Comparisons of In Vivo and In Vitro Opioid Effects of Newly Synthesized 14-Methoxycodeine-6-O-sulfate and Codeine-6-O-sulfate


 , ,
, ,
Abstract
:1. Introduction
2. Results
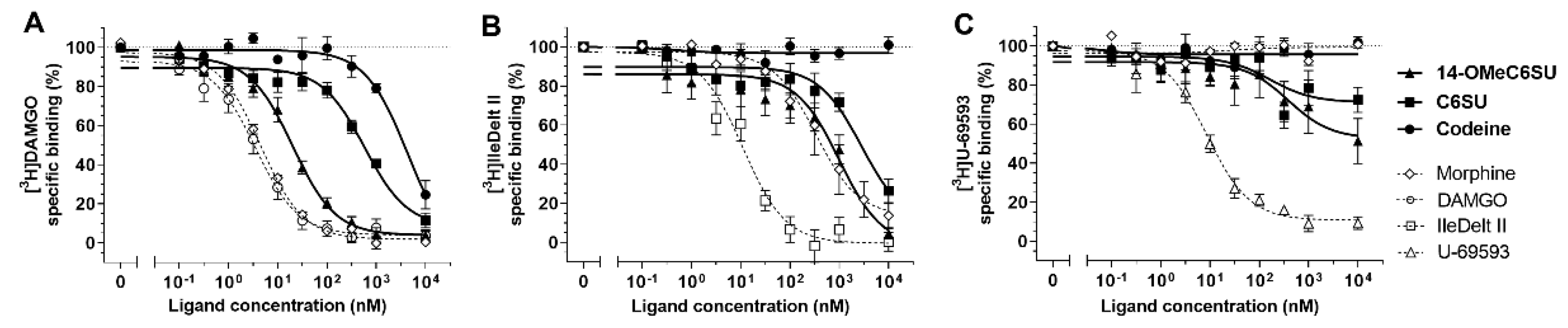
2.1. Receptor Binding Assays
2.1.1. 14-OMeC6SU Displayed High Affinity and MOR Selectivity in Radioligand Competition Binding Assay
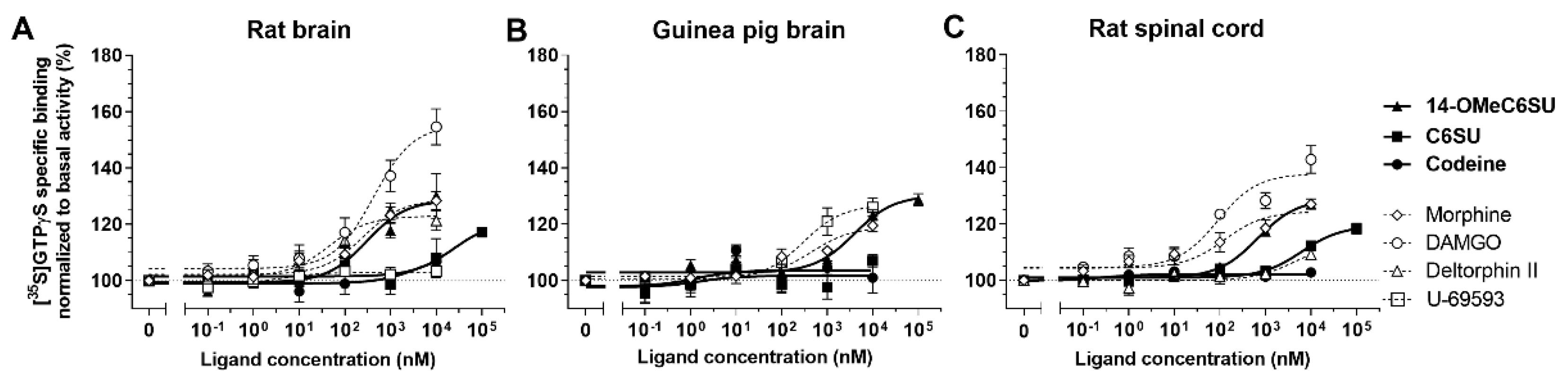
2.1.2. 14-OMeC6SU Shows Strong Agonist Activity in [35S]GTPγS Binding Assay
2.2. 14-OMeC6SU Is a Full Agonist in MVD and RVD
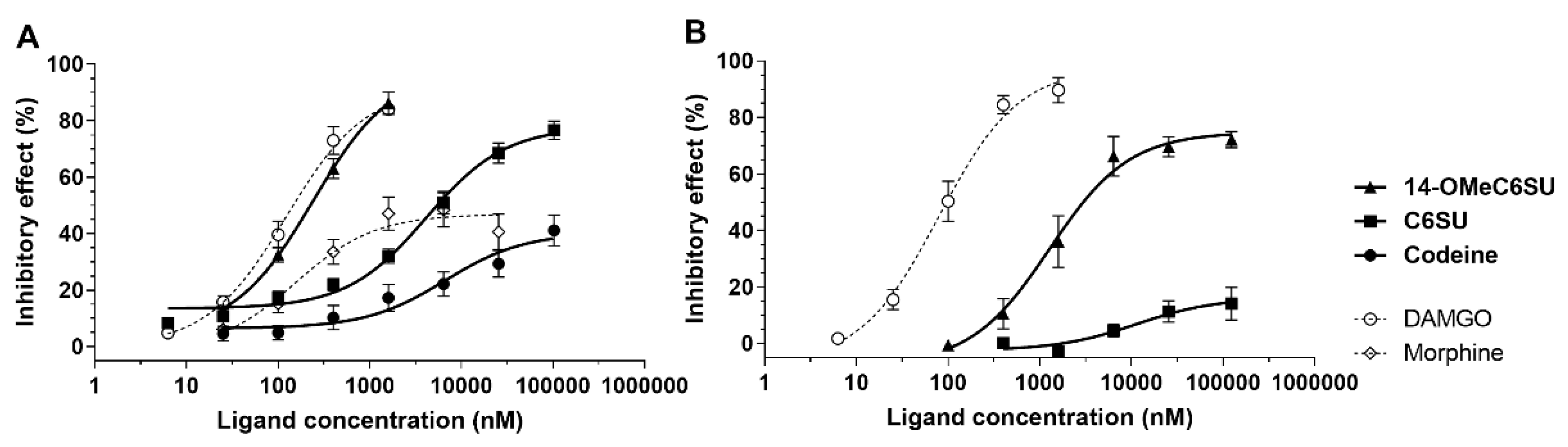
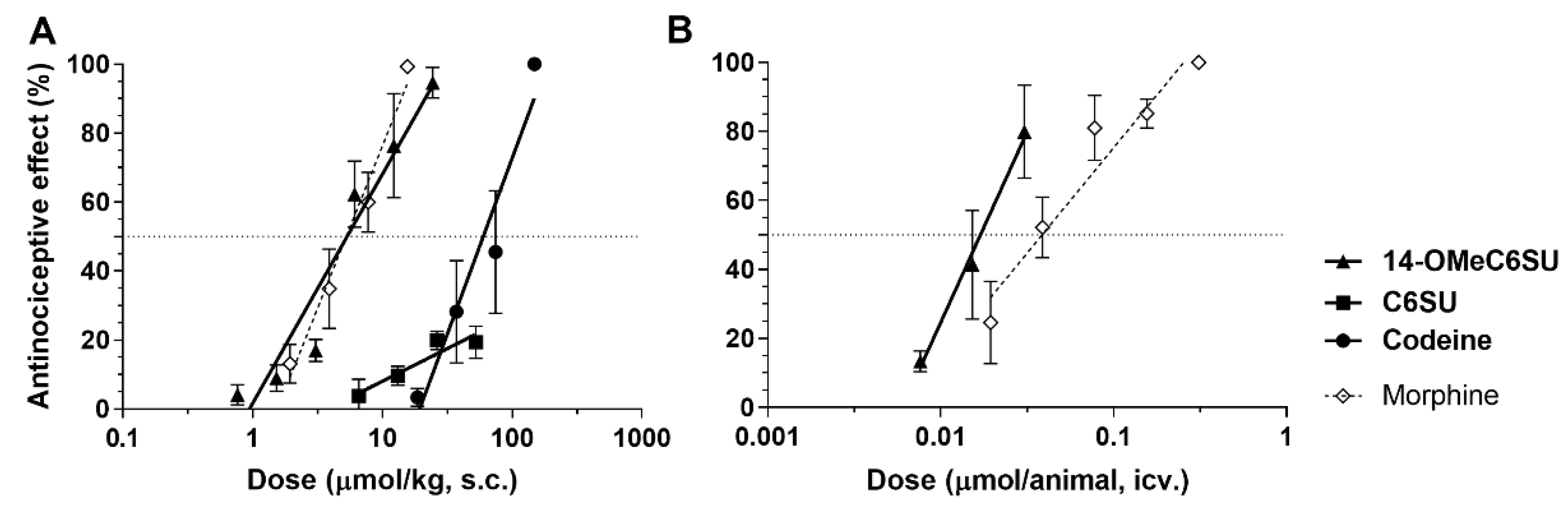
2.3. 14-OMeC6SU Produces Antinociceptive Effect in Rat Tail-flick Assay
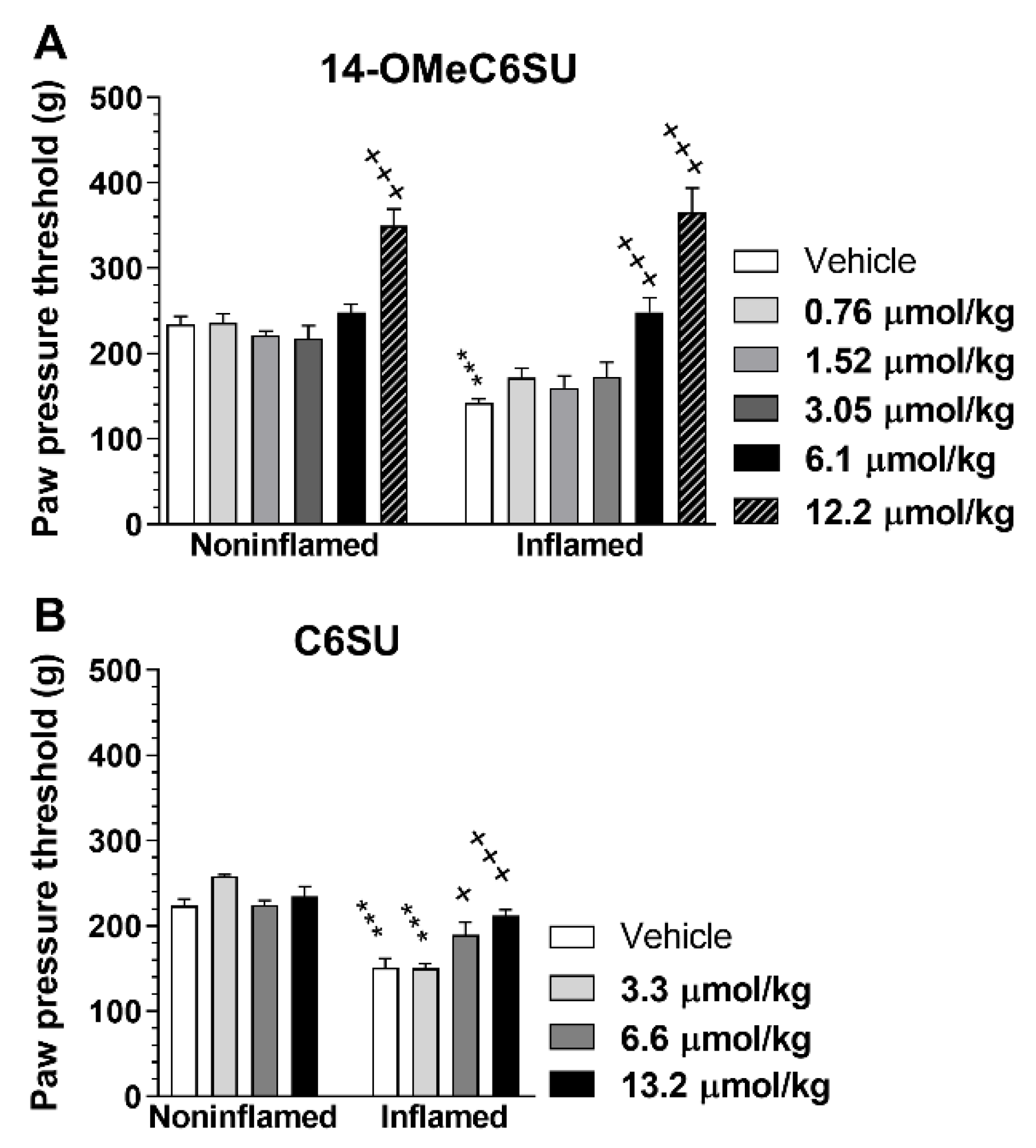
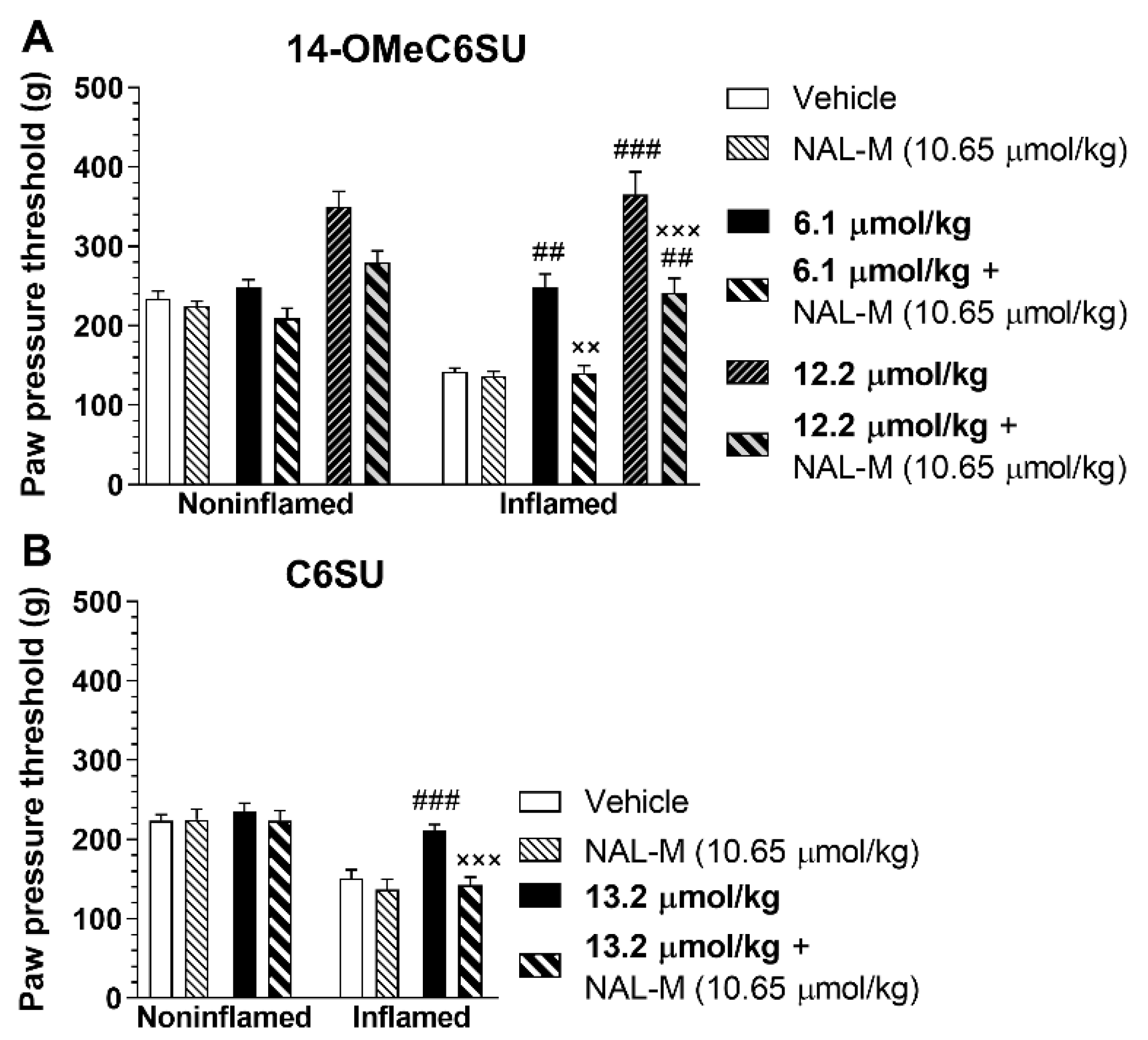
2.4. 14-OMeC6SU (in Certain Doses) and C6SU Possess Peripheral Antinociceptive Effects after Systemic Administration in Rats with CFA-Induced Inflammatory Pain
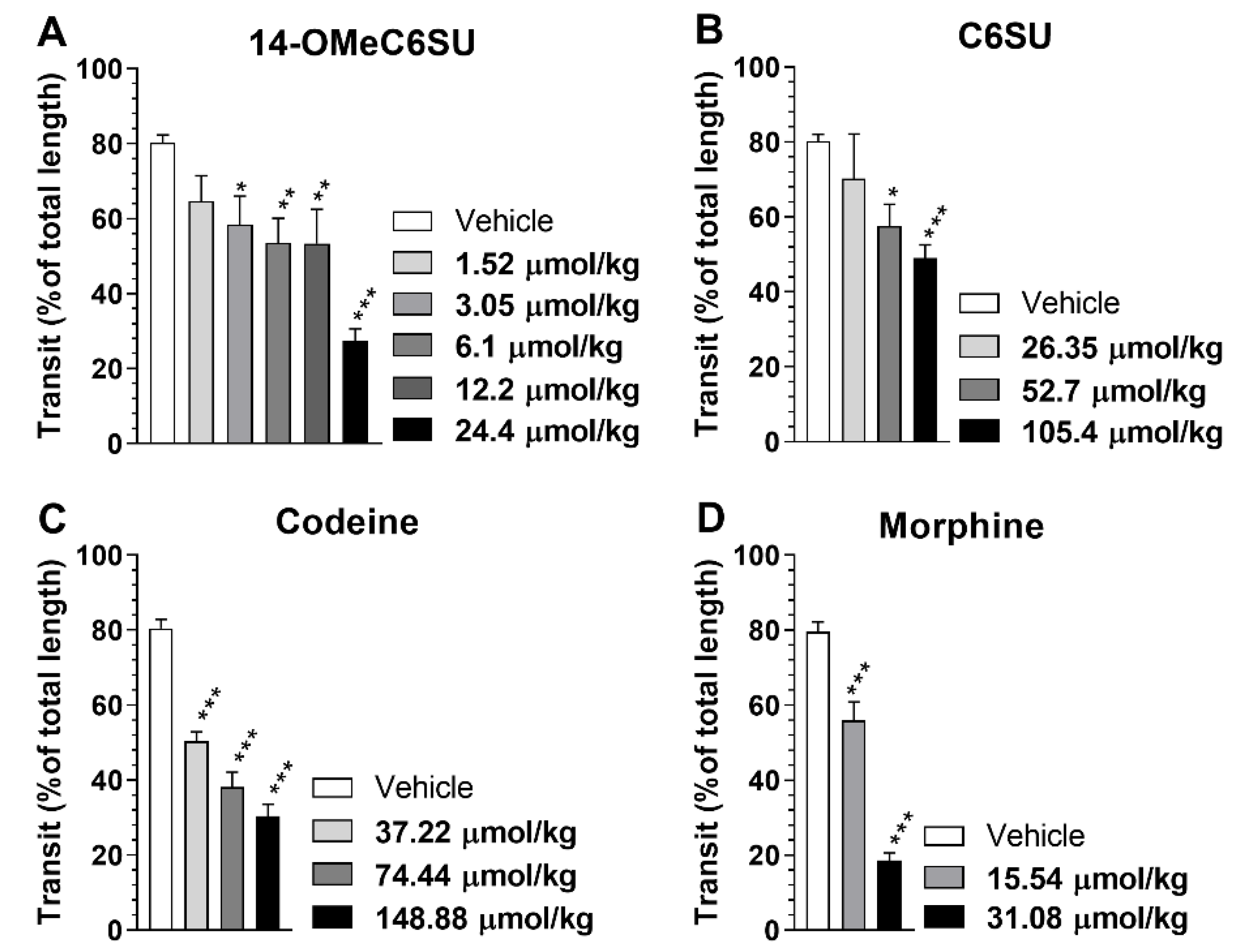
2.5. Inhibitory Effect of Systemic 14-OMeC6SU on Gastrointestinal Transit in Rats
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Chemicals
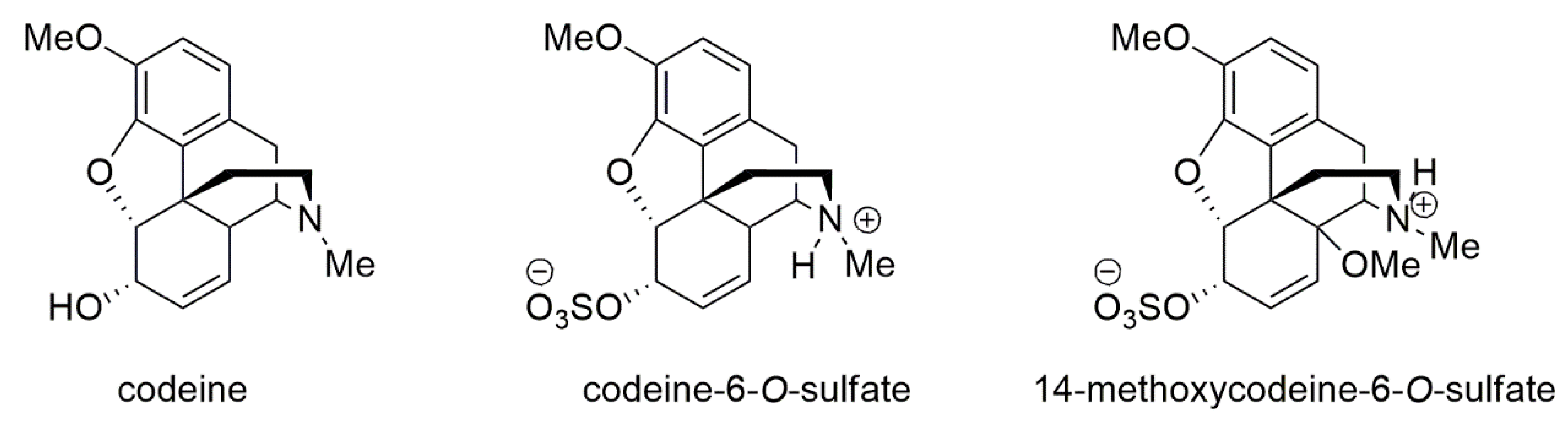
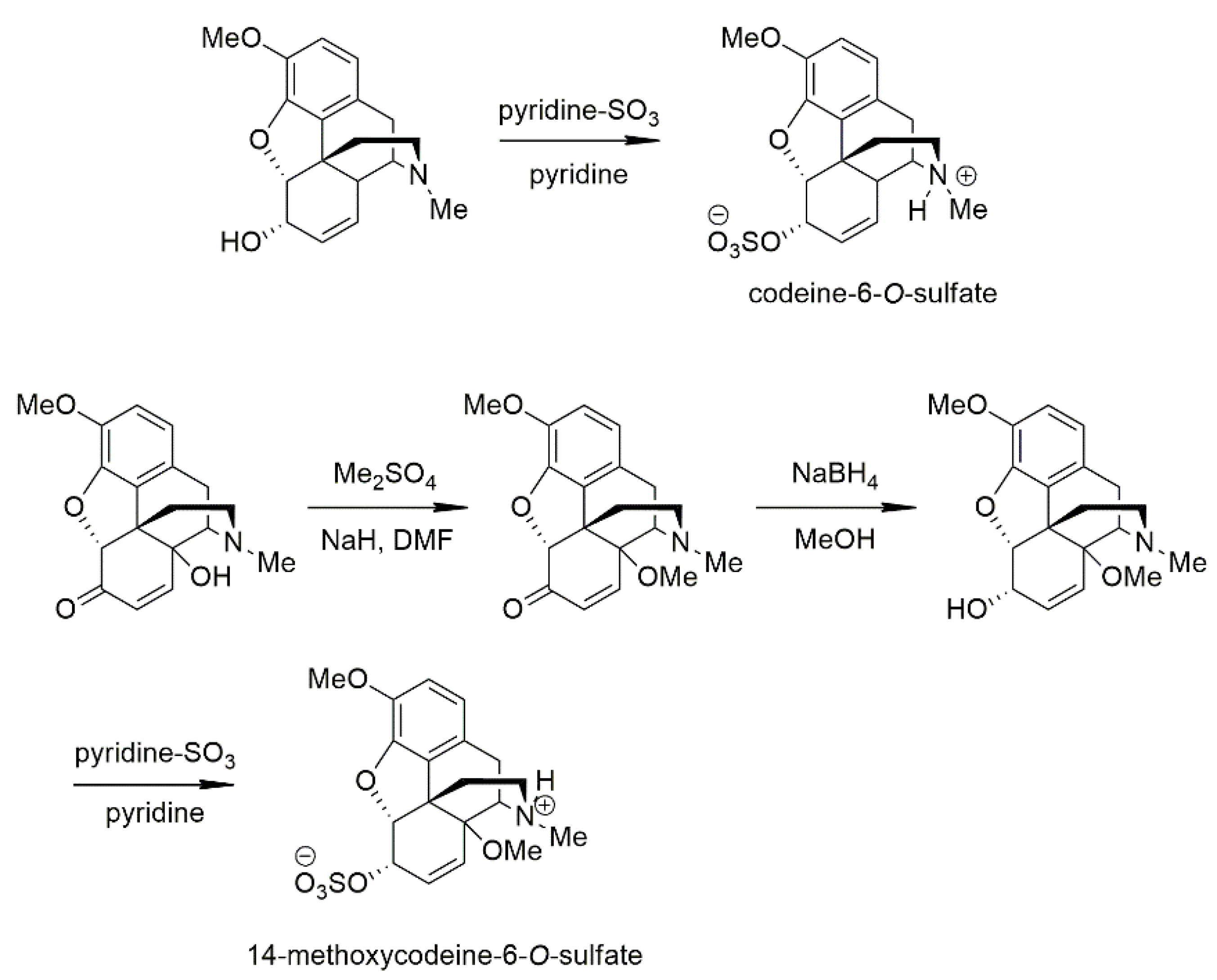
4.3. Chemistry
4.3.1. Synthesis of the Studied Compounds
4.3.2. Codeine-6-O-sulfate
4.3.3. 14-Methoxycodeine-6-O-sulfate
4.4. Receptor Binding Assays
4.4.1. Membrane Preparations
4.4.2. Radioligand Competition Binding Assays
4.4.3. Functional [35S]GTPγS Binding Assays
4.5. Isolated Organs
4.5.1. Mouse vas Deferens (MVD)
4.5.2. Rat vas Deferens (RVD)
4.5.3. Experimental Paradigms of MVD and RVD
4.6. Thermal Acute Pain Model (Tail-flick Test)
4.7. Inflammatory Pain Model (CFA-Evoked Hyperalgesia)
4.8. Determination the Effect of Test Compounds on Gastrointestinal Transit
4.9. Data Analysis
4.9.1. Receptor Binding Assays
4.9.2. MVD and RVD Bioassays
