
Human Serum Albumin Labelled with Sterically-Hindered Nitroxides as Potential MRI Contrast Agents

 , , , , , and
, , , , , and
Abstract
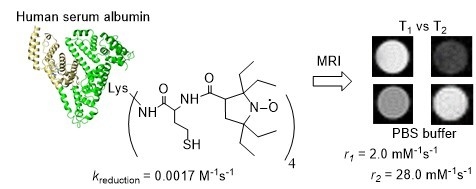
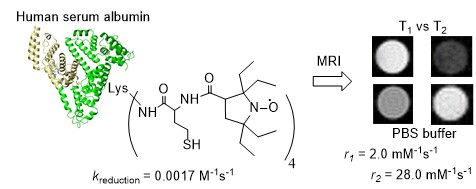
:
1. Introduction
2. Results
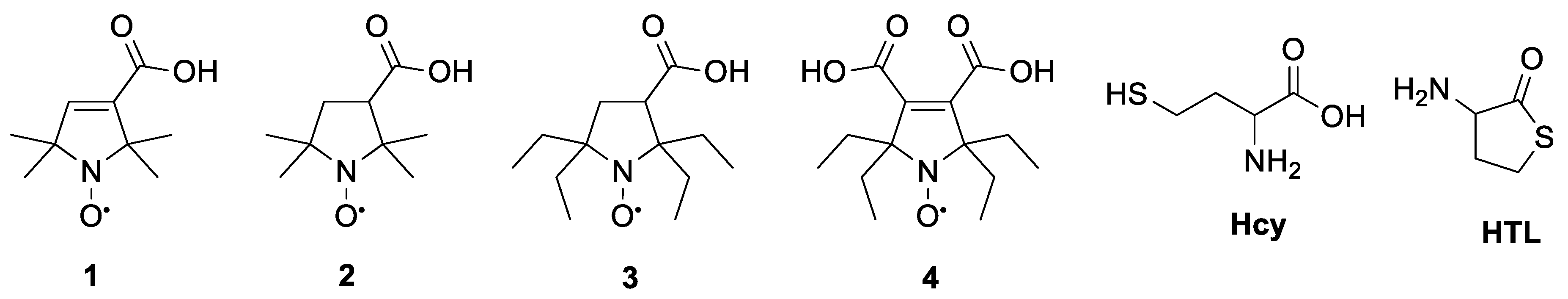
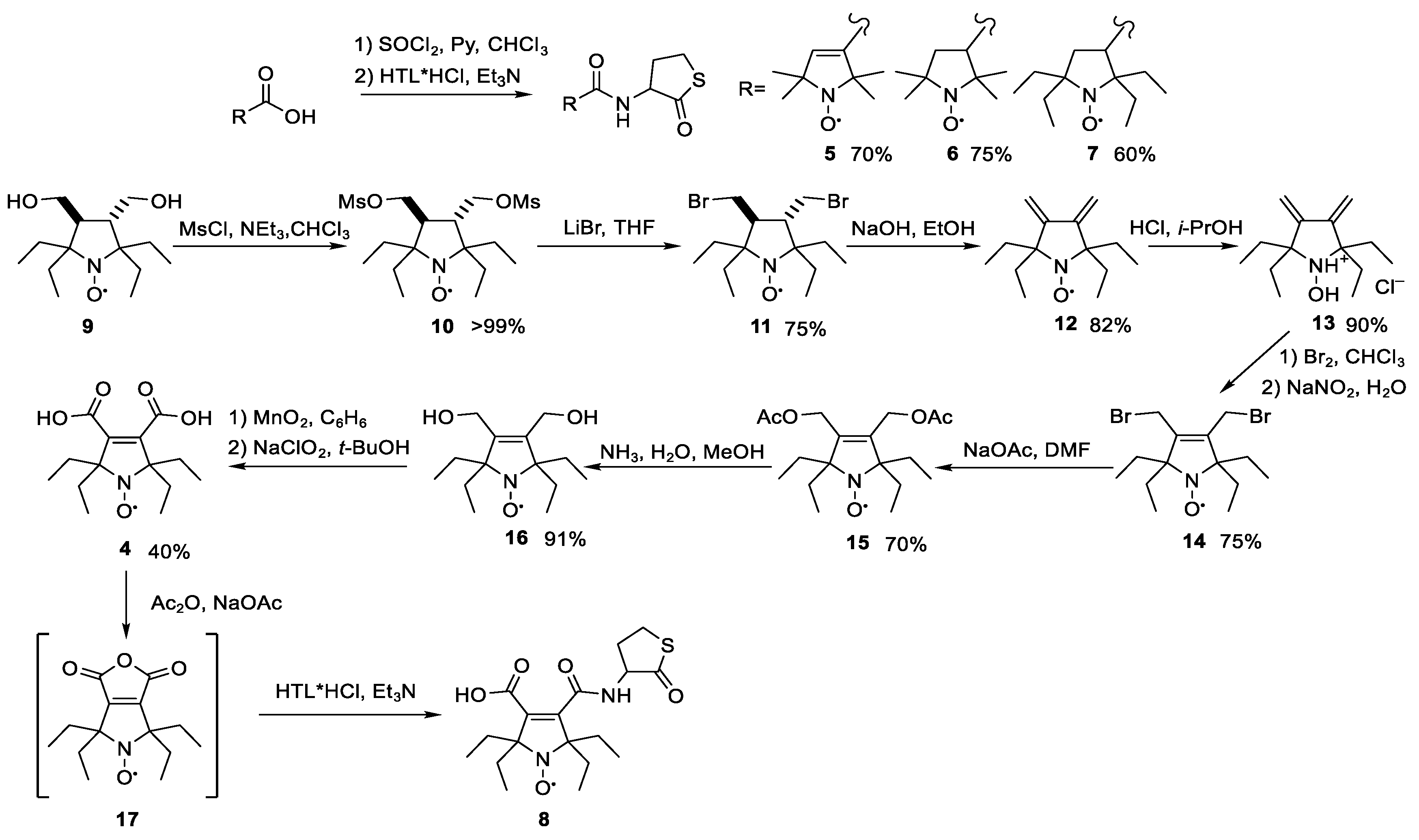
2.1. Synthesis of N-Substituted Homocysteine Thiolactone Derivatives
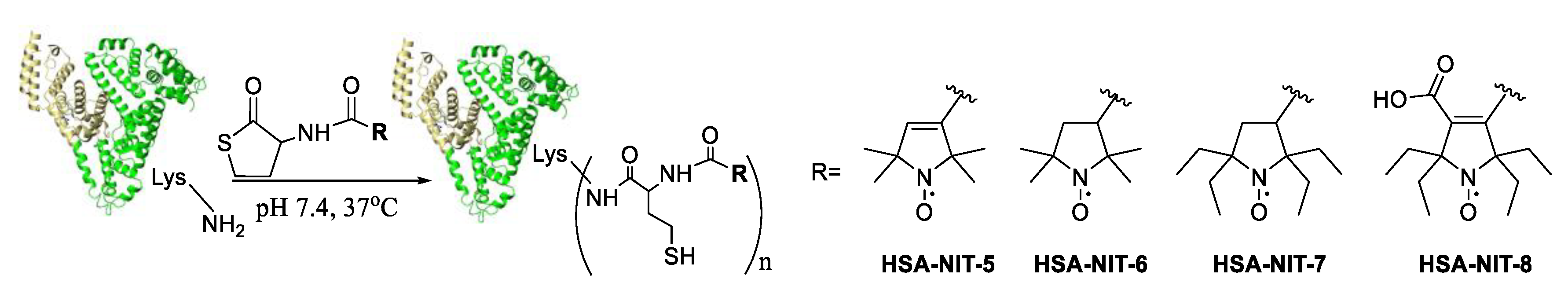
2.2. Synthesis and Characterization of HSA-Nitroxide Conjugates
2.3. Identification of N-Homocysteinylation Sites in HSA-NIT Conjugates
2.4. Reduction Rates of HSA-NIT Conjugates
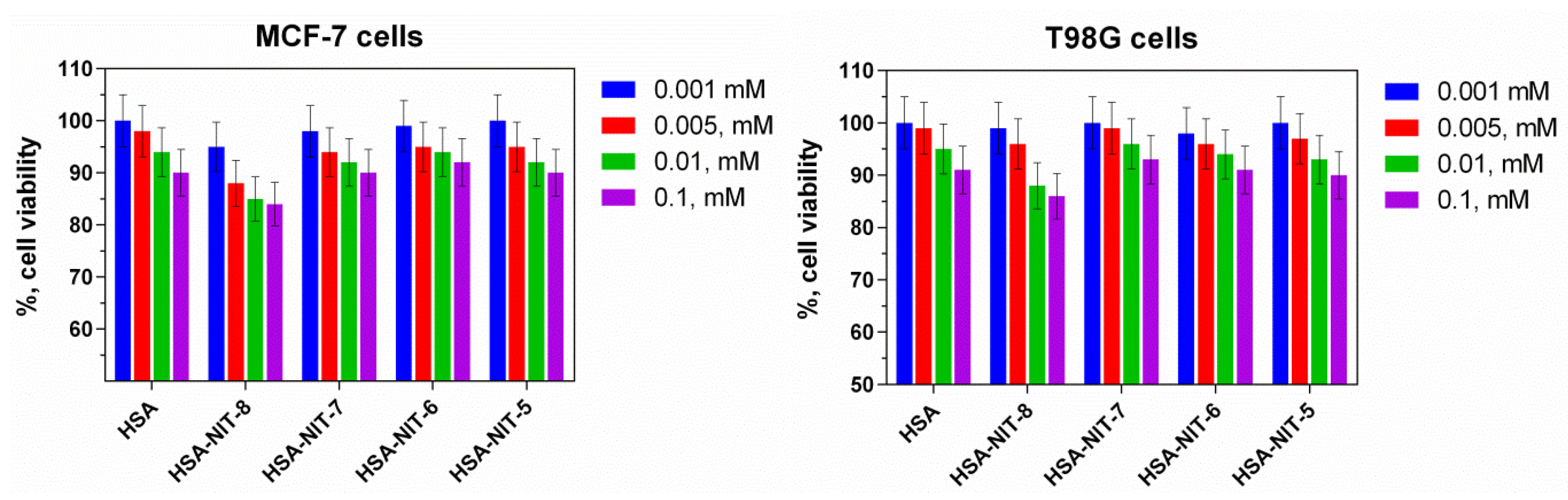
2.5. Cytotoxicity of HSA-NIT Conjugates
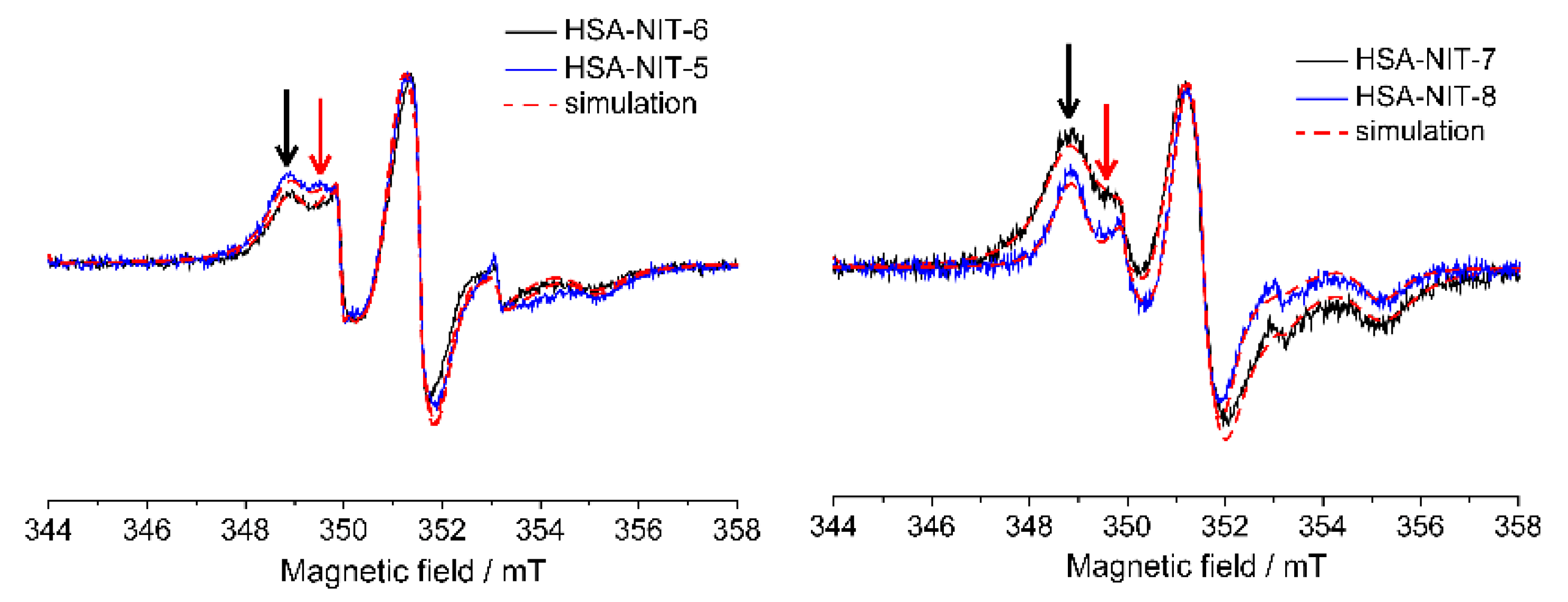
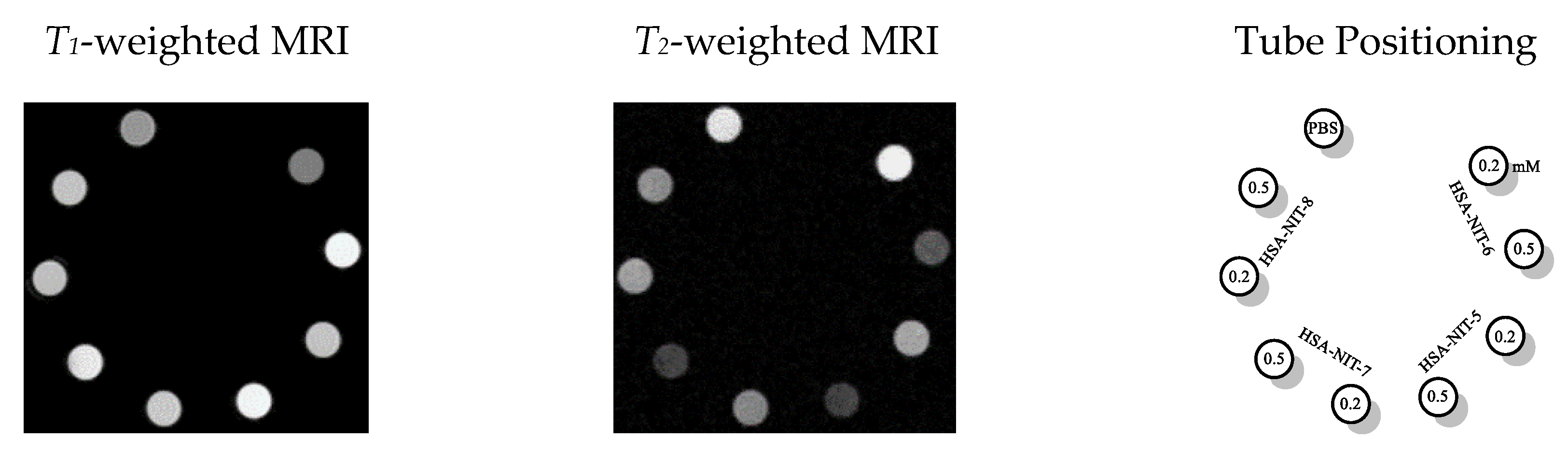
2.6. Characterization of HSA-NIT Magnetic Properties
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wahsner, J.; Gale, E.M.; Rodríguez-Rodríguez, A.; Caravan, P. Chemistry of MRI contrast agents: Current challenges and new frontiers. Chem. Rev. 2019, 119, 957–1057. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Lee, G.H.; Chang, Y. Gadolinium as an MRI contrast agent. Future Med. Chem. 2018, 10, 639–661. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Meade, T.J. Molecular MR Imaging with Gd(III)-based Agents: Challenges and Key Advances. J. Am. Chem. Soc. 2019, 141, 17025–17041. [Google Scholar] [CrossRef] [PubMed]
- Akakuru, O.U.; Iqbal, M.Z.; Saeed, M.; Liu, C.; Paunesku, T.; Woloschak, G.; Hosmane, N.S.; Wu, A. The Transition from Metal-Based to Metal-Free Contrast Agents for T1 Magnetic Resonance Imaging Enhancement. Bioconjug. Chem. 2019, 30, 2264–2286. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, H.S.; Morcos, S.K.; Almén, T.; Bellin, M.F.; Bertolotto, M.; Bongartz, G.; Clement, O.; Leander, P.; Heinz-Peer, G.; Reimer, P.; et al. Nephrogenic systemic fibrosis and gadolinium-based contrast media: Updated ESUR Contrast Medium Safety Committee guidelines. Eur. Radiol. 2013, 23, 307–318. [Google Scholar] [CrossRef]
- Pasquini, L.; Napolitano, A.; Visconti, E.; Longo, D.; Romano, A.; Tomà, P.; Espagnet, M.C.R. Gadolinium-Based Contrast Agent-Related Toxicities. CNS Drugs 2018, 32, 229–240. [Google Scholar] [CrossRef]
- Ramalho, J.; Ramalho, M. Gadolinium Deposition and Chronic Toxicity. Magn. Reson. Imaging Clin. N. Am. 2017, 25, 765–778. [Google Scholar] [CrossRef]
- Malikova, H.; Holesta, M. Gadolinium contrast agents – are they really safe? J. Vasc. Access 2017, 18, s1–s7. [Google Scholar] [CrossRef]
- Swaminathan, S.; Horn, T.D.; Pellowski, D.; Abul-Ezz, S.; Bornhorst, J.A.; Viswamitra, S.; Shah, S.V. Nephrogenic systemic fibrosis, gadolinium, and iron mobilization. N. Engl. J. Med. 2007, 357, 720–722. [Google Scholar] [CrossRef]
