The Influence of Halogenated Hypercarbon on Crystal Packing in the Series of 1-Ph-2-X-1,2-dicarba-closo-dodecaboranes (X = F, Cl, Br, I) †

 , and
, and
Abstract
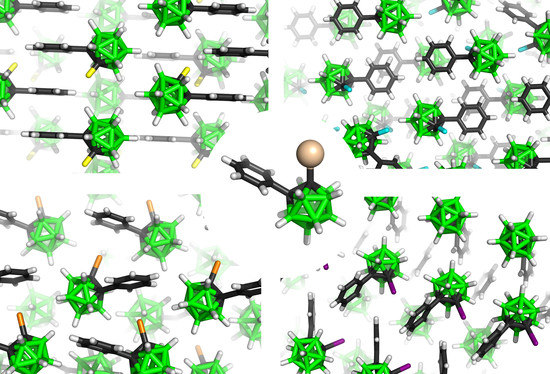
:
1. Introduction
2. Results and Discussion
2.1. Syntheses
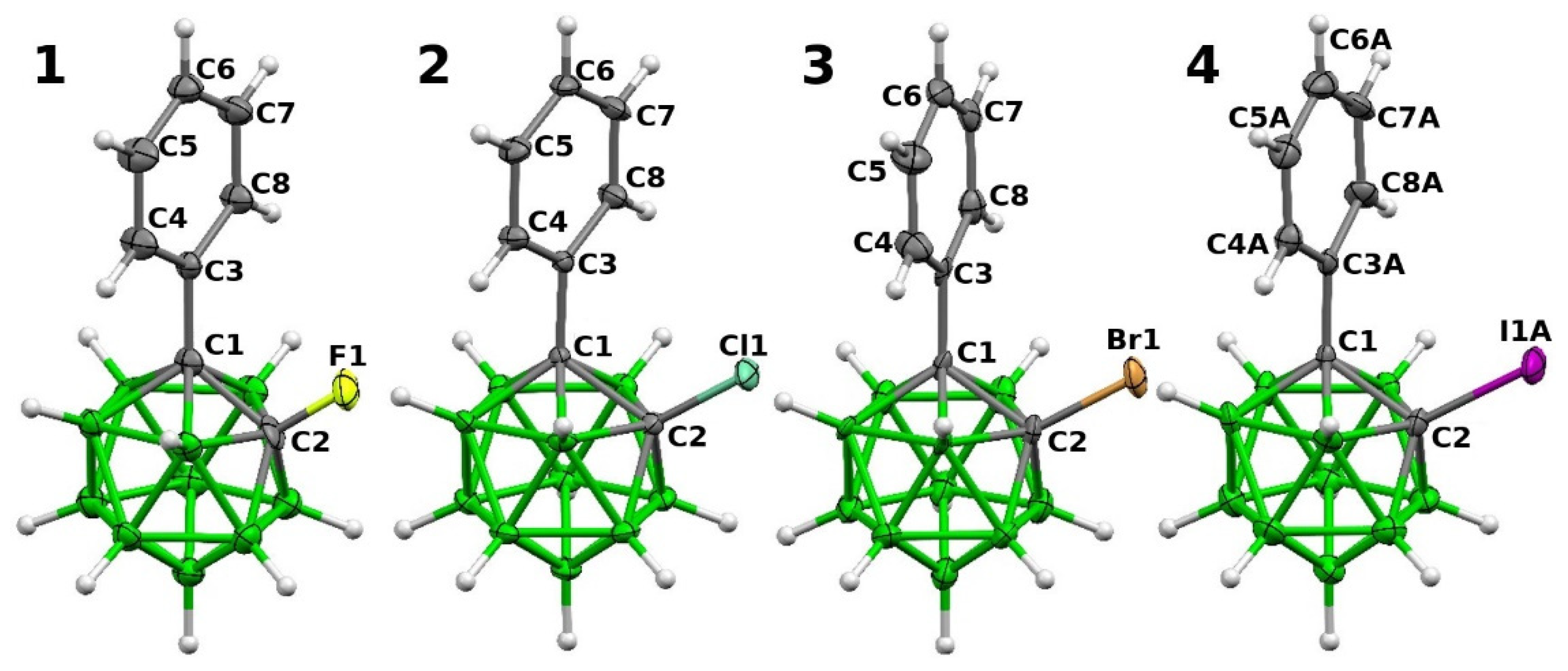
2.2. Structural Characterization
2.3. Computations
2.3.1. Heat of Formation (ΔHf298)
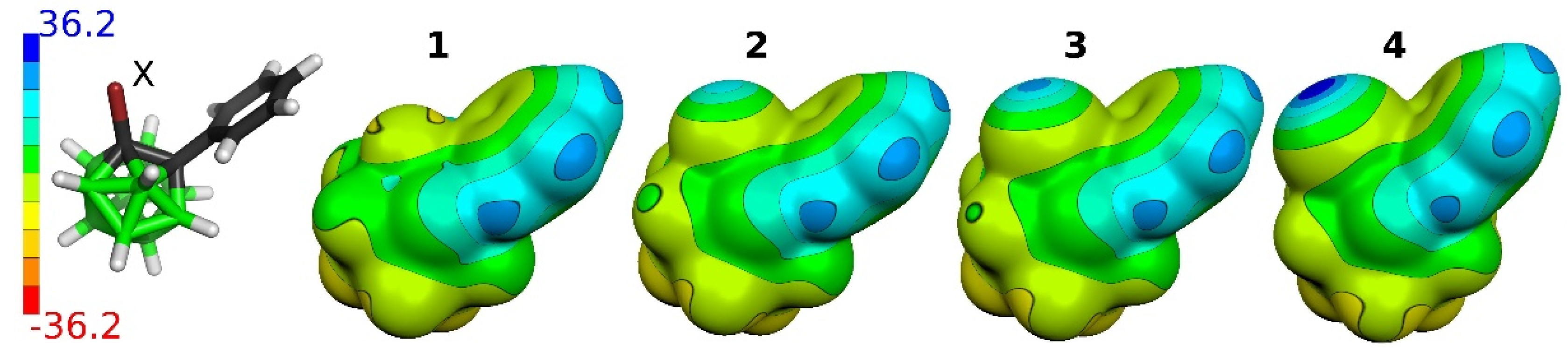
2.3.2. Electrostatic Potential (ESP)
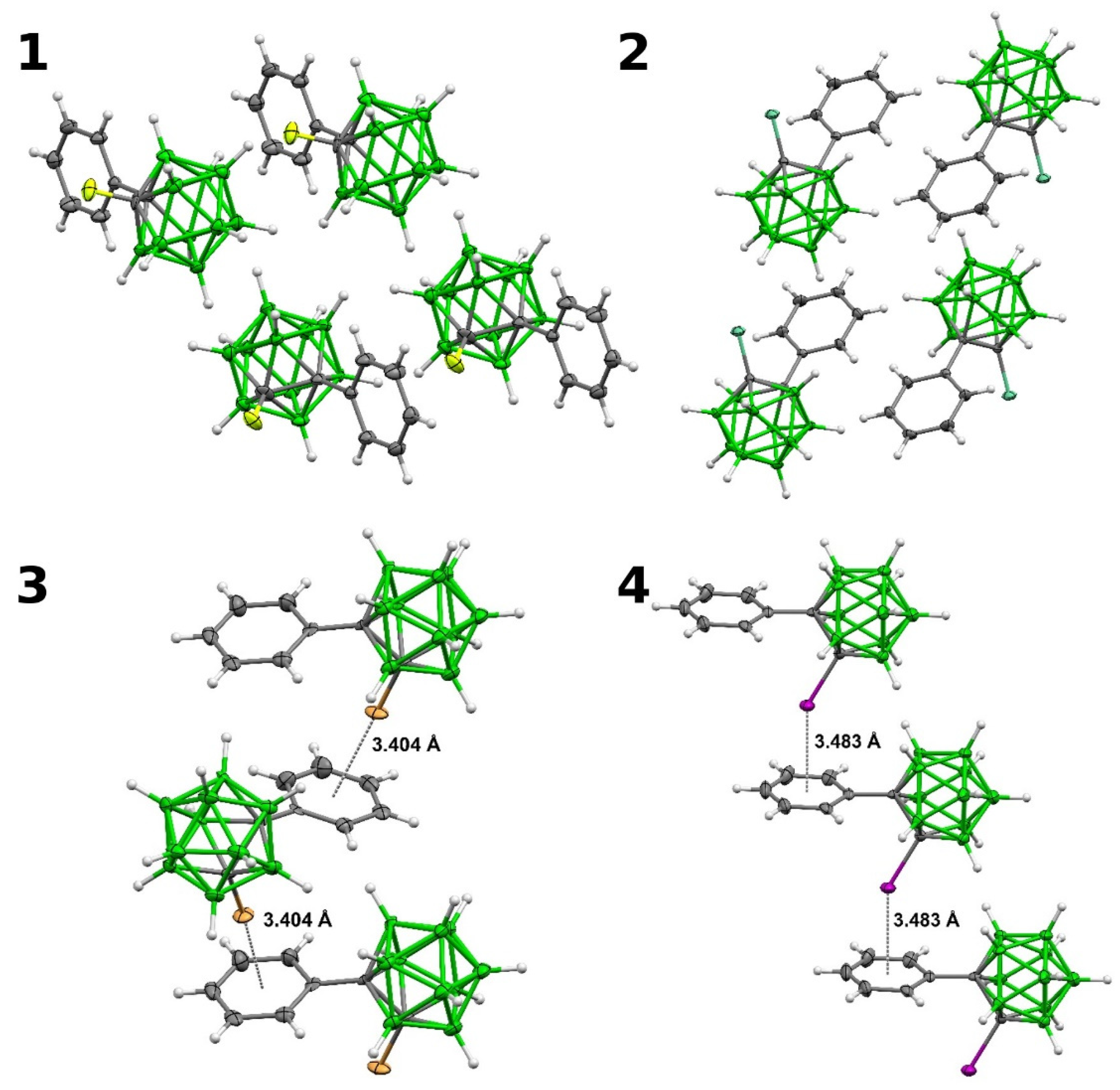
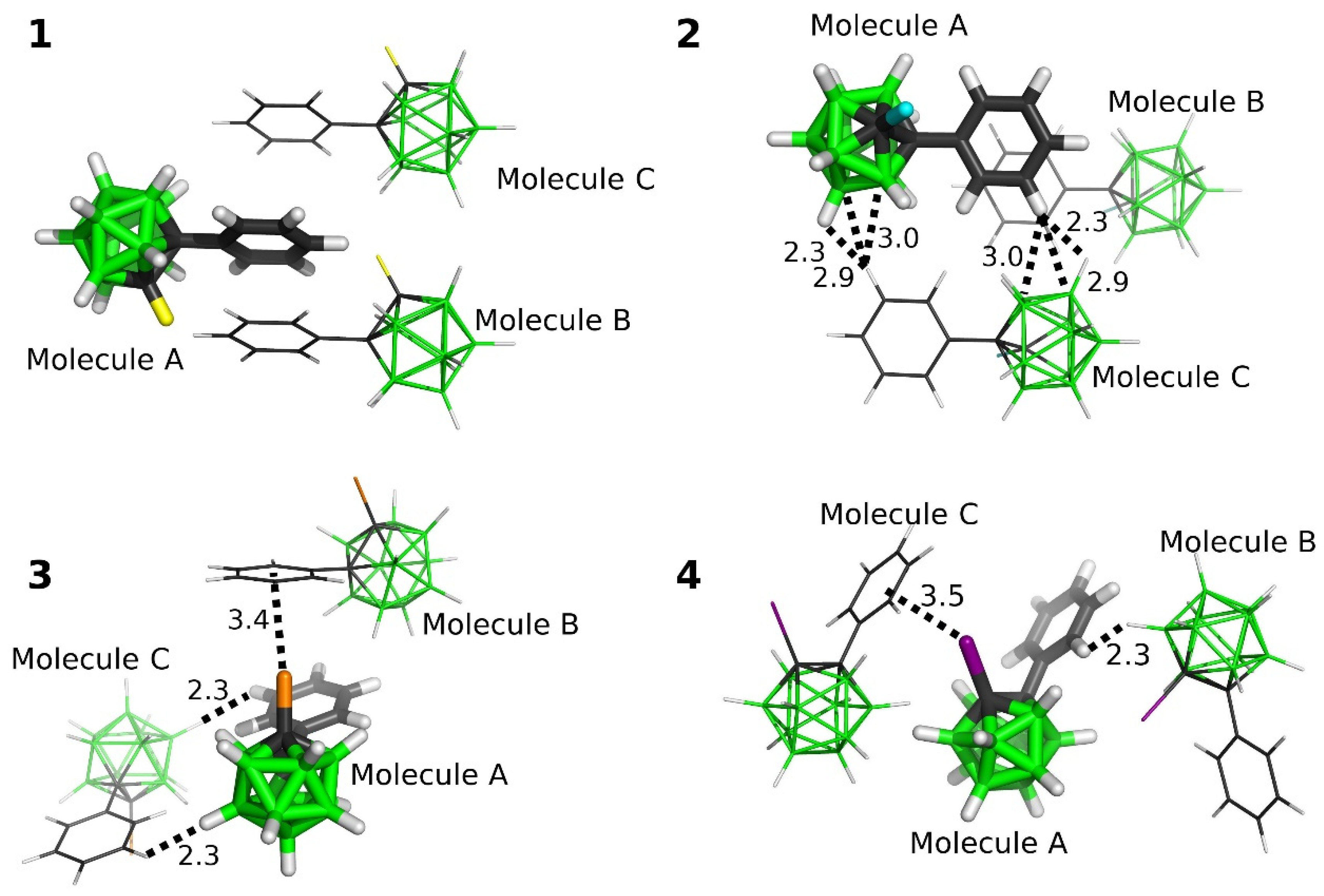
2.3.3. Interactions in the Single Crystals of 1–4
2.4. Cambridge Structural Database (CSD) search
3. Materials and Methods
3.1. X-Ray Crystallography
3.2. Computations
3.2.1. Electrostatic Potential (ESP)
3.2.2. Heat of Formation (ΔHf298)
3.2.3. Interaction Energy
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hnyk, D.; Všetečka, V.; Drož, L.; Exner, O. Charge Distribution within 1,2-Dicarba-closo-dodecaborane: Dipole Moments of its Phenyl Derivatives. Collect. Czech. Chem. Commun. 2001, 66, 1375–1379. [Google Scholar] [CrossRef]
- Brain, P.T.; Cowie, J.; Donohoe, D.J.; Hnyk, D.; Rankin, D.W.H.; Reed, D.; Reid, B.D.; Robertson, H.E.; Welch, A.J. 1-Phenyl-1,2-dicarba-closo-dodecaborane, 1-Ph-1,2-closo-C2B10H11. Synthesis, Characterization, and Structure in the Gas Phase by Electron Diffraction, in the Crystalline Phase at 199 K by X-ray Diffraction, and by ab Initio Computations. Inorg. Chem. 1996, 35, 1701–1708. [Google Scholar] [PubMed]
- Hnyk, D.; Všetečka, V.; Drož, L. Charge Distribution within 1-Ph-2-X-1,2-dicarba-closo-dodecaboranes, (X = F, Cl, Br, I): A Dipole Moment and Computational Study. J. Mol. Struct. 2010, 978, 246–249. [Google Scholar] [CrossRef]
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen Bonding: The σ-Hole. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef]
- Kolář, M.; Hostaš, J.; Hobza, P. The strength and directionality of a halogen bond are co-determined by the magnitude and size of the σ-hole. Phys. Chem. Chem. Phys. 2014, 16, 23279–23280. [Google Scholar] [CrossRef]