4.9.3. Rat Tail-flick Test, Gastrointestinal Transit and CFA-Evoked Hyperalgesia Tests
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Burford, N.; Wang, D.; Sadee, W. G-protein coupling of mu-opioid receptors (OP3): Elevated basal signalling activity. Biochem. J. 2000, 537, 531–537. [Google Scholar] [CrossRef]
- Koneru, A.; Satyanarayana, S.; Rizwan, S. Endogenous opioids: Their physiological role and receptors. Glob. J Pharmacol. 2009, 3, 149–153. [Google Scholar]
- Perret, D.; Luo, Z. Targeting voltage-gated calcium channels for neuropathic pain management. Neurotherapeutics 2009, 6, 679–692. [Google Scholar] [CrossRef] [Green Version]
- Khalefa, B.I.; Mousa, S.A.; Shaqura, M.; Lackó, E.; Hosztafi, S.; Riba, P.; Schäfer, M.; Ferdinandy, P.; Fürst, S.; Al-Khrasani, M. Peripheral antinociceptive efficacy and potency of a novel opioid compound 14-O-MeM6SU in comparison to known peptide and non-peptide opioid agonists in a rat model of inflammatory pain. Eur. J. Pharmacol. 2013, 713, 54–57. [Google Scholar] [CrossRef]
- Lackó, E.; Riba, P.; Giricz, Z.; Váradi, A.; Cornic, L.; Balogh, M.; Király, K.; Csekő, K.; Mousa, S.A.; Hosztafi, S.; et al. New Morphine Analogs Produce Peripheral Antinociception within a Certain Dose Range of Their Systemic Administration. J. Pharmacol. Exp. Ther. 2016, 359, 171–181. [Google Scholar] [CrossRef] [Green Version]
- Fürst, S.; Riba, P.; Friedmann, T.; Tímar, J.; Al-Khrasani, M.; Obara, I.; Makuch, W.; Spetea, M.; Schütz, J.; Przewlocki, R.; et al. Peripheral versus central antinociceptive actions of 6-amino acid-substituted derivatives of 14-O-methyloxymorphone in acute and inflammatory pain in the rat. J. Pharmacol. Exp. Ther. 2005, 312, 609–618. [Google Scholar] [CrossRef] [Green Version]
