Role of Indole Scaffolds as Pharmacophores in the Development of Anti-Lung Cancer Agents


Department of Biology, University of Massachusetts, Amherst, MA 01003, USA
*
Author to whom correspondence should be addressed.
Molecules 2020, 25(7), 1615; https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25071615
Submission received: 13 February 2020
/
Revised: 24 March 2020
/
Accepted: 30 March 2020
/
Published: 1 April 2020
(This article belongs to the Special Issue Development of Potential Drugs Based on Small Molecules)
Abstract
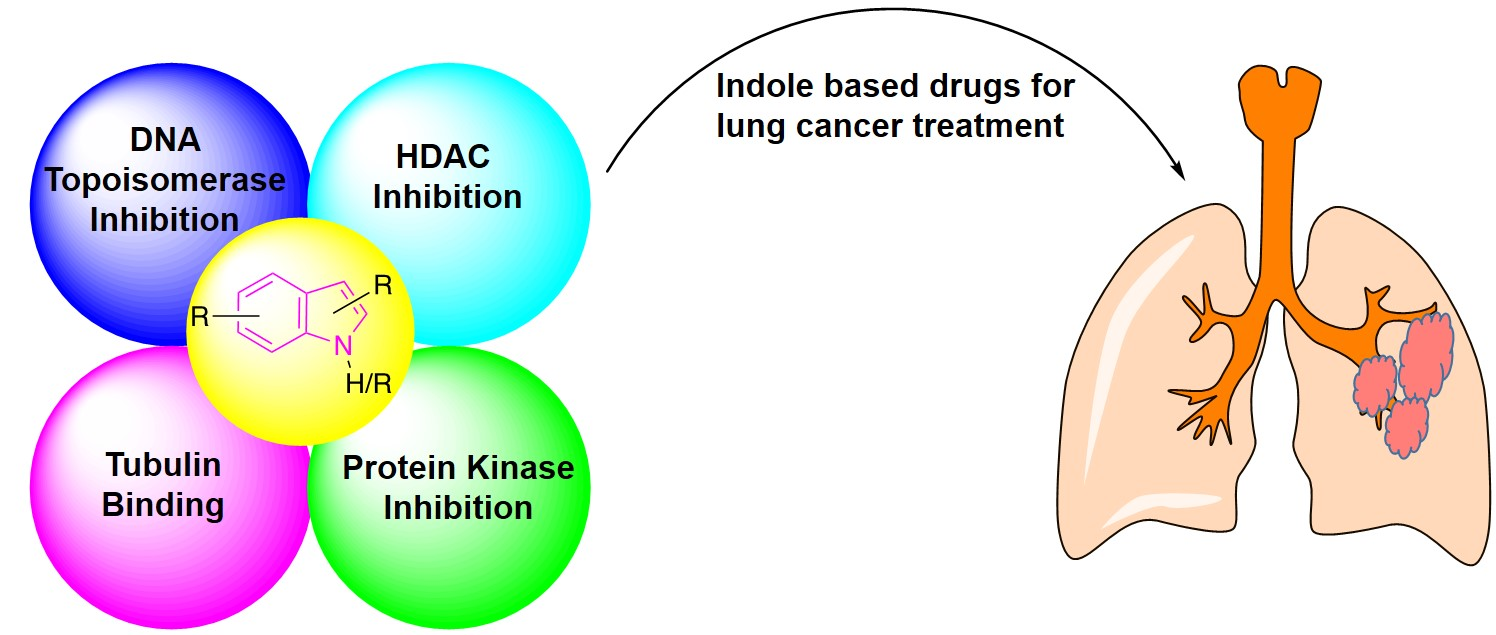
:Lung cancer is the leading cause of death in men and women worldwide, affecting millions of people. Between the two types of lung cancers, non-small cell lung cancer (NSCLC) is more common than small cell lung cancer (SCLC). Besides surgery and radiotherapy, chemotherapy is the most important method of treatment for lung cancer. Indole scaffold is considered one of the most privileged scaffolds in heterocyclic chemistry. Indole may serve as an effective probe for the development of new drug candidates against challenging diseases, including lung cancer. In this review, we will focus on discussing the existing indole based pharmacophores in the clinical and pre-clinical stages of development against lung cancer, along with the synthesis of some of the selected anti-lung cancer drugs. Moreover, the basic mechanism of action underlying indole based anti-lung cancer treatment, such as protein kinase inhibition, histone deacetylase inhibition, DNA topoisomerase inhibition, and tubulin inhibition will also be discussed.

