
Unveiling the Different Chemical Reactivity of Diphenyl Nitrilimine and Phenyl Nitrile Oxide in [3+2] Cycloaddition Reactions with (R)-Carvone through the Molecular Electron Density Theory

 ,
,  ,
,
Abstract
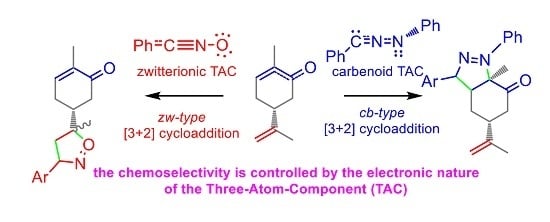
:
1. Introduction
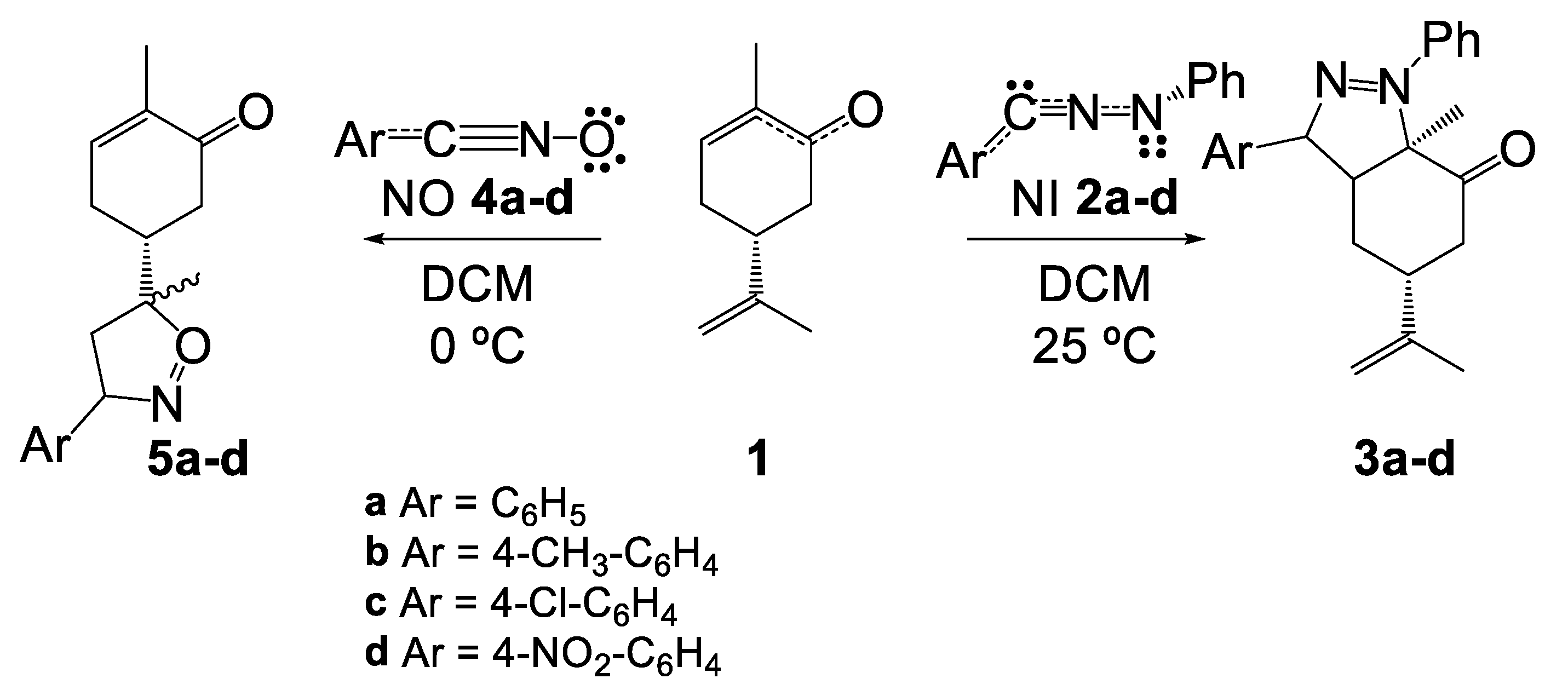
2. Results and Discussion
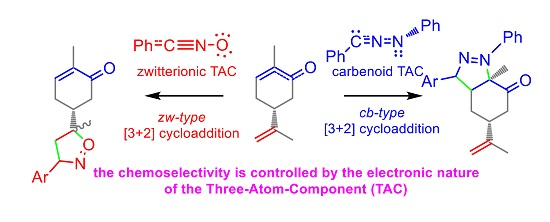
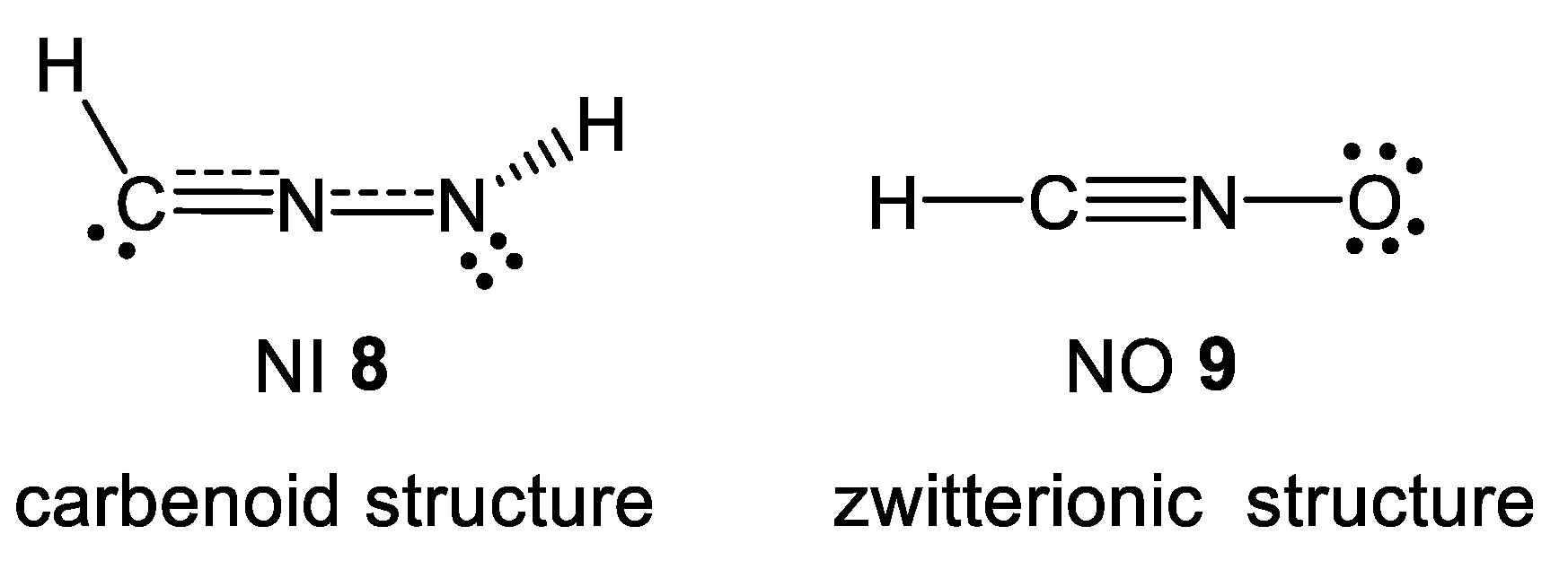
2.1. Topological Analysis of the Electron Localisation Function (ELF) and Natural Population Analysis (NPA) of Diphenyl-NI 2a, Phenyl-NO 4a and (R)-Carvone 1
2.2. Analysis of the Conceptual Density Functional (CDFT) Reactivity Indices of the Reagents
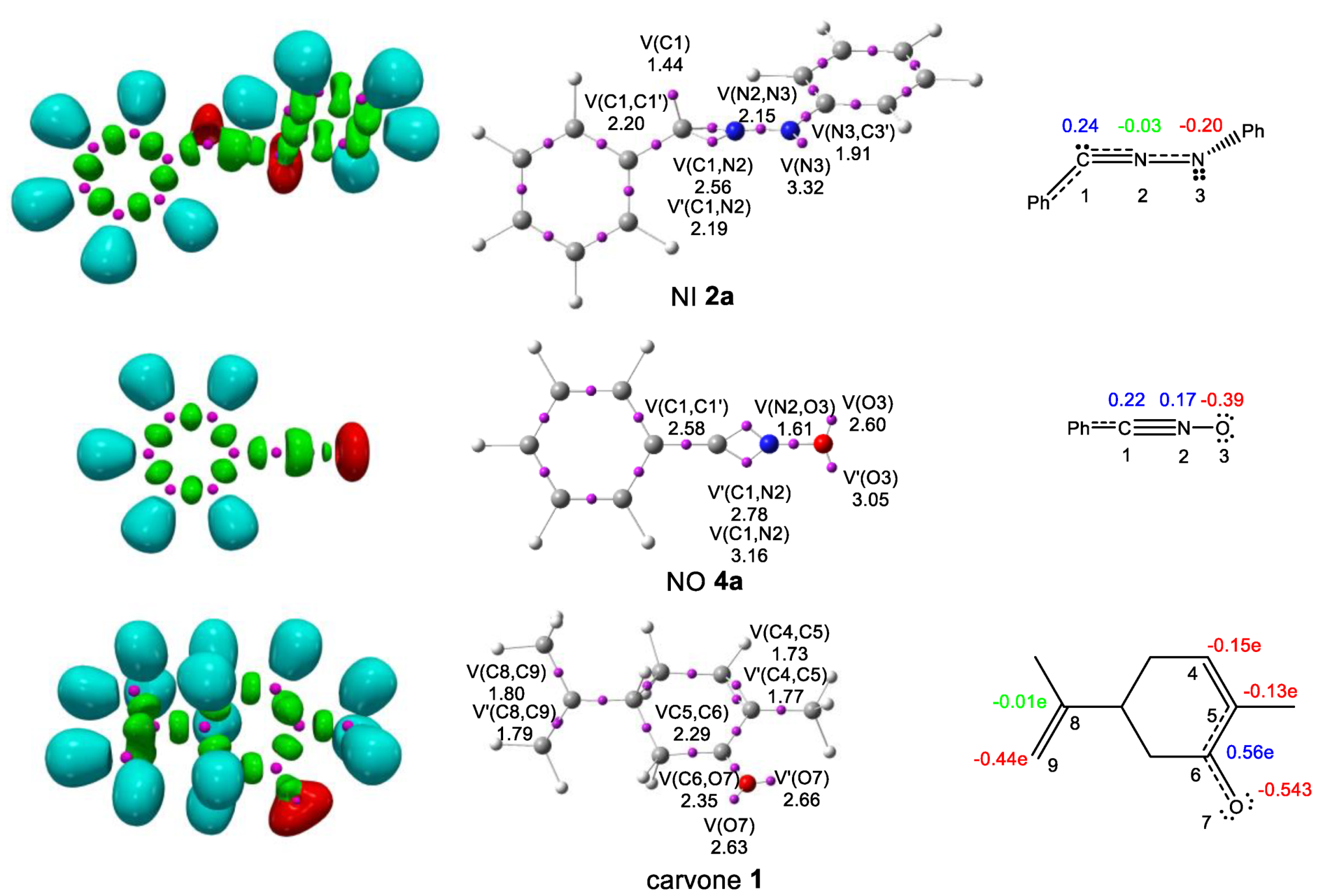
2.3. Study of the Reaction Paths Associated with the 32CA Reactions of Diphenyl NI 2a and Phenyl NO 4a with (R)-Carvone 1
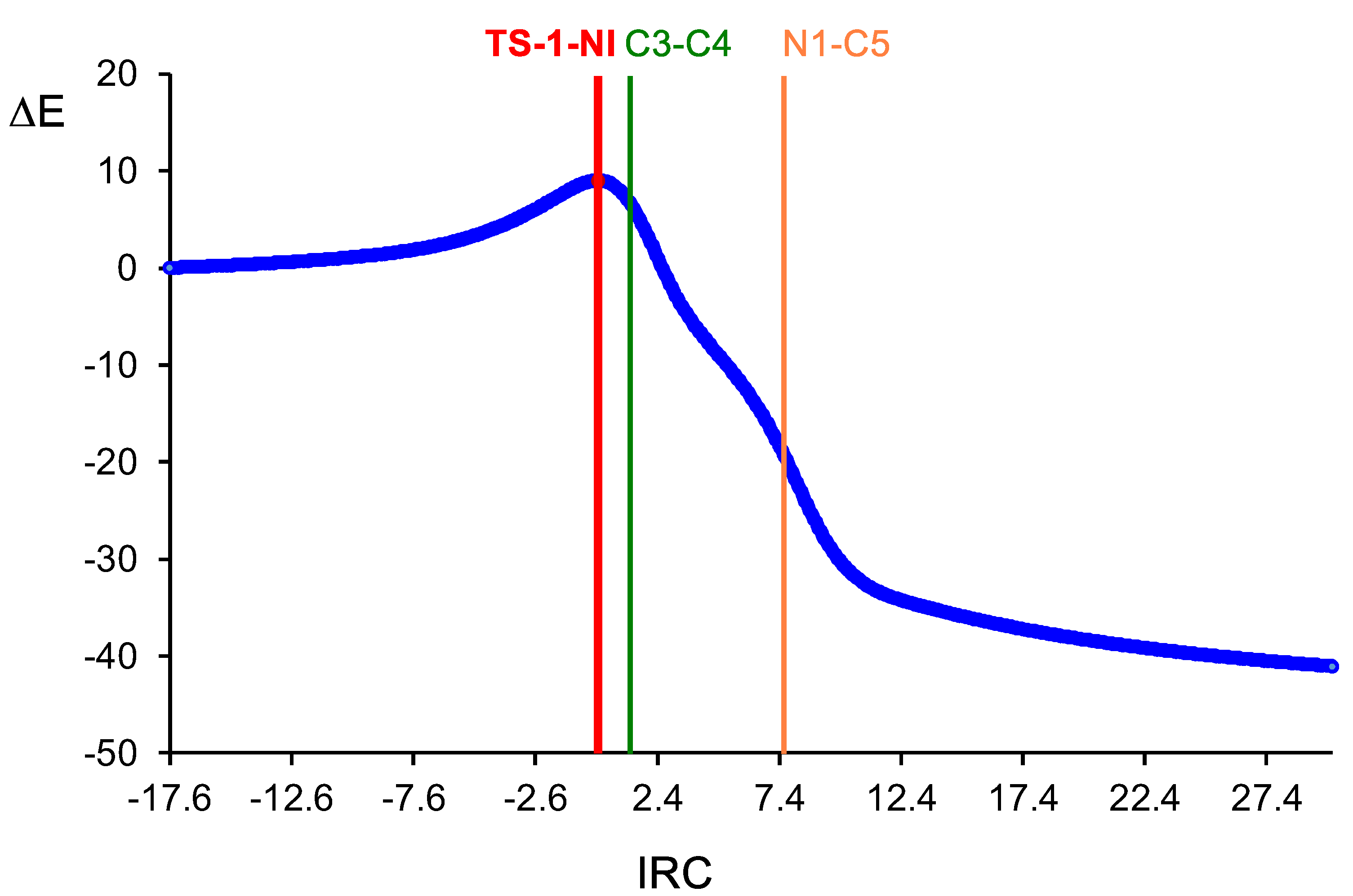
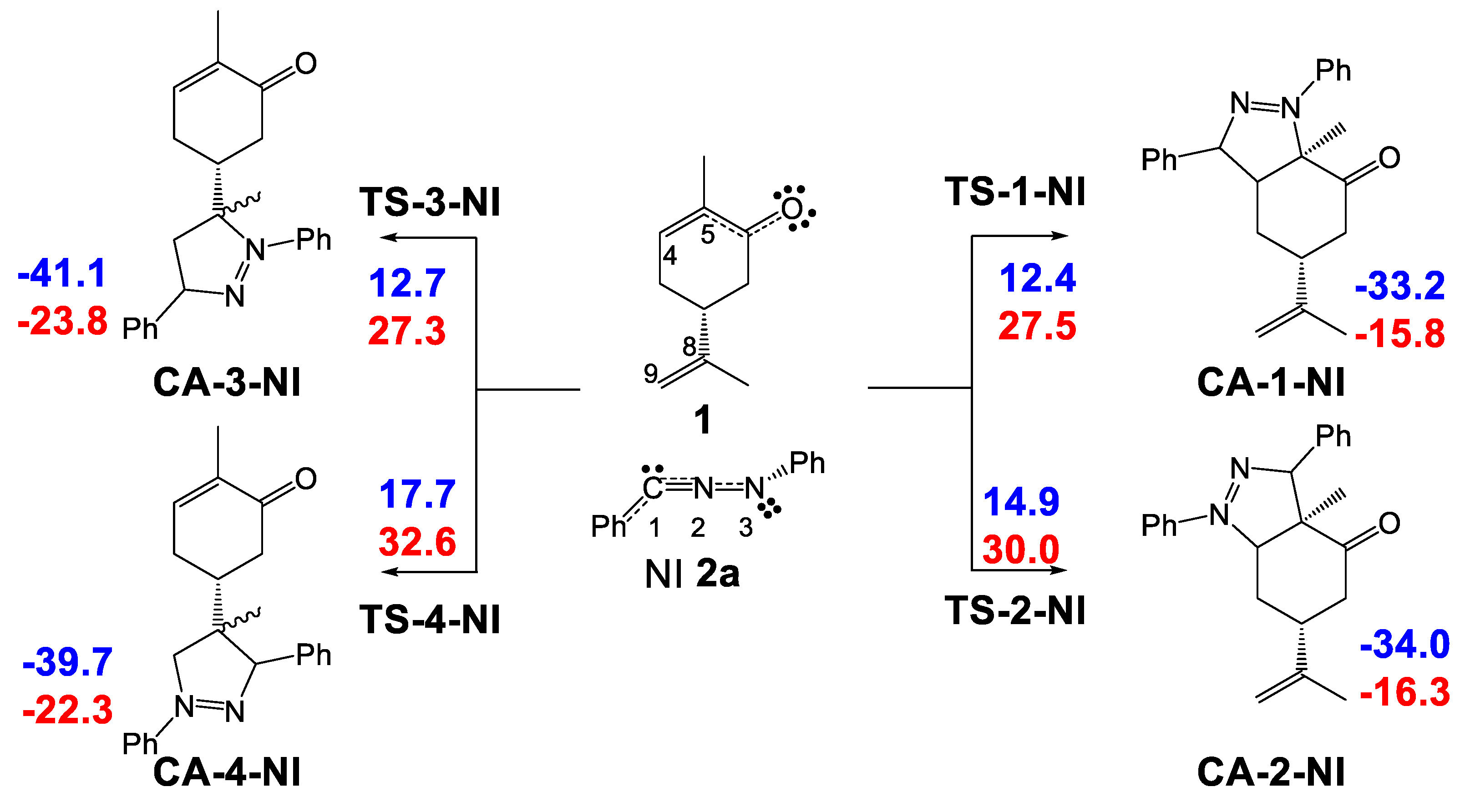
2.3.1. Study of the 32CA Reactions of Diphenyl NI 2a with (R)-Carvone 1
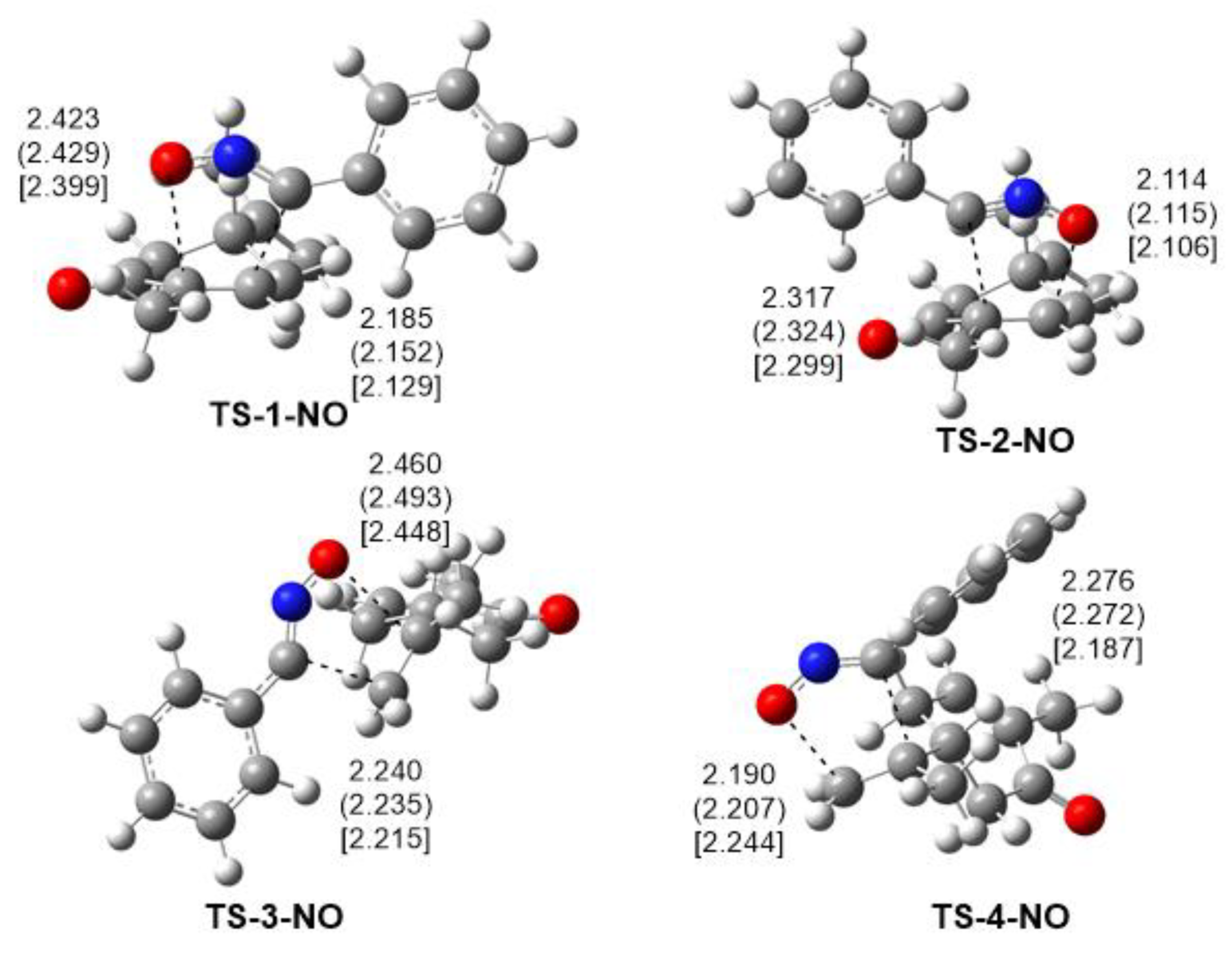
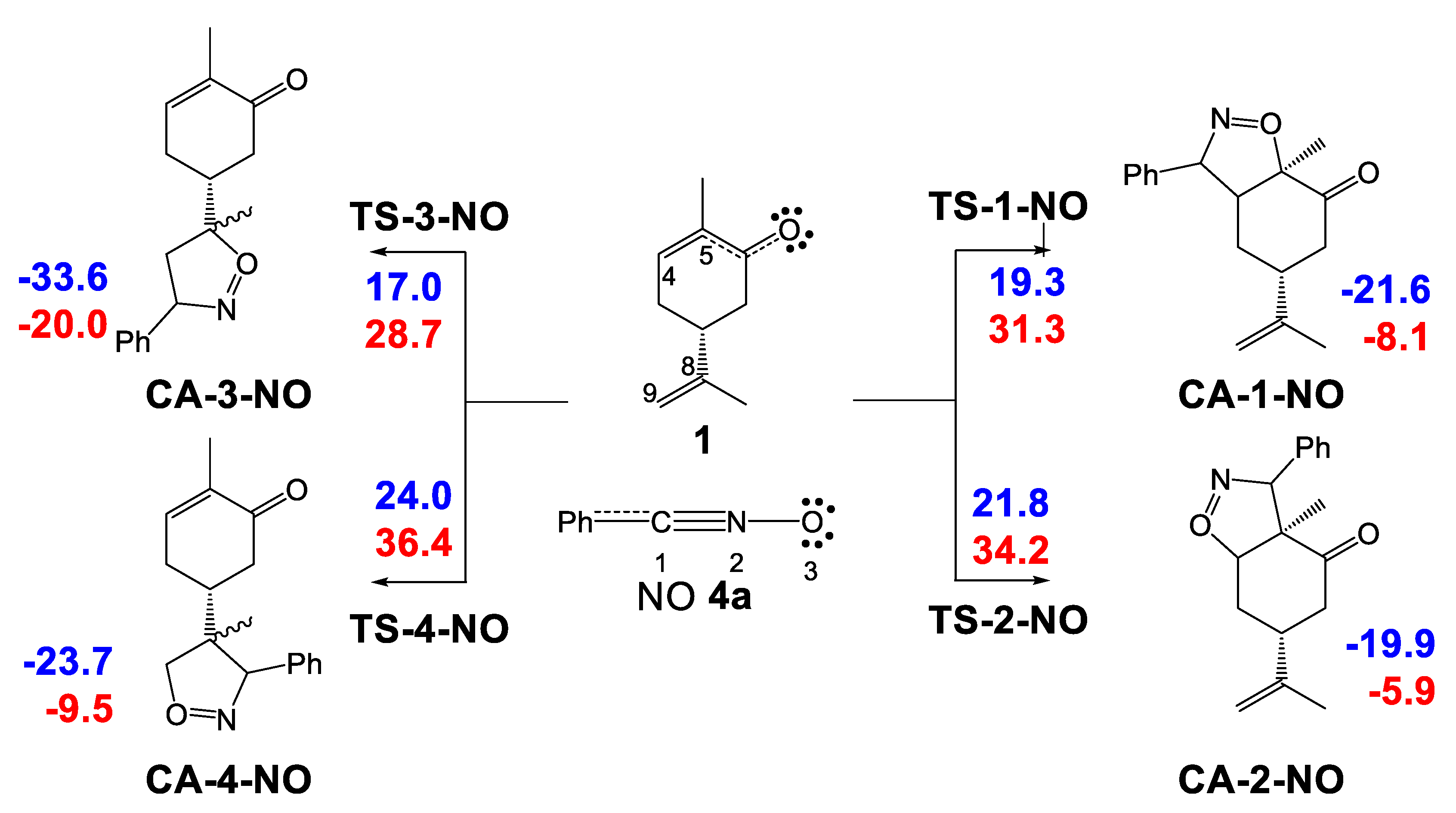
2.3.2. Study of the 32CA Reactions of Phenyl NO 4a with (R)-Carvone 1
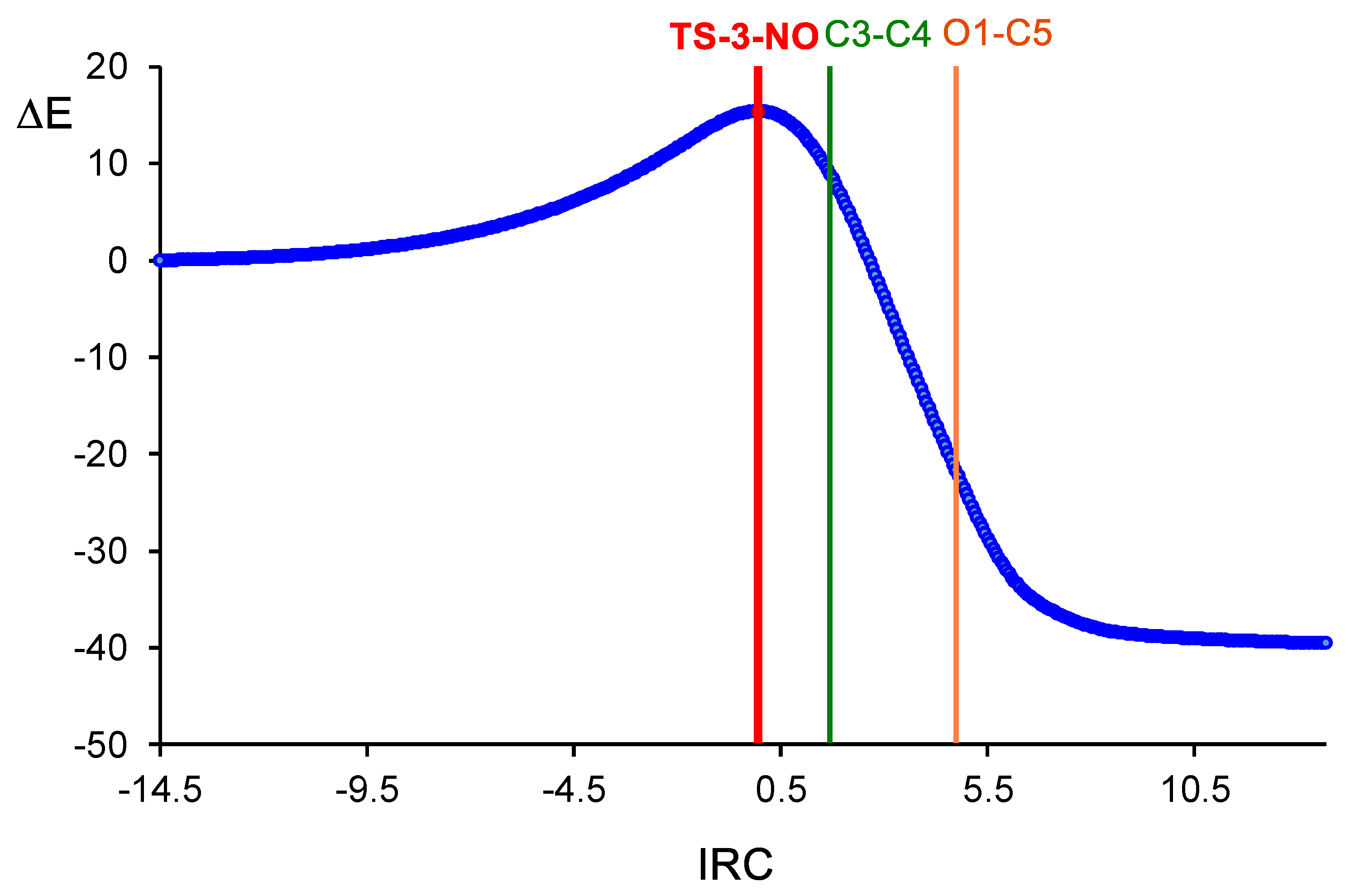
2.4. ELF Topological Analysis of the C−C and C−N Bond Formation along the Most Favourable Reaction Paths Associated with the 32CA Reaction of Diphenyl NI 2a and Phenyl NO 4a with (R)-Carvone 1
2.4.1. ELF Topological Analysis of the C−C and C−N Bond Formation along the Most Favourable Reaction Path Associated with the 32CA Reaction of Diphenyl NI 2a with (R)-Carvone 1
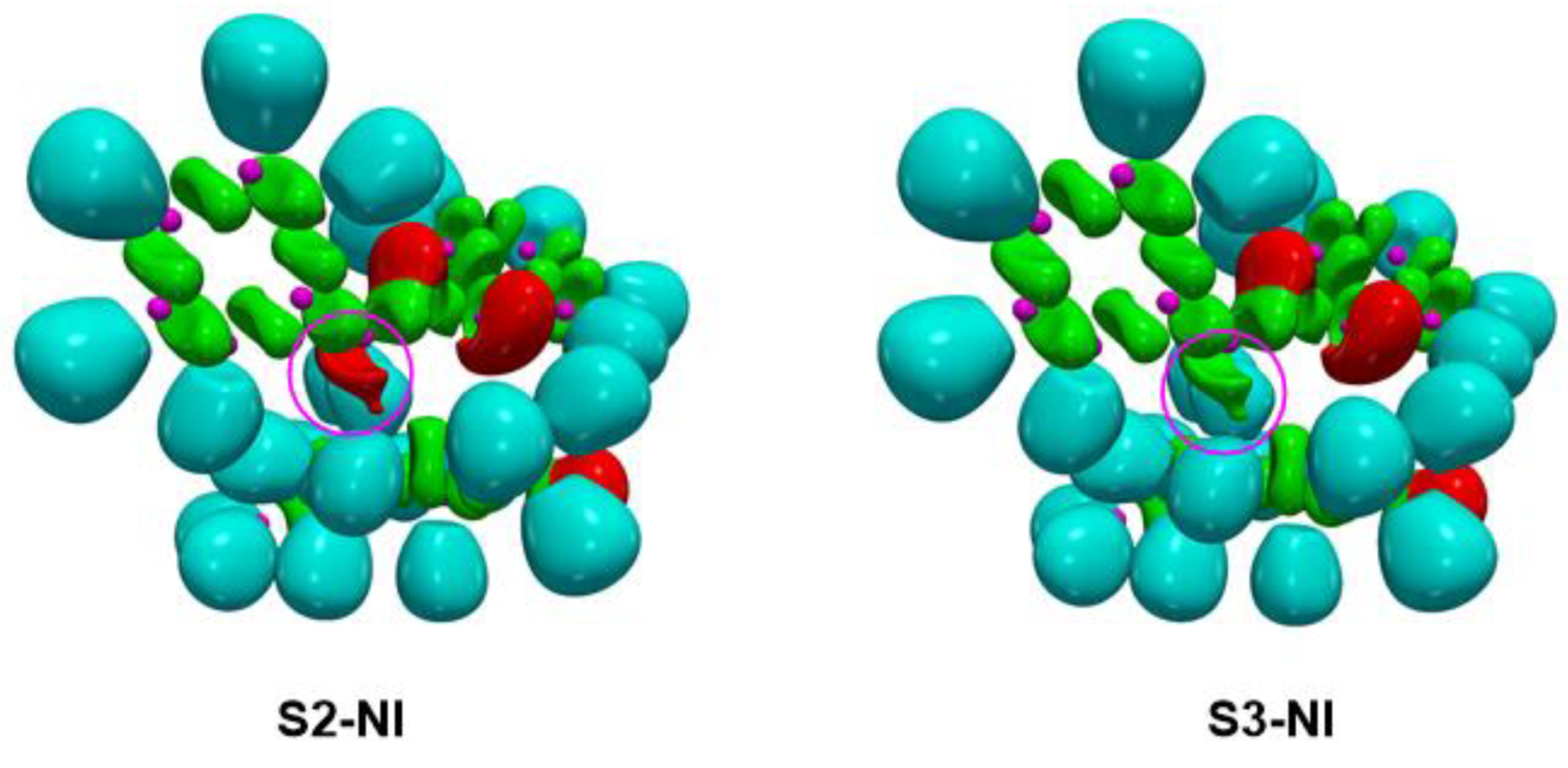
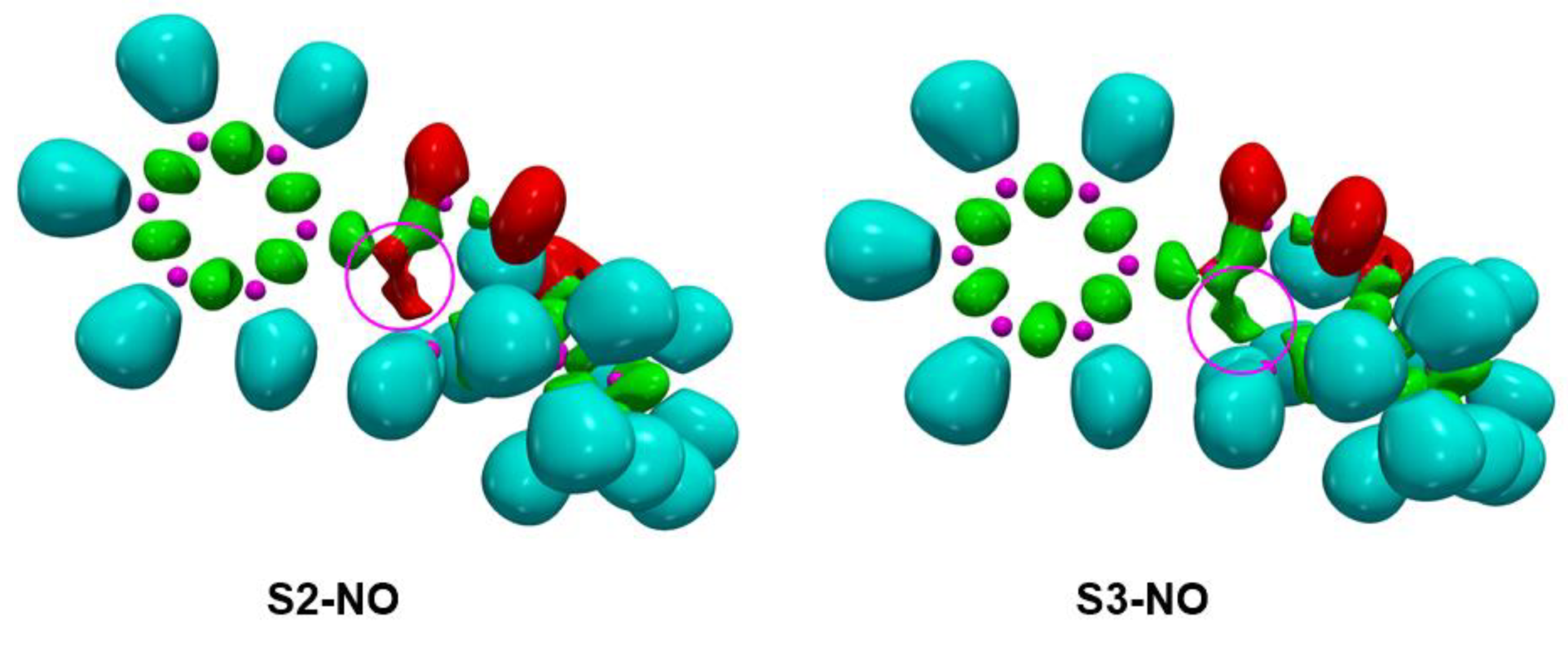
2.4.2. ELF Topological Analysis of the C−C and C−O Bond Formation along the Most Favourable Reaction Paths Associated with the 32CA Reaction of Phenyl NO 4a with (R)-Carvone 1