- Balogh, M.; Zádori, Z.S.; Lázár, B.; Karádi, D.; László, S.; Mousa, S.A.; Hosztafi, S.; Zádor, F.; Riba, P.; Schäfer, M.; et al. The Peripheral Versus Central Antinociception of a Novel Opioid Agonist: Acute Inflammatory Pain in Rats. Neurochem. Res. 2018, 43, 1250–1257. [Google Scholar] [CrossRef]
- Joris, J.L.; Dubner, R.; Hargreaves, K.M. Opioid analgesia at peripheral sites: A target for opioids released during stress and inflammation? Anesth. Analg. 1987, 66, 1277–1281. [Google Scholar] [CrossRef]
- Ferreira, S.H.; Nakamura, M. II - Prostaglandin hyperalgesia: The peripheral analgesic activity of morphine, enkephalins and opioid antagonists. Prostaglandins 1979, 18, 191–200. [Google Scholar] [CrossRef]
- McDougall, J.J. Peripheral analgesia: Hitting pain where it hurts. Biochim. Biophys. Acta Mol. Basis Dis. 2011, 1812, 459–467. [Google Scholar] [CrossRef] [Green Version]
- Kosterlitz, H.W.; Waterfield, A.A. In Vitro Models in the Study of Structure-Activity Relationships of Narcotic Analgesics. Annu. Rev. Pharmacol. 1975, 15, 29–47. [Google Scholar] [CrossRef]
- Mazak, K.; Noszal, B.; Hosztafi, S. Physicochemical and Pharmacological Characterization of Permanently Charged Opioids. Curr. Med. Chem. 2017, 24, 3633–3648. [Google Scholar] [CrossRef]
- Preechagoon, D.; Brereton, I.; Staatz, C.; Prankerd, R. Ester prodrugs of a potent analgesic, morphine-6-sulfate: Syntheses, spectroscopic and physicochemical properties. Int. J. Pharm. 1998, 163, 177–190. [Google Scholar] [CrossRef]
- Crooks, P.A.; Kottayil, S.G.; Al-Ghananeem, A.M.; Byrn, S.R.; Allan Butterfield, D. Opiate receptor binding properties of morphine-, dihydromorphine-, and codeine 6-O-sulfate ester congeners. Bioorg. Med. Chem. Lett. 2006, 16, 4291–4295. [Google Scholar] [CrossRef]
