
A Simple Zinc-Mediated Method for Selenium Addition to Michael Acceptors
by
 , ,
, ,
Francesca Giulia Nacca
1,2, 
Bonifacio Monti
1,
Eder João Lenardão
3 ,
,
Paul Evans
2 and
Claudio Santi
1,*
1
Group of Catalysis, Synthesis and Organic Green Chemistry, Department of Pharmaceutical Sciences University of Perugia Via del Liceo 1, 06123 Perugia, Italy
2
Centre for Synthesis and Chemical Biology, School of Chemistry University College Dublin, Dublin D04, N2E5, Ireland
3
LASOL–CCQFA, Universidade Federal de Pelotas—UFPel, P.O. Box 354, 96010-900 Pelotas, RS, Brazil
*
Author to whom correspondence should be addressed.
Molecules 2020, 25(9), 2018; https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25092018
Submission received: 29 March 2020
/
Revised: 20 April 2020
/
Accepted: 23 April 2020
/
Published: 26 April 2020
(This article belongs to the Special Issue Organoselenium Reagents and Their Applications)
Abstract
:In this work, we focused our attention on seleno-Michael type reactions. These were performed using zinc-selenolates generated in situ from diphenyl diselenide 1, 1,2-bis(3-phenylpropyl)diselenide 30, and protected selenocystine 31 via an efficient biphasic Zn/HCl-based reducing system. Alkenes with a variety of electron-withdrawing groups were investigated in order to gauge the scope and limitations of the process. Results demonstrated that the addition to acyclic α,β-unsaturated ketones, aldehydes, esters amides, and acids was effectively achieved and that alkyl substituents at the reactive β-centre can be accommodated. Similarly, cyclic enones undergo efficient Se-addition and the corresponding adducts were isolated in moderate to good yield. Vinyl sulfones, α,β-unsaturated nitriles, and chalcones are not compatible with these reaction conditions. A recycling experiment demonstrated that the unreacted Zn/HCl reducing system can be effectively reused for seven reaction cycles (91% conversion yield at the 7° recycling rounds).
1. Introduction
Organoselenium derivatives are widely utilized in the area of organic chemistry, this is due to their great versatility. The introduction of an organoselenium moiety can be easily obtained starting from diselenides that can be readily converted into electrophilic, nucleophilic, or radical species through oxidative, reductive, or homolytic cleavage of the selenium–selenium bond [1,2,3,4,5,6,7,8,9]. Researchers have taken advantage of this chemistry to synthesize a range of different bioactive compounds incorporating selenium [10,11,12]. Some selenium-containing molecules can behave as antioxidants, enzyme mimics and inhibitors, immunomodulators, cytoprotectors, antitumoral, anti-inflammatory, antihypertensive, and anti-infective agents [13,14,15,16]. Additionally, we have reported on the immunomodulatory and cytotoxic properties of selenium-containing compounds and their activity as inhibitors of several different enzymes and proteins, such as glutathione transferase (GSTp) and NCp7, which indicate interesting potential applications in anticancer and anti-HIV chemotherapy [17,18].
Nucleophilic selenolates are often generated in situ by reduction of the selenium–selenium bond in diselenides, or by the insertion of elemental selenium into organometallic species, such as Grignard reagents or organolithium derivatives [19,20,21]. The main drawbacks of these procedures are the use of inert conditions and the instability of the desired selenolates, which can rapidly oxidize to the corresponding diselenides. A variety of low-valent metals have been used for reductive cleavage of the Se–Se bond: Cd(0) [22], La(0) [23], In(0) [24], In(I) [25], Sm(0) [26,27], Sm(II) [28], Sn(0) [29], Yb(II) [30], Cu(II)/Sn(II) [31]. In addition, several different reducing agents, such as: NaBH4 [32], LiAlH4 [33], LiEt3BH [34], DIBAL [35], NaB(OMe)3H [36], H3PO2 [37], N2H4 [38], or Rongalite have all been used to good effect [39].
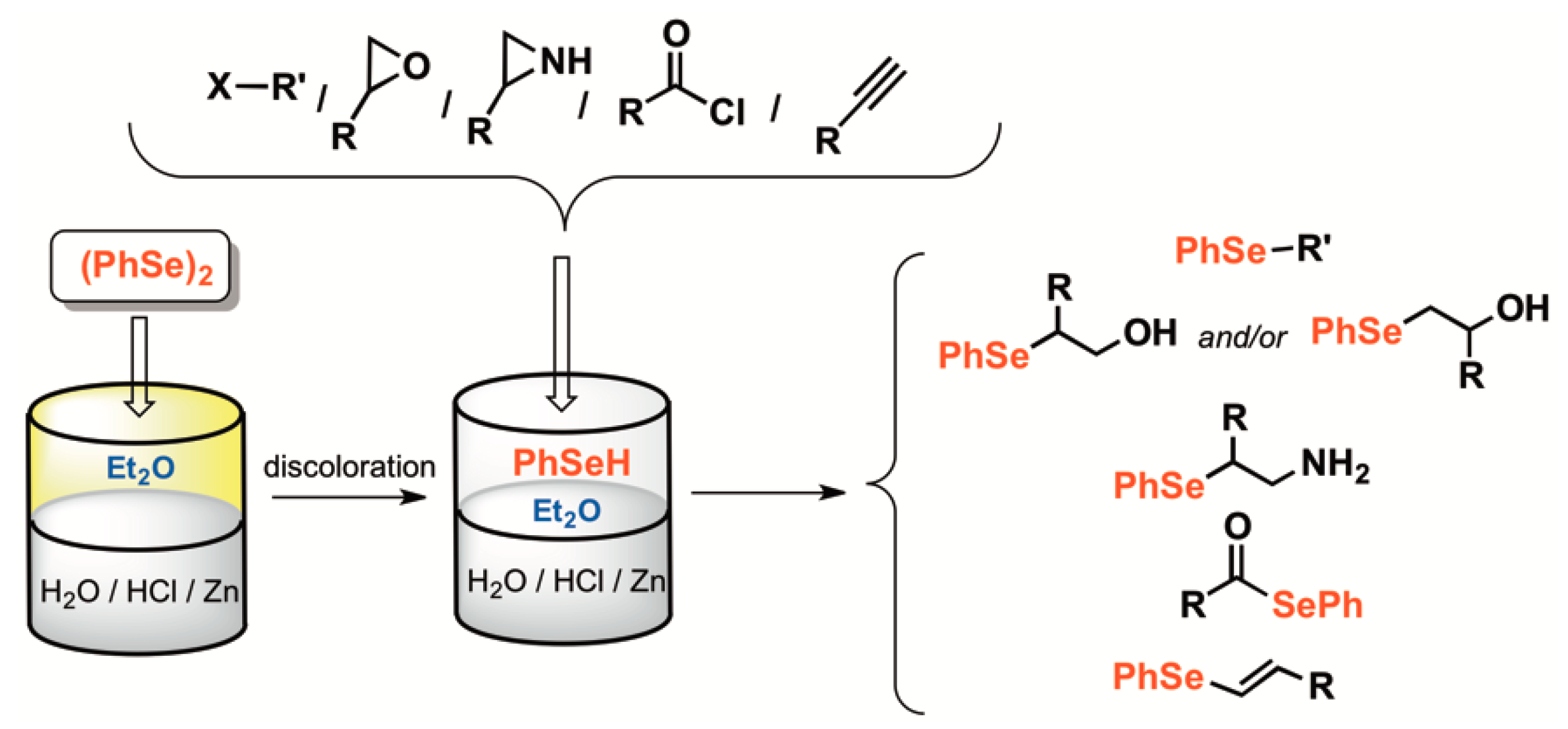
In order to obtain nucleophilic selenium species, we became interested in the applicability of zinc. The use of this metal in the S–S and Se–Se bond cleavages was already described, in combination with Lewis acids, such as aluminum chloride (AlCl3) [40,41], zirconium tetrachloride (ZrCl4) [42], RuCl3 [43] and TiCl4 [44], or simply basic [45] or acidic conditions [46]. The zinc insertion into Se-Halogen bond leading to the umpolung of the selenium atom and the formation of the first class of bench stable zinc selenolates, was deeply investigated, with a broad synthetic applicability [47,48,49,50,51,52,53,54,55]. Previously, it has been reported that this diselenide zinc-mediated reduction can be successfully coupled with chemistry aimed at derivatizing the sensitive, in situ formed selenol [56]. For instance, various epoxides [56], aziridines [57], alkyl and acyl halides [56,58] can engage in nucleophilic substitution processes and generate the corresponding organoselenide products in good to excellent yields (Scheme 1). A feature of this approach is that using a biphasic system (Et2O/HCl(aq)), the product partitions into the organic phase, facilitating the separation and the reuse of the unreacted zinc, as well as of the aqueous acidic medium. Indeed, often a simple solvent removal leads to the isolation of Se-derivatives without the requirement for additional purification. This method proved to be the best alternative to reduce S–S and Se–Se bonds of some non-natural peptides, as recently demonstrated by Flemer [59,60].