3. Conclusions
4. Computational Methods
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Al-Sehemi, A.; Bondock, S.; Ammar, Y.A. Transformations of naproxen into pyrazolecarboxamides: Search for potent anti-inflammatory, analgesic and ulcerogenic agents. Med. Chem. Res. 2014, 23, 827–838. [Google Scholar] [CrossRef]
- Airody, V.A.; Karabasanagouda, T.; Girisha, M. Synthesis of some new pyrazolines and isoxazoles carrying 4-methylthiophenyl moiety as potential analgesic and anti-inflammatory agents. Indian J. Chem. 2009, 48B, 430–437. [Google Scholar]
- Krishna, V.C.; Raja, S. Isoxazole–a potent pharmacophore. Int. J. Pharm. Pharm. Sci. 2017, 9, 13–24. [Google Scholar]
- Alegaon, S.G.; Alagawadi, K.R.; Garg, M.K.; Dushyant, K.; Vinod, D. 1,3,4-Trisubstituted pyrazole analogues as promising anti-inflammatory agents. Bioorg. Chem. 2014, 54, 51–59. [Google Scholar] [CrossRef]
- Lincy, J.; Mathew, G. Evalutation of in vivo and in vitro anti-inflamatory activity of novel isoxazole series. Eur. Int. J. Sci. Tech. 2016, 5, 35–42. [Google Scholar]
- Umesha, B.; Basavaraju, Y.B. Synthesis and characterization of novel benzo[d][1,3]dioxole gathered pyrazole derivatives and their antimicrobial evaluation. Med. Chem. Res. 2014, 23, 3744–3751. [Google Scholar] [CrossRef]
- Sagar, P.; Vilasrao, K.; Ramesh, B.; Sachin, S.M.; Vijay, P. Synthesis & Evaluation of isoxazole for their antimicrobial activity. Int. J. Comp. Adv. Pharmacol. 2017, 2, 19–26. [Google Scholar]
- Nagamallu, R.; Srinivasan, B.; Ningappa, M.B.; Kariyappa, A.K. Synthesis of novel coumarin appended bis(formylpyrazole) derivatives: Studies on their antimicrobial and antioxidant activities. Bioorg. Med. Chem. Lett. 2016, 26, 690–694. [Google Scholar] [CrossRef]