- Schmidhammer, H.; Spetea, M.; Windisch, P.; Schütz, J.; Riba, P.; Al-Khrasani, M.; Furst, S. Functionalization of the Carbonyl Group in Position 6 of Morphinan-6-ones. Development of Novel 6-Amino and 6-Guanidino Substituted 14-Alkoxymorphinans. Curr. Pharm. Des. 2014, 19, 7391–7399. [Google Scholar] [CrossRef]
- Al-Khrasani, M.; Spetea, M.; Friedmann, T.; Riba, P.; Király, K.; Schmidhammer, H.; Furst, S. DAMGO and 6β-glycine substituted 14-O-methyloxymorphone but not morphine show peripheral, preemptive antinociception after systemic administration in a mouse visceral pain model and high intrinsic efficacy in the isolated rat vas deferens. Brain Res. Bull. 2007, 74, 369–375. [Google Scholar] [CrossRef]
- Zuckerman, A.; Bolan, E.; de Paulis, T.; Schmidt, D.; Spector, S.; Pasternak, G.W. Pharmacological characterization of morphine-6-sulfate and codeine-6-sulfate. Brain Res. 1999, 842, 1–5. [Google Scholar] [CrossRef]
- Brown, C.E.; Roerig, S.C.; Burger, V.T.; Cody, R.B.; Fujimoto, J.M. Analgesic Potencies of Morphine 3- and 6-Sulfates After Intracerebroventricular Administration in Mice: Relationship to Structural Characteristics Defined by Mass Spectrometry and Nuclear Magnetic Resonance. J. Pharm. Sci. 1985, 74, 821–824. [Google Scholar] [CrossRef]
- Mori, M.; Oguri, K.; Yoshimura, H.; Shimomura, K.; Kamata, O.; Ueki, S. Chemical synthesis and analgesic effect of morphine ethereal sulfates. Life Sci. I. 1972, 11, 525–533. [Google Scholar] [CrossRef]
- Spetea, M.; Friedmann, T.; Riba, P.; Schütz, J.; Wunder, G.; Langer, T.; Schmidhammer, H.; Fürst, S. In vitro opioid activity profiles of 6-amino acid substituted derivatives of 14-O-methyloxymorphone. Eur. J. Pharmacol. 2004, 483, 301–308. [Google Scholar] [CrossRef]
- Holtman, J.R.; Crooks, P.A.; Johnson-Hardy, J.; Wala, E.P. Antinociceptive effects and toxicity of morphine-6-O-sulfate sodium salt in rat models of pain. Eur. J. Pharmacol. 2010, 648, 87–94. [Google Scholar] [CrossRef]