More recently, we have demonstrated that the soft-selenium-based nucleophile can efficiently add to a range of alkynes [61] and we collected some of these data in a recently published review article [62]. As an extension of these studies, in this current work, we report that this in situ formed a nucleophilic selenenylating mixture that can efficiently react with a range of Michael acceptors (Scheme 2).
2. Results and Discussion
Considering that conjugated systems, mainly aldehydes and ketones (EWG = CHO, COR) can be prone to reduction in the presence of Zn/HCl [63], new conditions were optimized with the removal of unreacted zinc after the discoloration of the organic phase, and before the addition of the substrates. Furthermore, to improve the “greenness” of the overall procedure, the organic phase (diethyl ether) was changed to ethyl acetate, because this solvent is easier to recycle, has a lower vapor pressure, and presents a series of other aspects (in terms of health and environmental impact), which have given it a recommended ranking by the CHEM21 selection guide [64].
Initially, a range of monosubstituted α,β-unsaturated alkenes presenting a single electron-withdrawing group were considered. As precedingly reported [56,58], the nucleophilic selenenylating mixture arises from the reduction of the diselenide through the oxidative insertion of zinc into the Se–Se bond affording the in situ formation of a selenolate [PhSeZnSePh] that, in the acidic biphasic system, is in equilibrium with the corresponding selenol. This process takes roughly 20 min, and experimentally the reduction progress can be visually determined by the loss of the yellow coloration in the organic phase, caused by diphenyl diselenide. The mixture was then decanted in order to remove residual unreacted zinc (used in excess) and the desired alkene was added. Following this protocol, a range of differently substituted alkenes underwent efficient conjugate addition and the adducts were isolated in moderate to good yield after stirring for 2 h (Table 1).
As shown in Table 1, Entry 1, methyl vinyl ketone 2 gave the adduct 11 in excellent yield. Similarly, acrolein 3, trans-but-2-enal 4, and trans-pent-2-enal 5 gave the corresponding phenylselenide adducts 12–14 in moderate to good yields (Table 1, Entries 2–4). The low yield in the case of 15 was primarily due to the limited stability of this Se-adduct during purification using silica-gel chromatography and not to a low conversion of the starting material. Next, we turned our attention to carboxylic acids and their derivatives. Methyl acrylate 7 and acrylic acid 8 gave ester 16 and acid 17 in good yields (Table 1, Entries 6 and 7). In the case of unsaturated amide 9 (Table 1, Entry 8), a low yield of Se-adduct 18 was isolated presumably due to poor conversion stemming from the reduced electrophilicity of the Michael-acceptor. Finally, under the conditions developed, pulegone 10 gave the adduct 19 in excellent yield as a mixture of diastereomers at the new chiral center (Table 1, Entry 9). Less electron-poor alkenes, such as phenyl vinyl sulfone, acrylonitrile, and chalcones did not prove to be suitable substrates and no conversions were observed under the conditions optimized for substrates 2–10.
The reaction between diphenyl diselenide 1 and a range of cyclic enones 20–24 was next considered (Table 2). As observed in Table 2, Entry 1, using cyclopentenone 20, the adduct 25 was isolated in excellent yield. Similarly, cyclohexenone 21 and cycloheptenone 22 gave the corresponding Se-adducts 26 and 27 in comparable yields (Table 2, Entries 2 and 3). The substituent in the γ-position in cyclopentenone 23 was compatible with this process and product 28 was isolated as a 75:25 mixture of undetermined diastereomers (Table 2, Entry 4). Finally, the natural product jacaranone 24, bearing two-potential sites for nucleophilic attack, was found to form in low yield (29%) mono-adduct 29 as a single undetermined diastereomer under the optimized conditions (Table 2, Entry 5).
Finally, the possibility of using alternative diselenides in the reaction with enones was considered. As shown in Table 3, both 1,2-bis(3-phenylpropyl)diselenide 30 and N,O-protected selenocystine 31 proved to generate the corresponding adducts in moderate to good yields.