1. Introduction
Lung cancer remains the deadliest disease on record, with approximately 2.09 million new cases reported each year. According to the World Health Organization, it is responsible for 1.7 million cancer deaths worldwide per year [1]. There are two major types of lung cancer: small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC). Among these two types of lung cancers, NSCLC is responsible for approximately 80%–90% of cancerous tumors [2,3]. NSCLC is further divided into three major histological subtypes: squamous-cell carcinoma, adenocarcinoma, and large-cell carcinoma. SCLC and squamous-cell carcinoma are strongly linked to smoking, while adenocarcinoma is the most common type of lung cancer in patients who have never smoked [4].
Treatment of lung cancer is commonly addressed by surgical removal of the affected area and by chemotherapy, which applies a combination of one or more drugs in conjunction with radiation therapy. However, multidrug resistance and the severe side effects associated with chemotherapy persists as one of the major challenges in the treatment of lung cancer.
1.1. Indole and its Pharmacological Significance
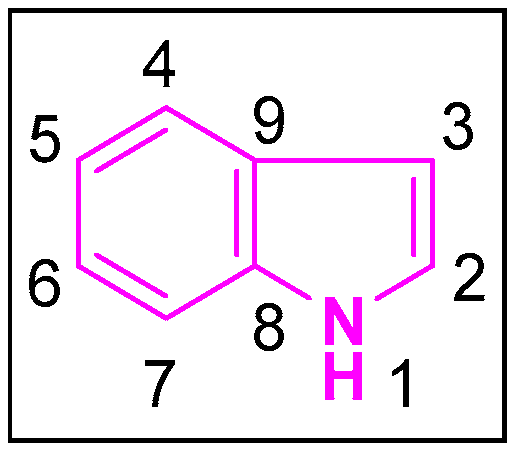
Indole is a pervasive and naturally occurring heterocyclic compound, the structure of which comprises of a six-membered benzene ring tethered to a five-membered pyrrole ring [5] (Figure 1). With its 10 π-electrons, indole follows Huckel’s rule of aromaticity. It is more prone to electrophilic substitution reactions similar to the benzene ring. Molecular orbital studies showed that the 3-position of indole is more reactive towards the electrophilic substitution reactions. Indoles can undergo N-substitution reactions under basic conditions owing to the slightly acidic nature of the—NH bond in indole [6]. The first synthesis of indole was accomplished in 1866 by Adolf von Baeyer [7]. Due to the diverse applications of the indole scaffold in both the biological and pharmacological fields, hundreds of synthetic schemes were published. The most prominent and widely used ones are those reported by Fisher [8], Bischler [9], Hemetsberger [10], Nenitzescu [11], Bartoli [12], and others [13,14].
1.2. Natural Abundance of Indoles
Naturally occurring indole derivatives, such as glucobrassicin and indole-3-carbinol, are commonly found in cruciferous vegetables like cabbage, broccoli, and sprouts [18]. Indole-3-carbinol and its dimeric product 3,3′-diindolylmethane have been found to induce apoptosis in cancer cells [18]. This is a property that has increasingly commanded the attention of researchers from around the world. To get the greatest benefits, indole rich sources belonging to the Brassica family are particularly recommended by the National Cancer Institute to reduce the risk of cancer [18].
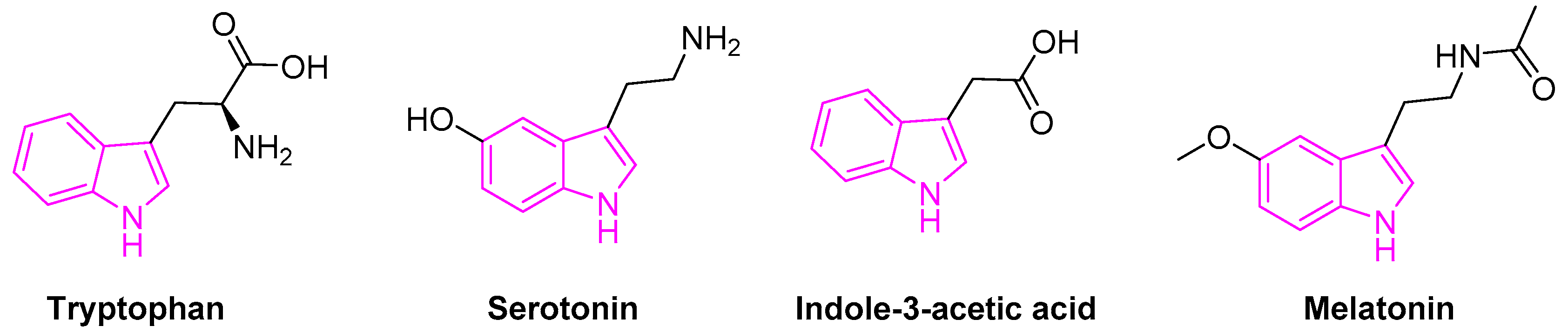
Tryptophan containing indole is an essential amino acid found in many natural resources. It is also a biosynthetic precursor for the tryptamine, 5-hydroxy-tryptophan, an immediate precursor of serotonin (Figure 2).
Serotonin (5-hydroxytryptamine) is an important neurotransmitter associated with the transmission of nerve impulses. It further serves as a precursor to melatonin, a neuro-hormone secreted by the pineal gland. Besides, melatonin is involved in several physiological processes beyond the circadian rhythm [19,20]. Indole-3-acetic acid is the most common and widely studied plant hormones of the auxin class (Figure 2) [21]. It is also the normal urinary tryptophan metabolite observed to be elevated in patients with various forms of liver disease [22].
1.3. Significance of Indole Based Drugs
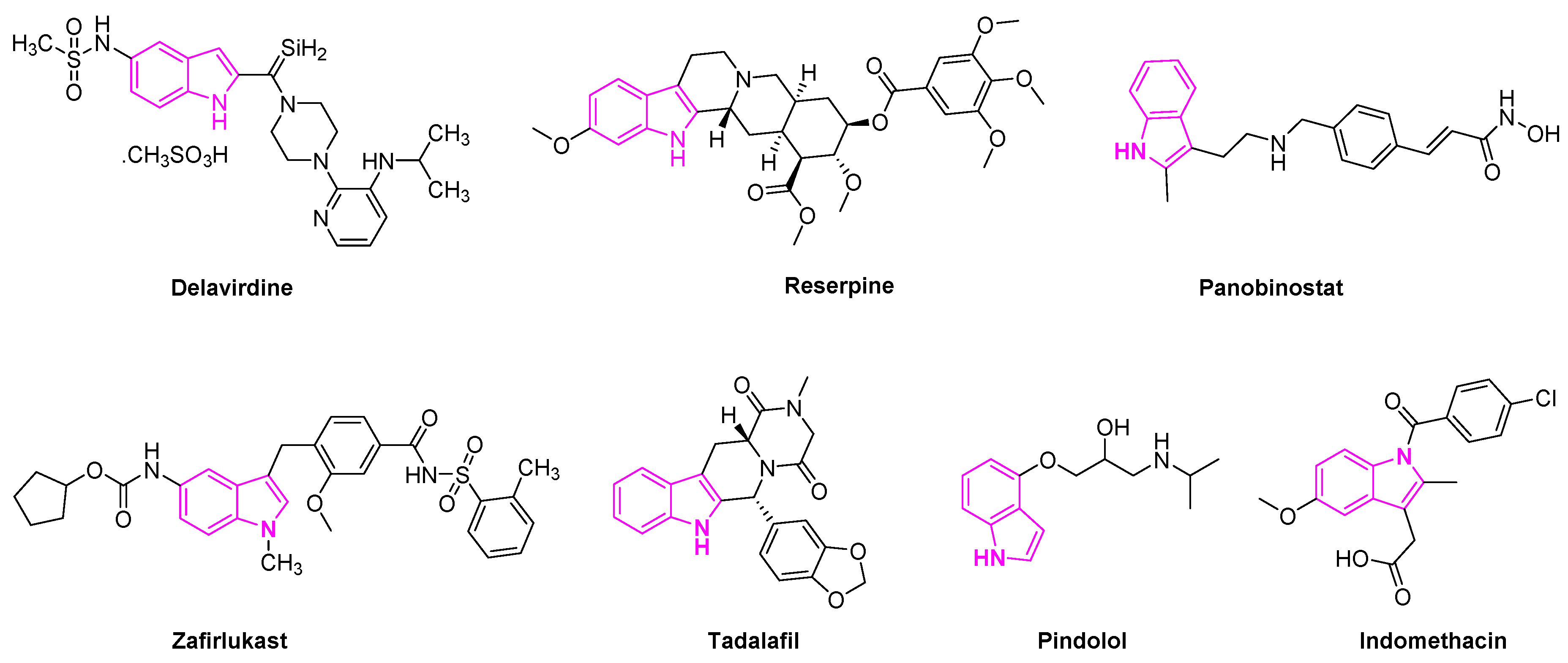
Delavirdine is an indole based anti-retroviral drug approved by the Food and Drug Administration (FDA) in 1997 for the treatment of HIV patients (Figure 3). The drug acts as a non-nucleoside reverse transcriptase inhibitor and an inhibitor of the cytochrome P450 enzyme CYP3A4 [23]. Reserpine is a known indole alkaloid derived from the Rauwolfia serpentina plant. It has been utilized as an anti-psychotic and anti-hypertensive agent to treat high blood pressure [24]. Panobinostat is an oral histone deacetylase inhibitor developed by Novartis and approved to treat multiple myeloma. The drug is currently in phase II trials to treat acute myeloid leukemia [25] and phase I and II clinical trials for the treatment of AIDS [26]. Zafirlukast is the first orally active indole-like leukotriene-receptor antagonist approved by the FDA to treat asthma (Figure 3) [27]. It is mostly used in combination with an inhaled steroid and/or long-acting bronchodilator. Tadalafil is another indole based drug approved for the treatment of erectile dysfunction in men [28]. Pindolol is an antagonist of the serotonin 5-HT1A receptor and it is used to treat hypertension and as an anti-depression agent [29,30]. Indomethacin is a non-steroidal anti-inflammatory drug applied to the treatment of severe osteoarthritis, rheumatoid arthritis, gouty arthritis, and ankylosing spondylitis [31] (Figure 3).
1.4. Methodology for Bibliographic Search
We used the following databases to survey the literature reporting indole scaffold usages for lung cancer in in vitro and in vivo models:
1. Scifinder website at https://scifinder.cas.org/.
2. PubMed website at https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/pubmed/.
In addition, we filtered the available articles based on the following criteria:
a. Articles must have chemical structures of indoles.
b. Articles must have biology data of indoles.
c. We included articles with bioactivity data of indoles specifically in lung cancer.
d. We excluded articles with bioactivity data of indoles other than lung cancer.
2. Indole Derivatives as Anti-Lung Cancer Agents
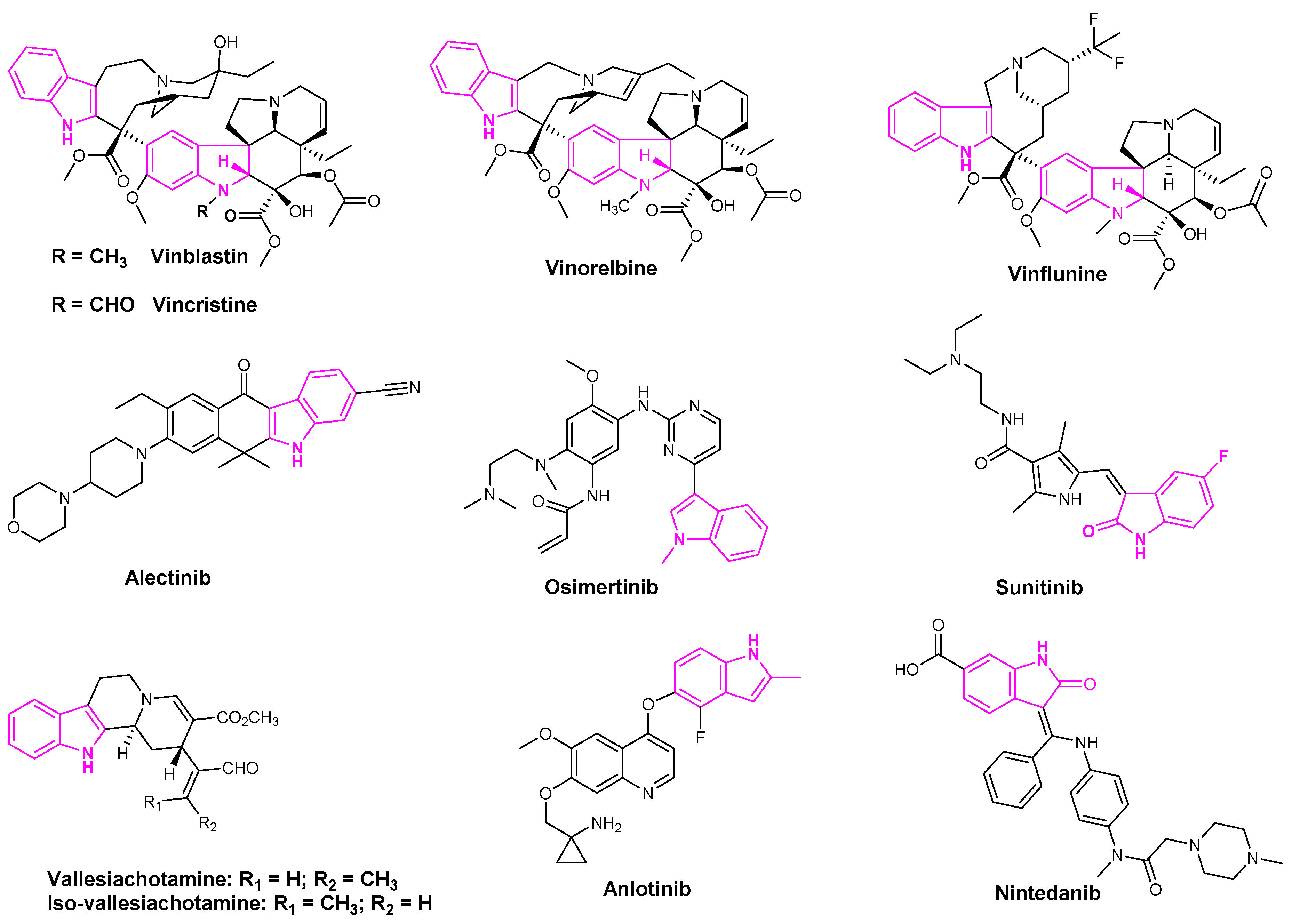
Indole based alkaloids share great historical popularity as anti-cancer drugs. The most prominent among them have been vincristine, vinblastine, vinflunine, and vinorelbine derived from the plant Catharanthus roseus [32] (Figure 4). With over 150 alkaloids, C. roseus is the most well-studied medicinal plant. Vinorelbine is the most commonly used drug to treat lung cancer and vinblastine in combination with cisplatin is used in the treatment of NSCLC [33]. The anti-cancer activity of these derivatives was attributed to their ability to dissolve the mitotic spindles and cell division resulting from the microtubule disruption [33].
Alectinib is a popular indole based oral drug developed by AstraZeneca. FDA approved the drug in 2015 for the treatment of crizotinib-resistant NSCLC [34]. Sunitinib is a multi-targeted receptor tyrosine kinase inhibitor that was approved in the first-line treatment of advanced renal cell carcinoma and gastrointestinal stromal tumors [35]. Osimertinib was also approved by the FDA in 2015 to treat metastatic EGFRT790M mutation-positive NSCLC patients [36]. Osimertinib and sunitinib are the indole based drugs, marketed for the treatment of NSCLC, advanced renal cell carcinoma, and gastrointestinal stromal tumors, respectively [36] (Figure 4). Mishra et al. recently isolated seven indole-based alkaloids and studied the anti-proliferative activities of their isolated compounds. They observed that aglycone indole based alkaloids, namely vallesiachotamine and iso-vallesiachotamine, exhibit anti-tumor activity with The half maximal inhibitory concentration (IC50) values of 4.24 μM and 3.79 μM, respectively, against H1299 human lung cancer cells (Figure 4) [37]. Anlotinib is a novel oral multi-target tyrosine kinase inhibitor, used for the third-line treatment of advanced lung cancer [38].
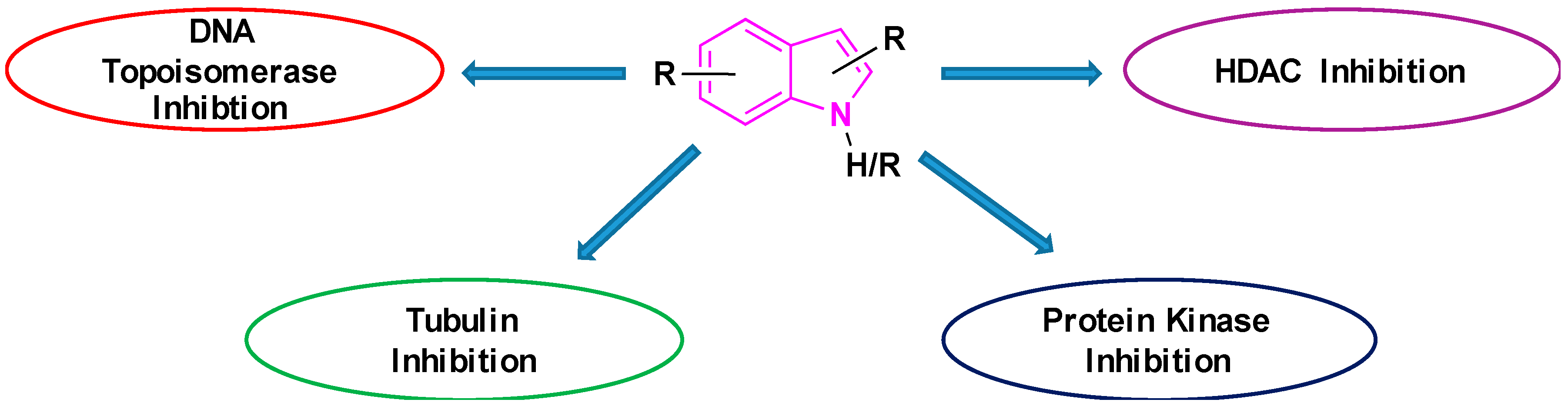
The potency of indoles as anti-cancer agents is exemplified by (i) various indole derivatives, such as indole-3-carbinol, indole-3-carboxaldehyde; (ii) functionalized indoles, such as diaryl-indoles, indolyl chalcones, indolyl azoles, and (iii) bisindole derivatives [39]. Several studies cast light on the anti-cancer efficacy of indole derivatives [40,41,42,43]. Besides, several reviews were submitted in recent years exploring the biological and pharmacological applications of indole derivatives as anticancer agents [39,44,45,46,47]. Though many comprehensive reviews were published, focusing on indole derivatives as anti-cancer agents, so far to our knowledge, there are no reports specifically focused on the efficacy of indole-based compounds in combatting lung cancer. Therefore, we are seeking to fill this gap by presenting a brief review focused on the development of indole-based derivatives as anti-lung cancer agents. In this paper, we included work between 2000 and 2019, of indole-based derivatives in the clinical and subclinical stages of development to combat lung cancer. As it is not possible to include the synthesis of all indole based anti-lung cancer agents, we are reporting herein the synthesis of some of the selected derivatives. We apologize in advance for possible neglect of any relevant work including synthesis on the subject. Basic mechanisms by which indole derivatives exhibit anti-lung cancer properties involve the induction of apoptosis, microtubule inhibition, protein kinase inhibition, histone deacetylase inhibition, and DNA topoisomerase inhibition (Figure 5). In the following sections, we will discuss several derivatives of indoles, developed and understudy for their prospective use against lung cancer, along with their mechanism and synthesis of some of the selected derivatives.
2.1. Indole Derivatives as Kinase Inhibitors
Protein tyrosine kinases are a large multi-gene family comprising key signaling molecules and, therefore, are attractive drug targets. There are two types of tyrosine kinases: (a) receptor kinases, (b) non-receptor kinases. Receptor kinases comprising transmembrane, an extracellular domain, and an intracellular domain. Non-receptor kinases comprise only an intracellular domain with no extracellular or transmembrane. Both kinases play critical roles in cellular signaling, cell growth, cell division, cellular metabolism, and apoptosis. Studies have confirmed that any kind of upregulation or overexpression of the receptors, or any mutation in these receptors, can lead to lung cancer [48,49,50]. Therefore, indole based small molecule inhibitors targeting the kinase receptors have become a promising option for drug discovery scientists in their attempt to battle lung cancer.
2.2. Targeting Akt Signaling Pathway
Nearly 51% of the NSCLC patient samples and 74% of the NSCLC cell lines show activation of phosphatidylinositol 3-kinase/Akt/mTOR pathway [51,52]. Protein kinase B/Akt regulates several key cellular processes such as cell division, cell proliferation, angiogenesis, and cell survival. It activates signaling pathways downstream of tyrosine kinases and dysregulation of these signaling pathways has been observed in several human cancers. Because of the biological roles of Akt signaling pathways, several categories of inhibitors targeting these kinases were developed for NSCLC treatment [53,54].
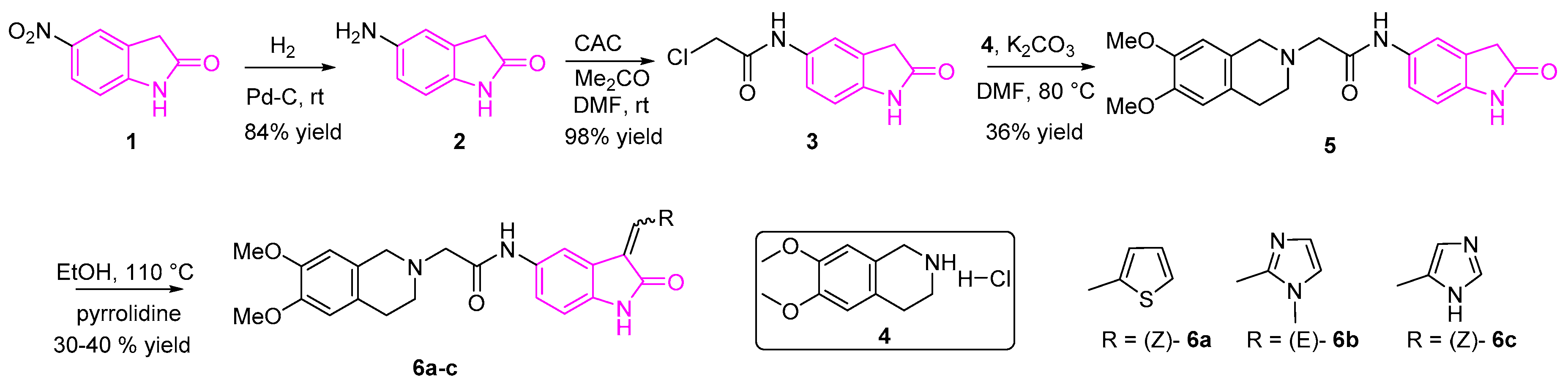
Nesi et al. designed and synthesized a novel set of 2-oxindole-derivatives ((6a–c), (OXIDs)) targeting serine/threonine kinases, 3-phosphoinositide-dependent protein kinase-1 and/or Akt pathways [55]. They revealed the significance of the oxindole core along with the thiophene substituent, in the 3rd position in exerting the antitumor activity. In their studies, they found that the compounds 6a-c showed six-fold higher antitumor activity than Akt inhibitor perifosine and were more potent than sunitinib with 2.5 to 10-fold higher anti-cancer activity. IC50 values ranged from submicromolar to micromolar. These studies revealed the potential of oxindole core in the further development of future therapeutic agents to treat NSCLC [55].
The synthesis of OXIDs is depicted in Scheme 1 [55]. Briefly, compound 2 (5-amino-2-oxindole) was synthesized by the Palladium on carbon (Pd/C) catalyzed hydrogenation of compound 1, followed by condensation with the 2-chloro-acetylchloride resulting in compound 3. The subsequent reaction of intermediate 3 with compound 4 in the presence of potassium carbonate afforded derivative 5, followed by the Knoevenagel reaction involving the corresponding heterocycles resulting in the desired OXIDs compounds (6a–c).
2.3. Epidermal Growth Factor (EGFR) Inhibition
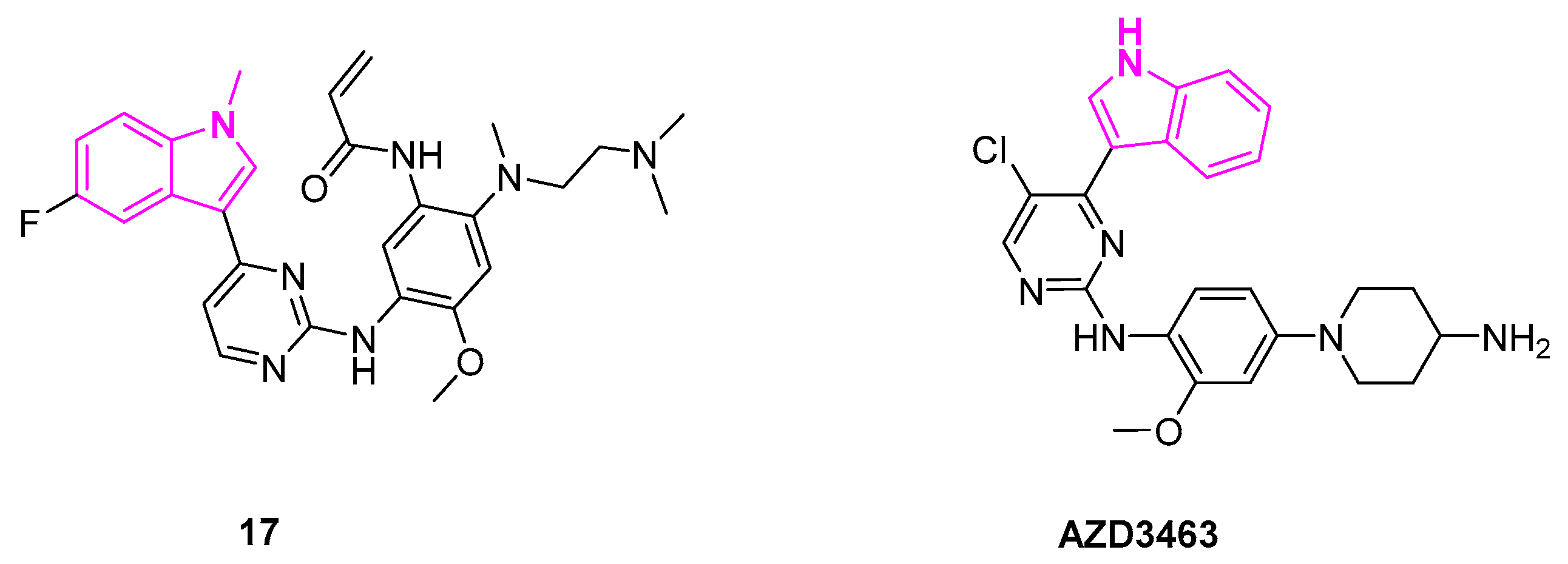