- Lacko, E.; Varadi, A.; Rapavi, R.; Zador, F.; Riba, P.; Benyhe, S.; Borsodi, A.; Hosztafi, S.; Timar, J.; Noszal, B.; et al. A novel µ-opioid receptor ligand with high in vitro and in vivo agonist efficacy. Curr Med Chem 2012, 19, 4699–4707. [Google Scholar] [CrossRef]
- Zádor, F.; Balogh, M.; Váradi, A.; Zádori, Z.S.; Király, K.; Szűcs, E.; Varga, B.; Lázár, B.; Hosztafi, S.; Riba, P.; et al. 14-O-Methylmorphine: A Novel Selective Mu-Opioid Receptor Agonist with High Efficacy and Affinity. Eur. J. Pharmacol. 2017, 814, 264–273. [Google Scholar] [CrossRef] [Green Version]
- Chidambaran, V.; Sadhasivam, S.; Mahmoud, M. Codeine and opioid metabolism: Implications and alternatives for pediatric pain management. Curr. Opin. Anaesthesiol. 2017, 30, 349–356. [Google Scholar] [CrossRef]
- Hennies, H.H.; Friderichs, E.; Schneider, J. Receptor binding, analgesic and antitussive potency of tramadol and other selected opioids. Arzneimittelforschung. 1988, 38, 877–880. [Google Scholar]
- Neil, A. Affinities of some common opioid analgesics towards four binding sites in mouse brain. Naunyn. Schmiedebergs. Arch. Pharmacol. 1984, 328, 24–29. [Google Scholar] [CrossRef]
- Mignat, C.; Wille, U.; Ziegler, A. Affinity profiles of morphine, codeine, dihydrocodeine and their glucuronides at opioid receptor subtypes. Life Sci. 1995, 56, 793–799. [Google Scholar] [CrossRef]
- Chen, Z.R.; Irvine, R.J.; Somogyi, A.A.; Bochner, F. Mu receptor binding of some commonly used opioids and their metabolites. Life Sci. 1991, 48, 2165–2171. [Google Scholar] [CrossRef]
- Peechakara, B.V.; Gupta, M. Codeine; StatPearls Publishing: Treasure Island, FL, USA, 2019. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/books/NBK526029/ (accessed on 17 March 2020).