- Abakumov, M.A.; Semkina, A.S.; Skorikov, A.S.; Vishnevskiy, D.A.; Ivanova, A.V.; Mironova, E.; Davydova, G.A.; Majouga, A.G.; Chekhonin, V.P. Toxicity of iron oxide nanoparticles: Size and coating effects. J. Biochem. Mol. Toxicol. 2018, 32, 1–6. [Google Scholar] [CrossRef]
- Singh, N.; Jenkins, G.J.S.; Asadi, R.; Doak, S.H. Potential toxicity of superparamagnetic iron oxide nanoparticles (SPION). Nano Rev. 2010, 1, 5358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallabani, N.V.S.; Singh, S. Recent advances and future prospects of iron oxide nanoparticles in biomedicine and diagnostics. 3 Biotech 2018, 8, 1–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tirotta, I.; Dichiarante, V.; Pigliacelli, C.; Cavallo, G.; Terraneo, G.; Bombelli, F.B.; Metrangolo, P.; Resnati, G. 19F Magnetic Resonance Imaging (MRI): From Design of Materials to Clinical Applications. Chem. Rev. 2015, 115, 1106–1129. [Google Scholar] [CrossRef]
- Jirak, D.; Galisova, A.; Kolouchova, K.; Babuka, D.; Hruby, M. Fluorine polymer probes for magnetic resonance imaging: Quo vadis? Magn. Reson. Mater. Physics Biol. Med. 2019, 32, 173–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, H.V.T.; Chen, Q.; Paletta, J.T.; Harvey, P.; Jiang, Y.; Zhang, H.; Boska, M.D.; Ottaviani, M.F.; Jasanoff, A.; Rajca, A.; et al. Nitroxide-Based Macromolecular Contrast Agents with Unprecedented Transverse Relaxivity and Stability for Magnetic Resonance Imaging of Tumors. ACS Cent. Sci. 2017, 3, 800–811. [Google Scholar] [CrossRef]
- Dharmarwardana, M.; Martins, A.F.; Chen, Z.; Palacios, P.M.; Nowak, C.M.; Welch, R.P.; Li, S.; Luzuriaga, M.A.; Bleris, L.; Pierce, B.S.; et al. Nitroxyl Modified Tobacco Mosaic Virus as a Metal-Free High- Relaxivity MRI and EPR Active Superoxide Sensor. Physiol. Behav. 2017, 176, 139–148. [Google Scholar] [CrossRef]
- Bye, N.; Hutt, O.E.; Hinton, T.M.; Acharya, D.P.; Waddington, L.J.; Moffat, B.A.; Wright, D.K.; Wang, H.X.; Mulet, X.; Muir, B.W. Nitroxide-loaded hexosomes provide MRI contrast in vivo. Langmuir 2014, 30, 8898–8906. [Google Scholar] [CrossRef]
- M Davis, R.; B Mitchell, J.; C Krishna, M. Nitroxides as Cancer Imaging Agents. Anti-Cancer Agents Med. Chem. 2012, 11, 347–358. [Google Scholar] [CrossRef]
- Nguyen, H.V.T.; Detappe, A.; Gallagher, N.M.; Zhang, H.; Harvey, P.; Yan, C.; Mathieu, C.; Golder, M.R.; Jiang, Y.; Ottaviani, M.F.; et al. Triply Loaded Nitroxide Brush-Arm Star Polymers Enable Metal-Free Millimetric Tumor Detection by Magnetic Resonance Imaging. ACS Nano 2018, 12, 11343–11354. [Google Scholar] [CrossRef]
- Soikkeli, M.; Horkka, K.; Moilanen, J.O.; Timonen, M.; Kavakka, J.; Heikkinen, S. Synthesis, stability and relaxivity of teepo-met: An organic radical as a potential tumour targeting contrast agent for magnetic resonance imaging. Molecules 2018, 23, 1034. [Google Scholar] [CrossRef] [Green Version]
