboxylation of carboxylic-acids—Preparation of alkanes, alkenes, and alcohols. J. Org. Chem. 1983, 48, 3575–3577. [Google Scholar] [CrossRef]
- Huang, W.H.; Wang, M.L.; Yue, H. Conversion of N-acyl amino acids into imides via oxidative decarboxylation induced by Ag+/Cu2+/S2O82- in water. Synthesis 2008, 1342–1344. [Google Scholar] [CrossRef]
- Bi, H.P.; Chen, W.W.; Liang, Y.M.; Li, C.J. A novel iron-catalyzed decarboxylative Csp3-Csp2 coupling of proline derivatives and naphthol. Org. Lett. 2009, 11, 3246–3249. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Wang, Z.Q.; Zhang, J.J.; Yu, L.T.; Tan, J.J. Cobalt-catalyzed decarboxylative acetoxylation of amino acids and arylacetic acids. Org. Lett. 2015, 17, 4476–4478. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Seidel, D. Nontraditional reactions of azomethine ylides: Decarboxylative three-component couplings of alpha-amino acids. J. Am. Chem. Soc. 2010, 132, 1798–1799. [Google Scholar] [CrossRef] [PubMed]
- Bi, H.P.; Zhao, L.; Liang, Y.M.; Li, C.J. The copper-catalyzed decarboxylative coupling of the sp3-hybridized carbon atoms of alpha-amino acids. Angew. Chem. Int. Edit. 2009, 48, 792–795. [Google Scholar] [CrossRef]
- Firouzabadi, H.; Iranpoor, N.; Ghaderi, A.; Ghavami, M. Cerium(IV) oxide as a neutral catalyst for aldehyde-induced decarboxylative coupling of L-proline with triethyl phosphite and nitromethane. Tetrahedron Lett. 2012, 53, 5515–5518. [Google Scholar] [CrossRef]
- Rahman, M.; Mukherjee, A.; Kovalev, I.S.; Kopchuk, D.S.; Zyryanov, G.V.; Tsurkan, M.V.; Majee, A.; Ranu, B.C.; Charushin, V.N.; Chupakhin, O.N.; et al. Recent advances on diverse decarboxylative reactions of amino acids. Adv. Synth. Catal. 2019, 361, 2161–2214. [Google Scholar] [CrossRef]
- Zervas, L.; Winitz, M.; Greenstein, J.P. Studies on arginine peptides.1. Intermediates in the synthesis of N-terminal and C-terminal arginine peptides. J. Org. Chem. 1957, 22, 1515–1521. [Google Scholar] [CrossRef]
- Vaidyanathan, R.; Kalthod, V.G.; Ngo, D.P.; Manley, J.M.; Lapekas, S.P. Amidations using N,N′-carbonyldiimidazole: Remarkable rate enhancement by carbon dioxide. J. Org. Chem. 2004, 69, 2565–2568. [Google Scholar] [CrossRef]
- Kiyokawa, K.; Watanabe, T.; Fra, L.; Kojima, T.; Minakata, S. Hypervalent iodine(III)-mediated decarboxylative ritter-type amination leading to the production of alpha-tertiary amine derivatives. J. Org. Chem. 2017, 82, 11711–11720. [Google Scholar] [CrossRef]
- Richards, J.C.; Spenser, I.D. H-2 NMR-spectroscopy as a probe of the stereochemistry of enzymic reactions at prochiral centers—Diamine oxidase. Tetrahedron 1983, 39, 3549–3568. [Google Scholar] [CrossRef]
- Nagao, Y.; Miyasaka, T.; Seno, K.; Fujita, E.; Shibata, D.; Doi, E. Utilization of sulfur-containing leaving group.6. Peptide-bond formation, chemoselective acylation of amino-acids, and crosslinking reaction between amino-acids utilizing a functional 5-membered heterocycle, 1,3-thiazolidine-2-thione. J. Chem. Soc.-Perkin Trans. 1984, 1, 2439–2446. [Google Scholar] [CrossRef]
- Sahakitpichan, P.; Disadee, W.; Ruchirawat, S.; Kanchanapoom, T. L-(N-trans-cinnamoyl)-arginine, an acylamino acid from Glinus oppositifolius (L.) Aug. DC. Molecules 2010, 15, 6186–6192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahindra, A.; Bagra, N.; Wangoo, N.; Khan, S.I.; Jacob, M.R.; Jain, R. Discovery of short peptides exhibiting high potency against cryptococcus neoformans. ACS Med. Chem. Lett. 2014, 5, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Rowley, G.L.; Greenleaf, A.L.; Kenyon, G.L. Specificity of creatine kinase-new glycocyamines and glycocyamine analogs related to creatine. J. Am. Chem. Soc. 1971, 93, 5542–5551. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 |
 |