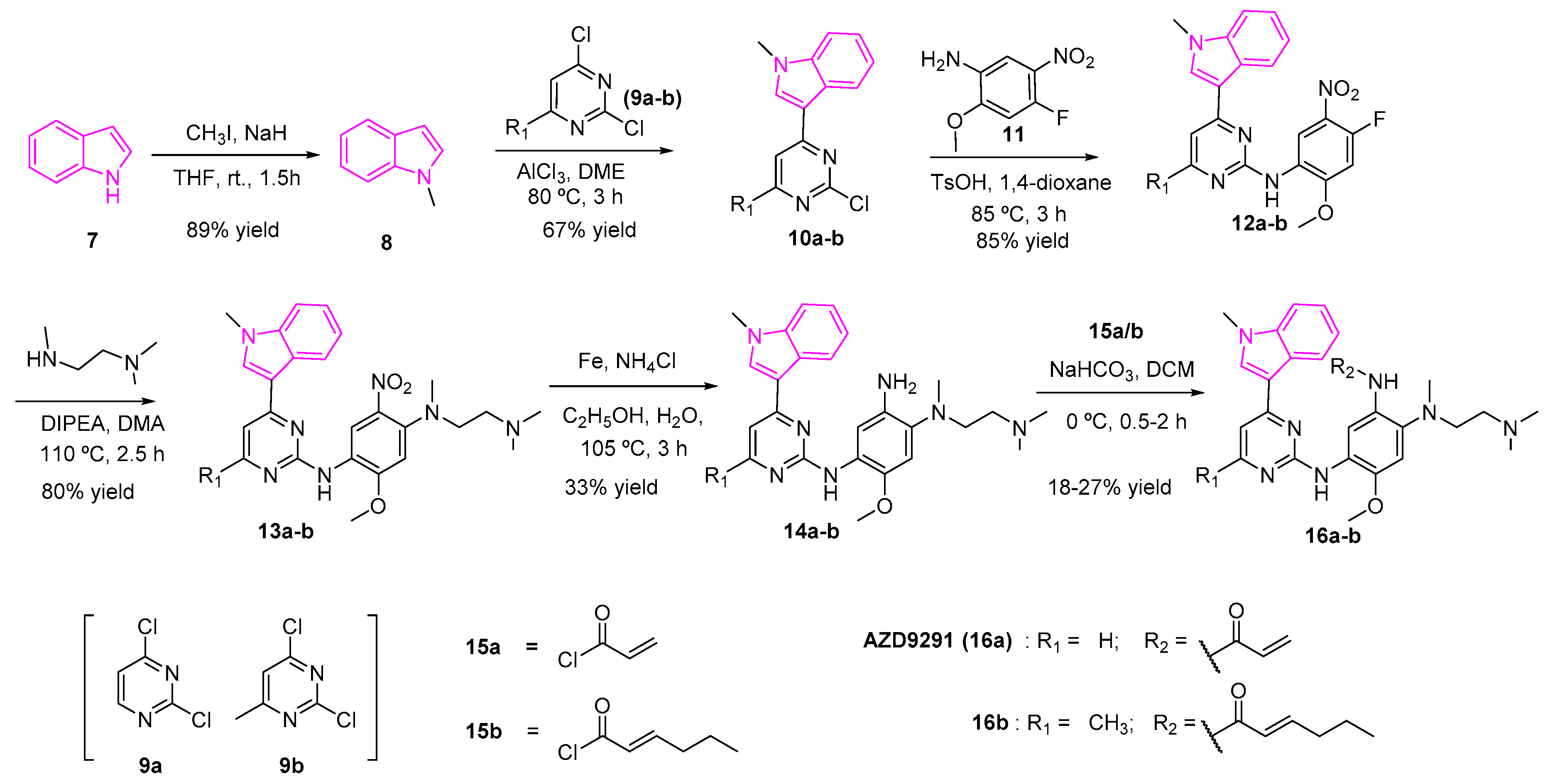
Osimertinib (AZD9291, 16a) is an FDA-approved drug to treat non-small lung cancer as an EGFR inhibitor [36]. However, AZD9291 showed low specificity to EGFR along with severe side effects and cardiotoxicity at high doses (Scheme 2). A structure-activity relationship study of AZD9291 reported by Zhao et al. showed that the fluorinated analog (17, Figure 6) improved selectivity towards EGFRT790M/L858R double mutants with IC50 as low as 8 nM and a 200-fold increase in the selectivity towards wild type EGFR. It was theorized that the fluorinated indole (17) plays an important role in enhancing the binding interaction with T790M active binding domain of EGFR and reducing the demethylation ability of the indole ring, which in turn, can enhance the anti-metabolism ability of AZD9291 derivatives. The fluorinated derivative of AZD9291 (17), however, resulted in the loss of kinase activity. Next, the authors designed compound 16b by the aid of molecular docking studies to obtain an optimized derivative without fluorine substitution. They showed that 16b can serve as a potent EGFRT790M/L858R inhibitor [56]. The synthesis of compounds 16a and 16b is shown in Scheme 2 [56,57].
The first step of the synthesis involved the methylation of indole (7) using methyl iodide and sodium hydride to furnish the methylated indole (8). A mixture of the 2,4-dichloro-pyrimidine (9a) or 2,4-dichloro-6-methyl-pyrimidine (9b) in dimethoxyethane and AlCl3 with 1-methylindole (8) yielded compound 10a or 10b respectively. A nucleophilic aromatic substitution reaction between the amino compound 11 and the chloro pyrimidine 10a/10b in generated the fluoro intermediate (12a/12b). The reaction of the fluoro compounds (12a/12b) with N,N,N-trimethylethane-l,2-diamine in diisopropylethylamine and dimethylacetamide gave the nitro intermediates 13a/13b, which was subsequently reduced to the amines 14a/14b. The latter amines were treated with acryloyl chloride to afford the desired final products (16a/16b) (Scheme 2).
2.4. Anaplastic Lymphoma Kinase Inhibition
Anaplastic lymphoma kinase (ALK) belongs to the family of tyrosine kinases and is regarded as a potential target for the treatment of neuroblastomas as ALK gene mutations and overexpression is frequently observed in neuroblastoma patients. AZD3463 is an ALK inhibitor designed by Astrazeneca to overcome the acquired resistance towards crizotinib (Figure 6). Crizotinib is an FDA-approved drug for the treatment of NSCLC [58]. It is further known to be a potent second-generation ALK (anaplastic lymphoma kinase) inhibitor, which proved effective against a broad spectrum of ALK rearrangements and ALK mutations [59].
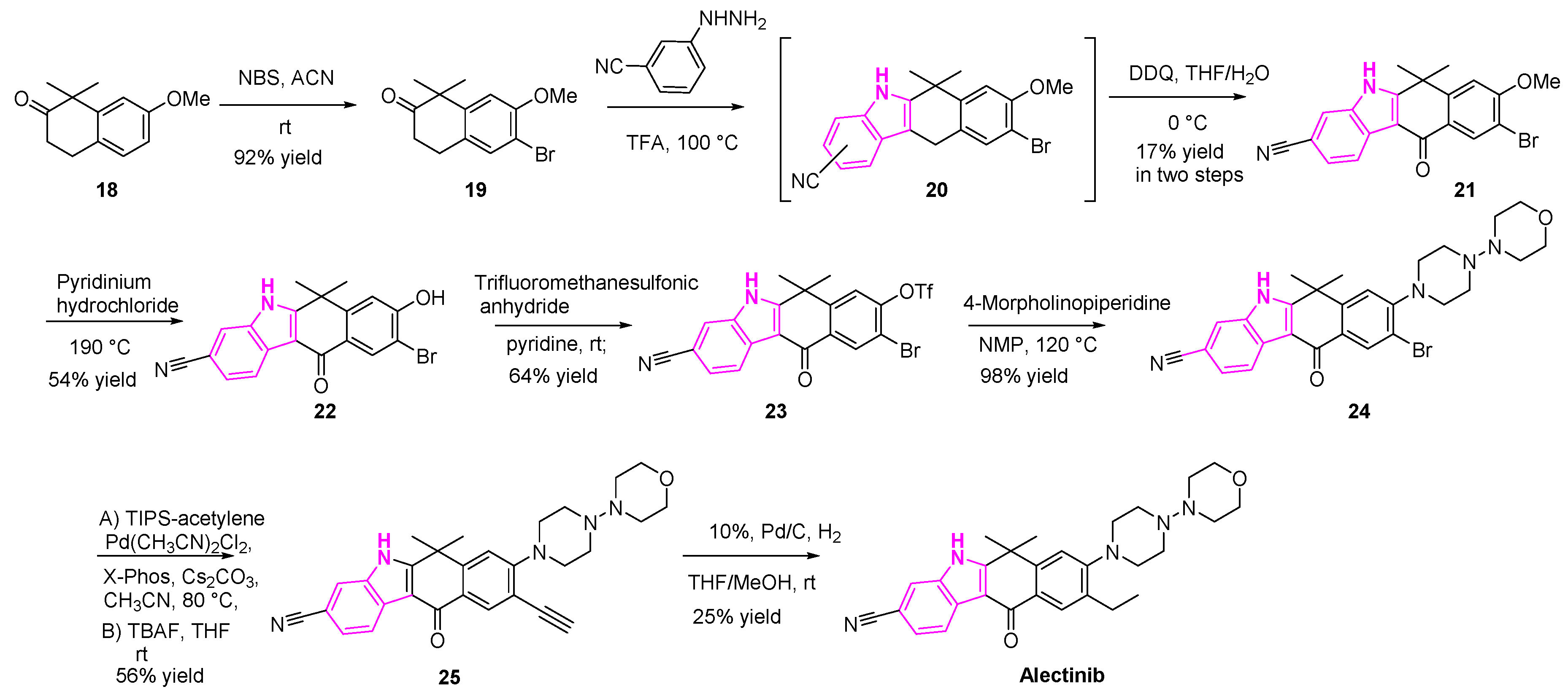
Alectinib, as mentioned earlier, is an FDA approved drug for the treatment of NSCLC (Scheme 3). It is a highly selective and potent ALK inhibitor. Synthesis of alectinib was accomplished in eight steps, as shown in Scheme 3 [60,61].
The bromination of compound 18 with N-bromosuccinimide, followed by the Fischer indole synthesis, resulted in the regioisomeric mixture (1:1, 1-CN- and 3-CN-derivatives) of compound 20. The desired isomer was isolated and the 2,3-Dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) oxidation was conducted, resulting in the ketone 21. Demethylation of compound 21 with pyridinium hydrochloride generated the alcohol (22). The resulting alcohol (22) was then triflated and then reacted with 4-morpholinopiperidine to provide the bromo compound (24). The bromo intermediate (24) was then subjected to Sonogashira coupling with acetylene, followed by hydrogenation of the terminal alkyne generated the desired compound (alectinib), as shown in Scheme 3.
2.5. Protein Kinase C Inhibition
Protein kinase C (PKC) is a family of structurally related serine-threonine kinases that can be activated by G-protein coupled receptors [62]. PKC is implicated in several cellular functions and the regulation of survival signals in various cell types and, therefore, PKC has become a drug target for the development of anti-cancer drugs. The activation of PKC by various oncogenes can lead to the malignant transformation and abnormal expression of PKC isoforms, as detected in the head, neck, and other types of cancers.
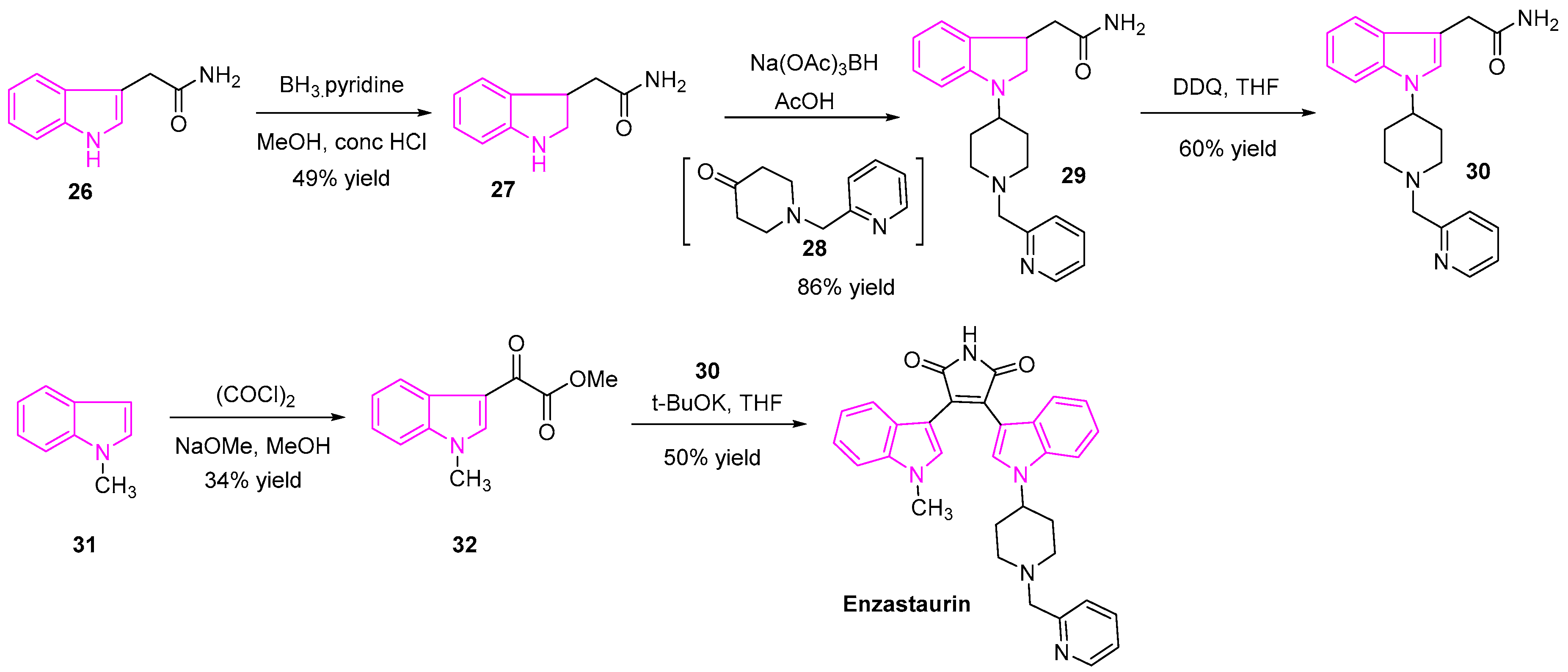
Enzastaurin is a popular anti-tumor agent, which showed promising results with T-cell and B-cell lymphoma, multiple myeloma and other solid tumor malignancies (Scheme 4). Enzastaurin is a potent inhibitor of PKC and PI3K/AKT pathway. It is found to effectively inhibit tumor cell growth in both in vitro and mouse xenograft models [63]. Nakajima and his team showed that enzastaurin displayed cell growth inhibition with IC50 of 3–10 µmol/L, as exhibited in a panel of both SCLC and NSCLC cell lines [64,65]. Besides, they found that enzastaurin can suppress tumor-induced angiogenesis in A549 NSCLC xenografts [66]. The drug is undergoing clinical trials to evaluate its efficacy in the treatment of NSCLC and several other types of cancers [67].
The synthesis of enzastaurin is shown in Scheme 4 [65]. Briefly, the reduction of indole-3-acetamide (26) with BH3.pyridine gave the indolin-3-acetamide (27) followed by reductive amination with the ketone (28) resulted in the intermediate 29. Compound 30 was obtained by oxidation of compound 29 using DDQ. On the other hand, the indole-3-glyoxylate ester (32) was synthesized from 1-methylindole (31) and oxalyl chloride. Finally, condensation of the amide intermediate (30) with indole-3-glyoxylate ester (32) in the presence of a base generated enzastaurin [67] (Scheme 4).
2.6. GSK Inhibition
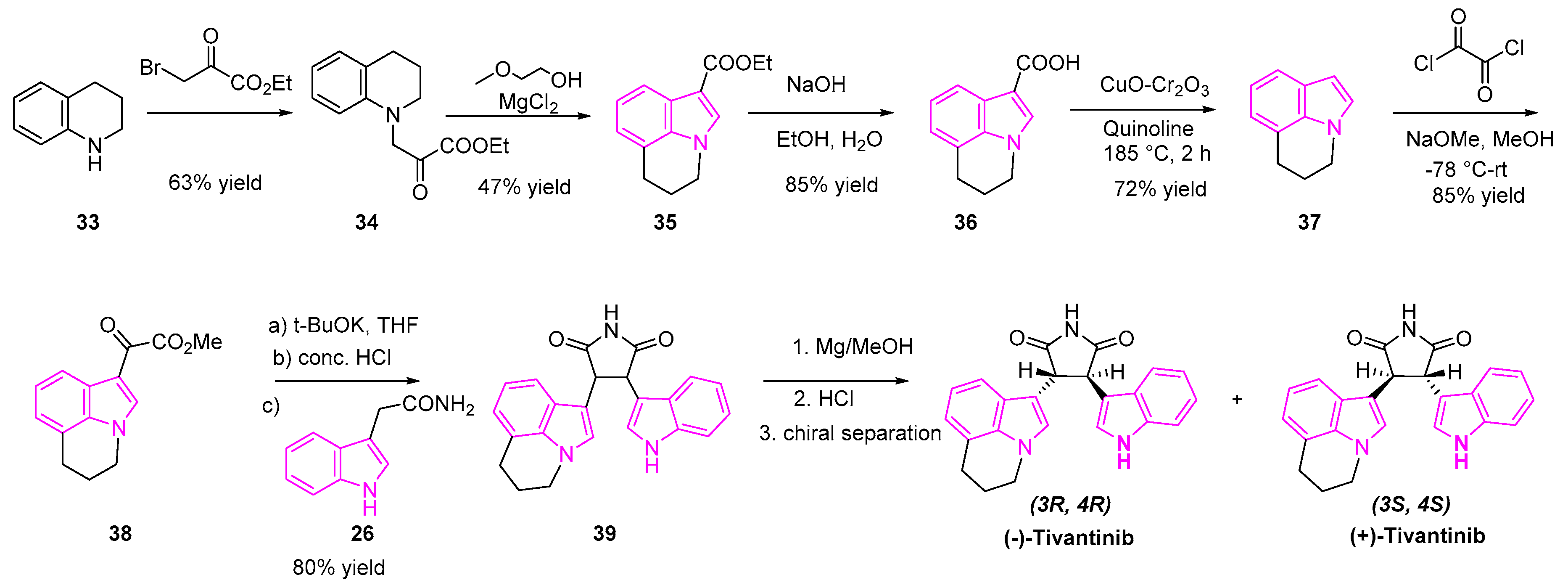
Tivantinib is an oral, selective indole based small molecule MET inhibitor, being evaluated in advanced clinical trials for the treatment of NSCLC (Scheme 5). Early reports showed that tivantinib is a potent and highly selective inhibitor of receptor tyrosine kinase (cMET). However, further investigation led by Rix et al. revealed glycogen synthetase kinase (GSK)3α and GSK3β to be novel tivantinib targets. In their detailed study of mechanism, they found that tivantinib acts by the simultaneous inhibition of GSK3α and GSK3β leading to apoptosis in lung cancer cells [68]. Their experiments demonstrated that (−)-tivantinib was more potent than (+)-tivantinib in the inhibition of GSK3α and GSK3β. The latest investigation by Scagliotti et al. has shown that a combination of tivantinib and erlotinib can enhance treatment efficacy in patients with previously treated, EGFR mutant, non-squamous NSCLC, as compared to erlotinib alone [69].
The synthesis of tivantinib is shown in Scheme 5 [69,70]. The reaction of the tetrahydroquinoline (33) with the bromo-ethylpyruvate yielded the ketoester 34, which was transformed to the ester indole 35 under anhydrous magnesium chloride treatment. Ester 35 was hydrolyzed to carboxylic acid intermediate 36 followed by decarboxylation reaction using copper chromite and quinoline afforded the indole 37, which was treated with oxalyl chloride and methanol to obtain the ketoester 38. Condensation of the ketoester 38 and indole-3-acetamide (26) generated the intermediate 39 as a racemic mixture of tivantinib. Preparative chiral HPLC purification yielded two optically pure diastereoisomers of (3R, 4R) (−)-tivantinib and (3S, 4S) (+)-tivantinib (Scheme 5).
2.7. Inhibition of Multiple Receptor Kinases
Anlotinib is a novel inhibitor targeting multiple receptor tyrosine kinases, namely vascular endothelial growth factor receptor type 2 and 3 (VEGF 2 and 3), the platelet-derived growth factor b (PDGFb), and the fibroblast growth factor 2 (FGF-2) [71] (Figure 4). Studies revealed that VEGF is a critical growth factor in tumor angiogenesis. Along with PDGF and FGF-2, it stimulates proliferation, migration, survival, and new vessel formation of endothelial cells in tumors. Studies showed that inhibition of these angiogenic receptor tyrosine kinases can be very effective in the treatment of tumors. Notably, the inhibitory effect of anlotinib was much more pronounced than with sunitinib, sorafenib, and nintedanib suggesting it as a potent anti-lung cancer drug. Anlotinib has emerged as a potential third-line treatment in advanced NSCLC patients [72].
Nintedanib is an oral, small molecule multiple kinase inhibitor (Figure 4). This drug in combination with docetaxel was approved worldwide for the treatment of NSCLC [73]. It functions as a tyrosine kinase inhibitor targeting three angiokinase inhibitors including VEGF (1–3), PDGF receptors (α/β) and fibroblast growth factors (1–3). Both in vitro and in vivo studies in human NSCLC xenografts have confirmed that by the inhibition of both AKT and mitogen-activated protein kinases signaling pathways, nintedanib resulted in the inhibition of cell proliferation, apoptosis in endothelial cells, and the cell types involved in angiogenesis [74].
2.8. Tubulin Inhibition
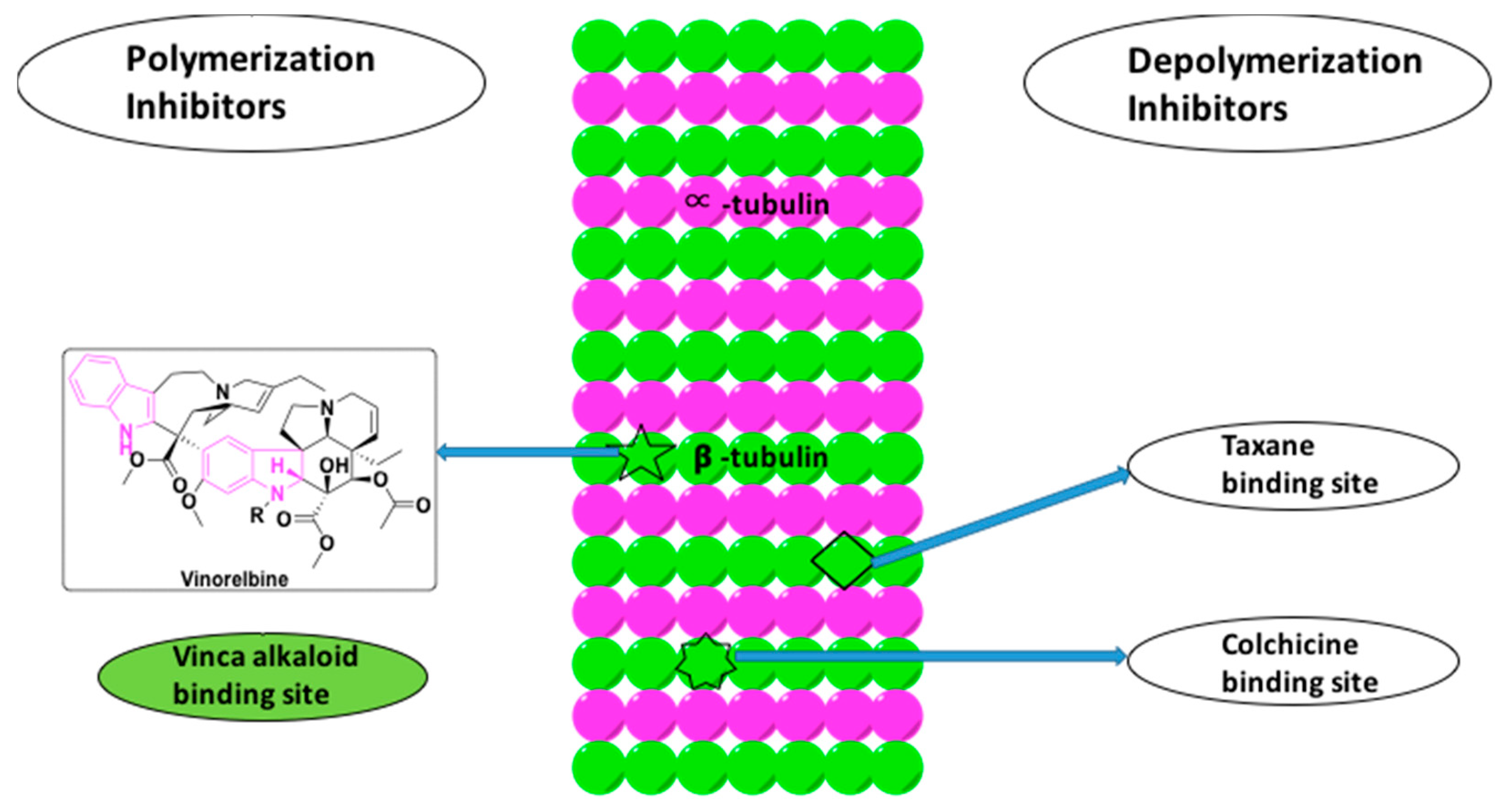
Tubulin is a globular protein comprising α and β-heterodimers, which polymerize to form the biopolymers of microtubules. Being the third major component of the cytoskeleton, microtubules play a critical role in cell division, in addition to aiding in both inter and intra-cellular movement and maintaining cell shape [75]. As microtubules play a key role in cell division, they are attractive targets for the development of anti-cancer agents.
Tubulin inhibitors interfere with the polymerization and depolymerization of the microtubules. They are categorized as either tubulin stabilizing agents or destabilizing agents (Figure 7). While the tubulin stabilizing agents ultimately lead to the polymerization of microtubules by inhibiting depolymerization, the destabilizing agents function by inhibiting polymerization, thereby shortening the microtubules [76]. The indole based alkaloids and their derivatives, such as vincristine, vinblastine, vinorelbine, vinflunine, vindesine belong to the class of polymerization inhibitors that bind to the β-subunit of tubulin at a distinct region called as the vinca-binding site, as shown in Figure 7 [76].
Vinorelbine is a semi-synthetic indole based alkaloid extracted from C. roseus (Figure 7). It is a known microtubule inhibitor with selectivity towards mitotic microtubules. It disrupts the mitotic spindle formation and ultimately prevents cell division. Vinorelbine is used in combination chemotherapy with cisplatin and cetuximab to treat NSCLC [77]. Besides vinorelbine, vinflunine was also shown to have the potential to treat the NSCLC in the adjuvant chemotherapy with platinum-based drugs (Figure 4) [78]. Among the other counterparts, such as vincristine, vinblastine, and vinorelbine, vinflunine is found to have higher activities in vivo with low drug resistance. Therefore, vinflunine has evolved to be a more potent drug in lung cancer treatment [33].
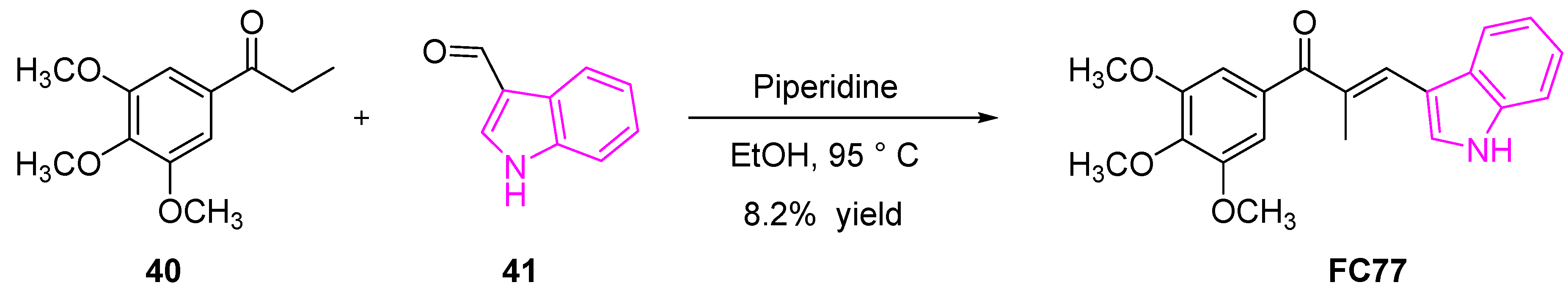
Cong et al. reported an indole-chalcone derivative (FC77, Scheme 6) that can arrest cancer cell growth by binding to tubulin, exhibiting a similar mechanism of action as colchicine. FC77 exhibited a potent growth inhibitor of a majority of NCI-60 human cancer cell lines with GI50 ~ 6 μM [79]. The ability of FC77 to retain its cytotoxicity against multi-drug resistant cancer cell lines (with nanomolar GI values) and low cytotoxicity levels towards normal mobilized peripheral blood cells revealed that FC77 may be a potential therapeutic agent in treating multi-drug resistant cancers such as NSCLC. Moreover, the direct interaction of FC77 with tubulin and inhibition of microtubule dynamics with IC50 values of 3 nM compared to popular drugs such as paclitaxel and vincristine with IC50 values of 14 nM and 37 nM, respectively, makes it an attractive microtubule-targeting agent for the treatment of multidrug-resistant cancers [79].
The synthesis of FC77 was accomplished in a single step, as shown in Scheme 6 [79]. The condensation reaction between the acetophenone derivative (40) with the indole-3-carboxaldehyde (41) in the presence of piperidine afforded the target compound (FC77).
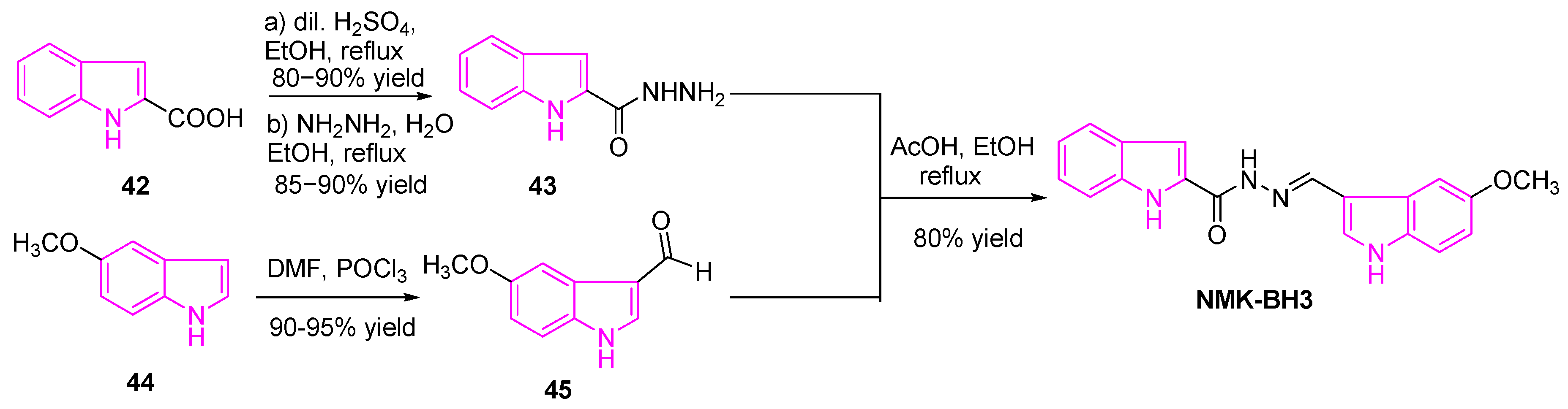