- Soikkeli, M.; Sievänen, K.; Peltonen, J.; Kaasalainen, T.; Timonen, M.; Heinonen, P.; Rönkkö, S.; Lehto, V.P.; Kavakka, J.S.; Heikkinen, S. Synthesis and in vitro phantom NMR and MRI studies of fully organic free radicals, TEEPO-glucose and TEMPO-glucose, potential contrast agents for MRI. RSC Adv. 2015, 5, 15507–15510. [Google Scholar] [CrossRef]
- Chan, H.C.; Sun, K.Q.; Magin, R.L.; Swartz, H.M. Potential of Albumin Labeled with Nitroxides as a Contrast Agent for Magnetic Resonance Imaging and Spectroscopy. Bioconjug. Chem. 1990, 1, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Shahrivarkevishahi, A.; Lumata, J.L.; Luzuriaga, M.A.; Hagge, L.M.; Benjamin, C.E.; Brohlin, O.R.; Parish, C.R.; Firouzi, H.R.; Nielsen, S.O.; et al. Chemical Science enhancement of metal-free magnetic resonance imaging contrast agents. Chem. Sci. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kocherginsky, N.; Swartz, H.M. Metabolism and Distribution of Nitroxides In Vivo. In Nitroxide Spin Labels: Reactions in Biology and Chemistry; CRC Press: Boca Raton, FL, USA, 1995; pp. 153–174. [Google Scholar]
- Teucher, M.; Zhang, H.; Bader, V.; Winklhofer, K.F.; García-Sáez, A.J.; Rajca, A.; Bleicken, S.; Bordignon, E. A new perspective on membrane-embedded Bax oligomers using DEER and bioresistant orthogonal spin labels. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef]
- Paletta, J.T.; Pink, M.; Foley, B.; Rajca, S.; Rajca, A. Synthesis and reduction kinetics of sterically shielded pyrrolidine nitroxides. Org. Lett. 2012, 14, 5322–5325. [Google Scholar] [CrossRef] [Green Version]
- Kirilyuk, I.A.; Polienko, Y.F.; Krumkacheva, O.A.; Strizhakov, R.K.; Gatilov, Y.V.; Grigor’ev, I.A.; Bagryanskaya, E.G. Synthesis of 2,5-bis(spirocyclohexane)-substituted nitroxides of pyrroline and pyrrolidine series, including thiol-specific spin label: An analogue of MTSSL with long relaxation time. J. Org. Chem. 2012, 77, 8016–8027. [Google Scholar] [CrossRef]
- Soule, B.P.; Hyodo, F.; Matsumoto, K.I.; Simone, N.L.; Cook, J.A.; Krishna, M.C.; Mitchell, J.B. The chemistry and biology of nitroxide compounds. Free Radic. Biol. Med. 2007, 42, 1632–1650. [Google Scholar] [CrossRef] [Green Version]
- Rajca, A.; Wang, Y.; Boska, M.; Paletta, J.T.; Olankitwanit, A.; Swanson, M.A.; Mitchell, D.G.; Eaton, S.S.; Eaton, G.R.; Rajca, S. Organic Radical Contrast Agents for Magnetic Resonance Imaging. J. Am. Chem. Soc. 2012, 134, 15724–15727. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Kang, N.; Xu, T.; Wang, D.; Ren, L.; Guo, X. Core-shell hybrid upconversion nanoparticles carrying stable nitroxide radicals as potential multifunctional nanoprobes for upconversion luminescence and magnetic resonance dual-modality imaging. Nanoscale 2015, 7, 5249–5261. [Google Scholar] [CrossRef]