| Entry | Oxidant | Additive (equiv.) | Solvent | T (°C) | Time (h) | (3a) Relative Amount (%) a | (4a) Relative Amount (%) a | (6a) Relative Amount (%) a | (7) Relative Amount (%) a |
|---|---|---|---|---|---|---|---|---|---|
| 1 | DIB/I2 (2:0.5) | - | CH2Cl2 | r.t. | 48 | >90 | vestigial | - | - |
| 2 f | DIB/I2 (2:0.5) | - | DMF | r.t. | 18 | 100 | - | - | - |
| 3 | DIB/I2 (4:1) | - | CH3CN | reflux | 56 | >90 | vestigial | - | - |
| 4 f | DIB/I2 (4:1) | - | CH2Cl2 | r.t. | 1.5 | 100 | - | - | - |
| 5 | DIB/I2 (4:1) | - | CH2Cl2 | reflux | 48 | 92 | 8 | - | - |
| 6 g | DIB/I2 (4:1) | - | DMSO | r.t. | 48 | - | - | - | - |
| 7 f | DIB/I2 (2:0.5) | - | AcOH/CH2Cl2 (3:1) | reflux | 72 | - | 59 e | - | - |
| 8 b | Ag(I)/S2O82− (0.1:1.2) | - | H2O | r.t. | 3 | 53 | 39 | 8 | - |
| 9 b,g | Ag(I)/S2O82− (0.2:1.2) | - | DMF | r.t. | >24 | - | - | - | - |
| 10 b,g | Ag(I)/S2O82− (0.15:1.2) | - | AcOH | r.t. | 96 | - | - | - | - |
| 11 b,f | Ag(I)/S2O82− (0.15:1.5) | - | MeOH | r.t. | 48 | 100 | - | - | - |
| 12 b | Ag(I)/S2O82− (0.15:1.5) | - | AcOH:H2O (1:1) | r.t. | 20 | - | 35 | 39 | 26 |
| 13 c | Ag(I)/S2O82− (0.15:1.5) | - | H2O | r.t. | 3 | - | 69 | 17 | 11 |
| 14 g | Ag(I)/S2O82− (0.15:3) | CuSO4 (0.2) | H2O | r.t. | 3 | - | - | - | - |
| 15 c | Ag(I)/S2O82− (0.15:1.5) | CuSO4 (1) | H2O | r.t. | 3 | - | 89 | 7 | 4 |
| 16 | Ag(I)/S2O82− (0.15:1.5) | CuSO4 (1) | H2O | 60 | 0.5 | - | 83 | 8 | 5 |
| 17 d | Ag(I)/S2O82− (0.15:1.5) | CuSO4 (1) | H2O | 60 | 0.5 | - | 72 e | - | - |
| From l-arginine derivatives (3) |
 |
| From carbamimidoyl-l-proline (2) |
 |
| Entry | Oxidant | Additive | Solvent | T (°C) | Time (h) | (9) a | (10) a | (4a) Yield (%) |
|---|---|---|---|---|---|---|---|---|
| 1 | Ag(I)/S2O82− (0.3:3) | - | ACN/H2O | r.t. | 5.5 | ✓ b | ✓ a | - |
| 2 | Ag(I)/S2O82− (0.3:3) | - | ACN/H2O | r.t. | 72 | ✓ b | ✓ a | - |
| 3 | Ag(I)/S2O82− (0.3:3) | - | AcOH | r.t. | 72 | ✓ b | ✓ a | - |
| 4 | Ag(I)/S2O82− (0.3:3) | - | DMA | r.t. | 72 | ✓ b | ✓ a | - |
| 5 | Ag(I)/S2O82− (0.3:3) | - | ACN/H2O | 80 | 6 | ✓ b | ✓ a | - |
| 6 | Ag(I)/S2O82− (0.3:3) | - | DMA | r.t. | 6 | - | - | - |
| 7 | Ag(I)/S2O82− (0.15:1) | CuSO4 | H2O | r.t. | 15 | ✓ a | - | - |
| 8 | Ag(I)/S2O82− (0.15:1) | CuSO4 | H2O | reflux | 2 | ✓ a | - | ✓ b |
| 9 d | Ag(I)/S2O82− (0.15:1) | H2O | reflux | 2 | ✓ b | ✓ a | - | |
| 10 d | Ag(I)/S2O82− (0.15:1) | CuSO4 | H2O | reflux | 2 | ✓a | - | - |
| 11 e | Ag(I)/S2O82− (0.15:1) | CuSO4 | MeOH | reflux | 16 | - | - | - |
| 12 | Ag(I)/S2O82− (0.15:1) | CuSO4 | ACN | reflux | 6 | - | ✓ b | 9 |
| 13 | Ag(I)/S2O82− (0.15:1) | CuSO4/TsOH (20 mol%) | H2O | reflux | 16 | - | - | ✓ a |
| 14 | Ag(I)/S2O82− (0.15:1) | CuSO4/Amberlite 15 | H2O | 60 °C | 16 | - | - | 56 c |
| 15 | Ag(I)/S2O82− (0.15:1) | CuSO4/Amberlite 15 | H2O | 60 °C | 24 | - | - | 50 c |
| 16 | Ag(I)/S2O82− (0.15:1) | CuSO4/Amberlite 15 | H2O | 60 °C | 48 | - | - | 33 c |
| 17 | Ag(I)/S2O82− (0.15:1) | CuSO4/Amberlite 15 | H2O | 60 °C | 72 | - | - | 22 c |
| 18 | Ag(I)/S2O82− (0.15:1) | CuSO4/Amberlite 15 | H2O | 60 °C | 6 | - | - | 18 c |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rippel, R.; Pinheiro, L.; Lopes, M.; Lourenço, A.; Ferreira, L.M.; Branco, P.S. Synthetic Approaches to a Challenging and Unusual Structure—An Amino-Pyrrolidine Guanine Core. Molecules 2020, 25, 797. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25040797
Rippel R, Pinheiro L, Lopes M, Lourenço A, Ferreira LM, Branco PS. Synthetic Approaches to a Challenging and Unusual Structure—An Amino-Pyrrolidine Guanine Core. Molecules. 2020; 25(4):797. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25040797
Chicago/Turabian StyleRippel, Rafael, Luís Pinheiro, Mónica Lopes, Ana Lourenço, Luísa M. Ferreira, and Paula S. Branco. 2020. "Synthetic Approaches to a Challenging and Unusual Structure—An Amino-Pyrrolidine Guanine Core" Molecules 25, no. 4: 797. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25040797