Das et al. recently synthesized a series of six bis(indolyl)-hydrazide−hydrazones (NMK-BH compounds), in order to develop novel and efficient microtubule-targeting chemotherapeutic agents against lung cancer. They achieved this by linking the two indole rings with an active pharmacophore linker, as shown in Scheme 7. Cytotoxicity studies of these compounds against human lung adenocarcinoma A-549 cells revealed that NMK-BH3 with the electron-donating 5-OMe group and the indole rings tethered to the C-2 position of the indole-A was found to be more potent than other combinations. The mechanism of action of NMK-BH3 in NSCLC (A549 cells) revealed the disruption of the mitotic spindle and depolymerization of the interphase microtubule network ultimately led to apoptotic cell death. This is signified by the overexpression of Bax, p53, caspase 3, and caspase 9 and decrease in the Bcl-2 expression [80]. Their experiments revealed that NMK-BH3 was more sensitive to human lung adenocarcinoma A549 cells with IC50 of ~2 μM, while highly resistant to normal human lung fibroblasts (WI38) with IC50 of 48.5 μM (24 fold).
The synthesis of NMK-BH3 was achieved as shown in Scheme 7 [80]. A solution of indole-2-carboxylic acid (42) was refluxed in ethanol with a catalytic amount of dilute sulfuric acid, to obtain the corresponding ethyl ester intermediate, which was treated with hydrazine hydrate to obtain the intermediate 43. On the other hand, the preparation of the indole-3-carboxaldehyde 45, via treatment of the methoxy indole 44 with phosphorous oxychloride and anhydrous DMF. Finally, condensation of hydrazide intermediate 43 and indole-3-carboxaldehyde 45 in the presence of a catalytic amount of glacial acetic acid, and the mixture was refluxed at 80 °C for 5 h. The resulting mixture was then cooled to collect the desired target (NMK-BH3).
2.9. Inhibition of DNA-Topoisomerases
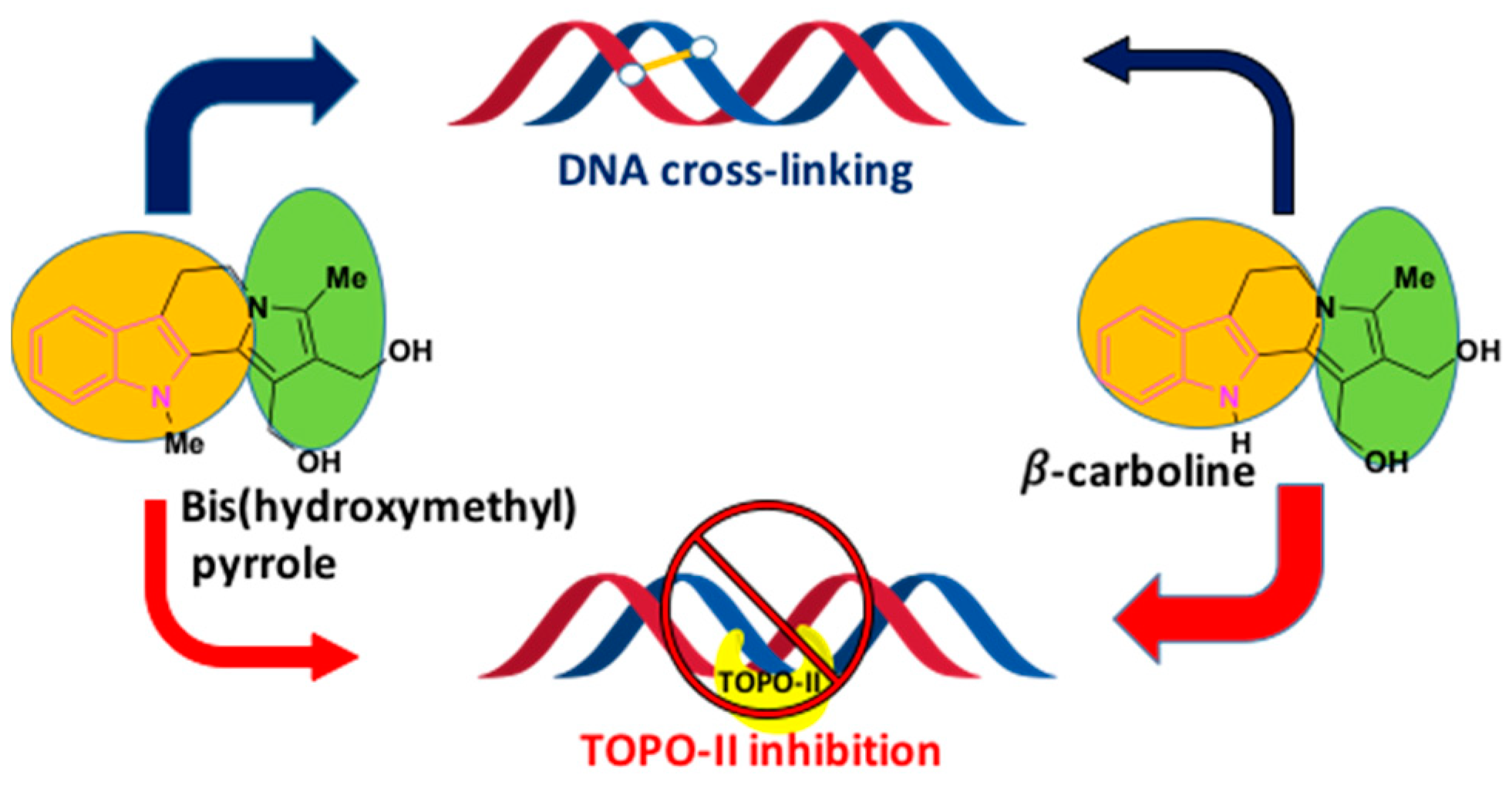
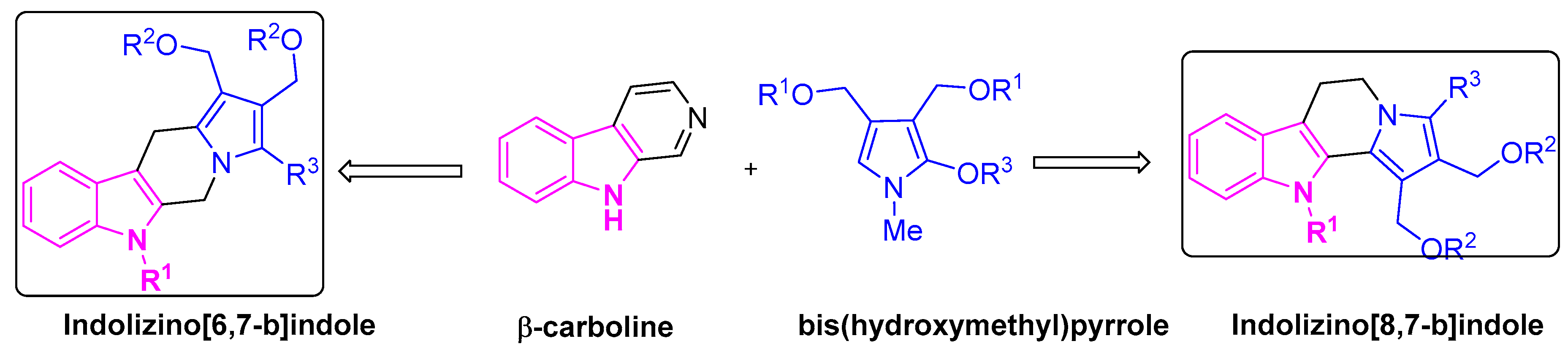
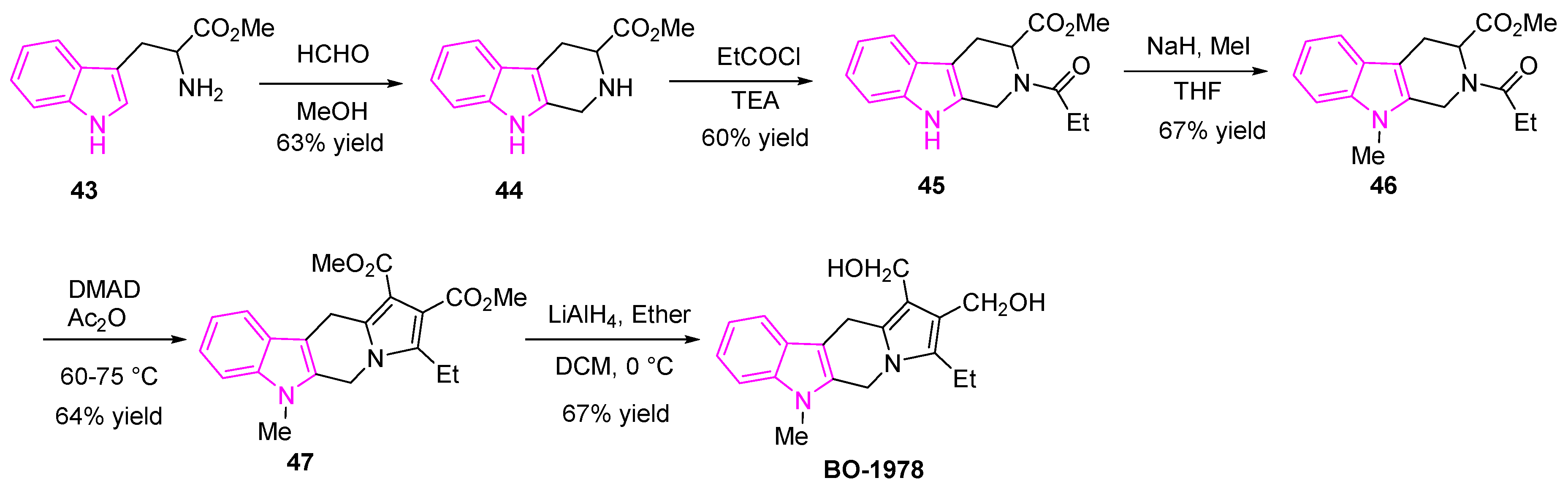
Owing to the drug resistance and acute toxicity associated with lung cancer treatment, the combination of two or more pharmacophores has become an attractive strategy in drug discovery and has evolved as an effective tool in treating lung cancer patients. To overcome the multi-drug resistance in NSCLC patients, Chen et al. adapted the above strategy and developed hybrid molecules to improve drug efficacy. They designed and synthesized a series of indolizino[6,7-b]indoles, incorporating two pharmacophores: β-carboline group and a bis(hydroxymethyl)pyrrole group with topoisomerase (Topo) I/II inhibition and DNA cross-linking abilities respectively [81,82] (Figure 8). The hybrid derivative (BO-1978) was found to be effective in the suppression of lung adenocarcinoma A549 cells through the inhibition of Topo I/II and the induction of DNA cross-links resulting in DNA damage and a resulting cell cycle arrest. The hybrid derivative (BO-1978) was synthesized as shown in Scheme 8 [81,82].
The tetrahydro-β-carboline 44 was synthesized by adding formaldehyde to L-tryptophan methyl ester hydrochloride (43). The reaction of compound 44 with propionyl chloride in triethylamine provided the amide (45). The nitrogen of the indole intermediate (45) was methylated using sodium hydride and methyl iodide to generate the compound 46. The reaction of 46 with dimethyl acetylenedicarboxylate (DMAD) in acetic anhydride yielded the diester 47, which was further reduced with lithium aluminum hydride to afford the desired compound (BO-1978) (Scheme 8).
The hybrid derivative compound (BO-1978) showed the synergistic effect of both the pharmacophores, which incorporates the anti-proliferative activity and Topo I and Topo II inhibition of β-carboline and the potent anti-tumor activity of bis-hydroxy-methylpyrrole, which is known to induce the DNA cross-linking (Figure 8).
In the same front, Chang et al. developed a series of novel indolizino[8,7-b]indole hybrids (Figure 9) [83].
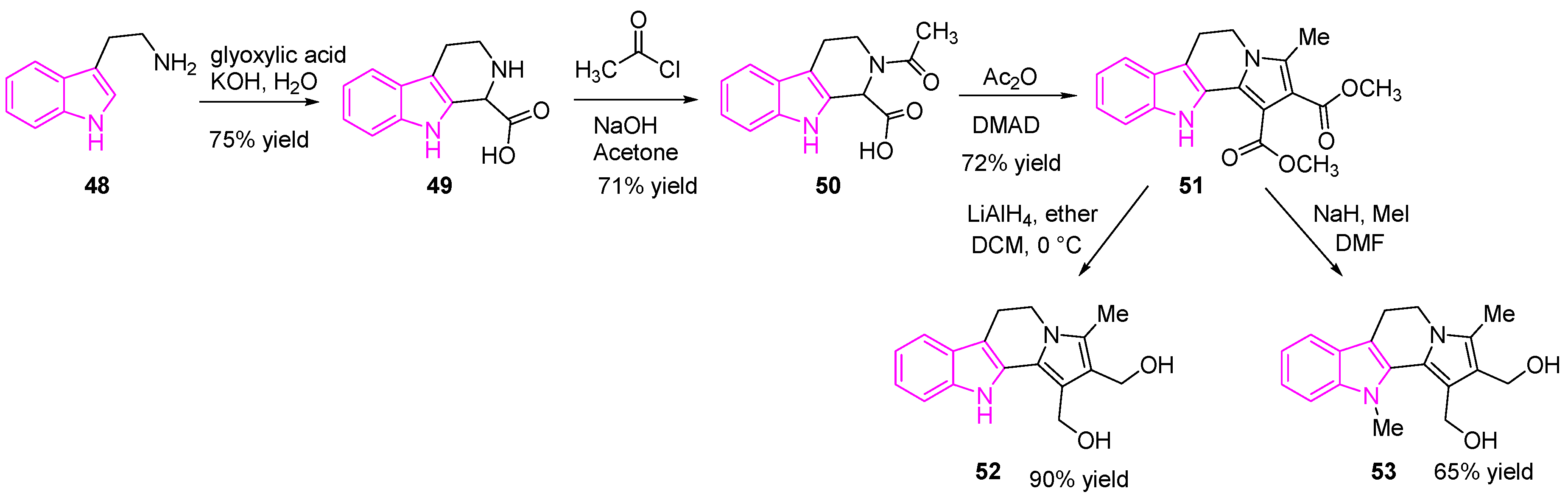
As shown in Figure 8, the substituent on the indole nitrogen is found to play a key role in determining the activity of the hybrid molecule. The presence of –Me group (53) makes them potent DNA cross-linking agents, whereas –NH derivatives (52) are potent TOPO-II inhibitors. The resulting compounds were most sensitive to SCLC (H526) cells both in vitro and in vivo studies. One of the compounds 52, was found to have potent Topo II inhibitory activity with significant tumor suppression in nude mice bearing SCLC H526 xenografts (Scheme 9). Compound 52 was shown to be more potent than cisplatin and etoposide and possessed the comparable potency of irinotecan [83].
The synthesis of compounds 52 and 53 is shown in Scheme 9 [83]. Briefly, the reaction of tryptamine hydrochloride (48) with an aqueous solution of glyoxalic acid and potassium hydroxide generated the tetrahydro-β-carboline intermediate (49). Compound 50 was prepared by the addition of acetyl chloride to 49, then DMAD was added to compound 50 in acetic anhydride generated the tetracyclic diesters (51). Finally, the reduction of 51 with lithium aluminum hydride afforded the desired tetracyclic diol (52), while the treatment of intermediate 51 with sodium hydride and methyl iodide resulted in desired N-methyl-tetracyclic diol (53).
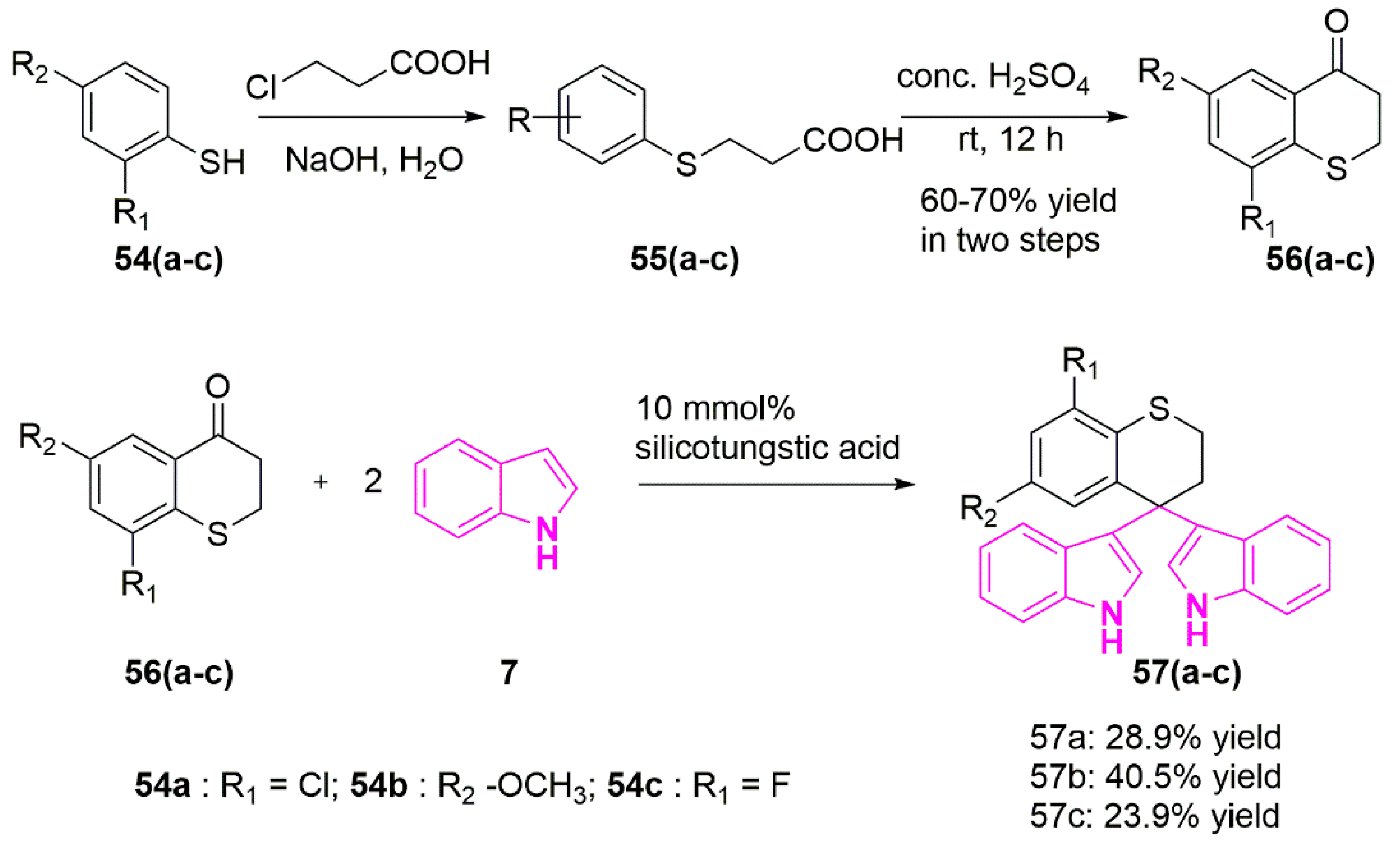
In an attempt to develop effective Topo-II inhibitors as potent anti-cancer agents, Song et al. synthesized a novel series of bisindolylalkanes analogs such as 3,3′-(thiochroman-4,4-diyl)bis(1H-indole) and tested against a variety of human cancer cell lines (Scheme 10). Notable among them, compounds 57(a-c) were found to be potent against A549 cells with IC50 values of ~9 µg/mL [84].
The synthetic scheme of compounds 57(a–c) was shown in Scheme 10 [84]. The first step involved the condensation of substituted benzene thiols 54(a–c) with 3-chloropropanoic acid and sodium hydroxide in water, followed by treatment of corresponding intermediate 55(a–c) with concentrated sulfuric acid to obtain the intermediates 56(a–c). A mixture of indole (7) and substituted thiochroman-4-ones 56(a–c), in the presence of silicotungstic acid was refluxed in ethanol yielded the desired compounds (57(a–c)).
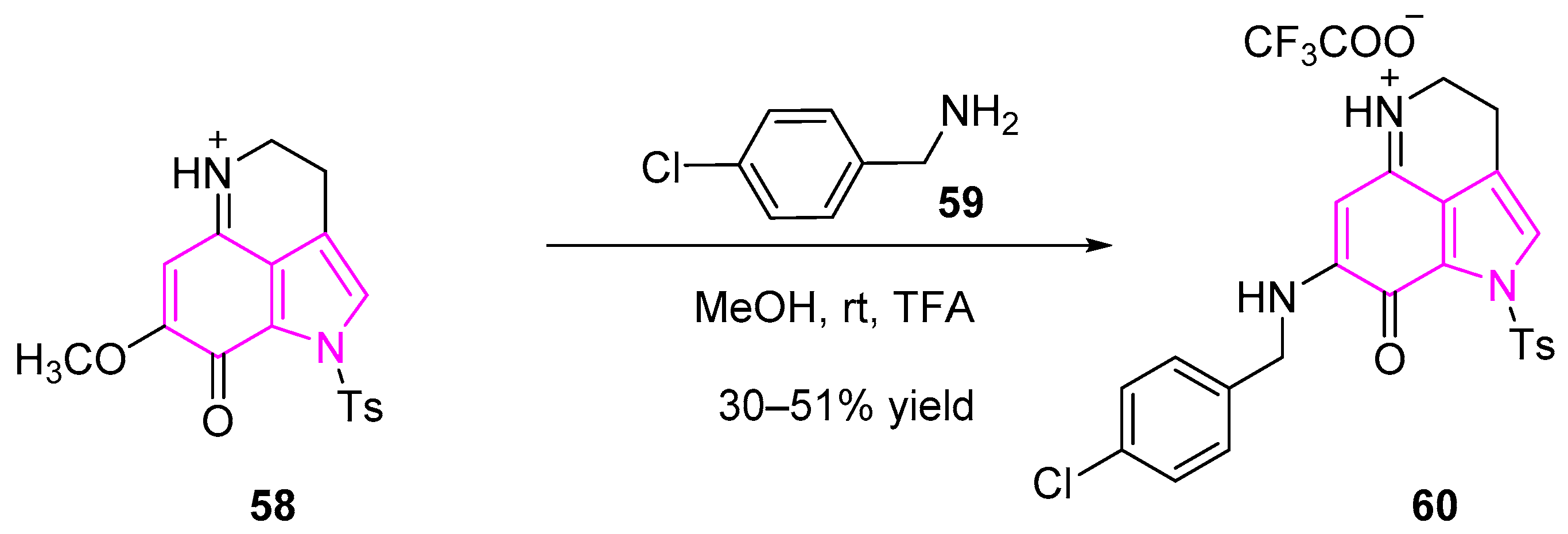
Makaluvamine is an indole based alkaloid metabolite isolated from a marine sponge Zyzzya. This compound showed significant cytotoxic activity and DNA topoisomerase II inhibition [85]. Nadkarni et al. synthesized a series of makaluvamine analogs and studied their biological activities against various human lung cancer cell lines (Scheme 11). One of the compounds (60) was found to be most potent against human lung cancer cell lines of both human adenocarcinoma (A549) and carcinoma (H1299) cells with IC50 of 0.3 and 0.58 µM, respectively. Further studies revealed that the compound induced both apoptosis and S-phase cell cycle arrest [86].
The synthesis of makaluvamine 60 is represented in Scheme 11 [86]. Compound 60 was synthesized in one-step synthesis from the tricyclic compound 58. Condensation of compound 58 with 4-chloro-benzylamine (59) in anhydrous methanol followed by treatment with trifluoroacetic acid (TFA) afforded the desired makaluvamine compound (60).
2.10. Histone Deacetylase (HDAC) Inhibitors
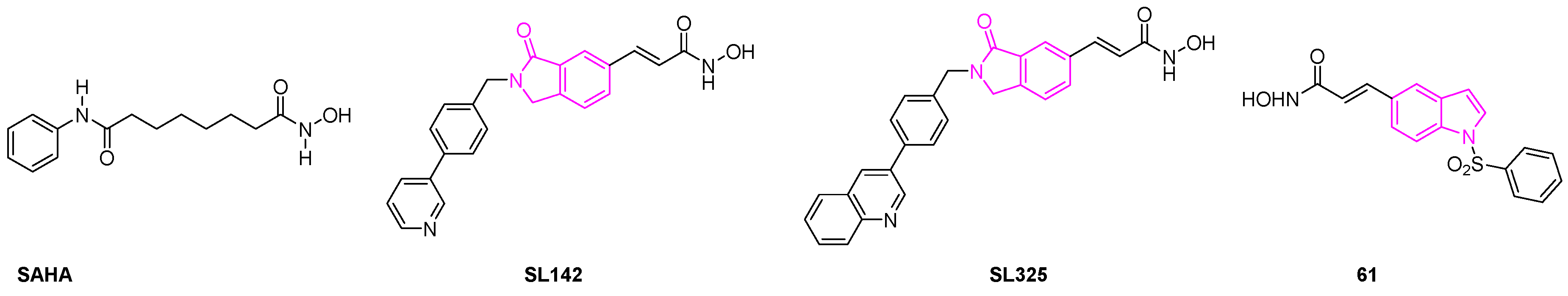
HDAC enzymes are involved in a variety of cellular processes such as cell proliferation, cell differentiation, and apoptosis [87]. Research studies show that the dysregulation of HDAC can lead to several types of tumor malignancies. HDAC inhibitors have, thus, evolved as a new class of anti-cancer agents because of their efficacy to arrest cell growth, cell differentiation, and cell death by apoptosis [88,89]. In this direction, Han et al. investigated the anti-cancer activity of A549 and H441 lung cancer cells using two novel cyclic amide-bearing hydroxamic acid-based HDAC inhibitors SL142 and SL325. Their studies revealed that both derivatives showed more potent cell growth inhibition and cell death than the hydroxamic acid-based HDAC inhibitor suberoylanilide hydroxamic acid (SAHA) (Figure 10). Besides, these compounds can also induce significant caspase activity and subG0/G1 population in human epithelial (H441) and adenocarcinoma (A549) lung cancer cells, thereby demonstrating that they are more effective than SAHA [90,91].
A new class of 1-arylsulfonyl-5-N-hydroxyacrylamide indoles was reported by Lai et al., as potent histone deacetylase inhibitors. Further investigation revealed that the lead compound 61 demonstrated GI50 of 1 μM against the A549 cell lines. They demonstrated that the N-hydroxyacrylamide group located at the C-5 position of indole ring was more potent than other derivatives. When the group tested the efficacy of the compounds on a human xenograft model in nude mice bearing A549 lung cancer cell lines, compound 61 was found to be more effective than SAHA (Figure 10). The authors found that there was a 47% tumor growth delay (32% tumor growth inhibition) by oral administration with compound 61 daily at 200 mg/ kg showing its efficacy as a potential anti-lung cancer agent [92].
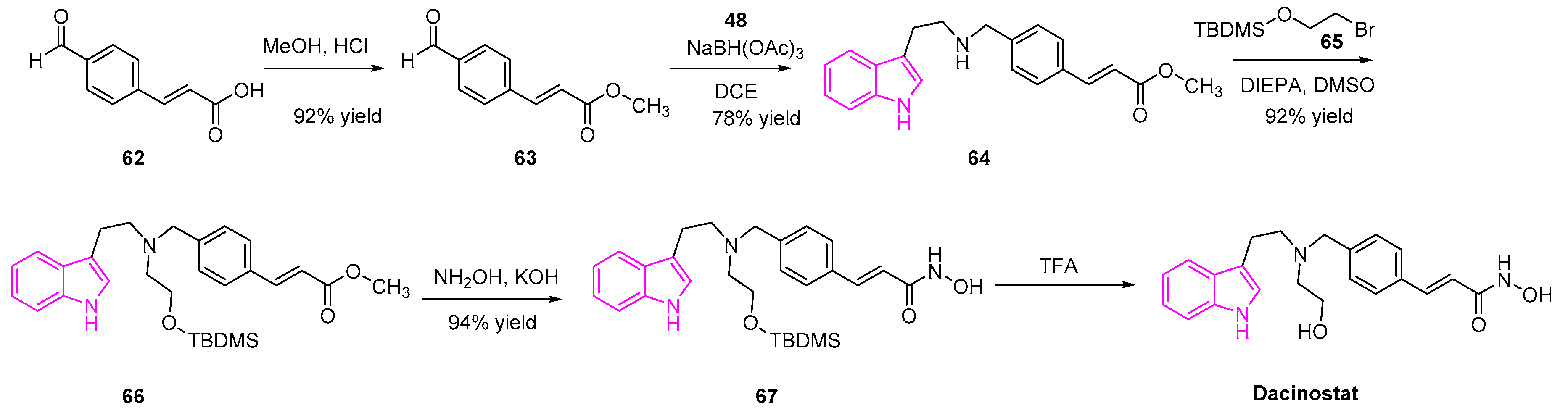
Dacinostat (NVPLAQ824) is an HDAC inhibitor introduced by Novartis in 2003 and is used as an anti-cancer agent (Scheme 12) [93]. Dacinostat was found to inhibit an HDAC enzyme isolated from H129 cell lysates with a potency of 32 nM and human lung cancer cells (H1299) with a potency of 150 nM, respectively. Besides, it was also found to be potent in the in vivo studies, in subject mice, against human lung carcinoma (A549 and HCT116) sub-cutaneous xenografts. Detailed studies of the mechanism of action showed that dacinostat acts on the A549 and HCT116 cells by G2-M arrest with a significant sub-G1 population, showing that the cells were subjected to apoptosis [94,95,96,97].
The synthesis of dacinostat is shown in the Scheme 12 [98]. Briefly, esterification of 4-formyl-cinnamic acid followed by reductive amination with tryptamine 48, generated the condensed indole 64. The resulting intermediate 64 was alkylated with 2-(tert-butyldimethylsilyloxy-ethylbromide (65) afforded the tertiary amine 66. The methyl ester of 66 was transformed to hydroxamic acid 67, followed by deprotection yielded the desired dacinostat (Scheme 12).
2.11. Miscellaneous Indole Derivatives in Anti-Lung Cancer Treatment
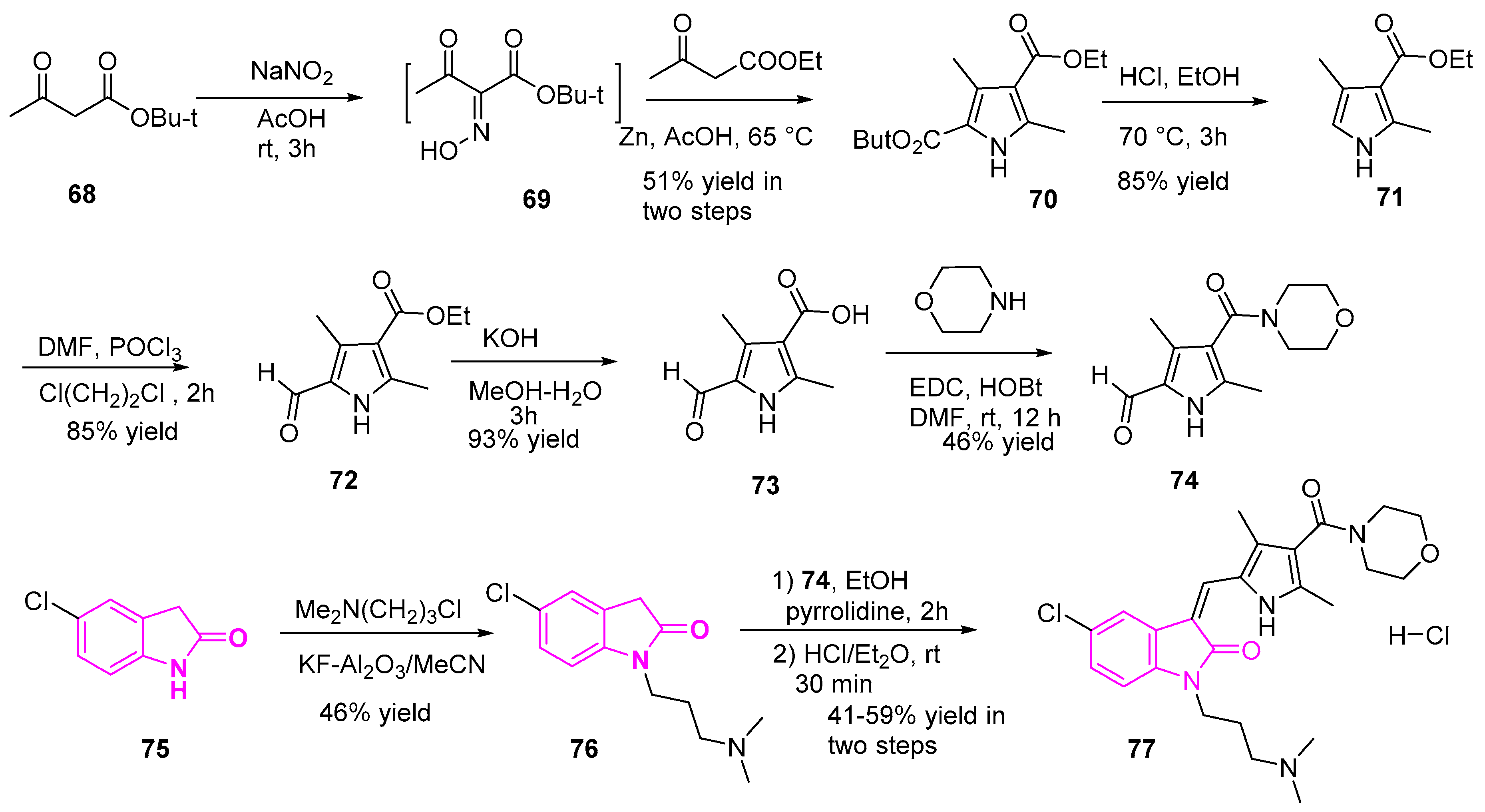
Lv et al. designed and synthesized a series of novel 1-(3-dimethyl-aminopropyl)indolin-2-one derivatives. Here the most active derivative was found to be more potent than sunitinib against NSCLC (A549) cells. These compounds were tested for their antiproliferative activity against lung adenocarcinoma (A549) cells and they found that all the compounds showed potent activity. Among all generated compounds, compound 77 (Scheme 13) proved to be most potent with IC50 of 1.10–1.47 µM, which is 1.8–6 fold higher than sunitinib [99].
A representation of the synthesis of the most potent compound 77 is shown in Scheme 13 [99,100]. Briefly, the reaction of tert-butyl acetoacetate 68 with sodium nitrite in acetic acid generated the oxime 69. The resulting oxime (69) was reacted with ethyl acetoacetate and zinc powder in acetic acid yielded the pyrrole diesters (70). Ester hydrolysis of 70, followed by monodecarboxylation reaction in the presence of conc. HCl and ethanol yielded the ethyl ester 71. Then subsequent treatment of 71 with phosphorus oxychloride yielded the aldehyde 72. The resulting intermediate (72) was hydrolyzed, followed by amide bond formation in the presence of morpholine and coupling reagents (EDC/HOBt) yielded compound 74. On the other hand, compound 76 was obtained through the treatment of intermediate 75 with 3-chloro-N,N-dimethyl-propan-1-amine in the presence of KF-alumina. Finally, Knoevenagel condensation of the intermediates 76 and 74, followed by HCl treatment generated the HCl salt of the desired target (77) (Scheme 13).
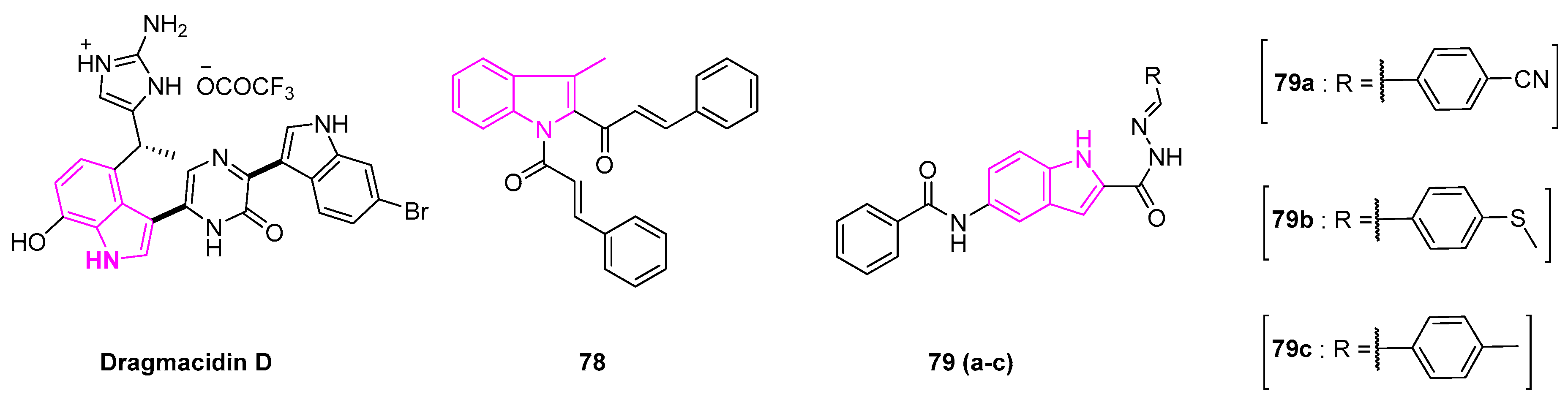
Dragmacidin is a cytotoxic bisalkaloid, isolated from a marine sponge Dragmacidin sp. This compound proved to be a potent antitumor agent. It showed IC50 value of 1–10 μg/mL against human lung cell lines (A549) in the in vitro studies (Figure 11) [101].
Recently, Zhao et al. reported a series of indolyl chalcones that can inhibit lung cancer cell growth. Of all the derivatives tested, they found compound 78 (Figure 11) to be effective in the induction of tumor apoptosis. It inhibited the growth of A549 cells by increasing the reactive oxygen species (ROS) levels and by activation of the The nuclear factor erythroid 2–related factor 2 (Nrf-2) pathway [102]. In the in vivo studies, compound 78 exhibited anti-growth activity of the tumor in an avian embryo model, showing the potential of the compound as the Nrf-2 activator in cancer therapy.