- Ahmad, A.; Ahmad, A.; Varsheney, H.; Rauf, A.; Rehan, M.; Subbarao, N.; Khan, A.U. Designing and synthesis of novel antimicrobial heterocyclic analogs of fatty acids. Eur. J. Med. Chem. 2013, 70, 887–900. [Google Scholar] [CrossRef]
- Ren, J.; Wang, S.; Ni, H.; Yao, R.; Liao, C.; Ruan, B. Synthesis, Characterization and Antitumor Activity of Novel Ferrocene-Based Amides Bearing Pyrazolyl Moiety. J. Inorg. Organomet. Polym. 2015, 25, 419–426. [Google Scholar] [CrossRef]
- Hamama, W.S.; Ibrahim, M.E.; Zoorob, H.H. Synthesis and Biological Evaluation of Some Novel Isoxazole Derivatives. J. Heterocycl. Chem. 2017, 54, 341–346. [Google Scholar] [CrossRef]
- Fioravanti, R.; Desideri, N.; Biava, M.; Droghini, P.; Atzori, E.M.; Ibba, C.; Collu, G.; Sanna, G.; Delogu, I.; Loddo, R. N-((1,3-Diphenyl-1H-pyrazol-4-yl)methyl)anilines: A novel class of anti-RSV agents. Bioorg. Med. Chem. Lett. 2015, 25, 2401–2404. [Google Scholar] [CrossRef]
- Li, F.; Hu, Y.; Wang, Y.; Ma, C.; Wang, J. Expeditious Lead Optimization of Isoxazole-Containing Influenza A Virus M2-S31N Inhibitors Using the Suzuki–Miyaura Cross-Coupling Reaction. J. Med. Chem. 2017, 60, 1580–1590. [Google Scholar] [CrossRef] [Green Version]
- Houk, K.N.; Sims, J.; Watts, C.R.; Luskus, L.J. Origin of reactivity, regioselectivity, and periselectivity in 1,3-dipolar cycloadditions. J. Am. Chem. Soc. 1973, 95, 7301–7315. [Google Scholar] [CrossRef]
- Rai, N.S.; Kalluraya, B.; Lingappa, B.; Shenoy, S.; Puranic, V.G. Convenient access to 1,3,4-trisubstituted pyrazoles carrying 5-nitrothiophene moiety via 1,3-dipolar cycloaddition of sydnones with acetylenic ketones and their antimicrobial evaluation. Eur. J. Med. Chem. 2008, 43, 1715–1720. [Google Scholar] [CrossRef]