- Nielsen, S.; MacDonald, T.; Johnson, J.L. Identifying and treating codeine dependence: A systematic review. Med. J. Aust. 2018, 208, 451–461. [Google Scholar] [CrossRef] [Green Version]
- Moore, R.A.; McQuay, H.J. Prevalence of opioid adverse events in chronic non-malignant pain: Systematic review of randomised trials of oral opioids. Arthritis Res. Ther. 2005, 7, R1046–R1051. [Google Scholar] [CrossRef] [Green Version]
- Benyamin, R.; Trescot, A.M.; Datta, S.; Buenaventura, R.; Adlaka, R.; Sehgal, N.; Glaser, S.E.; Vallejo, R. Opioid complications and side effects. Pain Physician. 2008, 11, S105–S120. [Google Scholar]
- Vowles, K.E.; McEntee, M.L.; Julnes, P.S.; Frohe, T.; Ney, J.P.; van der Goes, D.N. Rates of opioid misuse, abuse, and addiction in chronic pain. Pain 2015, 156, 569–576. [Google Scholar] [CrossRef] [Green Version]
- Thomasy, S.M.; Moeller, B.C.; Stanley, S.D. Comparison of opioid receptor binding in horse, guinea pig, and rat cerebral cortex and cerebellum. Vet. Anaesth. Analg. 2007, 34, 351–358. [Google Scholar] [CrossRef]
- Balogh, M.; Zádor, F.; Zádori, Z.S.; Shaqura, M.; Király, K.; Mohammadzadeh, A.; Varga, B.; Lázár, B.; Mousa, S.A.; Hosztafi, S.; et al. Efficacy-Based Perspective to Overcome Reduced Opioid Analgesia of Advanced Painful Diabetic Neuropathy in Rats. Front. Pharmacol. 2019, 10, 347. [Google Scholar] [CrossRef]
- Schmidhammer, H.; Aeppli, L.; Atwell, L.; Fritsch, F.; Jacobson, A.E.; Nebuchla, M.; Sperk, G. Synthesis and biological evaluation of 14-alkoxymorphinans. 1. Highly potent opioid agonists in the series of (-)-14-methoxy-N-methylmorphinan-6-ones. J. Med. Chem. 1984, 27, 1575–1579. [Google Scholar] [CrossRef]
- Khalefa, B.I.; Shaqura, M.; Al-Khrasani, M.; Fürst, S.; Mousa, S.A.; Schäfer, M. Relative contributions of peripheral versus supraspinal or spinal opioid receptors to the antinociception of systemic opioids. Eur. J. Pain 2012, 16, 690–705. [Google Scholar] [CrossRef]
- Stein, C. Targeting pain and inflammation by peripherally acting opioids. Front. Pharmacol. 2013, 4, 123. [Google Scholar] [CrossRef] [Green Version]
- Spetea, M.; Schmidhammer, H. Recent advances in the development of 14-alkoxy substituted morphinans as potent and safer opioid analgesics. Curr. Med. Chem. 2012, 19, 2442–2457. [Google Scholar] [CrossRef]
- Al-Khrasani, M.; Orosz, G.; Kocsis, L.; Farkas, V.; Magyar, A.; Lengyel, I.; Benyhe, S.; Borsodi, A.; Rónai, A.Z. Receptor constants for endomorphin-1 and endomorphin-1-ol indicate differences in efficacy and receptor occupancy. Eur. J. Pharmacol. 2001, 421, 61–67. [Google Scholar] [CrossRef]
- Koch, T.; Widera, A.; Bartzsch, K.; Schulz, S.; Brandenburg, L.O.; Wundrack, N.; Beyer, A.; Grecksch, G.; Höllt, V. Receptor Endocytosis Counteracts the Development of Opioid Tolerance. Mol. Pharmacol. 2005, 67, 280–287. [Google Scholar] [CrossRef] [Green Version]
- Stein, C.; Machelska, H.; Binder, W.; Schäfer, M. Peripheral opioid analgesia. Curr. Opin. Pharmacol. 2001, 1, 62–65. [Google Scholar] [CrossRef] [Green Version]
- Stein, C.; Lang, L.J. Peripheral mechanisms of opioid analgesia. Curr. Opin. Pharmacol. 2009, 9, 3–8. [Google Scholar] [CrossRef]
- Schäfer, M.; Imai, Y.; Uhl, G.R.; Stein, C. Inflammation enhances peripheral μ-opioid receptor-mediated analgesia, but not μ-opioid receptor transcription in dorsal root ganglia. Eur. J. Pharmacol. 1995, 279, 165–169. [Google Scholar] [CrossRef]
- Bianchi, G.; Fiocchi, R.; Tavani, A.; Manara, L. Quaternary narcotic antagonists’ relative ability to prevent antinociception and gastrointestinal transit inhibition in morphine-treated rats as an index of peripheral selectivity. Life Sci. 1982, 30, 1875–1883. [Google Scholar] [CrossRef]
- Lewanowitsch, T.; Irvine, R.J. Naloxone methiodide reverses opioid-induced respiratory depression and analgesia without withdrawal. Eur. J. Pharmacol. 2002, 445, 61–67. [Google Scholar] [CrossRef]
- Riba, P.; Ben, Y.; Nguyen, T.M.-D.; Furst, S.; Schiller, P.W.; Lee, N.M. [Dmt(1)]DALDA is highly selective and potent at mu opioid receptors, but is not cross-tolerant with systemic morphine. Curr. Med. Chem. 2002, 9, 31–39. [Google Scholar] [CrossRef]
- Holzer, P. Opioid receptors in the gastrointestinal tract. Regul. Pept. 2009, 155, 11–17. [Google Scholar] [CrossRef] [Green Version]
- Mehta, N.; O’Connell, K.; Giambrone, G.; Baqai, A.; Diwan, S. Efficacy of Methylnaltrexone for the Treatment of Opiod-Induced Constipation: A Meta-Analysis and Systematic Review. Postgrad. Med. 2016, 128, 282–289. [Google Scholar] [CrossRef]