As shown in Table 3, Entries 1–2, Se-adducts 34–35, obtained starting from 1,2-bis(3-phenylpropyl)diselenide 30, were isolated in good yields whereas the reactivity of the nucleophilic selenenylating mixture prepared starting from the N,O-protected selenocystine 31 is appreciably reduced affording the target compounds 35, 36, and 37 only in moderate yields (Entries 3, 4, and 5). Nevertheless, despite the reduced reactivity, it is worth mentioning that in these latter cases the Boc protecting group was not unduly affected by the acidic media. In the cases in which the conjugate addition of the organoselenium moiety generates a new chiral center in the presence of a preformed one in the substrate and or in the reagent (Entries 2, 4, and 5), a moderated stereoselectivity was obtained. In these cases, the reaction afforded a mixture of diastereomers from which, only in one case, was it possible to isolate the major one as a stereochemically undetermined product 34. In the other two cases, probably due to their high polarity, it was not possible to separate the diastereomers 36 and 37 by flash chromatography (Table 3, Entries 4–6, respectively).
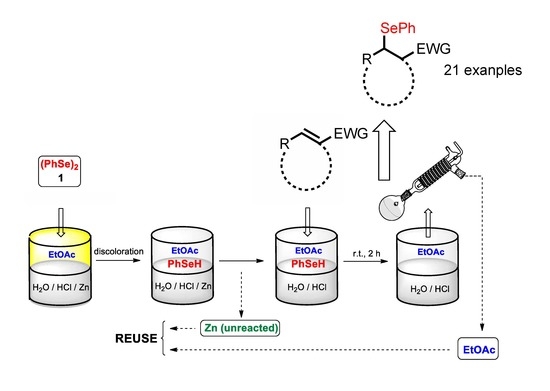
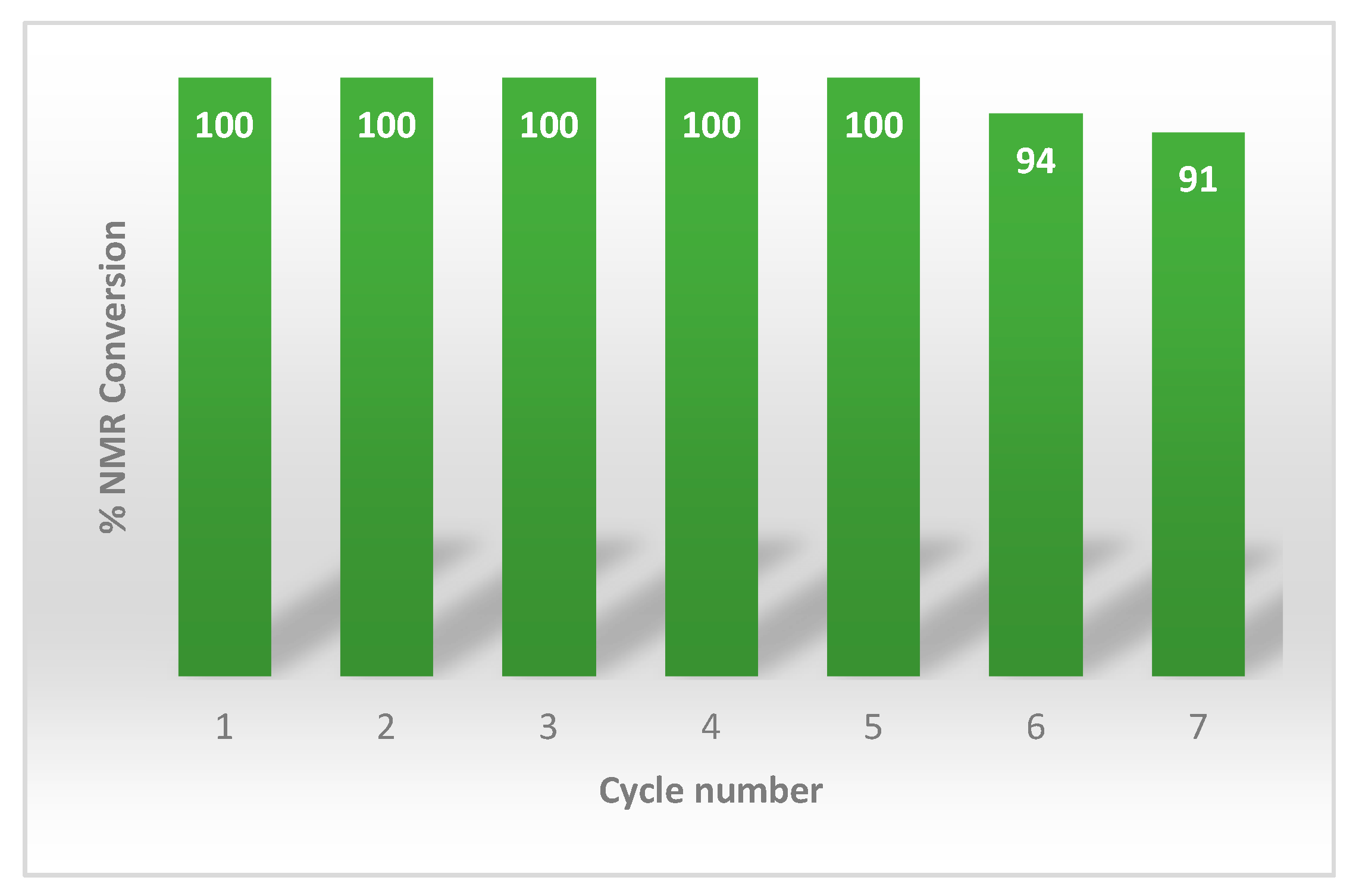
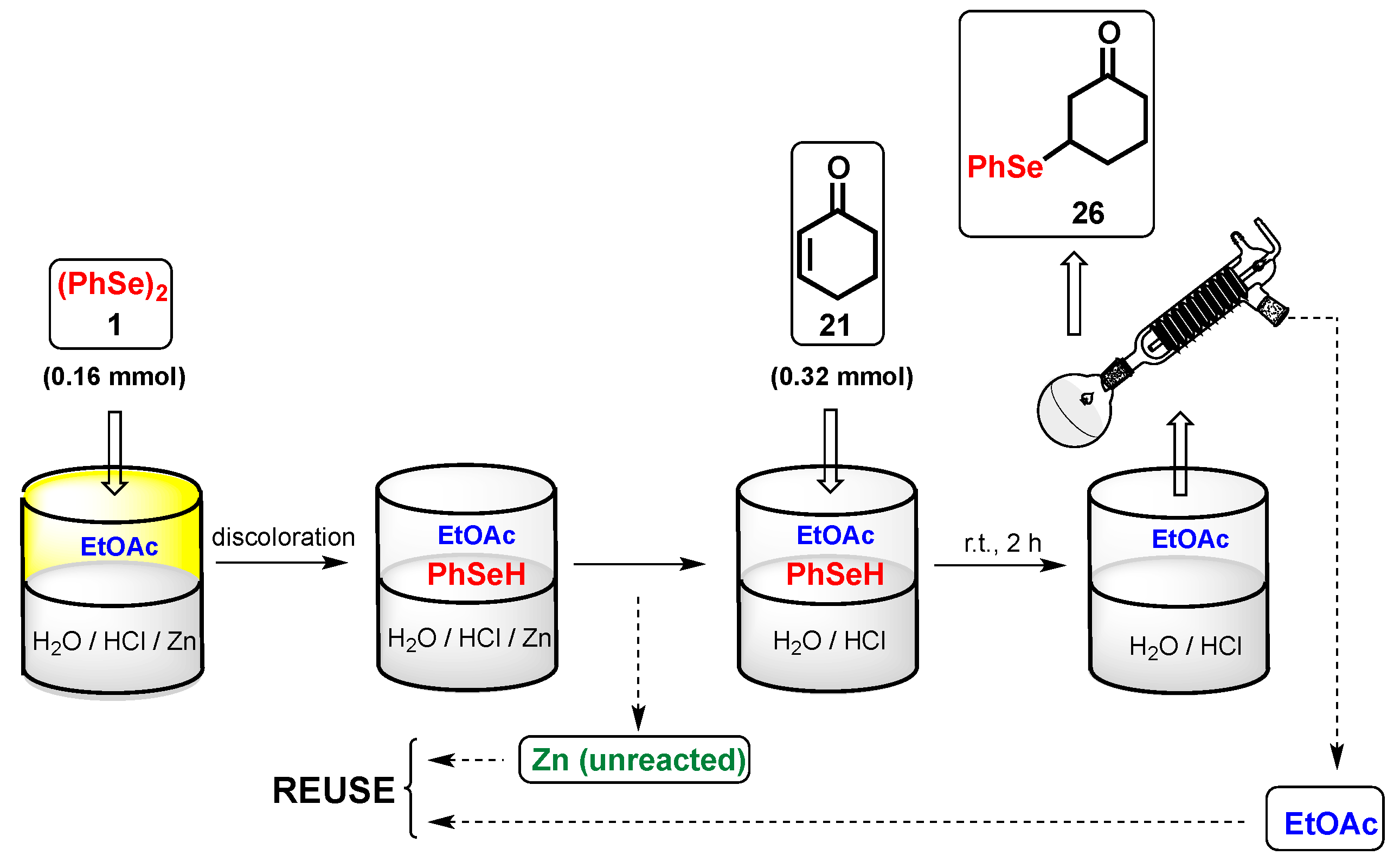
The recyclability of the biphasic medium and of the unreacted zinc was investigated in the reaction of diphenyl diselenide 1 with cyclohexanone 21 (in a 0.04 M solution of HCl10%v/v/EtOAc) as a general model (Scheme 3). After the reaction, the superior organic phase containing the adduct 26 was separated, and part of the organic solvent was recovered by distillation. The hydrochloric aqueous solution and the unreacted zinc were reused for a subsequent cycle, by the addition of fresh EtOAc, PhSeSePh 1, and after discoloration, a new amount of cyclohexanone 21 was added. As shown in Figure 1, the conversion for the first five cycles (evaluated by 1H-NMR spectroscopy) was found to be quantitative and it only decreased, slightly, following the 6th and 7th cycles.
3. Materials and Methods