- Gamez, P. The anion-π interaction: Naissance and establishment of peculiar supramolecular bond. Inorg. Chem. Front. 2014, 1, 35–43. [Google Scholar] [CrossRef]
- Prasanna, M.D.; Guru Rown, T.N. C-halogen•••π interactions and their influence on molecular conformation and crystal packing: A database study. Cryst. Eng. 2000, 3, 135–154. [Google Scholar] [CrossRef]
- Youn, I.S.; Kim, D.Y.; Cho, W.J.; Madridejos, J.M.L.; Lee, H.M.; Kolaski, M.; Lee, J.; Baig, C. Halogen-π Interactions between Benzene and X2/CX4 (X = Cl, Br): Assessment of Various Density Functionals with Respect to CCSD(T). J. Phys Chem. A 2016, 120, 9305–9314. [Google Scholar] [CrossRef]
- Sun, H.; Horatscheck, A.; Martos, V.; Bartetzko, M.; Uhrig, U.; Lentz, D.; Schmieder, P.; Nazare, M. Direct Experimental Evidence for Halogen-Aryl π Interactions in Solution from Molecular Torsion Balances. Angew. Chem. Int. Ed. 2017, 56, 6454–6458. [Google Scholar] [CrossRef] [PubMed]
- Fanfrlík, J.; Přáda, A.; Padělková, Z.; Pecina, A.; Macháček, J.; Lepšík, M.; Holub, J.; Růžička, A.; Hnyk, D.; Hobza, P. The Dominant Role of Chalcogen Bonding in the Crystal Packing of 2D/3D Aromatics. Angew. Chem. Int. Ed. 2014, 53, 10139–10142. [Google Scholar] [CrossRef] [PubMed]
- Macháček, J.; Plešek, J.; Holub, J.; Hnyk, D.; Všetečka, V.; Císařová, I.; Kaupp, M.; Štíbr, B. New Route to 1-Thia-closo-dodecaborane(11), closo-1-SB11H11, and its Halogenation Reactions. The Effect of the Halogen on the Dipole Moments and the NMR Spectra and the Importance of Spin−Orbit Coupling for the 11B Chemical Shifts. Dalton Trans. 2006, 1024–1029. [Google Scholar]
- Fanfrlík, J.; Holub, J.; Růžičková, Z.; Řezáč, J.; Lane, P.D.; Wann, D.A.; Hnyk, D.; Růžička, A.; Hobza, P. Competition between Halogen, Hydrogen and Dihydrogen Bonding in Brominated Carboranes. ChemPhysChem 2016, 17, 3373–3376. [Google Scholar]
- McGrath, T.D.; Welch, A.J. Steric Effects in Heteroboranes. IV. 1-Ph-2-Br-1,2-closo-C2B10H10. Acta Cryst. C51 1995, 649–651. [Google Scholar] [CrossRef]