- Alvaradejo, G.G.; Nguyen, H.V.-T.; Harvey, P.; Gallagher, N.M.; Le, D.; Ottaviani, M.F.; Jasanoff, A.; Delaittre, G.; Johnson, J.A. Polyoxazoline-Based Bottlebrush and Brush-Arm Star Polymers via ROMP: Syntheses and Applications as Organic Radical Contrast Agents. Physiol. Behav. 2017, 176, 139–148. [Google Scholar] [CrossRef]
- Sleep, D.; Cameron, J.; Evans, L.R. Albumin as a versatile platform for drug half-life extension. Biochim. Biophys. Acta 2013, 1830, 5526–5534. [Google Scholar] [CrossRef] [PubMed]
- Caspersen, M.B.; Kuhlmann, M.; Saxton, M.J.; Cameron, J. Albumin-based drug delivery using cysteine 34 chemical conjugates – important considerations and requirements. Ther. Deliv 2017, 8, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Tormyshev, A.V.; Chubarov, A.; Krumkacheva, O.; Trukhin, D.; Rogozhnikova, O.; Spitsina, A.; Kuzhelev, A.; Koval, V.; Fedin, M.; Bowman, M.; et al. A Methanethiosulfonate Derivative of OX063 Trityl: A Promising and Efficient Reagent for SDSL of Proteins. Chem. A Eur. J. 2020, 26, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Krumkacheva, O.A.; Timofeev, I.O.; Politanskaya, L.V.; Polienko, Y.F.; Tretyakov, E.V.; Rogozhnikova, O.Y.; Trukhin, D.V.; Tormyshev, V.M.; Chubarov, A.S.; Bagryanskaya, E.G.; et al. Triplet Fullerenes as Prospective Spin Labels for Nanoscale Distance Measurements by Pulsed Dipolar EPR. Angew. Chemie Int. Ed. 2019, 58, 13271–13275. [Google Scholar] [CrossRef]
- Spicer, C.D.; Davis, B.G. Selective chemical protein modification. Nat. Commun. 2014, 5, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matos, M.J.; Oliveira, B.L.; Martínez-Sáez, N.; Guerreiro, A.; Cal, P.M.S.D.; Bertoldo, J.; Maneiro, M.M.; Perkins, E.; Howard, J.; Deery, M.J.; et al. Chemo- and Regioselective Lysine Modification on Native Proteins. J. Am. Chem. Soc. 2018, 140, 4004–4017. [Google Scholar] [CrossRef]
- Asano, S.; Patterson, J.T.; Gaj, T.; Barbas, C.F. Site-selective labeling of a lysine residue in human serum albumin. Angew. Chemie Int. Ed. 2014, 53, 11783–11786. [Google Scholar] [CrossRef]
- Chubarov, A.S.; Shakirov, M.M.; Koptyug, I.V.; Sagdeev, R.Z.; Knorre, D.G.; Godovikova, T.S. Synthesis and characterization of fluorinated homocysteine derivatives as potential molecular probes for 19F magnetic resonance spectroscopy and imaging. Bioorg. Med. Chem. Lett. 2011, 21, 4050–4053. [Google Scholar] [CrossRef]
- Lisitskiy, V.A.; Khan, H.; Popova, T.V.; Chubarov, A.S.; Zakharova, O.D.; Akulov, A.E.; Shevelev, O.B.; Zavjalov, E.L.; Koptyug, I.V.; Moshkin, M.P.; et al. Multifunctional human serum albumin-therapeutic nucleotide conjugate with redox and pH-sensitive drug release mechanism for cancer theranostics. Bioorganic Med. Chem. Lett. 2017, 27, 3925–3930. [Google Scholar] [CrossRef]