Reactions were conducted in closed vials (6 mL) and were stirred with a Teflon-coated magnetic stirring bar. Solvents and reagents were used as received. The analytical thin layer chromatography (TLC) was performed on silica gel 60 F254 precoated aluminum foil sheets (Merck, Darmstadt, Germany) and visualized by UV irradiation (Spectroline® UV light, Sigma-Aldrich, St Louis, Missouri, USA) or by use of a KMnO4 stain (Merck, Darmstadt, Germany) Silica gel Kiesinger 60 (70–230 mesh) was used for flash column chromatography. NMR spectroscopic (Bruker, Fällanden, Switzerland) experiments were obtained at 25 °C on Bruker DPX spectrometers operating at the specified frequencies. 1H and 13C chemical shifts (δ) are reported in parts per million (ppm) and they are relative to TMS 0.0 ppm and/or the solvent peak of CDCl3 at δ 7.26 and δ 77.00 ppm in 1H and 13C NMR spectra, respectively. Data are reported as follows: Chemical shift (multiplicity, coupling constants, number of hydrogen atoms, where applicable, and assignment where possible). Abbreviations are as follows: S (singlet), d (doublet), t (triplet), q (quartet), p (pentet), dd (doublet of doublet), ddd (doublet of doublet of doublet), dddd (doublet of doublet of doublet of doublet), dt (doublet of triplet), ddt (doublet of doublet of triplet), tt (triplet of triplet), m (multiplet), and bs (broad signal). Coupling constants (J) are quoted in Hertz (Hz) to the nearest 0.1 Hz. High resolution mass spectra were carried out on a VG analytical 70-E mass spectrometer (VG Analytical Ltd., Manchester, UK) under an electrospray ionization condition (ESI). IR spectra were recorded on a Bruker Alpha FTIR spectrometer (Billerica, MA, USA). Optical rotation measurements were recorded using a Perkin-Elmer Model 343 polarimeter at 589 nm (Perkin Elmer’s, Waltham, MA, USA) and are given in units of 10−1degcm2g−1. Melting points are uncorrected and were recorded using a Gallenkamp electrothermal melting point apparatus (Cole-Parmer Ltd., Staffordshire, UK). Alkenes 2–10 and 20–22 are commercially available and were used without further purification; alkenes 23 and 24 (with OTBS and NHBoc substituents) were synthesized as described [67,68]. Jacaranone 25 was prepared by oxidation of the methyl 4-hydroxyphenylacetic acid.
Diphenyl diselenide 1 is commercially available, whereas diselenide 32 was prepared according to the procedure reported in the literature [69]. Selenocystine is commercially available and it was N- and O-protected, according to the procedure reported in the literature [70] in order to obtain compound 33.