- Pergolizzi, J.V.; Raffa, R.B.; Pappagallo, M.; Fleischer, C.; Pergolizzi, J.; Zampogna, G.; Duval, E.; Hishmeh, J.; LeQuang, J.A.; Taylor, R.; et al. Peripherally acting μ-opioid receptor antagonists as treatment options for constipation in noncancer pain patients on chronic opioid therapy. Patient Prefer. Adherence 2017, 11, 107–119. [Google Scholar] [CrossRef] [Green Version]
- Kobylecki, R.J.; Carling, R.W.; Lord, J.A.; Smith, C.F.; Lane, A.C. Common anionic receptor site hypothesis: Its relevance to the antagonist action of naloxone. J. Med. Chem. 1982, 25, 116–120. [Google Scholar] [CrossRef]
- Currie, A.C.; Gillon, J.; Newbold, G.T.; Spring, F.S. 157. Some reactions of 14-hydroxycodeine. J. Chem. Soc. 1960, 773–781. [Google Scholar] [CrossRef]
- Sawa, Y.K.; Horiuchi, M.; Tanaka, K. Elimination of the 4-hydroxyl group of the alkaloids related to morphine—IX: Synthesis of 3-methoxy-n-methylisomorphinan derivatives. Tetrahedron 1968, 24, 255–260. [Google Scholar] [CrossRef]
- Váradi, A.; Gergely, A.; Béni, S.; Jankovics, P.; Noszál, B.; Hosztafi, S. Sulfate esters of morphine derivatives: Synthesis and characterization. Eur. J. Pharm. Sci. 2011, 42, 65–72. [Google Scholar] [CrossRef]
- Benyhe, S.; Farkas, J.; Tóth, G.; Wollemann, M. Met5-enkephalin-Arg6-Phe7, an endogenous neuropeptide, binds to multiple opioid and nonopioid sites in rat brain. J. Neurosci. Res. 1997, 48, 249–258. [Google Scholar] [CrossRef]
- Zádor, F.; Kocsis, D.; Borsodi, A.; Benyhe, S. Micromolar concentrations of rimonabant directly inhibits delta opioid receptor specific ligand binding and agonist-induced G-protein activity. Neurochem. Int. 2014, 67, 14–22. [Google Scholar] [CrossRef]
- Frey, K.A.; Albin, R.L. Receptor Binding Techniques. Curr. Protoc. Neurosci. 1997, 1, 1–4. [Google Scholar] [CrossRef]
- Strange, P.G. Use of the GTPγS ([35S]GTPγS and Eu-GTPγS) binding assay for analysis of ligand potency and efficacy at G protein-coupled receptors. Br. J. Pharmacol. 2010, 161, 1238–1249. [Google Scholar] [CrossRef] [Green Version]
- Selley, D.E.; Sim, L.J.; Xiao, R.; Liu, Q.; Childers, S.R. mu-Opioid receptor-stimulated guanosine-5’-O-(gamma-thio)-triphosphate binding in rat thalamus and cultured cell lines: Signal transduction mechanisms underlying agonist efficacy. Mol. Pharmacol. 1997, 51, 87–96. [Google Scholar] [CrossRef] [Green Version]
- Traynor, J.; Nahorski, S. Modulation by mu-opioid agonists of guanosine-5’-O-(3-[35S]thio)triphosphate binding to membranes from human neuroblastoma SH-SY5Y cells. Mol. Pharmacol. 1995, 47, 848–854. [Google Scholar]
- Rónai, A.Z.; Gráf, L.; Székely, J.I.; Dunai-Kovács, Z.; Bajusz, S. Differential behaviour of LPH-(61-91)-peptide in different model systems: Comparison of the opioid activities of LPH-(61-91)-peptide and its fragments. FEBS Lett. 1977, 74, 182–184. [Google Scholar] [CrossRef] [Green Version]
- Kosterlitz, H.W.; Watt, A.J. Kinetic parameters of narcotic agonists and antagonists, with particular reference to N-allylnoroxymorphone (naloxone). Br. J. Pharmacol. Chemother. 1968, 33, 266–276. [Google Scholar] [CrossRef] [Green Version]
- Fürst, Z.; Búzás, B.; Friedmann, T.; Schmidhammer, H.; Borsodi, A. Highly potent novel opioid receptor agonist in the 14-alkoxymetopon series. Eur. J. Pharmacol. 1993, 236, 209–215. [Google Scholar] [CrossRef]
- Rittner, H.L.; Brack, A.; Machelska, H.; Mousa, S.A.; Bauer, M.; Schäfer, M.; Stein, C. Opioid Peptide–expressing Leukocytes. Anesthesiology 2001, 95, 500–508. [Google Scholar] [CrossRef]
- Mousa, S.A.; Cheppudira, B.P.; Shaqura, M.; Fischer, O.; Hofmann, J.; Hellweg, R.; Schafer, M. Nerve growth factor governs the enhanced ability of opioids to suppress inflammatory pain. Brain 2007, 130, 502–513. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Prusoff, W.H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar]
- Litchfield, J.; Wilcoxon, F. A simplified method of evaluating dose-effectexperiments. J. Pharmacol. Exp. Ther. 1949, 96, 99–113. [Google Scholar]
Sample Availability: Upon reasonable request and considering the amount of material in hand, 14-methoxycodeine-6-O-sulfate and codeine-6-O-sulfate can be distributed in a timely fashion by Dr. Hosztafi. However, as supplies are finite, we cannot guarantee every request can be accommodated. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ki ± S.E.M. (nM) | Selectivity Ratio | |||||
|---|---|---|---|---|---|---|
| Compounds | [3H]DAMGO (μ) 1 | [3H]IleDelt II (δ) 1 | [3H]U-69593 (κ) 2 | δ/μ | κ/μ | δ/κ |
| 14-OMeC6SU | 3.37 ± 0.48 ** (n = 6) | 345.52 ± 132.52 (n = 5) | 245.57 ± 215.01 (n = 5) | 102.5 | 72.9 | 1.4 |