- Holub, J.; Vrána, J.; Růžička, A.; Růžičková, Z.; Fanfrlík, J.; Hnyk, D. Thiaboranes on Both Sides of the Icosahedral Barrier: Retaining and Breaking the Barrier with Carbon Functionalities. ChemPlusChem 2019, 84, 822–827. [Google Scholar] [CrossRef] [PubMed]
- Hnyk, D.; Wann, D.A.; Holub, J.; Samdal, S.; Rankin, D.W.H. Why is the Antipodal Effect in closo-1-SB9H9 so Large? A Possible Explanation Based on the Geometry from the Concerted Use of Gas Electron Diffraction and Computational Methods. Dalton Trans. 2011, 40, 5734–5737. [Google Scholar] [PubMed] [Green Version]
- Hnyk, D.; Rankin, D.W.H.; Robertson, H.E.; Hofmann, M.; Schleyer, P.v.R.; Bühl, M. Molecular Structure of 1,2-Dicarba-closo-decaborane(10) As Studied by the Concerted Use of Electron Diffraction and ab Initio Calculations. Inorg. Chem. 1994, 33, 4781–4786. [Google Scholar] [CrossRef]
- Aubert, E.; Espinossa, E.; Nicolas, I.; Jeanin, O.; Fourmigue, M. Toward a reverse hierarchy of halogen bonding between bromine and iodine. Faraday Discuss. 2017, 203, 389–406. [Google Scholar] [CrossRef] [Green Version]
- Ciancaleoni, G.; Belpassi, L. Disentanglement of orthogonal hydrogen and halogen bonds via natural orbital for chemical valence: A charge displacement analysis. J. Comput. Chem. 2020, 1–9. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Cryst. 2016, B72, 171–179. [Google Scholar] [CrossRef]
- Batsanov, S.S. Van der Waals Radii of Elements. Inorganic Materials 2001, 37, 871–885. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Popov, A.A.; Miller, S.M.; Anderson, O.P.; Strauss, S.H. Synthesis and structure of Ag(1-Me-12-SiPh3-CB11F10): Selective F12 substitution in 1-Me-CB11F11− and the first Ag(arene)4+ tetrahedron. Inorg. Chem. 2007, 46, 8505–8507. [Google Scholar] [CrossRef]
- Ivanova, S.M.; Ivanov, S.V.; Miller, S.M.; Anderson, O.P.; Solntsev, K.A.; Strauss, S.H. Mono-, Di-, Tri-, and Tetracarbonyls of Copper(I), Including the Structures of Cu(CO)2(1-Bn-CB11F11) and [Cu(CO)4][1-Et-CB11F11]. Inorg. Chem. 1999, 38, 3756–3757. [Google Scholar] [CrossRef]
- Romanato, P.; Duttwyler, S.; Linden, A.; Baldridge, K.K.; Siegel, J.S. Competition between π-arene and lone-pair halogen coordination of silylium ions? J. Am. Chem. Soc. 2011, 133, 11844–11846. [Google Scholar] [CrossRef] [PubMed]