- Oubella, A.; Ait Itto, M.Y.; Auhmani, A.; Riahi, A.; Robert, A.; Daran, J.-C.; Morjani, H.; Parish, C.A.; Esseffar, M. Diastereoselective synthesis and cytotoxic evaluation of new isoxazoles and pyrazoles with monoterpenic skeleton. J. Mol. Struct. 2019, 1198, 126924. [Google Scholar] [CrossRef]
- Ess, D.H.; Houk, K.N. Distortion/Interaction Energy Control of 1,3-Dipolar Cycloaddition Reactivity. J. Am. Chem. Soc. 2007, 129, 10646–10647. [Google Scholar] [CrossRef]
- Ess, D.H.; Houk, K.N. Theory of 1,3-Dipolar Cycloadditions: Distortion/Interaction and Frontier Molecular Orbital Models. J. Am. Chem. Soc. 2008, 130, 10187–10198. [Google Scholar] [CrossRef]
- Bickelhaupt, F.M. Understanding reactivity with Kohn–Sham molecular orbital theory: E2–SN2 mechanistic spectrum and other concepts. J. Comput. Chem. 1999, 20, 114–128. [Google Scholar] [CrossRef]
- Domingo, L.R. Molecular electron density theory: A modern view of reactivity in organic chemistry. Molecules 2016, 21, 1319. [Google Scholar] [CrossRef]
- Ríos-Gutiérrez, M.; Domingo, L.R. Unravelling the mysteries of the [3+2] cycloaddition reactions. Eur. J. Org. Chem. 2019, 267–282. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. A Molecular electron density theory study of the reactivity and selectivities in [3 + 2] cycloaddition reactions of C,N-dialkyl nitrones with ethylene derivatives. J. Org. Chem. 2018, 83, 2182–2197. [Google Scholar] [CrossRef]
- Ríos-Gutiérrez, M.; Domingo, L.R. The Carbenoid-Type Reactivity of Simplest Nitrile Imine from a Molecular Electron Density Theory perspective. Tetrahedron 2019, 75, 1961–1967. [Google Scholar] [CrossRef]