General Procedure for the Se-Michael-Type Addition
Diselenides (1, 32, and 33) (0.16 mmol) were introduced to a biphasic system composed of EtOAc (2 mL) and HCl (10% v/v, 2 mL). Then, 1.6 mmol (10 equiv.) of zinc powder (or turnings) were added. The vial was closed and the mixture was vigorously stirred (approx. 800 rpm) at room temperature until the discoloration of the organic layer (approx. 20 min). After that, zinc was removed and the olefin (0.32 mmol) was added to the liquid. The reaction was stirred at room temperature for an additional 2 h. The ethyl acetate was separated and the resultant aqueous phase extracted with EtOAc (3 × 2 mL). The organic layers were combined, washed with brine, dried over Na2SO4, filtered, and the solvent was removed under reduced pressure.
In the recycling, experiment diselenide 1 (0.16 mmol) was introduced to a biphasic system composed of EtOAc (4 mL) and HCl (10% v/v, 4 mL). Then, 1.6 mmol (10 equiv.) of zinc turnings were added. The vial was closed and the mixture was vigorously stirred (approx. 800 rpm) at room temperature until the discoloration of the organic layer (approx. 20 min). After that, zinc was removed and recovered and 0.32 mmol of 21 was added to the liquid. The reaction was stirred at room temperature for an additional 2 h. The ethyl acetate was separated from the aqueous layer that was recovered. The organic phase was distilled and partially recovered to obtain a crude that was analyzed by proton NMR for the conversion yield. Recovered acidic water, EtOAc, and unreacted zinc mixed with small amounts of fresh solvents to reach the original volumes (4 + 4 mL) were reused for seven subsequent cycles.
4-(Phenylselanyl)butan-2-one (11) [71]: Isolated as a yellow oil in 91% yield (0.066 g) without purification. 1H NMR (200 MHz, CDCl3) δ: 7.60–7.45 (m, 2 H, CH), 7.30–7.20 (m, 3 H, CH), 3.02 (t, J = 6.7 Hz, 2 H, CH2), 2.80 (t, J = 6.7 Hz, 2 H, CH2), 2.18 (s, 3 H, CH3) ppm; 13C NMR (50.31 MHz, CDCl3) δ = 207.1, 132.7, 129.6, 129.1, 127.0, 44.0, 29.9, 20.4 ppm.
3-(Phenylselanyl)propanal (12) [72]: Isolated as a yellow oil in 44% yield (0.030 g) after flash column chromatography, eluent cyclohexane/ethyl acetate (9:1). 1H NMR (500 MHz, CDCl3) δ: 9.74 (bs, 1 H, CH), 7.51–7.48 (m, 2 H, CH), 7.29–7.25 (m, 3 H, CH), 3.09 (t, 2 H, J = 7.1 Hz, CH2), 2.85 (dt, 2 H, J = 0.9 and 7.1 Hz, CH2) ppm; 13C-NMR (125.77 MHz, CDCl3) δ = 200.6, 133.3, 129.2, 129.1, 127.4, 44.2, 18.9 ppm.
3-(Phenylselanyl)butanal (13) [73]: Isolated as a yellow oil in 70% yield (0.051 g) after flash column chromatography, eluent petroleum ether/ethyl acetate (95:5).1H NMR (200 MHz, CDCl3) δ = 9.67–9.66 (m, 1 H, CH), 7.60–7.45 (m, 2 H, CH), 7.30–7.20 (m, 3 H, CH), 3.65 (sextet, J = 7.0 Hz, 1 H, CH), 2.73 (ddd, J = 1.8, 7.0, and 13.5 Hz, 1 H, CH2), 2.63 (ddd, J = 1.7, 7.0, and 13.5 Hz, 1 H, CH2), 1.39 (d, J = 7.0 Hz, 3 H, CH3) ppm; 13C NMR (50.31 MHz, CDCl3) δ = 200.8, 135.6, 129.2, 128.2, 128.0, 51.0, 31.7, 22.1 ppm.
3-(Phenylselanyl)pentanal (14) [74]: Isolated as a yellow oil in 60% yield (0.030 g) after flash column chromatography, eluent petroleum ether/ethyl acetate (95:5). 1H NMR (200 MHz, CDCl3) δ = 9.74–9.72 (m, 1 H, CH), 7.60–7.45 (m, 2 H, CH), 7.30–7.20 (m, 3 H, CH), 3.50 (p, J = 6.8 Hz, 1 H, CH), 2.78–2.71 (m, 2 H, CH2), 1.76–1.61 (m, 2 H, CH2), 1.10 (t, J = 7.3 Hz, 3 H, CH3) ppm; 13C NMR (50.31 MHz, CDCl3) δ = 201.0, 135.6, 129.0, 128.0, 127.6, 48.8, 40.0, 28.4, 12.3 ppm.
3-Methyl-3-(phenylselanyl)butanal (15) [74]: Isolated as a yellow oil in 22% yield (0.017 g) due to the limited stability during silica gel column chromatography, eluent cyclohexane/ethyl acetate (9:1). 1H NMR (400 MHz, CDCl3) δ = 9.86 (t, J = 2.7 Hz, 1 H, CH), 7.67–7.55 (m, 2 H, CH), 7.41–7.29 (m, 3 H, CH), 2.55 (d, J = 2.7 Hz, 2 H, CH2), 1.51 (s, 6 H, CH3) ppm. 13C NMR (100 MHz, CDCl3) δ = 201.8, 138.3, 129.1, 129.0, 126.9, 55.1, 41.9, 30.0 ppm.
Methyl 3-(phenylselanyl)propanoate (16) [44]: Isolated as a yellow oil in 79% yield (0.097 g) as pure compound without purification. 1H NMR (200 MHz, CDCl3) δ = 7.60–7.45 (m, 2 H, CH), 7.30–7.20 (m, 3 H, CH), 3.72 (s, 3 H, CH3), 3.15 (t, 2 H, J = 6.5 Hz, CH2), 2.74 (t, 2 H, J = 6.5 Hz, CH2) ppm; 13C NMR (50.31 MHz, CDCl3) δ = 172.6, 133.3, 131.5, 129.2, 127.3, 51.8, 35.1, 21.8 ppm.
3-(Phenylselanyl)propanoic acid (17) [75]: Isolated as a yellow oil in 60% yield (0.140 g) and purified by crystallization from petroleum ether and ethyl acetate. 1H NMR (200 MHz, CDCl3) δ = 7.66–7.56 (m, 2 H, CH), 7.36–7.25 (m, 3 H, CH), 3.18 (t, 2 H, J = 7.0 Hz, CH2), 2.86 (t, 2 H, J = 7.0 Hz, CH2) ppm; 13C-NMR (CDCl3, 50.31 MHz): δ = 178.6, 133.3, 131.3, 129.1, 127.3, 35.0, 21.5 ppm.