Hu et al. synthesized a series of indole derivatives with varying substituents at 2 and 5 positions. The resulting compounds exhibited excellent anti-proliferative activity on lung cancer cell lines (H460 and A549) (Figure 11). Notably, compound 79a exhibited a high level of potency against A549 cells, with the IC50 value of 0.50 ± 0.06 μM, while compounds 79b and 79c exhibited IC50 values of 0.16 ± 0.05 μM and 0.48 ± 0.05 μM, respectively. Further detailed studies revealed that the compounds induced apoptosis by the inhibition of phosphorylation of Ser2 of the carboxyl-terminal domain of RNA polymerase II, where RNA polymerase II is essential for messenger RNA (mRNA) synthesis [103].
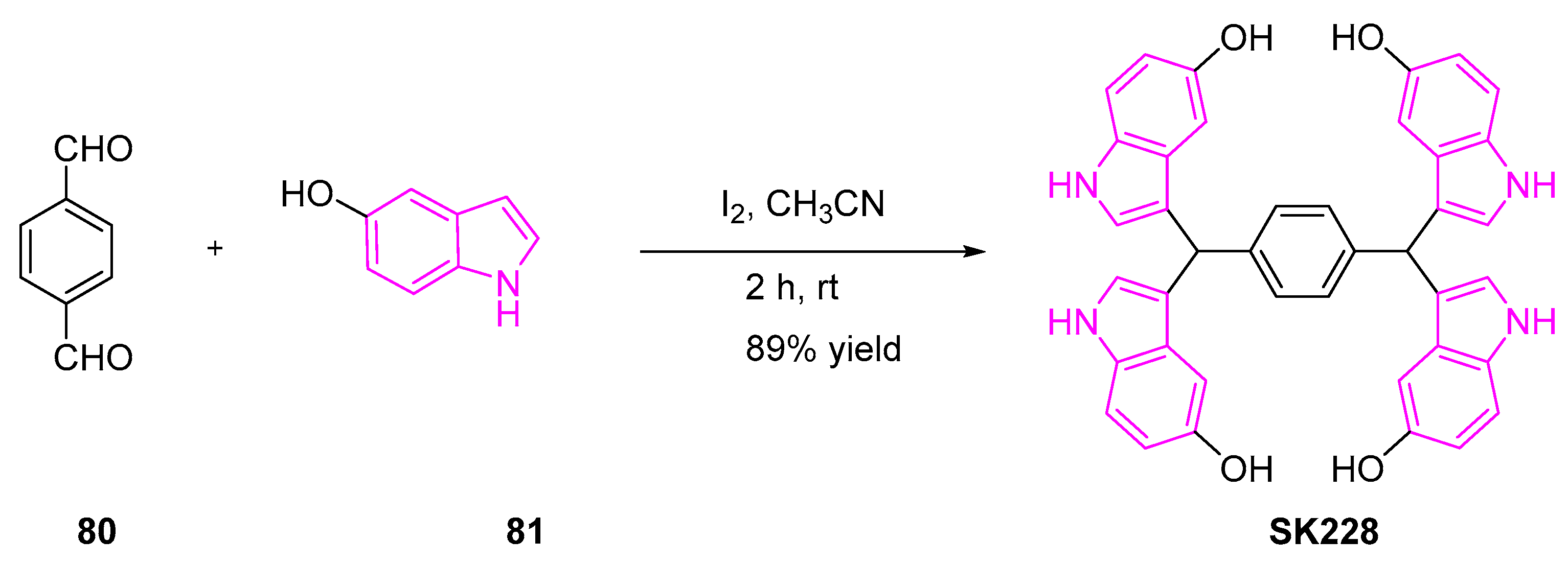
Huang et al. further identified a novel indole derivative, SK228 that can effectively inhibit lung cancer cell lines by ROS production and induce cell death by apoptosis. To evaluate the potency of SK228 against various human cancer cell lines, the authors treated the cells with varying concentrations of SK228 for 48 h. The resulting IC50 values were found to be 3.4 and 0.3 μM for A549 and H1299 cells respectively [104,105]. Further investigation of the mechanism of action showed that SK228 enhanced mitochondrial ROS production and inflicted DNA damage on cancer cells, ultimately leading to intrinsic mitochondria-dependent mitosis.
SK228 was synthesized in a single step, as shown in Scheme 14 [105]. A mixture of terephthalaldehyde (80) and 5-hydroxy-indole (81) in the presence of iodine afforded the desired tetraindoles product (SK228).
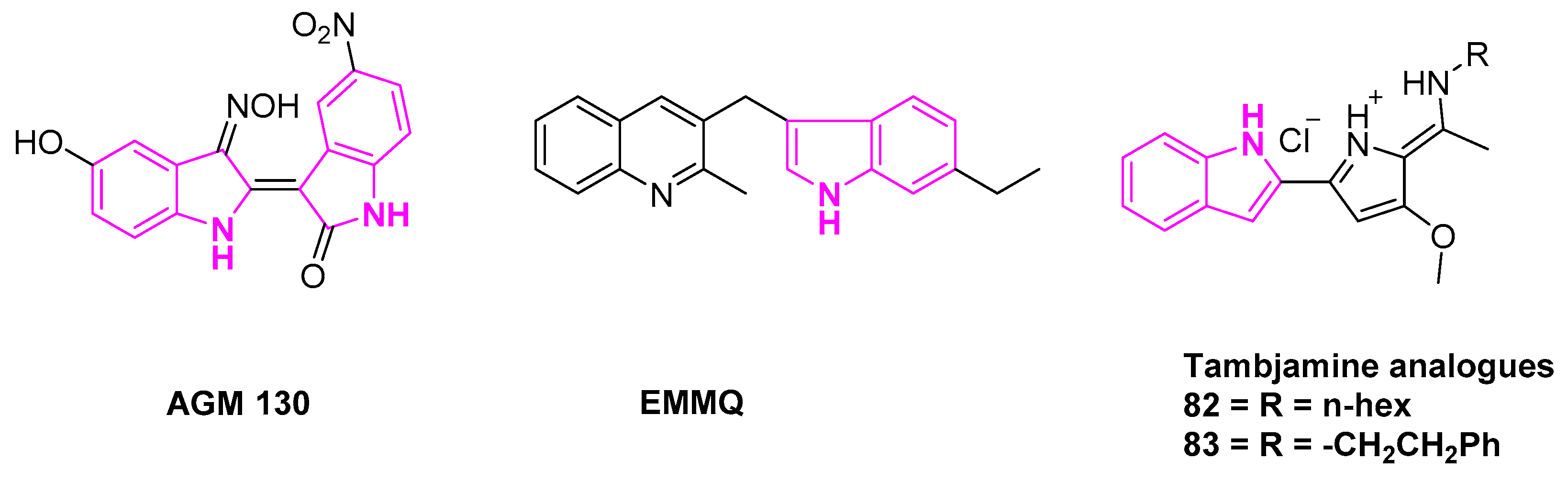
Kim et al. synthesized a novel indirubin-3′-oxime derivative AGM130 with excellent anti-tumor activity and inhibition of cancer growth in A549 cells (Figure 12). Further investigation of the activity on A549 cells demonstrated that AGM130 induced caspase-dependent apoptosis via the mitochondria-mediated intrinsic pathway [106].
Liu et al. reported a novel indolyl quinoline moiety 3-((7- ethyl-1H-indol-3-yl)-methyl)-2-methyl-quinoline (EMMQ) that can arrest cell-growth in NSCLC (A549 and H460) cells (Figure 12). Cell growth inhibition studies after 48 h of dose-dependent treatment indicated EMMQ IC50 values of 8 μM in both the A549 and H460 cell lines. This process involved the disruption of the mitochondrial membrane potential, DNA damage and ultimately apoptotic cell death [107].
To note, tambjamines are indole based marine alkaloids, known to be highly efficient transmembrane anion transporters in the model liposomes. They have widespread pharmacological applications (Figure 12). Studies have shown that these alkaloids can induce apoptosis in several cancer cell lines, rendering IC50 values in the micromolar range. Manresa et al. reported highly active indole based tambjamine derivatives, investigating their molecular mechanism of action. The results revealed that compounds 82 and 83 can induce apoptosis in lung cancer cells, in both in vitro and in vivo studies. Cytotoxicity is induced by the ROS activated stress kinase pathway. This occurs through the activation of P38 mitogen-activated protein kinase [108,109].
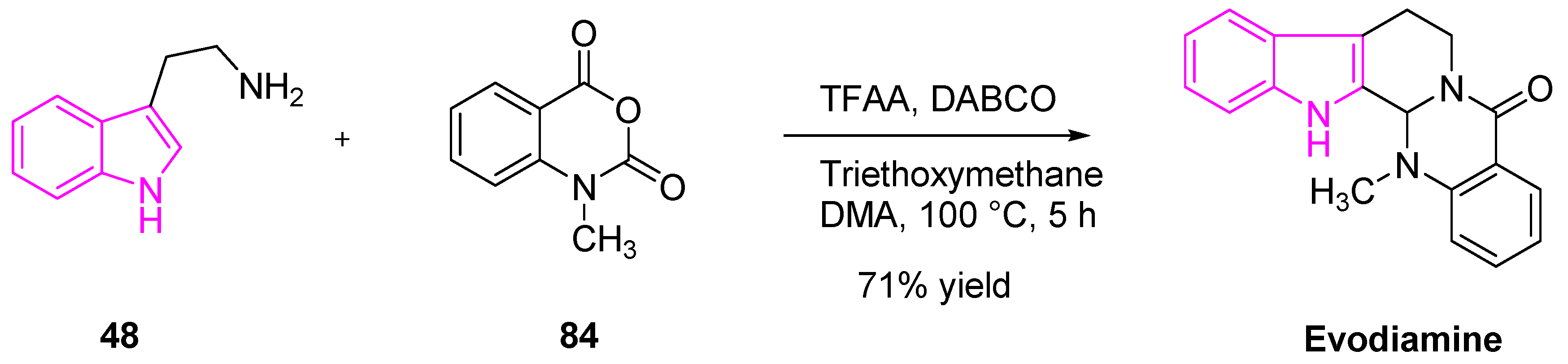
Evodiamine is an indole-containing alkaloid, isolated from a Chinese medicinal herb Evodia rutaecarpa (Wu-Chu-Yu). It has numerous pharmacological applications such as anti-inflammatory, anti-obesity, anti-viral, anti-tumor, and anti-nociceptive effects (Scheme 15). Studies have shown that evodiamine is capable of strong anti-tumor and cytotoxic activity against various human cancer cell lines (gastric, colon, liver, lung, and breast). Detailed mechanistic studies by Zou et al. revealed that evodiamine can exert the anti-tumor activity by the inhibition of Metadherin, which was identified as a novel oncogene that is overexpressed in many tumor malignancies [110]. Zou and his team demonstrated that the treatment of NSCLC cells (A549) with evodiamine results in an increased level of Bax expression. This is followed by a decrease of Bcl-2 and MTDH, which substantiated the apoptotic mechanism of action [111,112,113].
Evodiamine was synthesized as shown in Scheme 15 [114]. The reaction of tryptamine (48) with N-methylisatoic anhydride (84) in the presence of trifluoroacetic anhydride, 1,4-diazabicyclo[2.2. 2]octane (DABCO), triethoxymethane, and dimethylacetamide afforded the desired product of evodiamine (Scheme 15).
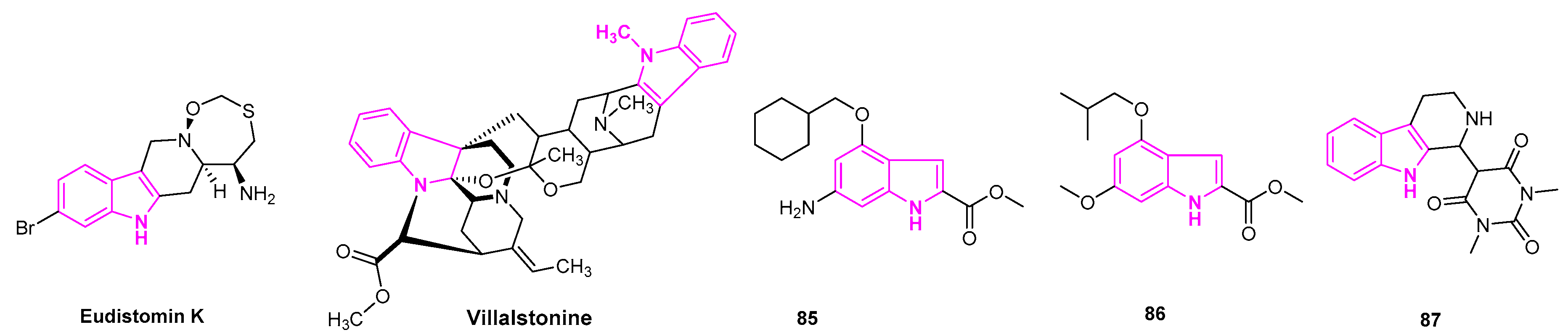
An indole based alkaloid, Eudistomin K isolated from the Caribbean ascidian Eudistoma olivaceum was found to be potent against human lung cancer (A-549) cell lines. It is reported to inhibit the P-388 tumor cell line with IC50 of 0.01 µg/mL (Figure 13) [115].
Bisindolylalkanes comprise two monomeric indole alkaloid units, derived from several natural terrestrial and marine sources. They are known for their biological, pharmacological, and medicinal properties. Villalstonine is derived from the Alstonia species, which is commonly found in the tropical and subtropical regions. It was evaluated for its anti-cancer activity against human NSCLC cell lines, adenocarcinoma (MOR-P), and large cell carcinoma (COR-L23) cell lines. Villalstonine was demonstrated to be potent in cytotoxic activity with IC50 of 5 µM and 2.5 µM in MOR-P and COR-L23 cells, respectively (Figure 13) [116].
Ji et al. reported a novel class of indole-2-carboxylate derivatives, evaluating their anti-cancer activity. They found that some compounds exhibited excellent anti-proliferative activity against HepG2, A549, and MCF7 cells. Most importantly, compounds 85 and 86 showed more potency than etoposide and pyrroloquinoline quinone drugs with IC50 ranging from 3.4 to 24 µM (Figure 13). When compounds 85 and 86 were subjected to a Western blot analysis of poly ADP ribose polymerase (PARP), both compounds triggered ROS production and induced PARP cleavage in A549 cells. This study was done in a dose-dependent manner, indicating the induction of apoptosis. The results showed that these substances can be promising lead compounds for the development of anti-lung cancer drugs [117].
Skouta et al. designed and synthesized a series of tetrahydro-β-carbolines derivatives. These compounds were tested for their efficacy against a variety of human cancer cell lines, including the lung cancer cells (H1299). They found that derivatives of tetrahydro-ß-carboline and barbituric acid moieties with no spacer showed modest activity with LC50 of 32.8 µM in the H1299 cell lines. Those analogs with a carbon spacer were either weakly active or non-selective. Compound 87 was found to be the most potent than any of the derivatives. It was observed to induce caspase-independent cell death selectively in cancerous cells [118].
3. Conclusions
Overall, indole based compounds have emerged as a powerful probe for scientists in the ongoing battle against such life-threatening diseases, such as lung cancer. Significant advances in this area are evidenced in the number of indole-based compounds entering the pre-clinical and clinical stages. More advances are expected to meet the challenges associated with the accompanying chemotherapy. With a better understanding of the drug resistance mechanism in lung cancer patients, coupled with the multi-drug therapy advances offered by the indole-based derivatives, better and more promising strategies for combatting lung cancer can be expected in the near future.
Author Contributions
Conceptualization, J.D. and R.S.; Methodology, n/a; Software, n/a.; Validation, n/a.; Formal Analysis, n/a; Investigation, n/a; Resources, n/a.; Data Curation, n/a.; Writing-Original Draft Preparation, J.D.; Writing-Review & Editing, J.D. and R.S.; Visualization, J.D. and R.S.; Supervision, R.S.; Project Administration, n/a.; Funding Acquisition, R.S. All authors have read and agreed to the published version of the manuscript.
Funding
This specific review received no external funding.
Acknowledgments
The authors thank: (i) Lung Cancer Research Foundation, (ii) the Department of Chemistry, and (iii) the Department Biology at the University of Massachusetts (UMASS) for financial support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bray, F.; Ferlay, J.; Soerjomataram, I. Global. GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries, CA. Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstraw, P.; Chansky, K.; Crowley, J.; Rami-Porta, R.; Asamura, H.; Eberhardt, W.E.E.; Nicholson, A.G.; Groome, P.; Mitchell, A.; Bolejack, V.; et al. The IASLC Lung Cancer Staging Project: Proposals for Revision of the TNM Stage Groupings in the Forthcoming (Eighth) Edition of the TNM Classification for Lung Cancer. J. Thorac. Oncol. 2016, 11, 39–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, G.S.; Baldwin, D.R. Recent Advances in the Management of Lung Cancer. Clin. Med. Lond. Engl. 2018, 18, s41–s46. [Google Scholar] [CrossRef]
- Hakim, M.; Broza, Y.Y.; Barash, O.; Peled, N.; Phillips, M.; Amann, A.; Haick, H. Volatile Organic Compounds of Lung Cancer and Possible Biochemical Pathways. Chem. Rev. 2012, 112, 5949–5966. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi Ziarani, G.; Moradi, R.; Ahmadi, T.; Lashgari, N. Recent Advances in the Application of Indoles in Multicomponent Reactions. RSC Adv. 2018, 8, 12069–12103. [Google Scholar] [CrossRef] [Green Version]
- Sundberg, R.J. Electrophilic Substitution Reactions of Indoles. In Heterocyclic Scaffolds II: Reactions and Applications of Indoles; Gribble, G.W., Ed.; Springer: Berlin/Heidelberg, Germany, 2010; pp. 47–115. [Google Scholar] [CrossRef]
- Baeyer, A.; Emmerling, A. Synthese des Indols. Berichte Dtsch. Chem. Ges. 1869, 2, 679–682. [Google Scholar] [CrossRef]
- Fischer, E.; Hess, O. Synthese von Indolderivaten. Berichte Dtsch. Chem. Ges. 1884, 17, 559–568. [Google Scholar] [CrossRef] [Green Version]
- Bischler, A.; Napieralski, B. Zur Kenntniss einer neuen Isochinolinsynthese. Berichte Dtsch. Chem. Ges. 1893, 26, 1903–1908. [Google Scholar] [CrossRef] [Green Version]
- Hemetsberger, H.; Knittel, D. Synthese und Thermolyse von α-Azidoacrylestern. Mon. Fur Chem. 1972, 103, 194–204. [Google Scholar] [CrossRef]
- Nenitzescu, C.D. Derivatives of 2-Methyl-5-Hydroxyindole. Bull. Soc. Chim. Rom. 1929, 11, 37–43. [Google Scholar]
- Bartoli, G.; Palmieri, G.; Bosco, M.; Dalpozzo, R. The Reaction of Vinyl Grignard Reagents with 2-Substituted Nitroarenes: A New Approach to the Synthesis of 7-Substituted Indoles. Tetrahedron Lett. 1989, 30, 2129–2132. [Google Scholar] [CrossRef]
- Yamagishi, M.; Nishigai, K.; Ishii, A.; Hata, T.; Urabe, H. Facile Preparation of Indoles and 1,2-Benzothiazine 1,1-Dioxides: Nucleophilic Addition of Sulfonamides to Bromoacetylenes and Subsequent Palladium-Catalyzed Cyclization. Angew. Chem. Int. Ed. 2012, 51, 6471–6474. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Hartwig, J.F. Palladium-Catalyzed Amination of Aromatic C−H Bonds with Oxime Esters. J. Am. Chem. Soc. 2010, 132, 3676–3677. [Google Scholar] [CrossRef] [PubMed]
- Li Petri, G.; Cascioferro, S.; El Hassouni, B.; Carbone, D.; Parrino, B.; Cirrincione, G.; Peters, G.J.; Diana, P.; Giovannetti, E. Biological Evaluation of the Antiproliferative and Anti-Migratory Activity of a Series of 3-(6-Phenylimidazo[2,1-b][1,3,4]Thiadiazol-2-Yl)-1 H -Indole Derivatives Against Pancreatic Cancer Cells. Anticancer Res. 2019, 39, 3615–3620. [Google Scholar] [CrossRef]
- Cascioferro, S.; Attanzio, A.; Di Sarno, V.; Musella, S.; Tesoriere, L.; Cirrincione, G.; Diana, P.; Parrino, B. New 1,2,4-Oxadiazole Nortopsentin Derivatives with Cytotoxic Activity. Mar. Drugs 2019, 17, 35. [Google Scholar] [CrossRef] [Green Version]
- Carbone, A.; Parrino, B.; Cusimano, M.; Spanò, V.; Montalbano, A.; Barraja, P.; Schillaci, D.; Cirrincione, G.; Diana, P.; Cascioferro, S. New Thiazole Nortopsentin Analogues Inhibit Bacterial Biofilm Formation. Mar. Drugs 2018, 16, 274. [Google Scholar] [CrossRef] [Green Version]
- Higdon, J.; Delage, B.; Williams, D.; Dashwood, R. Cruciferous Vegetables and Human Cancer Risk: Epidemiologic Evidence and Mechanistic Basis. Pharmacol. Res. 2007, 55, 224–236. [Google Scholar] [CrossRef] [Green Version]
- Hornedo-Ortega, R.; Da Costa, G.; Cerezo, A.B.; Troncoso, A.M.; Richard, T.; Garcia-Parrilla, M.C. In Vitro Effects of Serotonin, Melatonin, and Other Related Indole Compounds on Amyloid-β Kinetics and Neuroprotection. Mol. Nutr. Food Res. 2018, 62, 1700383. [Google Scholar] [CrossRef]