- Ndassa, I.M.; Adjieufack, A.I.; Ketcha, J.M.; Berski, S.; Ríos-Gutiérrez, M.; Domingo, L.R. Understanding the reactivity and regioselectivity of [3+2] cycloaddition reactions between substituted nitrile oxides and methyl acrylate. A molecular electron density theory study. Int. J. Quantum Chem. 2017, 117, 25451. [Google Scholar] [CrossRef]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular-systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Silvi, B.; Savin, A. Classification of chemical bonds based on topological analysis of electron localization functions. Nature 1994, 371, 683–686. [Google Scholar] [CrossRef]
- Huisgen, R. 1,3-Dipolar Cycloadditions. Proc. Chem. Soc. 1961, 357–396. [Google Scholar]
- Geerlings, P.; De Proft, F.; Langenaeker, W. Conceptual density functional theory. Chem. Rev. 2003, 103, 1793–1873. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the conceptual density functional indices to organic chemistry reactivity. Molecules 2016, 21, 748. [Google Scholar] [CrossRef] [Green Version]
- Parr, R.G.; Pearson, R.G. Absolute hardness: Companion parameter to absolute electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Parr, R.G.; Szentpaly, L.V.; Liu, S. Electrophilicity index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Domingo, L.R.; Chamorro, E.; Pérez, P. Understanding the reactivity of captodative ethylenes in polar cycloaddition reactions. A theoretical study. J. Org. Chem. 2008, 73, 4615–4624. [Google Scholar] [CrossRef]
- Domingo, L.R. A new C-C bond formation model based on the quantum chemical topology of electron density. RSC Adv. 2014, 4, 32415–32428. [Google Scholar] [CrossRef] [Green Version]
- Krokidis, X.; Noury, S.; Silvi, B. Characterization of Elementary Chemical Processes by Catastrophe Theory. J. Phys. Chem. A 1997, 101, 7277–7282. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. How does the global electron density transfer diminish activation energies in polar cycloaddition reactions? A Molecular Electron Density Theory study. Tetrahedron 2017, 73, 1718–1724. [Google Scholar] [CrossRef]