- Shoji, Y.; Tanaka, N.; Mikami, K.; Uchiyama, M.; Fukushima, T. A two-coordinate boron cation featuring C-B+-C bonding. Nat. Chem. 2014, 6, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Geis, V.; Guttsche, K.; Knapp, C.; Scherer, H.; Uzun, R. Synthesis and characterization of synthetically useful salts of the weakly-coordinating dianion [B12Cl12]2−. Dalton Trans. 2009, 2687–2694. [Google Scholar] [CrossRef] [PubMed]
- Kordts, N.; Kunzler, S.; Rathjen, S.; Sieling, T.; Groekappenberg, H.; Schmidtmann, M.; Muller, T. Silyl Chalconium Ions: Synthesis, Structure and Application in Hydrodefluorination Reactions. Chem. Eur. J. 2017, 23, 10068–10079. [Google Scholar] [CrossRef]
- Douvris, C.; Reed, C.A. Increasing the Reactivity of Vaska’s Compound. Oxidative Addition of Chlorobenzene at Ambient Temperature. Organometallics 2008, 27, 807–810. [Google Scholar]
- Binder, H.; Kellner, R.; Vaas, K.; Hein, M.; Baumann, F.; Wanner, M.; Winter, R.; Kaim, W.; Honle, W.; Grin, Y.; et al. The closo-cluster triad: B9X9, [B9X9](•−), and [B9X9](2−) with tricapped trigonal prisms (X = Cl, Br, I). Syntheses, crystal and electronic structures. Z. Anorg. Allg. Chem. 1999, 625, 1059–1072. [Google Scholar] [CrossRef]
- Gu, W.; McCulloch, B.J.; Reibenspies, J.H.; Ozerov, O.V. Improved methods for the halogenation of the [HCB11H11]− anion. Chem. Commun. 2010, 46, 2820–2822. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Fuku-en, S.; Sugawara, S.; Kojima, S.; Yamamoto, Y. Demethylation of an Allene Bearing Two Dimethoxythioxanthene Groups by Oxidation via a Vinyl Cation Intermediate. Aust. J. Chem. 2010, 63, 1638. [Google Scholar] [CrossRef]
- Rifat, A.; Mahon, M.F.; Weller, A.S. Dehydrogenation of cyclohexenes to cyclohexadienes by [(PPh3)2Rh]+. The isolation of an intermediate in the dehydrogenation of cyclohexane to benzene: Crystal structure of [(η4-C6H8)Rh(PPh3)2][closo-CB11H6Br6]. J. Organomet. Chem. 2003, 667, 1–4. [Google Scholar] [CrossRef]