| C6SU | 96.91 ± 20.3 * (n = 5) | 968.28 ± 471.38 (n = 6) | N.D.3 (n = 5) | 9.9 | - | - |
| Codeine | 736.74 ± 319.63 (n = 4) | N.D. 4 (n = 4) | N.D. 4 (n = 4) | - | - | - |
| Morphine 5 | 0.76 ± 0.08 (n = 7) | 114.21 ± 44.83 (n = 6) | N.D. 4 (n = 4) | 150.3 | - | - |
| Homologous ligand6 | 0.59 ± 0.12 (n = 5) | 3.81 ± 0.88 (n = 6) | 5.51 ± 0.97 (n = 5) | - | - | - |
| Compounds | Tissue Samples (n) | Emax ± S.E.M. (%) | EC50 ± S.E.M. (nM) |
|---|---|---|---|
| 14-OMeC6SU | Rat brain (7) | 128.6 ± 3.69 | 301.1 ± 196.6 |
| Guinea pig brain (6) | 130.2 ± 2.72 | >1000 | |
| Rat spinal cord (5) | 128.8 ± 1.51 | 674.2 ± 157.9 | |
| C6SU | Rat brain (5) | 121 ± 7.27 | >10000 |
| Guinea pig brain (5) | 101.7 ± 1.97 | N.D.1 | |
| Rat spinal cord (5) | 119.3 ± 1.81 | >1000 | |
| Codeine | Rat brain (5) | 109.7 ± 11.48 | N.D.1 |
| Guinea pig brain (5) | 103.6 ± 2.04 | N.D.1 | |
| Rat spinal cord (4) | 102.1 ± 0.49 | N.D.1 | |
| Morphine | Rat brain (7) 2 | 128.8 ± 2.66 | 250 ± 131.7 |
| Guinea pig brain (5) 3 | 119 ± 1.99 | 461.6 ± 250.1 | |
| Rat spinal cord (7) 2 | 124.4 ± 2.09 | 126 ± 72.37 | |
| DAMGO (μ) | Rat brain (7) | 155.3 ± 4.98 | 427 ± 201.9 |
| Rat spinal cord (5) | 137.8 ± 2.65 | 90.14 ± 40.61 | |
| Delt II (δ) | Rat brain (7) | 122.9 ± 1.77 | 44.39 ± 23.51 |
| Rat spinal cord (3) | 113 ± 3.32 | >1000 | |
| U-69593 (κ) | Rat brain (4) | 103 ± 0.84 | N.D.1 |
| Guinea pig brain (7) | 126.8 ± 3.05 | 298.3 ± 176.2 |
| [35S]GTPγS Specific Binding ± S.E.M. (%) | ||
|---|---|---|
| Compounds | Rat Brain | Guinea Pig Brain |
| 14-OMeC6SU | 130.1 ± 7.99 (n = 7) | 128.7 ± 2.08 (n = 5) |
| +10 µM naloxone | 96.24 ± 4.51 ** (n = 5) | 95.84 ± 1.34 *** (n = 5) |
| Emax ± S.E.M. (%) | EC50 ± S.E.M. (nM) | |||
|---|---|---|---|---|
| Compounds (n) | MVD | RVD | MVD | RVD |
| 14-OMeC6SU (13; 6) | 98.31 ± 0.52 ***/###/+++ | 74.81 ± 2.74 ***/×××/$$$ | 239.2 ± 6.15 ***/### | >1000 |
| C6SU (10; 5) | 77.81 ± 3.11 ###/+++ | 16.15 ± 3.25 ××× | >1000 ##/+++/××× | >10000 |
| Codeine (5; 4) | 40.33 ± 4.69 | No effect | >1000 | N.D. 2 |
| Morphine (8; 6) 1 | 46.91 ± 3.23 | No effect | 154.5 ± 93.36 | N.D. 2 |
| DAMGO (8; 5) | 91.91 ± 2.97 | 97.57 ± 4.52 | 122.4 ± 19.55 | 79.81 ± 19.61 |
| Ke ± S.E.M. (nM) | ||||
|---|---|---|---|---|
| Naloxone (µ) | Naltrindole (δ) | nor-BNI (κ) | ||
| Compounds | MVD | RVD | MVD | MVD |
| 14-OMeC6SU | 2.08 ± 0.16 (n = 11) | 2.59 ± 0.53 (n = 4) | 4.01 ± 1.06 (n = 4) | 5.31 ± 1.09 (n = 4) |
| C6SU | 3.25 ± 0.77 (n = 10) | N.D.1 | 2.69 ± 0.93 (n = 4) | 3.05 ± 0.89 (n = 6) |
| DAMGO (µ) | 1.8 ± 0.32 (n = 5) | 1.87 ± 0.4 (n = 5) | N.D.1 | N.D. 1 |
| DPDPE (δ) | N.D.1 | N.D.1 | 0.63 ± 0.33 (n = 6) | N.D. 1 |
| U-69593 (κ) | N.D.1 | N.D.1 | N.D.1 | 0.33 ± 0.14 (n = 3) |
| ED50 (95% Confidence Limit) (µmol/kg) | ||
|---|---|---|
| Time After s.c. Administration (min) | ||
| Compounds | 30 | 60 |
| 14-OMeC6SU | 5.34 a (3.14–9.06) | 9.88 (7.47–13.06) |
| Codeine | 54.01 a (33.97–85.88) | 89.86 (61.54–131.2) |
| Morphine 1 | 6.87 a (4.59–10.27) | 14.17 (10.24–19.62) |
| ED50 (95% Confidence Limit) (µmol/animal) | ||||
|---|---|---|---|---|
| Time after i.c.v. Administration (min) | ED50 s.c./i.c.v. Ratio | |||
| Compounds | 10 | 20 | 30 | |
| 14-OMeC6SU | 0.017 a (0.011–0.026) | 0.018 (0.012–0.028) | - | 314.12 |
| Morphine | - | 0.0552 (0.032–0.095) | 0.039 a,2 (0.022–0.068) | 176.15 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zádor, F.; Mohammadzadeh, A.; Balogh, M.; Zádori, Z.S.; Király, K.; Barsi, S.; Galambos, A.R.; László, S.B.; Hutka, B.; Váradi, A.; et al. Comparisons of In Vivo and In Vitro Opioid Effects of Newly Synthesized 14-Methoxycodeine-6-O-sulfate and Codeine-6-O-sulfate. Molecules 2020, 25, 1370. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25061370
Zádor F, Mohammadzadeh A, Balogh M, Zádori ZS, Király K, Barsi S, Galambos AR, László SB, Hutka B, Váradi A, et al. Comparisons of In Vivo and In Vitro Opioid Effects of Newly Synthesized 14-Methoxycodeine-6-O-sulfate and Codeine-6-O-sulfate. Molecules. 2020; 25(6):1370. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25061370
Chicago/Turabian StyleZádor, Ferenc, Amir Mohammadzadeh, Mihály Balogh, Zoltán S. Zádori, Kornél Király, Szilvia Barsi, Anna Rita Galambos, Szilvia B. László, Barbara Hutka, András Váradi, and et al. 2020. "Comparisons of In Vivo and In Vitro Opioid Effects of Newly Synthesized 14-Methoxycodeine-6-O-sulfate and Codeine-6-O-sulfate" Molecules 25, no. 6: 1370. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25061370