- Domingo, L.R.; Sáez, J.A.; Zaragozá, R.J.; Arnó, M. Understanding the Participation of Quadricyclane as NucleophileinPolar [2σ + 2σ + 2π] Cycloadditions toward Electrophilic π Molecules. J. Org. Chem. 2008, 73, 8791–8799. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. The role of exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Hehre, M.J.; Radom, L.; Schleyer, P.v.R.; Pople, J. Ab initio Molecular Orbital Theory; Wiley: New York, NY, USA, 1986. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. Hybrid meta density functional theory methods for thermochemistry, thermochemical kinetics, and noncovalent Interactions: The MPW1B95 and MPWB1K models and comparative assessments for hydrogen bonding and van der Waals interactions. J. Phys. Chem. A 2004, 108, 6908–6918. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinectics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Chai, J.-D.; Head-Gordon, M. Systematic optimization of long-range corrected hybrid density functionals. Chem. Phys. 2008, 128, 084106. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parameterization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar]
- Schlegel, H.B. Optimization of equilibrium geometries and transition structures. J. Comput. Chem. 1982, 3, 214–218. [Google Scholar] [CrossRef]
- Schlegel, H.B. Modern Electronic Structure Theory; Yarkony, D.R., Ed.; World Scientific Publishing: Singapore, 1994. [Google Scholar]
- Fukui, K. Formulation of the reaction coordinate. J. Phys. Chem. 1970, 74, 4161–4163. [Google Scholar] [CrossRef]