- Douvris, C.; Stoyanov, E.S.; Tham, F.S.; Reed, C.A. Isolating fluorinated carbocations. Chem. Commun. 2007, 1145–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moxham, G.L.; Douglas, T.M.; Brayshaw, S.K.; Kociok-Kohn, G.; Lowe, J.P.; Weller, A.S. The role of halogenated carborane monoanions in olefin hydrogenation catalysed by cationic iridium phosphine complexes. Dalton Trans. 2006, 5492–5505. [Google Scholar] [CrossRef] [PubMed]
- Bruker. SAINT (Version 8.38A) in APEX3 (Version 2018.1-0); Bruker AXS Inc.: Madison, WI, USA, 2018. [Google Scholar]
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Cryst. 2015, 48, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. SHELXT-Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision, D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Flűkiger, P.; Lűthi, H.P.; Portmann, S.; Weber, J. MOLEKEL 4.3; Swiss Center for Scientific Computing: Manno, Switzerland, 2000. [Google Scholar]
- Portmann, S.; Luthi, H.P. MOLEKEL: An Interactive Molecular Graphic Tool. CHIMIA Int. J. Chem. 2000, 54, 766–770. [Google Scholar]
- Řezáč, J. Cuby: An Integrative Framework for Computational Chemistry. J. Comput. Chem. 2016, 37, 1230–1237. [Google Scholar] [CrossRef]
- Ahlrichs, R.; Bar, M.; Haser, M.; Horn, H.; Kolmel, C. Electronic Structure Calculations on Workstation Computers: The Program System Turbomole. Chem. Phys. Lett. 1989, 162, 165–169. [Google Scholar] [CrossRef]
- Hostaš, J.; Řezáč, J. Accurate DFT-D3 Calculations in a Small Basis Set. J. Chem. Theory Comput. 2017, 13, 3575–3585. [Google Scholar] [CrossRef]
- Řezáč, J.; Hobza, P. Advanced Corrections of Hydrogen Bonding and Dispersion for Semiempirical Quantum Mechanical Methods. J. Chem. Theory Comput. 2012, 8, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Jeziorski, B.; Moszynski, R.; Szalewicz, K. Perturbation Theory Approach to Intermolecular Potential Energy Surfaces of van der Waals Complexes. Chem. Rev. 1994, 94, 1887–1930. [Google Scholar] [CrossRef]
- Parker, T.M.; Burns, L.A.; Parrish, R.M.; Ryno, A.G.; Sherrill, C.D. Levels of Symmetry Adapted Perturbation Theory (SAPT). I. Efficiency and Performance for Interaction Energies. J. Chem. Phys. 2014, 140, 094106. [Google Scholar] [PubMed]