- Sikora, M.; Marczak, Ł.; Kubalska, J.; Graban, A.; Jakubowski, H. Identification of N-homocysteinylation sites in plasma proteins. Amino Acids 2014, 46, 235–244. [Google Scholar] [CrossRef]
- Marczak, L.; Sikora, M.; Stobiecki, M.; Jakubowski, H. Analysis of site-specific N-homocysteinylation of human serum albumin in vitro and in vivo using MALDI-ToF and LC-MS/MS mass spectrometry. J. Proteomics 2011, 74, 967–974. [Google Scholar] [CrossRef]
- Sikora, M.; Marczak, Ł.; Twardowski, T.; Stobiecki, M.; Jakubowski, H. Direct monitoring of albumin lysine-525 N-homocysteinylation in human serum by liquid chromatography/mass spectrometry. Anal. Biochem. 2010, 405, 132–134. [Google Scholar] [CrossRef] [PubMed]
- Präbst, K.; Engelhardt, H.; Ringgeler, S.; Hübner, H. Basic Colorimetric Proliferation Assays: MTT, WST, and Resazurin. In Cell Viability Assays. Methods in Molecular Biology; Humana Press: New York, NY, USA, 2017; pp. 1–17. [Google Scholar]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Jakubowski, H. Molecular basis of homocysteine toxicity in humans. Cell. Mol. Life Sci. 2004, 61, 470–487. [Google Scholar] [CrossRef] [PubMed]
- Chubarov, A.S.; Zakharova, O.D.; Koval, O.A.; Romaschenko, A.V.; Akulov, A.E.; Zavjalov, E.L.; Razumov, I.A.; Koptyug, I.V.; Knorre, D.G.; Godovikova, T.S. Design of protein homocystamides with enhanced tumor uptake properties for 19F magnetic resonance imaging. Bioorg. Med. Chem. 2015, 23, 6943–6954. [Google Scholar] [CrossRef]
- Sibrian-Vazquez, M.; Escobedo, J.O.; Lim, S.; Samoei, G.K.; Strongin, R.M. Homocystamides promote free-radical and oxidative damage to proteins. Proc. Natl. Acad. Sci. USA 2010, 107, 551–554. [Google Scholar] [CrossRef] [Green Version]
- Frank, D.; Espeel, P.; Claessens, S.; Mes, E.; Du Prez, F.E. Synthesis of thiolactone building blocks as potential precursors for sustainable functional materials. Tetrahedron 2016, 6–15. [Google Scholar] [CrossRef]
- Espeel, P.; Prez, F. One-pot double modification of polymers based on thiolactone chemistry. Adv. Polym. Sci. 2015, 269, 105–132. [Google Scholar]
- Dobrynin, S.A.; Khoroshunova, Y.V.; Kirilyuk, I.A. Synthesis of 2,2,5,5-tetraethyl-3-carboxypyrrolidine 1-oxyl. RU. Patent No. 2,702,331, 25 July 2019. [Google Scholar]
- Dichtl, A.; Seyfried, M.; Schoening, K.U. A novel method for the synthesis of N-alkoxyamines starting from nitroxide radicals and ketones. Synlett 2008, 2008, 1877–1881. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Dobrynin, S.A.; Glazachev, Y.I.; Gatilov, Y.V.; Chernyak, E.I.; Salnikov, G.E.; Kirilyuk, I.A. Synthesis of 3,4-Bis(hydroxymethyl)-2,2,5,5-tetraethylpyrrolidin-1-oxyl via 1,3-Dipolar Cycloaddition of Azomethine Ylide to Activated Alkene. J. Org. Chem. 2018, 83, 5392–5397. [Google Scholar] [CrossRef]
- Kálai, T.; Bognár, B.; Zsolnai, D.; Berente, Z.; Hideg, K. Synthesis of nitroxide-annulated carbocycles and heterocycles. Synthesis 2012, 44, 3655–3660. [Google Scholar] [CrossRef]