- González, C.; Schlegel, H.B. Reaction path following in mass-weighted internal coordinates. J. Phys. Chem. 1990, 94, 5523–5527. [Google Scholar] [CrossRef]
- González, C.; Schlegel, H.B. Improved algorithms for reaction path following: Higher-order implicit algorithms. J. Chem. Phys. 1991, 95, 5853–5860. [Google Scholar] [CrossRef] [Green Version]
- Tomasi, J.; Persico, M. Molecular interactions in solution: And overview of methods based on continuous distributions of the solvent. Chem. Rev. 1994, 94, 2027–2094. [Google Scholar] [CrossRef]
- Simkin, B.Y.; Sheikhet, I.I. Quantum Chemical and Statistical Theory of Solutions—Computational Approach; Ellis Horwood: London, UK, 1995. [Google Scholar]
- Cossi, M.; Barone, V.; Cammi, R.; Tomasi, J. Ab initio study of solvated molecules: A new implementation of the polarizable continuum model. Chem. Phys. Lett. 1996, 255, 327–335. [Google Scholar] [CrossRef]
- Cances, E.; Mennucci, B.; Tomasi, J. A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to isotropic and anisotropic dielectrics. J. Chem. Phys. 1997, 107, 3032–3041. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M.; Tomasi, J. Geometry optimization of molecular structures in solution by the polarizable continuum model. J. Comput. Chem. 1998, 19, 404–417. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A. 03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Noury, S.; Krokidis, X.; Fuster, F.; Silvi, B. Computational tools for the electron localization function topological analysis. Comput. Chem. 1999, 23, 597–604. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView, Version 6.0; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Ahrens, J.; Geveci, B.; Law, C. ParaView: An End-User Tool for Large Data Visualization, Visualization Handbook; Elsevier: Amsterdam, The Netherlands, 2005; ISBN 978-0123875822. [Google Scholar]