- Stewart, J.P. Optimization of parameters for semiempirical methods IV: Extension of MNDO, AM1, and PM3 to more main group elements. J. Mol. Model. 2004, 10, 155–164. [Google Scholar] [CrossRef]
- Turney, J.M.; Simmonett, A.C.; Parrish, R.M.; Hohenstein, E.G.; Evangelista, F.; Fermann, J.T.; Mintz, B.J.; Burns, L.A.; Wilke, J.J.; Abrams, M.L.; et al. Psi4: An Open-Source Ab Initio Electronic Structure Program. WIREs Comput. Mol. Sci. 2012, 2, 556–565. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 1–4 are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | B9 | B12 | B4, B5 | B7, B11 | B3, B6 | B8, B10 |
|---|---|---|---|---|---|---|
| 1 | −6.6 (−7.1) | −11.2 (−11.3) | −12.6 (−14.0) | −13.6 (−15.6) | −14.6 (−16.1) | −14.6 (−16.4) |
| 2 | −4.6 (−5.1) | −6.4 (−6.8) | −10.2 (−11.6) | −10.2 (−11.6) | −10.7 (−12.3) | −11.8 (−13.5) |
| 3 | −4.2 (−4.8) | −5.3 (−6.0) | −9.2 (−11.2) | −10.8 (−12.0) | −10.8 (−12.8) | −10.8 (−13.1) |
| 4 | −3.2 (−4.4) | −3.7 (−4.5) | −7.8 (−11.2) | −9.4 (−11.6) | −9.4 (−12.6) | −10.2 (−12.8) |
| Compound | ΔHf298 |
|---|---|
| 12-vertex series | |
| 1-Ph-2-F-closo-1,2-C2B10H10 (1) | 2.7 |
| 1-Ph-2-Cl-closo-1,2-C2B10H10 (2) | 45.7 |
| 1-Ph-2-Br-closo-1,2-C2B10H10 (3) | 57.0 |
| 1-Ph-2-I-closo-1,2-C2B10H10 (4) | 64.9 |
| 10-vertex series | |
| 1-Ph-2-F-closo-1,2-C2B8H8 | 30.9 |
| 1-Ph-2-Cl-closo-1,2-C2B8H8 | 73.2 |
| 1-Ph-2-Br-closo-1,2-C2B8H8 | 84.6 |
| 1-Ph-2-I-closo-1,2-C2B8H8 | 92.6 |
| Compound | ΣΔE2 (1st Layer) | ΔEMB (1st Layer) | ΣΔE2 (2nd Layer) | Total |
|---|---|---|---|---|
| 1-Ph-2-F-closo-1,2-C2B10H10 (1) | −50.74 | 5.04 | −2.69 | −48.39 |
| 1-Ph-2-Cl-closo-1,2-C2B10H10 (2) | −52.29 | 5.51 | −4.60 | −51.38 |
| 1-Ph-2-Br-closo-1,2-C2B10H10 (3) | −53.72 | 4.05 | −3.58 | −53.25 |
| 1-Ph-2-I-closo-1,2-C2B10H10 (4) | −56.31 | 3.45 | −2.37 | −55.23 |
| Motif | DFT-D3 | SAPT0 | ||||
|---|---|---|---|---|---|---|
| Total | Eelec | Eind | Edisp | Eexch | ||
| 1-Ph-2-F-closo-1,2-C2B10H10 (1) | ||||||
| A•••B | −6.93 | −7.20 | −1.92 (15.8%) | −0.57 (4.7%) | −9.65 (79.5%) | 4.94 |