- Kalai, T.; Balog, M.; Jekő, J.; Hideg, K. Synthesis and reactions of a symmetric paramagnetic pyrrolidine diene. Synthesis 1999, 973–980. [Google Scholar] [CrossRef]
- Janatova, J.; Fuller, J.K.; Hunter, M.J. The heterogeneity of bovine albumin with respect to sulfhydryl and dimer content. J. Biol. Chem. 1968, 243, 3612–3622. [Google Scholar] [PubMed]
- Sterling, H.J.; Prell, J.S.; Cassou, C.A.; Williams, E.R. Protein conformation and supercharging with DMSO from aqueous solution. J. Am. Soc. Mass Spectrom. 2011, 22, 1178–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Abreu Costa, L.; Ottoni, M.H.F.; Dos Santos, M.G.; Meireles, A.B.; De Almeida, V.G.; De Fátima Pereira, W.; De Avelar-Freitas, B.A.; Brito-Melo, G.E.A. Dimethyl sulfoxide (DMSO) decreases cell proliferation and TNF-α, IFN-, and IL-2 cytokines production in cultures of peripheral blood lymphocytes. Molecules 2017, 22, 1789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yiannios, C.N.; Karabinos, J.V. Oxidation of Thiols by Dimethyl Sulfoxide. J. Org. Chem. 1963, 28, 3246–3248. [Google Scholar] [CrossRef]
- Papanyan, Z.K. Interaction of L-cysteine with dimethyl sulfoxide in mild conditions. Proc. Yerevan State Univ. 2013, 2, 11–14. [Google Scholar]
- Perła-Kaján, J.; Twardowski, T.; Jakubowski, H.; Perla-Kajan, J.; Twardowski, T.; Jakubowski, H. Mechanisms of homocysteine toxicity in humans. Amino Acids 2007, 32, 561–572. [Google Scholar] [CrossRef]
- Jakubowski, H. Homocysteine in Protein Structure/Function and Human Disease; Springer: Wien, Austria, 2013. [Google Scholar]
- Perczel, A.; Hollósi, M.; Tusnády, G.; Fasman, G.D. Convex constraint analysis: A natural deconvolution of circular dichroism curves of proteins. Protein Eng. 1991, 4, 669–679. [Google Scholar] [CrossRef]
- Varkovitzky, R.L. Assimilation, accommodation, and overaccommodation: An examination of information processing styles in female victims of adolescent and adult sexual assault. ProQuest Diss. Theses 2012, 1, 218. [Google Scholar]
- Paoli, P.; Sbrana, F.; Tiribilli, B.; Caselli, A.; Pantera, B.; Cirri, P.; De Donatis, A.; Formigli, L.; Nosi, D.; Manao, G.; et al. Protein N-homocysteinylation induces the formation of toxic amyloid-like protofibrils. J. Mol. Biol. 2010, 400, 889–907. [Google Scholar] [CrossRef] [PubMed]
- Willard, L.; Ranjan, A.; Zhang, H.; Monzavi, H.; Boyko, R.F.; Sykes, B.D.; Wishart, D.S. VADAR: A web server for quantitative evaluation of protein structure quality. Nucleic Acids Res. 2003, 31, 3316–3319. [Google Scholar] [CrossRef] [PubMed]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Søndergaard, C.R.; Olsson, M.H.M.; Rostkowski, M.; Jensen, J.H. Improved treatment of ligands and coupling effects in empirical calculation and rationalization of pKa values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [Google Scholar] [CrossRef]