- Ayachit, U. The ParaView Guide: A Parallel Visualization Application; Kitware: Clifton Park, NY, USA, 2015; ISBN 978-1930934306. [Google Scholar]
Sample Availability: Samples of the compounds are not available from the authors. |












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| μ | η | ω | N | |
|---|---|---|---|---|
| diphenyl NI 2a | −3.36 | 3.78 | 1.50 | 3.87 |
| phenyl NO 4a | −3.83 | 5.02 | 1.46 | 2.78 |
| (R)-carvone 1 | −3.84 | 5.23 | 1.41 | 2.67 |
| simplest NI 8 | −3.55 | 5.87 | 1.07 | 2.64 |
| simplest NO 9 | −3.40 | 7.94 | 0.73 | 1.75 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ríos-Gutiérrez, M.; Domingo, L.R.; Esseffar, M.; Oubella, A.; Ait Itto, M.Y. Unveiling the Different Chemical Reactivity of Diphenyl Nitrilimine and Phenyl Nitrile Oxide in [3+2] Cycloaddition Reactions with (R)-Carvone through the Molecular Electron Density Theory. Molecules 2020, 25, 1085. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25051085
Ríos-Gutiérrez M, Domingo LR, Esseffar M, Oubella A, Ait Itto MY. Unveiling the Different Chemical Reactivity of Diphenyl Nitrilimine and Phenyl Nitrile Oxide in [3+2] Cycloaddition Reactions with (R)-Carvone through the Molecular Electron Density Theory. Molecules. 2020; 25(5):1085. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25051085
Chicago/Turabian StyleRíos-Gutiérrez, Mar, Luis R. Domingo, M’hamed Esseffar, Ali Oubella, and My Youssef Ait Itto. 2020. "Unveiling the Different Chemical Reactivity of Diphenyl Nitrilimine and Phenyl Nitrile Oxide in [3+2] Cycloaddition Reactions with (R)-Carvone through the Molecular Electron Density Theory" Molecules 25, no. 5: 1085. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25051085