| A•••C | −6.58 | −7.15 | −2.56 (19.5%) | −0.68 (5.2%) | −9.87 (75.3%) | 5.96 |
| 1-Ph-2-Cl-closo-1,2-C2B10H10 (2) | ||||||
| A•••B | −7.10 | −7.82 | −3.44 (21.5%) | −0.91 (5.7%) | −11.63 (72.8%) | 8.15 |
| A•••C | −5.43 | −4.64 | −1.68 (15.9%) | −1.03 (9.8 %) | −7.87 (74.4%) | 5.95 |
| 1-Ph-2-Br-closo-1,2-C2B10H10 (3) | ||||||
| A•••B1 | −6.91 | −7.10 | −4.80 (27.6%) | −1.27 (7.3%) | −11.30 (65.1%) | 10.23 |
| A•••C2 | −5.67 | −5.27 | −2.19 (22.6%) | −0.73 (7.5%) | −6.75 (69.8%) | 4.40 |
| 1-Ph-2-I-closo-1,2-C2B10H10 (4) | ||||||
| A•••B | −5.98 | − | − | − | − | − |
| A•••C | −5.79 | − | − | − | − | − |
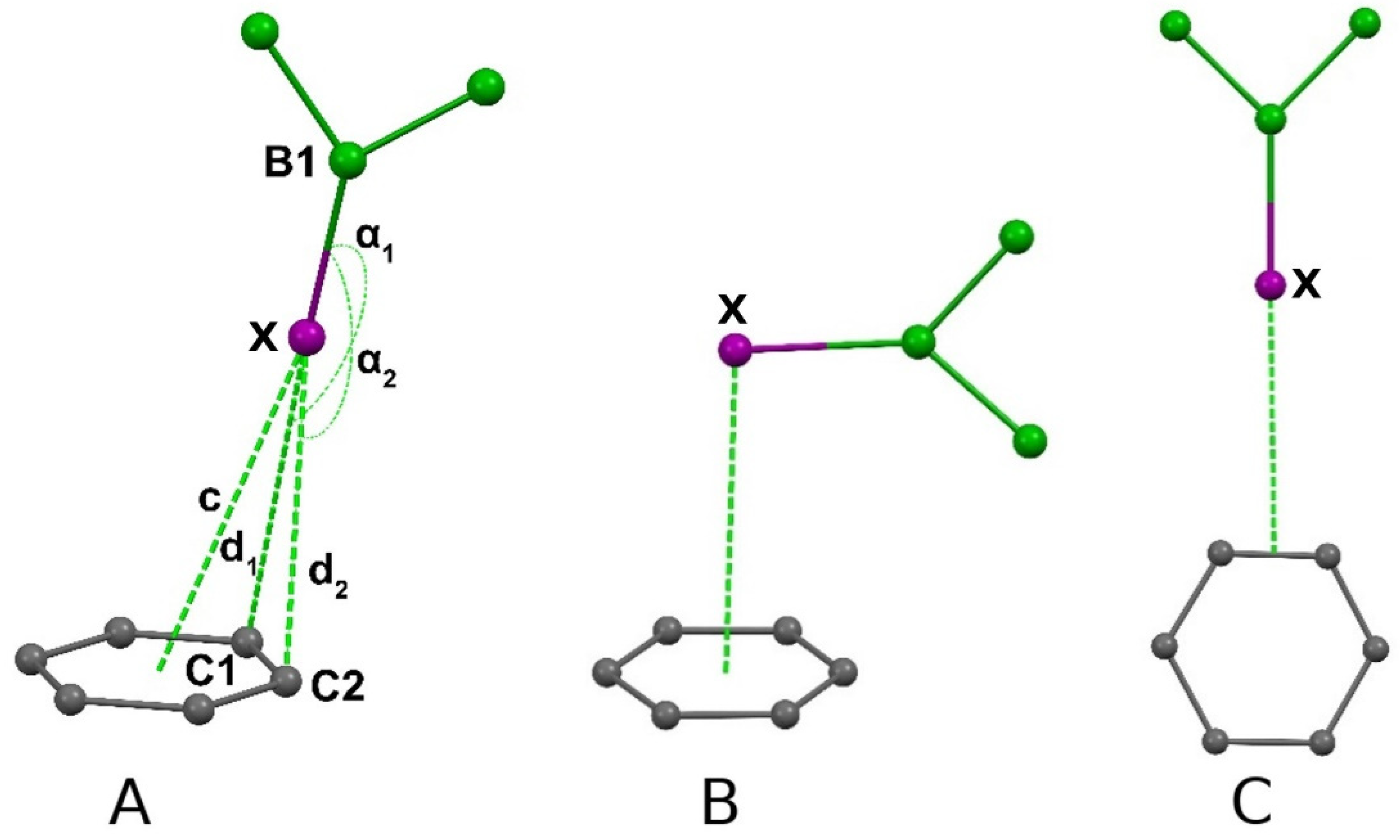
| X | Restraints, Å | Number of Structures | Angles (B-Hal-C) α1, α2,° | d1, d2, Å | c, Å |
|---|---|---|---|---|---|
| F | 2.8 < (d1, d2) < 3.1 | 4 | 99.08–168.80 | 2.871–3.090 | 3.235–4.013 |
| Cl | 3.1 < (d1, d2) < 3.5 | 19 | 96.77–169.76 | 3.156–3.497 | 3.172–4.287 |
| Br | 3.1 < (d1, d2) < 3.6 | 10 | 104.44–171.01 | 3.329–3.586 | 3.567–4.476 |
| I | 3.1 < (d1, d2) < 3.8 | 4 | 140.21–176.25 | 3.437–3.795 | 3.416–4.078 |
| Compound | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| Chemical formula | C8H15B10F | C8H15B10Cl | C8H15B10Br | C8H15B10I |
| Formula weight | 238.30 | 254.75 | 299.21 | 346.20 |
| Temperature/K | 150(2) | 150(2) | 150(2) | 150(2) |
| Crystal system | Monoclinic | Monoclinic | Orthorhombic | Monoclinic |
| Space group | P21/m | P21/n | Pbca | P21 |
| a/Å | 8.6564(9) | 7.2920(4) | 10.257(3) | 12.0950(8) |
| b/Å | 7.5229(7) | 23.9912(14) | 11.448(3) | 7.2033(5) |
| c/Å | 10.5576(11) | 7.7979(5) | 24.132(7) | 16.9983(14) |
| α/° | 90 | 90 | 90 | 90 |
| β/° | 106.168(3) | 93.397(2) | 90 | 90.453(3) |
| γ/° | 90 | 90 | 90 | 90 |
| Volume/Å | 660.33(12) | 1361.80(14) | 2833.8(14) | 1480.91(19) |
| Z | 2 | 4 | 8 | 4 |
| ρcalc g/cm3 | 1.199 | 1.243 | 1.403 | 1.553 |
| μ/mm−1 | 0.066 | 0.248 | 2.870 | 2.133 |
| F(000) | 244 | 520 | 1184 | 664 |
| Crystal size/mm3 | 0.989 × 0.504 × 0.386 | 0.414 × 0.225 × 0.148 | 0.342 × 0.192 × 0.150 | 0.753 × 0.416 × 0.343 |