- Palmieri, A.; Petrini, M. Tryptophol and Derivatives: Natural Occurrence and Applications to the Synthesis of Bioactive Compounds. Nat. Prod. Rep. 2019, 36, 490–530. [Google Scholar] [CrossRef]
- Fu, S.-F.; Wei, J.-Y.; Chen, H.-W.; Liu, Y.-Y.; Lu, H.-Y.; Chou, J.-Y. Indole-3-Acetic Acid: A Widespread Physiological Code in Interactions of Fungi with Other Organisms. Plant Signal. Behav. 2015, 10, e1048052. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Sun, W.; Zhang, A.; Wang, X. Metabolite Profiling and Biomarkers Analysis of Jaundice Syndrome-Related Animal Models. In Chinmedomics; Elsevier: Boston, MA, USA, 2015; pp. 109–145. [Google Scholar] [CrossRef]
- Delavirdine Gets FDA Approval. Food and Drug Administration. AIDS Alert 1997, 12, 70. [Google Scholar]
- Lobay, D. Rauwolfia in the Treatment of Hypertension. Integr. Med. Encinitas Calif. 2015, 14, 40–46. [Google Scholar]
- Yee, A.J.; Raje, N.S. Panobinostat and Multiple Myeloma in 2018. Oncologist 2018, 23, 516–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasmussen, T.A.; Tolstrup, M.; Brinkmann, C.R.; Olesen, R.; Erikstrup, C.; Solomon, A.; Winckelmann, A.; Palmer, S.; Dinarello, C.; Buzon, M.; et al. Panobinostat, a Histone Deacetylase Inhibitor, for Latent-Virus Reactivation in HIV-Infected Patients on Suppressive Antiretroviral Therapy: A Phase 1/2, Single Group, Clinical Trial. Lancet HIV 2014, 1, e13–e21. [Google Scholar] [CrossRef]
- Kelloway, J.S. Zafirlukast: The First Leukotreene-Receptor Antagonist Approved for the Treatment of Asthma. Ann. Pharmacother. 1997, 31, 1012–1021. [Google Scholar] [CrossRef] [PubMed]
- Frajese, G.V.; Pozzi, F.; Frajese, G. Tadalafil in the Treatment of Erectile Dysfunction; an Overview of the Clinical Evidence. Clin. Interv. Aging 2006, 1, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhou, X.; Zhu, D.; Chen, J.; Qin, B.; Zhang, Y.; Wang, X.; Yang, D.; Meng, H.; Luo, Q.; et al. Is Pindolol Augmentation Effective in Depressed Patients Resistant to Selective Serotonin Reuptake Inhibitors? A Systematic Review and Meta-Analysis: PINDOLOL IN SSRI-RESISTANT DEPRESSION. Hum. Psychopharmacol. Clin. Exp. 2015, 30, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Blier, P.; Bergeron, R. The Use of Pindolol to Potentiate Antidepressant Medication. J. Clin. Psychiatry 1998, 59, 16–23, discussion 24-5. [Google Scholar]
- Ferreira, S.H.; Moncada, S.; Vane, J.R. Indomethacin and Aspirin Abolish Prostaglandin Release from the Spleen. Nat. New Biol. 1971, 231, 237–239. [Google Scholar] [CrossRef]
- Heijden, R.; Jacobs, D.; Snoeijer, W.; Hallard, D.; Verpoorte, R. The Catharanthus Alkaloids:Pharmacognosy and Biotechnology. Curr. Med. Chem. 2004, 11, 607–628. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, S.-H.; Guo, X.-L. New Insights into Vinca Alkaloids Resistance Mechanism and Circumvention in Lung Cancer. Biomed. Pharm. Biomed. Pharm. 2017, 96, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Tomasini, P.; Egea, J.; Souquet-Bressand, M.; Greillier, L.; Barlesi, F. Alectinib in the Treatment of ALK-Positive Metastatic Non-Small Cell Lung Cancer: Clinical Trial Evidence and Experience with a Focus on Brain Metastases. Ther. Adv. Respir. Dis. 2019, 13. [Google Scholar] [CrossRef] [PubMed]
- Le Tourneau, C.; Raymond, E.; Faivre, S. Sunitinib: A Novel Tyrosine Kinase Inhibitor. A Brief Review of Its Therapeutic Potential in the Treatment of Renal Carcinoma and Gastrointestinal Stromal Tumors (GIST). Ther. Clin. Risk Manag. 2007, 3, 341–348. [Google Scholar] [CrossRef] [Green Version]
- Greig, S.L. Osimertinib: First Global Approval. Drugs 2016, 76, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Mishra, D.P.; Khan, M.A.; Yadav, D.K.; Rawat, A.K.; Singh, R.K.; Ahamad, T.; Hussain, M.K.; Saquib, M.; Khan, M.F. Monoterpene Indole Alkaloids from Anthocephalus Cadamba Fruits Exhibiting Anticancer Activity in Human Lung Cancer Cell Line H1299. Chem. Sel. 2018, 3, 8468–8472. [Google Scholar] [CrossRef]
- Shao, L.; Wang, W.; Song, Z.; Zhang, Y. The Efficacy and Safety of Anlotinib Treatment for Advanced Lung Cancer. Onco Targets Ther. 2019, 12, 6549–6554. [Google Scholar] [CrossRef] [Green Version]
- Dadashpour, S.; Emami, S. Indole in the Target-Based Design of Anticancer Agents: A Versatile Scaffold with Diverse Mechanisms. Eur. J. Med. Chem. 2018, 150, 9–29. [Google Scholar] [CrossRef]
- Leoni, A.; Locatelli, A.; Morigi, R.; Rambaldi, M. 2-Indolinone a Versatile Scaffold for Treatment of Cancer: A Patent Review (2008–2014). Expert Opin. Ther. Pat. 2016, 26, 149–173. [Google Scholar] [CrossRef]
- Kumar, N.M.; Kumar, D. Recent Developments on Synthetic Indoles as Potent Anticancer Agents. Chem. Biol. Interface 2013, 3, 276–303. [Google Scholar]
- De Sa Alves, F.; Barreiro, E.; Manssour Fraga, C. From nature to drug discovery: The indole scaffold as a ‘privileged structure. Mini-Rev. Med. Chem. 2009, 9, 782–793. [Google Scholar] [CrossRef]
- Sravanthi, T.V.; Manju, S.L. Indoles—A Promising Scaffold for Drug Development. Eur. J. Pharm. Sci. 2016, 91, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, N.; Kaushik, N.; Attri, P.; Kumar, N.; Kim, C.; Verma, A.; Choi, E. Biomedical Importance of Indoles. Molecules 2013, 18, 6620–6662. [Google Scholar] [CrossRef] [PubMed]
- Sunil, D.; Kamath, P. Multi-Target Directed Indole Based Hybrid Molecules in Cancer Therapy: An Up-To-Date Evidence-Based Review. Curr. Top. Med. Chem. 2017, 17, 959–985. [Google Scholar] [CrossRef] [PubMed]
- Chadha, N.; Silakari, O. Indoles as Therapeutics of Interest in Medicinal Chemistry: Bird’s Eye View. Eur. J. Med. Chem. 2017, 134, 159–184. [Google Scholar] [CrossRef] [PubMed]
- El-sayed, M.T.; Hamdy, N.A.; Osman, D.A.; Ahmed, K.M. Indoles as Anticancer Agents. Adv. Mod. Oncol. Res. 2015, 1, 20. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, R.I.; Gee, J.M.W.; Harper, M.E. EGFR and Cancer Prognosis. Eur. J. Cancer 2001, 37, 9–15. [Google Scholar] [CrossRef]
- Engelman, J.A. The Role of Phosphoinositide 3-Kinase Pathway Inhibitors in the Treatment of Lung Cancer. Clin. Cancer Res. 2007, 13, 4637s–4640s. [Google Scholar] [CrossRef] [Green Version]
- Brognard, J.; Clark, A.S.; Ni, Y.; Dennis, P.A. Akt/Protein Kinase B Is Constitutively Active in Non-Small Cell Lung Cancer Cells and Promotes Cellular Survival and Resistance to Chemotherapy and Radiation. Cancer Res. 2001, 61, 3986–3997. [Google Scholar]
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt Pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189. [Google Scholar] [CrossRef] [Green Version]
- Altomare, D.A.; Testa, J.R. Perturbations of the AKT Signaling Pathway in Human Cancer. Oncogene 2005, 24, 7455–7464. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.; Shcherba, M.; Pendurti, G.; Liang, Y.; Piperdi, B.; Perez-Soler, R. Targeting the PI3K/AKT/MTOR Pathway: Potential for Lung Cancer Treatment. Lung Cancer Manag. 2014, 3, 67–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Du, J.; Kwiatkowski, D.J. Molecular Dissection of AKT Activation in Lung Cancer Cell Lines. Mol. Cancer Res. 2013, 11, 282–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nesi, G.; Sestito, S.; Mey, V.; Ricciardi, S.; Falasca, M.; Danesi, R.; Lapucci, A.; Breschi, M.C.; Fogli, S.; Rapposelli, S. Synthesis of Novel 3,5-Disubstituted-2-Oxindole Derivatives As Antitumor Agents against Human Nonsmall Cell Lung Cancer. ACS Med. Chem. Lett. 2013, 4, 1137–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, B.; Xiao, Z.; Qi, J.; Luo, R.; Lan, Z.; Zhang, Y.; Hu, X.; Tang, Q.; Zheng, P.; Xu, S.; et al. Design, Synthesis and Biological Evaluation of AZD9291 Derivatives as Selective and Potent EGFRL858R/T790M Inhibitors. Eur. J. Med. Chem. 2019, 163, 367–380. [Google Scholar] [CrossRef]
- Sam, B.; Maurice, R.V.F.; Richard, A.W.; Vasantha, K.K.; Reddy, C.C.; Andiappan, M.; Heather, M.R. 2-(2,4,5-Substituted Anilino)Pyrimidine Derivatives as EGFR Modulators Useful for Treating Cancer. WO2013014448A1, 31 January 2013. [Google Scholar]
- Wang, Y.; Wang, L.; Guan, S.; Cao, W.; Wang, H.; Chen, Z.; Zhao, Y.; Yu, Y.; Zhang, H.; Pang, J.C.; et al. Novel ALK Inhibitor AZD3463 Inhibits Neuroblastoma Growth by Overcoming Crizotinib Resistance and Inducing Apoptosis. Sci. Rep. 2016, 6, 19423. [Google Scholar] [CrossRef]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the Transforming EML4–ALK Fusion Gene in Non-Small-Cell Lung Cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef]
- Kinoshita, K.; Asoh, K.; Furuichi, N.; Ito, T.; Kawada, H.; Hara, S.; Ohwada, J.; Miyagi, T.; Kobayashi, T.; Takanashi, K.; et al. Design and Synthesis of a Highly Selective, Orally Active and Potent Anaplastic Lymphoma Kinase Inhibitor (CH5424802). Bioorg. Med. Chem. 2012, 20, 1271–1280. [Google Scholar] [CrossRef]
- Kinoshita, K.; Kobayashi, T.; Asoh, K.; Furuichi, N.; Ito, T.; Kawada, H.; Hara, S.; Ohwada, J.; Hattori, K.; Miyagi, T.; et al. 9-Substituted 6,6-Dimethyl-11-Oxo-6,11-Dihydro-5 H -Benzo[ b ]Carbazoles as Highly Selective and Potent Anaplastic Lymphoma Kinase Inhibitors. J. Med. Chem. 2011, 54, 6286–6294. [Google Scholar] [CrossRef]
- Newton, A.C. Protein Kinase C: Structure, Function, and Regulation. J. Biol. Chem. 1995, 270, 28495–28498. [Google Scholar] [CrossRef] [Green Version]
- Herbst, R.S.; Oh, Y.; Wagle, A.; Lahn, M. Enzastaurin, a Protein Kinase C Selective Inhibitor, and Its Potential Application as an Anticancer Agent in Lung Cancer. Clin. Cancer Res. 2007, 13, 4641s–4646s. [Google Scholar] [CrossRef] [Green Version]
- Ann, M.M.; Bruce, W.K.; Rebecca, L.L.; Donald, T.; Larry, D.; Bruce, C.; Julia, H.C.; Jeremy, R.G. Enzastaurin (LY317615.HCl) Suppresses Signaling through the PKC and AKT Pathways, Inducing Apoptosis, Suppressing Tumor-Induced Angiogenesis and Reducing Growth of Human Cancer Xenografts. Cancer Res. 2006, 66, 314–315. [Google Scholar]
- Nakajima, E.; Helfrich, B.; Chan, D.; Zhang, Z.; Hirsch, F.R.; Chen, V.; Ma, D.; Bunn, P.A. Enzastaurin a Protein Kinase Cbeta-Selective Inhibitor, Inhibits the Growth of SCLC and NSCLC Cell Lines. J. Clin. Oncol. 2006, 24, 13138. [Google Scholar] [CrossRef]
- Crump, M.; Leppä, S.; Fayad, L.; Lee, J.J.; Di Rocco, A.; Ogura, M.; Hagberg, H.; Schnell, F.; Rifkin, R.; Mackensen, A.; et al. Randomized, Double-Blind, Phase III Trial of Enzastaurin Versus Placebo in Patients Achieving Remission After First-Line Therapy for High-Risk Diffuse Large B-Cell Lymphoma. J. Clin. Oncol. 2016, 34, 2484–2492. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Xu, L.; Gao, M.; Miller, K.D.; Sledge, G.W.; Zheng, Q.-H. [11C]Enzastaurin, the First Design and Radiosynthesis of a New Potential PET Agent for Imaging of Protein Kinase, C. Bioorg. Med. Chem. Lett. 2011, 21, 1649–1653. [Google Scholar] [CrossRef]
- Remsing Rix, L.L.; Kuenzi, B.M.; Luo, Y.; Remily-Wood, E.; Kinose, F.; Wright, G.; Li, J.; Koomen, J.M.; Haura, E.B.; Lawrence, H.R.; et al. GSK3 Alpha and Beta Are New Functionally Relevant Targets of Tivantinib in Lung Cancer Cells. ACS Chem. Biol. 2014, 9, 353–358. [Google Scholar] [CrossRef]
- Scagliotti, G.V.; Shuster, D.; Orlov, S.; von Pawel, J.; Shepherd, F.A.; Ross, J.S.; Wang, Q.; Schwartz, B.; Akerley, W. Tivantinib in Combination with Erlotinib versus Erlotinib Alone for EGFR-Mutant NSCLC: An Exploratory Analysis of the Phase 3 MARQUEE Study. J. Thorac. Oncol. 2018, 13, 849–854. [Google Scholar] [CrossRef] [Green Version]
- Li, C.J.; Ashwell, M.A.; Hill, J.; Moussa, M.M.; Munshi, N. Maleimide Derivatives, Pharmaceutical Compositions and Methods for Treatment of Cancer. EP1846406B9, 11 January 2012. [Google Scholar]
- Lin, B.; Song, X.; Yang, D.; Bai, D.; Yao, Y.; Lu, N. Anlotinib Inhibits Angiogenesis via Suppressing the Activation of VEGFR2, PDGFRβ and FGFR1. Gene 2018, 654, 77–86. [Google Scholar] [CrossRef]
- Cheng, Y.; Wang, Q.; Li, K.; Shi, J.; Liu, Y.; Wu, L.; Han, B.; Chen, G.; He, J.; Wang, J.; et al. 1738OOverall Survival (OS) Update in ALTER 1202: Anlotinib as Third-Line or Further-Line Treatment in Relapsed Small-Cell Lung Cancer (SCLC). Ann. Oncol. 2019, 30. [Google Scholar] [CrossRef]
- Reck, M.; Mellemgaard, A.; Novello, S.; Postmus, P.E.; Gaschler-Markefski, B.; Kaiser, R.; Buchner, H. Change in Non-Small-Cell Lung Cancer Tumor Size in Patients Treated with Nintedanib plus Docetaxel: Analyses from the Phase III LUME-Lung 1 Study. OncoTargets Ther. 2018, 11, 4573–4582. [Google Scholar] [CrossRef] [Green Version]