N,N-Dimethyl-3-(phenylselanyl)propanamide (18): Isolated as a yellow oil in 25% yield (0.02 g) after flash column chromatography, eluent cyclohexane/ethyl acetate (1:1). Rf = 0.29 (c-Hex/EtOAc 1:1); νmax = 3000, 2926, 1642, 1578, 1477, 1437, 1397, 1326, 1303, 1264, 1189, 1128, 1072, 1022 cm−1. 1H NMR (500 MHz, CDCl3) δ = 7.60–7.45 (m, 2 H, CH), 7.30–7.20 (m, 3 H, CH), 3.18 (t, 2 H, J = 7.6 Hz, CH2), 2.94 (s, 3 H, CH3), 2.92 (s, 3 H, CH3), 2.73 (t, 2 H, J = 7.6 Hz, CH2) ppm; 13C NMR (125.77 MHz, CDCl3) δ = 171.4, 132.6, 130.1, 129.0, 126.9, 37.0, 35.4, 34.2, 22.4 ppm. HRMS (ESI+) C11H15NONaSe (MNa+) calcd: 280.0217; found: 280.0230.
(R)-5-Methyl-2(R/S)-(2-(phenylselanyl)propan-2-yl)cyclohexanone (19): Isolated as a yellow oil and a mixture of two isomers (62/38) in 95% yield (0.031 g) after flash column chromatography, eluent petroleum ether/DCM 20:80. νmax = 3070, 3056, 2954, 2925, 2869, 1708, 1576, 1474, 1455, 1437, 1379, 1362, 1285, 1208, 1117, 1088, 1046, 1021 cm−1. For 1H NMR (400 MHz, CDCl3) and 13C NMR (100 MHz, CDCl3) of the mixture see Supplementary Materials. HRMS (ESI +) C16H22ONaSe (MNa+) calcd: 333.0735; found: 333.0734.
3-(Phenylselanyl)cyclopentanone (25) [76]: Isolated as a yellow oil in 93% yield (0.072 g) without purification. 1H NMR (200 MHz, CDCl3) δ = 7.70–7.50 (m, 2 H, CH), 7.30–7.20 (m, 3 H, CH), 3.89 (m, 1 H, CH), 2.70 (dd, J = 7.3 and 17.8 Hz, 1 H, CH2), 2.50–1.75 (m, 5 H, CH2) ppm; 13C NMR (50.31 MHz, CDCl3) δ = 216.9, 134.8, 129.1, 128.3, 128.0, 45.6, 37.4, 37.2, 30.0 ppm.
3-(Phenylselanyl)cyclohexanone (26) [71]: Isolated as a yellow oil in 88% yield (0.071 g) without purification. 1H NMR (200 MHz, CDCl3) δ = 7.70–7.50 (m, 2 H, CH), 7.30–7.20 (m, 3 H, CH), 3.45 (tt, J = 4.4 and 10.5 Hz, 1 H, CH), 2.74 (ddt, J = 4.4, 13.7 and 1.5 Hz, 1 H, CH2), 2.40 (dd, J = 10.5 and 13.7 Hz, 1 H, CH2), 2.36–2.06 (m, 4 H, CH2), 1.87–1.51 (m, 2 H, CH2) ppm; 13C NMR (50.31 MHz, CDCl3) δ = 208.8, 135.6, 129.1, 128.1, 127.4, 48.6, 40.8, 40.1, 31.9, 25.1 ppm.
3-(Phenylselanyl)cycloheptanone (27) [48]: Isolated as a yellow oil in 95% yield (0.082 g) without purification. 1H NMR (200 MHz, CDCl3) δ = 7.70–7.50 (m, 2 H, CH), 7.40–7.30 (m, 3 H, CH), 3.50 (dddd, J = 2.8, 4.0, 9.6, and 9.8 Hz, 1 H, CH), 2.85 (ddd, J = 0.9, 4.0, and 14.9 Hz, 1 H, CH2), 2.75 (dd, J = 9.6 and 14.9 Hz, 1 H, CH2), 2.55–2.35 (m, 2 H, CH2), 2.15–2.05 (m, 1 H, CH2) 1.86–1.44 (m, 5 H, CH2) ppm; 13C NMR (50.31 MHz, CDCl3) δ = 211.8, 135.0, 129.1, 128.8, 128.0, 50.3, 43.9, 38.8, 37.7, 29.1, 23.7 ppm.
tert-Butyl ((1R,2S/R-4-oxo-2-(phenylselanyl)cyclopentyl)carbamate (28): Obtained as a mixture of diastereomers (75/25) from which the major one was isolated as a yellow oil in 62% yield (0.05 g) after flash column chromatography, eluent petroleum spirit/ethyl acetate (95:5). Rf = 0.28 (c-Hex/EtOAc 5:1); νmax= 3370, 3327, 2976, 2930, 1747, 1721, 1675, 1512, 1473, 1435, 1391, 1366, 1156, 1018 cm−1. 1H NMR (400 MHz, CDCl3) δ = 7.70–7.50 (m, 2 H, CH), 7.40–7.20 (m, 3 H, CH), 4.77 (d, 1 H, J = 5.0 Hz, NH), 4.15–4.01 (m, 1 H, CH), 3.52 (q, 1 H, J = 8.0 Hz, CH), 2.81 (dd, 1 H, J = 7.6 and 15.1 Hz, CH2), 2.77 (dd, 1 H, J = 7.3 and 14.8 Hz, CH2), 2.3 (dd, 1 H, J = 9.1 and 19.5 Hz, CH2), 2.2 (dd, 1 H, J = 8.6 and 19.6 Hz, CH2), 1.4 (s, 9 H, CH3) ppm; 13C NMR (50.31 MHz, CDCl3) δ = 212.9, 155.1, 135.9, 129.1, 128.6, 126.4, 53.9, 45.2, 44.8, 42.2, 28.8 ppm. HRMS (ESI+) C16H21NO3NaSe (MNa+) calcd: 378.0584; found: 378.0585.