| Radiation type | MoKα (λ = 0.71073 Å) | MoKα (λ = 0.71073 Å) | MoKα (λ = 0.71073 Å) | MoKα (λ = 0.71073 Å) |
| 2θ range for data collection/° | 2.450 to 27.996 | 2.751 to 26.415 | 2.606 to 24.999 | 2.396 to 28.276 |
| Index ranges | −11 < = h < =11, −9 < = k < = 9, −13 < = l < = 13 | −9 < = h < = 9, −29 < = k < = 30, −9 < = l < = 9 | −12 < = h < = 12, −13 < = k < = 12, −28 < = l < = 28 | −14 < = h < = 16, −9 < = k < = 9, −22 < = l < = 22 |
| Reflections collected | 14986 | 33108 | 14758 | 21597 |
| Independent reflections | 1704 [R(int) = 0.0745] | 2794 [R(int) = 0.0694] | 2439 [R(int) = 0.1169] | 6917 [R(int) = 0.0499] |
| Data/restraints/parameters | 1704/12/115 | 2794/0/172 | 2439/264/172 | 6917/1/344 |
| Goodness-of-fit on F2 | 1.049 | 1.067 | 1.176 | 1.053 |
| Final R indexes [I > 2σ(I)] | R1 = 0.0620, wR2 = 0.1615 | R1 = 0.0479, wR2 = 0.1096 | R1 = 0.1237, wR2 = 0.2686 | R1 = 0.0370, wR2 = 0.0718 |
| Largest diff. peak/hole/e Å-3 | 0.673 and −0.393 | 0.282 and −0.307 | 1.683 and −1.521 | 1.635 and −1.431 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Havránek, M.; Samsonov, M.A.; Holub, J.; Růžičková, Z.; Drož, L.; Růžička, A.; Fanfrlík, J.; Hnyk, D. The Influence of Halogenated Hypercarbon on Crystal Packing in the Series of 1-Ph-2-X-1,2-dicarba-closo-dodecaboranes (X = F, Cl, Br, I). Molecules 2020, 25, 1200. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25051200
Havránek M, Samsonov MA, Holub J, Růžičková Z, Drož L, Růžička A, Fanfrlík J, Hnyk D. The Influence of Halogenated Hypercarbon on Crystal Packing in the Series of 1-Ph-2-X-1,2-dicarba-closo-dodecaboranes (X = F, Cl, Br, I). Molecules. 2020; 25(5):1200. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25051200
Chicago/Turabian StyleHavránek, Miroslav, Maksim A. Samsonov, Josef Holub, Zdeňka Růžičková, Ladislav Drož, Aleš Růžička, Jindřich Fanfrlík, and Drahomír Hnyk. 2020. "The Influence of Halogenated Hypercarbon on Crystal Packing in the Series of 1-Ph-2-X-1,2-dicarba-closo-dodecaboranes (X = F, Cl, Br, I)" Molecules 25, no. 5: 1200. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25051200