- Corrales, L.; Nogueira, A.; Passiglia, F.; Listi, A.; Caglevic, C.; Giallombardo, M.; Raez, L.; Santos, E.; Rolfo, C. Second-Line Treatment of Non-Small Cell Lung Cancer: Clinical, Pathological, and Molecular Aspects of Nintedanib. Front. Med. 2017, 4. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, D.A.; Mullins, R.D. Cell Mechanics and the Cytoskeleton. Nature 2010, 463, 485–492. [Google Scholar] [CrossRef] [Green Version]
- Naaz, F.; Haider, M.R.; Shafi, S.; Yar, M.S. Anti-Tubulin Agents of Natural Origin: Targeting Taxol, Vinca, and Colchicine Binding Domains. Eur. J. Med. Chem. 2019, 171, 310–331. [Google Scholar] [CrossRef] [PubMed]
- Adam, Z.; Coupková, H.; Kolek, V.; Kucera, M.; Loffelmann, L.; Martinez, A.; Nováková, M.; Průsa, P.; Reiterer, P.; Skalová, B. Vinorelbine and Cisplatin in the Treatment of Advanced Non-Small Cell Lung Cancer: Results of a Multicenter Czech Study. Acta Med. Austriaca 1995, 22, 120–124. [Google Scholar] [PubMed]
- Genova, C.; Alama, A.; Coco, S.; Rijavec, E.; Dal Bello, M.G.; Vanni, I.; Biello, F.; Barletta, G.; Rossi, G.; Grossi, F. Vinflunine for the Treatment of Non-Small Cell Lung Cancer. Expert Opin. Investig. Drugs 2016, 25, 1447–1455. [Google Scholar] [CrossRef] [PubMed]
- Cong, H.; Zhao, X.; Castle, B.T.; Pomeroy, E.J.; Zhou, B.; Lee, J.; Wang, Y.; Bian, T.; Miao, Z.; Zhang, W.; et al. An Indole–Chalcone Inhibits Multidrug-Resistant Cancer Cell Growth by Targeting Microtubules. Mol. Pharm. 2018, 15, 3892–3900. [Google Scholar] [CrossRef] [PubMed]
- Das Mukherjee, D.; Kumar, N.M.; Tantak, M.P.; Das, A.; Ganguli, A.; Datta, S.; Kumar, D.; Chakrabarti, G. Development of Novel Bis(Indolyl)-Hydrazide–Hydrazone Derivatives as Potent Microtubule-Targeting Cytotoxic Agents against A549 Lung Cancer Cells. Biochemistry 2016, 55, 3020–3035. [Google Scholar] [CrossRef] [PubMed]
- Chaniyara, R.; Tala, S.; Chen, C.-W.; Zang, X.; Kakadiya, R.; Lin, L.-F.; Chen, C.-H.; Chien, S.-I.; Chou, T.-C.; Tsai, T.-H.; et al. Novel Antitumor Indolizino[6,7- b ]Indoles with Multiple Modes of Action: DNA Cross-Linking and Topoisomerase I and II Inhibition. J. Med. Chem. 2013, 56, 1544–1563. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-W.; Wu, M.-H.; Chen, Y.-F.; Yen, T.-Y.; Lin, Y.-W.; Chao, S.-H.; Tala, S.; Tsai, T.-H.; Su, T.-L.; Lee, T.-C. A Potent Derivative of Indolizino[6,7-b]Indole for Treatment of Human Non–Small Cell Lung Cancer Cells. Neoplasia 2016, 18, 199–212. [Google Scholar] [CrossRef] [Green Version]
- Chang, S.-M.; Christian, W.; Wu, M.-H.; Chen, T.-L.; Lin, Y.-W.; Suen, C.-S.; Pidugu, H.B.; Detroja, D.; Shah, A.; Hwang, M.-J.; et al. Novel Indolizino[8,7-b]Indole Hybrids as Anti-Small Cell Lung Cancer Agents: Regioselective Modulation of Topoisomerase II Inhibitory and DNA Crosslinking Activities. Eur. J. Med. Chem. 2017, 127, 235–249. [Google Scholar] [CrossRef]
- Song, Y.-L.; Dong, Y.-F.; Yang, T.; Zhang, C.-C.; Su, L.-M.; Huang, X.; Zhang, D.-N.; Yang, G.-L.; Liu, Y.-X. Synthesis and Pharmacological Evaluation of Novel Bisindolylalkanes Analogues. Bioorg. Med. Chem. 2013, 21, 7624–7627. [Google Scholar] [CrossRef]
- Barrows, L.R.; Radisky, D.C.; Copp, B.R.; Swaffar, D.S.; Kramer, R.A.; Warters, R.L.; Ireland, C.M. Makaluvamines, Marine Natural Products, Are Active Anti-Cancer Agents and DNA Topo II Inhibitors. Anticancer Drug Des. 1993, 8, 333–347. [Google Scholar] [PubMed]
- Nadkarni, D.; Wang, F.; Wang, W.; Rayburn, E.; Ezell, S.; Murugesan, S.; Velu, S.; Zhang, R. Synthesis and In Vitro Anti-Lung Cancer Activity of Novel 1, 3, 4, 8- Tetrahydropyrrolo [4, 3, 2-de]Quinolin-8(1H)-o Ne Alkaloid Analogs. Med. Chem. 2009, 5, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Lane, A.A.; Chabner, B.A. Histone Deacetylase Inhibitors in Cancer Therapy. J. Clin. Oncol. 2009, 27, 5459–5468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richon, V.M.; O’Brien, J.P. Histone Deacetylase Inhibitors: A New Class of Potential Therapeutic Agents for Cancer Treatment. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2002, 8, 662–664. [Google Scholar]
- Johnstone, R.W. Histone-Deacetylase Inhibitors: Novel Drugs for the Treatment of Cancer. Nat. Rev. Drug Discov. 2002, 1, 287–299. [Google Scholar] [CrossRef]
- Shinji, C.; Maeda, S.; Imai, K.; Yoshida, M.; Hashimoto, Y.; Miyachi, H. Design, Synthesis, and Evaluation of Cyclic Amide/Imide-Bearing Hydroxamic Acid Derivatives as Class-Selective Histone Deacetylase (HDAC) Inhibitors. Bioorg. Med. Chem. 2006, 14, 7625–7651. [Google Scholar] [CrossRef]
- Han, S.; Fukazawa, T.; Yamatsuji, T.; Matsuoka, J.; Miyachi, H.; Maeda, Y.; Durbin, M.; Naomoto, Y. Anti-Tumor Effect in Human Lung Cancer by a Combination Treatment of Novel Histone Deacetylase Inhibitors: SL142 or SL325 and Retinoic Acids. PLoS ONE 2010, 5, e13834. [Google Scholar] [CrossRef]
- Lai, M.-J.; Huang, H.-L.; Pan, S.-L.; Liu, Y.-M.; Peng, C.-Y.; Lee, H.-Y.; Yeh, T.-K.; Huang, P.-H.; Teng, C.-M.; Chen, C.-S.; et al. Synthesis and Biological Evaluation of 1-Arylsulfonyl-5-(N-Hydroxyacrylamide)Indoles as Potent Histone Deacetylase Inhibitors with Antitumor Activity in Vivo. J. Med. Chem. 2012, 55, 3777–3791. [Google Scholar] [CrossRef] [Green Version]
- Remiszewski, S.W.; Sambucetti, L.C.; Bair, K.W.; Bontempo, J.; Cesarz, D.; Chandramouli, N.; Chen, R.; Cheung, M.; Cornell-Kennon, S.; Dean, K.; et al. N -Hydroxy-3-Phenyl-2-Propenamides as Novel Inhibitors of Human Histone Deacetylase with in Vivo Antitumor Activity: Discovery of (2 E)- N -Hydroxy-3-[4-[[(2-Hydroxyethyl)[2-(1 H -Indol-3-Yl)Ethyl]Amino]Methyl]Phenyl]-2-Propenamide (NVP-LAQ824). J. Med. Chem. 2003, 46, 4609–4624. [Google Scholar] [CrossRef]
- Catley, L.; Weisberg, E.; Tai, Y.-T.; Atadja, P.; Remiszewski, S.; Hideshima, T.; Mitsiades, N.; Shringarpure, R.; LeBlanc, R.; Chauhan, D.; et al. NVP-LAQ824 Is a Potent Novel Histone Deacetylase Inhibitor with Significant Activity against Multiple Myeloma. Blood 2003, 102, 2615–2622. [Google Scholar] [CrossRef]
- Atadja, P.; Gao, L.; Kwon, P.; Trogani, N.; Walker, H.; Hsu, M.; Yeleswarapu, L.; Chandramouli, N.; Perez, L.; Versace, R.; et al. Selective Growth Inhibition of Tumor Cells by a Novel Histone Deacetylase Inhibitor, NVP-LAQ824. Cancer Res. 2004, 64, 689–695. [Google Scholar] [CrossRef] [Green Version]
- Cuneo, K.C.; Fu, A.; Osusky, K.; Huamani, J.; Hallahan, D.E.; Geng, L. Histone Deacetylase Inhibitor NVP-LAQ824 Sensitizes Human Nonsmall Cell Lung Cancer to the Cytotoxic Effects of Ionizing Radiation: Anticancer. Drugs 2007, 18, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Ellis, L.; Bots, M.; Lindemann, R.K.; Bolden, J.E.; Newbold, A.; Cluse, L.A.; Scott, C.L.; Strasser, A.; Atadja, P.; Lowe, S.W.; et al. The Histone Deacetylase Inhibitors LAQ824 and LBH589 Do Not Require Death Receptor Signaling or a Functional Apoptosome to Mediate Tumor Cell Death or Therapeutic Efficacy. Blood 2009, 114, 380–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remiszewski, S.W. The Discovery of NVP-LAQ824: From Concept to Clinic. Curr. Med. Chem. 2003, 10, 2393–2402. [Google Scholar] [CrossRef] [PubMed]
- Lv, K.; Wang, L.-L.; Zhou, X.-B.; Liu, M.-L.; Liu, H.-Y.; Zheng, Z.-B.; Li, S. Synthesis and in Vitro Antitumor Activity of 1-(3-Dimethylamino)Propyl Indolin-2-One Derivatives. Med. Chem. Res. 2013, 22, 1723–1729. [Google Scholar] [CrossRef]
- Lv, K.; Wang, L.-L.; Liu, M.-L.; Zhou, X.-B.; Fan, S.-Y.; Liu, H.-Y.; Zheng, Z.-B.; Li, S. Synthesis and Antitumor Activity of 5-[1-(3-(Dimethylamino)Propyl)-5-Halogenated-2-Oxoindolin-(3Z)-Ylidenemethyl]-2,4-Dimethyl-1H-Pyrrole-3-Carboxamides. Bioorg. Med. Chem. Lett. 2011, 21, 3062–3065. [Google Scholar] [CrossRef] [PubMed]
- Kohmoto, S.; Kashman, Y.; McConnell, O.J.; Rinehart, K.L.; Wright, A.; Koehn, F. Dragmacidin, a New Cytotoxic Bis(Indole) Alkaloid from a Deep Water Marine Sponge, Dragmacidon sp. J. Org. Chem. 1988, 53, 3116–3118. [Google Scholar] [CrossRef]
- Zhao, X.; Dong, W.; Gao, Y.; Shin, D.-S.; Ye, Q.; Su, L.; Jiang, F.; Zhao, B.; Miao, J. Novel Indolyl-Chalcone Derivatives Inhibit A549 Lung Cancer Cell Growth through Activating Nrf-2/HO-1 and Inducing Apoptosis in Vitro and in Vivo. Sci. Rep. 2017, 7, 3919. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Wu, J.; Ao, M.; Wang, H.; Zhou, T.; Xue, Y.; Qiu, Y.; Fang, M.; Wu, Z. Synthesis, Structure-Activity Relationship Studies and Biological Evaluation of Novel 2,5-Disubstituted Indole Derivatives as Anticancer Agents. Chem. Biol. Drug Des. 2016, 88, 766–778. [Google Scholar] [CrossRef]
- Huang, S.-M.; Hsu, P.-C.; Chen, M.-Y.; Li, W.-S.; More, S.V.; Lu, K.-T.; Wang, Y.-C. The Novel Indole Compound SK228 Induces Apoptosis and FAK/Paxillin Disruption in Tumor Cell Lines and Inhibits Growth of Tumor Graft in the Nude Mouse. Int. J. Cancer 2012, 131, 722–732. [Google Scholar] [CrossRef]
- Li, W.-S.; Wang, C.-H.; Ko, S.; Chang, T.T.; Jen, Y.C.; Yao, C.-F.; More, S.V.; Jao, S.-C. Synthesis and Evaluation of the Cytotoxicities of Tetraindoles: Observation That the 5-Hydroxy Tetraindole (SK228) Induces G 2 Arrest and Apoptosis in Human Breast Cancer Cells. J. Med. Chem. 2012, 55, 1583–1592. [Google Scholar] [CrossRef] [PubMed]
- Ahn, M.-Y.; Kim, T.-H.; Kwon, S.-M.; Yoon, H.-E.; Kim, H.-S.; Kim, J.-I.; Kim, Y.-C.; Kang, K.-W.; Ahn, S.-G.; Yoon, J.-H. 5-Nitro-5′-Hydroxy-Indirubin-3′-Oxime (AGM130), an Indirubin-3′-Oxime Derivative, Inhibits Tumor Growth by Inducing Apoptosis against Non-Small Cell Lung Cancer in Vitro and in Vivo. Eur. J. Pharm. Sci. 2015, 79, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-Y.; Wu, P.-T.; Wang, J.-P.; Fan, P.-W.; Hsieh, C.-H.; Su, C.-L.; Chiu, C.-C.; Yao, C.-F.; Fang, K. An Indolylquinoline Derivative Promotes Apoptosis in Human Lung Cancer Cells by Impairing Mitochondrial Functions. Apoptosis 2015, 20, 1471–1482. [Google Scholar] [CrossRef] [PubMed]
- Manuel-Manresa, P.; Korrodi-Gregório, L.; Hernando, E.; Villanueva, A.; Martínez-García, D.; Rodilla, A.M.; Ramos, R.; Fardilha, M.; Moya, J.; Quesada, R.; et al. Novel Indole-Based Tambjamine-Analogues Induce Apoptotic Lung Cancer Cell Death through P38 Mitogen-Activated Protein Kinase Activation. Mol. Cancer Ther. 2017, 16, 1224–1235. [Google Scholar] [CrossRef] [Green Version]
- Hernando, E.; Soto-Cerrato, V.; Cortés-Arroyo, S.; Pérez-Tomás, R.; Quesada, R. Transmembrane Anion Transport and Cytotoxicity of Synthetic Tambjamine Analogs. Org. Biomol. Chem. 2014, 12, 1771–1778. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.; Liu, Y.; Qin, Z. Metadherin Contributes to Epithelial-Mesenchymal Transition and Paclitaxel Resistance Induced by Acidic Extracellular PH in Nasopharyngeal Carcinoma. Oncol. Lett. 2018, 3, 3858–3863. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.; Qin, X.; Xiong, H.; Zhu, F.; Chen, T.; Wu, H. Apoptosis of Human Non-Small-Cell Lung Cancer A549 Cells Triggered by Evodiamine through MTDH-Dependent Signaling Pathway. Tumor Biol. 2015, 36, 5187–5193. [Google Scholar] [CrossRef]
- Kan, S.-F.; Yu, C.-H.; Pu, H.-F.; Hsu, J.-M.; Chen, M.-J.; Wang, P.S. Anti-Proliferative Effects of Evodiamine on Human Prostate Cancer Cell Lines DU145 and PC3. J. Cell. Biochem. 2007, 101, 44–56. [Google Scholar] [CrossRef]
- Jiang, J.; Hu, C. Evodiamine: A Novel Anti-Cancer Alkaloid from Evodia Rutaecarpa. Molecules 2009, 14, 1852–1859. [Google Scholar] [CrossRef]
- Wang, Z.-X.; Xiang, J.-C.; Wang, M.; Ma, J.-T.; Wu, Y.-D.; Wu, A.-X. One-Pot Total Synthesis of Evodiamine and Its Analogues through a Continuous Biscyclization Reaction. Org. Lett. 2018, 20, 6380–6383. [Google Scholar] [CrossRef]
- Menna, M.; Fattorusso, E.; Imperatore, C. Alkaloids from Marine Ascidians. Molecules 2011, 16, 8694–8732. [Google Scholar] [CrossRef]
- Keawprdub, N.; Houghton, P.; Eno-Amooquaye, E.; Burke, P. Activity of Extracts and Alkaloids of Thai Alstonia Species Against Human Lung Cancer Cell Lines. Planta Med. 1997, 63, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Xue, S.; Zhan, Y.; Shen, J.; Wu, L.; Jin, J.; Wang, Z.; Li, Z. Design, Synthesis and Antiproliferative Activity of a Novel Class of Indole-2-Carboxylate Derivatives. Eur. J. Med. Chem. 2014, 83, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Skouta, R.; Hayano, M.; Shimada, K.; Stockwell, B.R. Design and Synthesis of Pictet–Spengler Condensation Products That Exhibit Oncogenic-RAS Synthetic Lethality and Induce Non-Apoptotic Cell Death. Bioorg. Med. Chem. Lett. 2012, 22, 5707–5713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Structure of indole.