Methyl-2-(1-hydroxy-4-oxo-6-(phenylselanyl)cyclohex-2-en-1-yl)acetate (29): Isolated as a colorless oil in 29% yield (0.031g) after flash column chromatography, eluent cyclohexane/ethyl acetate (2:1). Rf = 0.22 (c-Hex/EtOAc; 2:1). νmax = 3454, 3056, 2985, 2955, 2925, 2852, 1732, 1685, 1619, 1577, 1521,1437, 1354, 1265 cm-1. 1H NMR (500 MHz, CDCl3), δ = 7.64–7.57 (m, 2 H, CH), 7.36–7.28 (m, 3 H, CH), 6.91 (d, 1 H, J = 10.2 Hz, CH), 6.02 (d, 1 H, J = 10.2 Hz, CH), 4.18 (s, 1 H, OH), 3.74 (dd, 1 H, J = 4.3 and 14.3 Hz, CH), 3.75 (s, 3 H, CH3), 3.30 (d, 1 H, J = 16.2 Hz, CH), 3.11 (dd, 1 H, J = 10.0 and 17.0 Hz, CH), 2.89 (dd, 1 H, J = 4.3 and 17.0 Hz, CH), 2.80 (d, 1 H, J = 16.2 Hz, CH) ppm; 13C NMR (125.77 MHz, CDCl3): 196.8, 171.9, 149.3, 135.2, 129.4, 129.1, 128.3, 128.2, 70.0, 52.22, (52.24), 50.7, 42.6, 42.5 ppm. HRMS (ESI+) C15H16O4NaSe (MNa+) calcd: 363.0111; found: 363.0098.
3-((3-Phenylpropyl)selanyl)cyclohexanone (33): Isolated as a yellow oil in 65% yield (0.044 g) and purified by flash column chromatography, eluent cyclohexane/ethyl acetate (9:1). Rf = 0.28 (c-Hex/EtOAc 9:1); νmax = 3026, 2928, 2858, 2362, 1714, 1602, 1495, 1454, 1274, 1122, 1030 cm-1. 1H NMR (400 MHz, CDCl3) δ = 7.28–7.25 (m, 2 H, CH), 7.19–7.15 (m, 3 H, CH), 3.18 (tt, 1 H, J = 3.9 and 10.5 Hz, CH), 2.76 (ddt, 1 H, J = 1.6, 4.5 and 14.2 Hz, CH2), 2.70 (t, J = 7.4 Hz, 2 H, CH2), 2.60 (t, 2 H, J = 7.4 Hz, CH2), 2.47 (ddd, 1 H, J = 1.1, 10.9, and 14.2 Hz, CH2), 2.39–2.26 (m, 2 H, CH2), 2.19–2.06 (m, 2 H, CH2), 1.97 (p, 2 H, J = 7.6 Hz, CH2), 1.83–1.64 (m, 2 H, CH2) ppm; 13C NMR (100 MHz, CDCl3) δ = 208.9, 141.3, 128.4, 128.3, 125.9, 49.2, 40.9, 35.9, 35.8, 32.5, 32.4, 25.4, 22.5 ppm. HRMS (ESI+) C15H20ONaSe (MNa+) calcd: 319.0577; found: 319.0568.
(3S,4S/R)-3-((tert-Butyldimethylsilyl)oxy)-4-((3-phenylpropyl)selanyl)cyclopentanone (34): Isolated as a 89/11 mixture of undetermined diastereomers from which the major one has been isolated as a yellow oil in 49% yield (0.050 g) by flash column chromatography, eluent cyclohexane/ethyl acetate (95:5). Rf = 0.32 (c-Hex/EtOAc; 95:5); νmax = 3085, 3062, 3026, 2953, 2927, 2855, 1748, 1604, 1496, 1455, 1253, 1102, 1066, 1026 cm-1. 1H NMR (400 MHz, CDCl3) δ = 7.29–7.26 (m, 2 H, CH), 7.20–7.15 (m, 3 H, CH), 4.41 (dt, 1 H, J = 2.8 and 5.4 Hz, CH), 3.45–3.39 (m, 1 H, CH), 2.92–2.85 (m, 1 H, CH2), 2.76–2.52 (m, 5 H, CH2), 2.23–2.11 (m, 2 H, CH2), 2.09–1.93 (m, 2 H, CH2), 0.85 (s, 9 H, CH3), 0.06 (s, 3 H, CH3), 0.04 (s, 3 H, CH3) ppm; 13C NMR (100 MHz, CDCl3) δ = 215.0, 128.4, 128.3, 126.0, 125.91, 75.2, 46.1, 43.1, 41.0, 35.8, 31.9, 25.6, 23.7, −4.7, −4.8 ppm. HRMS (ESI+) C20H32O2NaSeSi (MNa+) calcd: 435.1234; found: 435.1235 [α]D = −17 (c = 0.5, CHCl3).
(2R)-Methyl 2-((tert-butoxycarbonyl)amino)-3-((3-oxobutyl)selanyl)propanoate (35): Isolated as a yellow oil in 30% yield (0.021 g) after flash column chromatography, eluent cyclohexane/ethyl acetate (3:1). Rf = 0.17 (c-Hex/EtOAc; 3:1); νmax = 3379, 3344, 2977, 2953, 2929, 1708, 1436, 1365, 1249, 1212, 1160, 1050, 1007 cm-1. 1H NMR (600 MHz, CDCl3) δ = 5.35 (d, 1 H, J = 6.5 Hz, NH), 4.59 (d, 1 H, J = 6.9 Hz, CH), 3.74 (s, 3 H, CH3), 3.05–2.98 (m, 2 H, CH2), 2.85–2.79 (m, 2 H, CH2), 2.74–2.70 (m, 2 H, CH2), 2.14 (s, 3 H, CH3), 1.43 (s, 9 H, CH3) ppm; 13C-NMR (125.77 MHz, CDCl3) δ = 206.7, 171.5, 154.9, 80.0, 53.5, 52.6, 44.4, 29.9, 28.3, 26.6, 17.5 ppm. HRMS (ESI+) C13H23NO5NaSe (MNa+) calcd: 376.0639; found: 376.0653 [α]D = +19 (c = 0.5, CHCl3).
Methyl 2-((tert-butoxycarbonyl)amino)-3-((3-oxocyclohexyl)selanyl)propanoate (36): Isolated as a yellow oil in 53% yield (0.050 g) as mixture of two inseparable diastereomers (66/34) after flash column chromatography, eluent cyclohexane/ethyl acetate (3:1). Rf = 0.23 (c-Hex/EtOAc; 3:1); νmax = 3358, 2976, 1742, 1704, 1500, 1436, 1392, 1365, 1346, 1248, 1215, 1048, 1007 cm−1. For 1H NMR (500 MHz, CDCl3) and 13C NMR (125.77 MHz, CDCl3) of the mixture, see Supplementary Materials. HRMS (ESI+) C15H25NO5NaSe (MNa+) calcd: 402.0796; found: 402.0779.