- Altenbach, C.; Marti, T.; Khorana, H.G.; Hubbell, W.L. Transmembrane protein structure: Spin labeling of bacteriorhodopsin mutants. Science 1990, 248, 1088–1092. [Google Scholar] [CrossRef]
- Stoll, S.; Schweiger, A. Easyspin: Simulating CW ESR spectra. Biol. Magn. Reson. 2007, 27, 299–321. [Google Scholar]
- Stoll, S.; Schweiger, A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 2006, 178, 42–55. [Google Scholar] [CrossRef]
- Zhurko, I.F.; Dobrynin, S.; Gorodetskii, A.A.; Glazachev, Y.I.; Rybalova, T.V.; Chernyak, E.I.; Asanbaeva, N.; Bagryanskaya, E.G.; Kirilyuk, I.A. 2-Butyl-2-tert-butyl-5,5-diethylpyrrolidine-1-oxyls: Synthesis and properties. Molecules 2020, 25, 845. [Google Scholar] [CrossRef] [Green Version]
- Jagtap, A.P.; Krstic, I.; Kunjir, N.C.; Hänsel, R.; Prisner, T.F.; Sigurdsson, S.T. Sterically shielded spin labels for in-cell EPR spectroscopy: Analysis of stability in reducing environment. Free Radic. Res. 2015, 49, 78–85. [Google Scholar] [CrossRef]
- Goldstein, S.; Merenyi, G.; Russo, A.; Samuni, A. The role of oxoammonium cation in the SOD-mimic activity of cyclic nitroxides. J. Am. Chem. Soc. 2003, 125, 789–795. [Google Scholar] [CrossRef]
- Sowers, M.A.; Mccombs, J.R.; Wang, Y.; Paletta, J.T.; Morton, S.W.; Dreaden, E.C.; Boska, M.D.; Ottaviani, M.F.; Hammond, P.T.; Rajka, A.; et al. Redox-responsive branched-bottlebrush polymers for in vivo MRI and fluorescence imaging Molly. Nat Commun. 2014, 5, 5460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popova, T.V.; Khan, H.; Chubarov, A.S.; Lisitskiy, V.A.; Antonova, N.M.; Akulov, A.E.; Shevelev, O.B.; Zavjalov, E.L.; Silnikov, V.N.; Ahmad, S.; et al. Biotin-decorated anti-cancer nucleotide theranostic conjugate of human serum albumin: Where the seed meets the soil? Bioorganic Med. Chem. Lett. 2018, 28, 260–264. [Google Scholar] [CrossRef] [PubMed]
- Larsen, M.T.; Kuhlmann, M.; Hvam, M.L.; Howard, K.A. Albumin-based drug delivery: Harnessing nature to cure disease. Mol. Cell. Ther. 2016, 4, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levitt, D.G.; Levitt, M.D. Human serum albumin homeostasis: A new look at the roles of synthesis, catabolism, renal and gastrointestinal excretion, and the clinical value of serum albumin measurements. Int. J. Gen. Med. 2016, 9, 229–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Sample Availability: Samples of the compounds 5, 6, 7 are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CD Spectral Analysis | SDS-PAGE Analysis | ||||
|---|---|---|---|---|---|
| HSA Type | n * | α-Helix, % | β-Sheet, % | Oligomer, % | Monomer, % |
| HSA | 0 | 55.0 | 4.5 | 16 | 84 |
| HSA-NIT-5 | 4.8 | 45.8 | 4.8 | 17 | 83 |
| HSA-NIT-6 | 3.9 | 46.5 | 5.6 | 18 | 82 |