Figure 2.
Indole scaffold in biologically important compounds.

Figure 3.
Indole scaffold in Food and Drug Administration (FDA) approved drugs.

Figure 4.
Indole containing anti-lung cancer drugs in clinical and pre-clinical stages.

Figure 5.
Basic mechanism of action for indole containing anti-lung cancer drugs.

Scheme 1.
Synthesis of 2-oxindole-derivatives (OXIDs) (6a–c) adapted from the reference [55].
Scheme 1.
Synthesis of 2-oxindole-derivatives (OXIDs) (6a–c) adapted from the reference [55].


Figure 6.
Structures of compounds 17 and AZD3463.


Scheme 4.
Synthesis of enzastaurin adapted from the reference [65].
Scheme 4.
Synthesis of enzastaurin adapted from the reference [65].


Figure 7.
Binding sites in tubulin.

Scheme 6.
Synthesis of FC77 adapted from the reference [79].
Scheme 6.
Synthesis of FC77 adapted from the reference [79].

Scheme 7.
Synthesis of NMK-BH3 adapted from the reference [80].
Scheme 7.
Synthesis of NMK-BH3 adapted from the reference [80].


Figure 8.
Biological significance of bis(hydroxymethyl)pyrrole and β-carboline.

Scheme 9.
Synthesis of compounds 52 and 53, adapted from the reference [83].
Scheme 9.
Synthesis of compounds 52 and 53, adapted from the reference [83].

Figure 9.
Design of indolizino[6,7-b]indole and indolizino[8,7-b]indole derivatives.

Scheme 10.
Synthesis of compounds 57(a–c) adapted from reference [84].
Scheme 10.
Synthesis of compounds 57(a–c) adapted from reference [84].

Scheme 11.
Synthesis of compound 60 adapted from reference [86].
Scheme 11.
Synthesis of compound 60 adapted from reference [86].

Figure 10.
Structure of compounds suberoylanilide hydroxamic acid (SAHA), SL142, SL325, and 61.

Scheme 12.
Synthesis of dacinostat adapted from the reference [98].
Scheme 12.
Synthesis of dacinostat adapted from the reference [98].


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 11.
Structures of dragmacidin, compounds 78, and 79(a–c).

Scheme 14.
Synthesis of SK228 adapted from the reference [105].
Scheme 14.
Synthesis of SK228 adapted from the reference [105].

Figure 12.
Structures of AGM130, EMMQ and tambjamine derivatives.

Scheme 15.
Synthesis of evodiamine adapted from the reference [114].
Scheme 15.
Synthesis of evodiamine adapted from the reference [114].

Figure 13.
Structures of eudistomin K, villalstonine, compounds 85, 86 and 87.

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Dhuguru, J.; Skouta, R. Role of Indole Scaffolds as Pharmacophores in the Development of Anti-Lung Cancer Agents. Molecules 2020, 25, 1615. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25071615
AMA Style
Dhuguru J, Skouta R. Role of Indole Scaffolds as Pharmacophores in the Development of Anti-Lung Cancer Agents. Molecules. 2020; 25(7):1615. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25071615
Chicago/Turabian StyleDhuguru, Jyothi, and Rachid Skouta. 2020. "Role of Indole Scaffolds as Pharmacophores in the Development of Anti-Lung Cancer Agents" Molecules 25, no. 7: 1615. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25071615