| HSA-NIT-7 | 3.9 | 46.2 | 5.1 | 17 | 83 |
| HSA-NIT-8 | 2.4 | 48.0 | 5.0 | 15 | 85 |
| Hcy-has | 3.3 | 44.0 | 8.0 | 41 | 59 |
| HSA Type | N-Hcy site (Lys residues) * | ||||||
|---|---|---|---|---|---|---|---|
| 525 | 212 | 205 | 159 | 137 | 12 | 4 | |
| HSA-NIT-5 | ++ | + | ++ | + | + | ++ | + |
| HSA-NIT-6 | ++ | + | ++ | + | + | + | 0 |
| HSA-NIT-7 | ++ | + | ++ | + | ++ | + | + |
| HSA-NIT-8 | ++ | + | ++ | + | + | + | ++ |
| Hcy-HSA [41,42,43] | ++ | ++ | + | + | ++ | + | + |
| PFT-Hcy-HSA [40] | ++ | 0 | ++ | 0 | ++ | 0 | 0 |
| Nitroxide Structure | Method | Nitroxide Structure | Method | |||||
|---|---|---|---|---|---|---|---|---|
| EPR | r1-meas. | EPR | r1-meas. | |||||
| init. | «fast» | «slow» | ||||||
 | 0.0010 | 0.0025 | 0.0006 | 0.0022 ± 0.0003 |  | - | - | |
 | 0.0017 | 0.005 | 0.0015 | 0.0018 ± 0.0003 |  | R=OH ~0.0013 [73] 0.002 [74] | R=OH 0.0023 | |
 | 0.056 | 0.13 | 0.053 | 0.008 ± 0.001 |  | R=OH 0.063 [26,29] 0.097 [27] R=NH2 0.30 [27] | R=OH n.d.** | |
 | 0.096 | 0.22 | 0.066 | 0.015 ± 0.003 |  | R=OH 0.22 [27] R=NH2 0.58 [27] | R=OH n.d.** | |
| HSA Type | t °C | r1, mM−1s−1 | r2, mM−1s−1 | n * | τcor (ns) ** | r1, mM−1s−1 | r2, mM−1s−1 | r2/r1 |
|---|---|---|---|---|---|---|---|---|
| per albumin molecule | per nitroxide radical | |||||||
| HSA-NIT-8 | 25 | 0.80 ±0.03 | 11.2 ±0.2 | 2.4 | 14/2.6 | 0.33 | 4.7 | 14.2 |
| HSA-NIT-7 | 25 | 1.99 ±0.05 | 27.9 ±0.5 | 3.9 | 14/2.7 | 0.51 | 7.2 | 14.1 |
| 37 | 1.51 ±0.03 | 20.6 ±0.1 | 3.9 | 0.39 | 5.3 | 13.6 | ||
| HSA-NIT-6 | 25 | 1.86 ±0.04 | 20.4 ±0.3 | 3.9 | 10/2.1 | 0.48 | 5.2 | 10.8 |
| 37 | 1.51 ±0.05 | 16.6 ±0.3 | 3.9 | 0.39 | 4.3 | 11.0 | ||
| HSA-NIT-5 | 25 | 2.24 ±0.08 | 27.7 ±0.2 | 4.8 | 10/1.9 | 0.47 | 5.8 | 12.3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dobrynin, S.; Kutseikin, S.; Morozov, D.; Krumkacheva, O.; Spitsyna, A.; Gatilov, Y.; Silnikov, V.; Angelovski, G.; Bowman, M.K.; Kirilyuk, I.; et al. Human Serum Albumin Labelled with Sterically-Hindered Nitroxides as Potential MRI Contrast Agents. Molecules 2020, 25, 1709. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25071709
Dobrynin S, Kutseikin S, Morozov D, Krumkacheva O, Spitsyna A, Gatilov Y, Silnikov V, Angelovski G, Bowman MK, Kirilyuk I, et al. Human Serum Albumin Labelled with Sterically-Hindered Nitroxides as Potential MRI Contrast Agents. Molecules. 2020; 25(7):1709. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25071709
Chicago/Turabian StyleDobrynin, Sergey, Sergei Kutseikin, Denis Morozov, Olesya Krumkacheva, Anna Spitsyna, Yurii Gatilov, Vladimir Silnikov, Goran Angelovski, Michael K. Bowman, Igor Kirilyuk, and et al. 2020. "Human Serum Albumin Labelled with Sterically-Hindered Nitroxides as Potential MRI Contrast Agents" Molecules 25, no. 7: 1709. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25071709