Methyl-2-((tert-butoxycarbonyl)amino)-3-(((2S)-2-((tert-butoxycarbonyl)amino)-4-oxocyclopentyl)selanyl)propanoate (37): Isolated as a white solid, mixture of diastereomers (55/45) in a 30% yield (0.046 g) after recrystallization from a concentrated DCM solution which was layered with c-Hex. Rf = 0.28 (c-Hex/EtOAc; 5:1). m.p. 114–116 °C. νmax = 3362, 2932, 1743, 1677, 1511, 1437, 1366, 1317, 1278, 1253, 1228, 1161, 1045, 1017 cm−1. For 1H NMR (600 MHz, CDCl3) and 13C NMR (150.9 MHz, CDCl3) of the mixture, see Supplementary Materials. HRMS (ESI+) C19H32N2O7NaSe (MNa+) calcd: 503.1272; found: 503.1271.
4. Conclusions
An efficient, fast, and safe protocol for seleno-Michael-type addition reactions has been reported. The method represents a good way to achieve the in situ reduction of diphenyl diselenide 1 using a biphasic Zn-EtOAc-HCl(aq) mixture. The resultant nucleophilic selenenylating mixture was shown to react with a range of Michael acceptors in order to provide the corresponding Se-adducts. Yields are often high and these were found to depend on the electrophilicity of the substrate. Alternative diselenides 32 and 33 were successfully used, affording Se-alkylated versions of protected selenocysteine.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/1420-3049/25/9/2018/s1, Figure of NMR spectra of all the synthesized compounds.
Author Contributions
Conceptualization, C.S., P.E., and E.J.L.; methodology, C.S., P.E., and E.J.L.; investigation, F.G.N. and B.M.; resources, C.S., P.E., and E.J.L.; data curation, C.S., P.E., and E.J.L., writing—original draft preparation, F.G.N. and B.M.; writing—review and editing, C.S., P.E., and E.J.L.; visualization, F.G.N. and B.M.; supervision, C.S., P.E., and E.J.L.; funding acquisition, C.S. and P.E. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
University of Perugia for Fondo per il sostegno della Ricerca di Base 2018, Project “Sviluppo di metodologie innovative per la sintesi efficiente di composti eterociclici, molecole drug-like e intermedi sintetici ad alto valore aggiunto” (C.S.) and “Mobilità Accordi Quadro”(B.M.); CAPES—Coordenação de Aperfeiçoamento de Pessoal de Nível Superior–Brasil–Finance Code 001, FAPERGS (PqG 17/2551-0000987-8), CNPq and FINEP (E.J.L.); the Eramus + trainership program and University College Dublin (F.G.N.) are acknowledged for their support. Clodagh Muldowney is thanked for the synthesis of 25. This work has been undertaken under the umbrella of the Selenium Sulfur Redox and Catalysis Network (SeSRedCat).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Nicolaou, K.C.; Petasis, N.A. Selenium in Natural Products Synthesis; CIS: Philadelphia, PA, USA, 1984. [Google Scholar]
- Paulmier, C. Selenium Reagents and Intermediates in Organic Synthesis; Pergamon: Oxford, UK, 1986. [Google Scholar]
- Patai, S.; Rappoport, Z. The Chemistry of Organic Selenium and Tellurium Compounds; Wiley: New York, NY, USA, 1986. [Google Scholar]
- Liotta, D. Organoselenium Chemistry; Wiley: New York, NY, USA, 1987. [Google Scholar]
- Back, T.G. Organoselenium Chemistry: A Practical Approach; Oxford University Press: Oxford, UK, 1999. [Google Scholar]
- Reich, H.J. Functional Group Manipulation Using Organoselenium Reagents. Acc. Chem. Res. 1979, 12, 22–30. [Google Scholar] [CrossRef]
- Liotta, D. New Organoselenium Methodology. Acc. Chem. Res. 1984, 17, 28–34. [Google Scholar] [CrossRef]
- Wirth, T. Organoselenium Chemistry–Modern Developments in Organic Synthesis, Topics in Current Chemistry; Spring: Heidelberg, Germany, 2000. [Google Scholar] [CrossRef]
- Mugesh, G.; Singh, H.B. Heteroatom-Directed Aromatic Lithiation: A Versatile Route to the Synthesis of Organochalcogen (Se, Te) Compounds. Acc. Chem. Res. 2002, 35, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Rigby, J.H.; Maharoof, U.S.M.; Mateo, M.E. Studies on the Narciclasine Alkaloids: Total Synthesis of (+)-Narciclasine and (+)-Pancratistatin. J. Am. Chem. Soc. 2000, 122, 6624–6628. [Google Scholar] [CrossRef]
- Knapp, S.; Zhao, D. Synthesis of the Sialidase Inhibitor Siastatin B. Org. Lett. 2000, 25, 4037–4040. [Google Scholar] [CrossRef]
- Treadwell, E.M.; Neighbors, J.D.; Wiemer, D.F. A Cascade Cyclization Approach to Schweinfurthin B. Org. Lett. 2002, 21, 3639–3642. [Google Scholar] [CrossRef]
- Hossain, S.U.; Sharma, A.K.; Ghosh, S.; Bhattacharya, S. Synthesis and Biological Evaluation of Novel Spiro 6-methoxytetralin-1,3′-pyrrolidine Based Organoselenocyanates Against Cadmium-induced Oxidative and Hepatic Damage in Mice. Eur. J. Med. Chem. 2010, 45, 3265–3273. [Google Scholar] [CrossRef]
- Jeong, L.S.; Choi, Y.N.; Tosh, D.K.; Choi, W.J.; Kim, H.O.; Choi, J.J. Design and Synthesis of Novel 2′,3′-dideoxy-4′-selenonucleosides as Potential Antiviral Agents. Bioorg. Med. Chem. 2008, 16, 9891–9897. [Google Scholar] [CrossRef]
- De Souza, D.; Mariano, D.O.; Nedel, F.; Schultze, E.; Campos, V.F.; Seixas, F.; da Silva, R.S.; Munchen, T.S.; Ilha, V.; Dornelles, L.; et al. New Organochalcogen Multitarget Drug: Synthesis and Antioxidant and Antitumoral Activities of Chalcogenozidovudine Derivatives. J. Med. Chem. 2015, 58, 3329–3339. [Google Scholar] [CrossRef]
- Pietrella, D. Antimicrobial activity of Organoselenium Compounds. In Organoselenium Chemistry: Between Synthesis and Biochemistry; Santi, C., Ed.; Bentham eBooks: Sharjah, UAE, 2014; pp. 328–344. [Google Scholar] [CrossRef] [Green Version]
- Legnaioli, S.; Piroddi, M.; Tidei, C.; Santi, C.; Galli, F. Targeting the GSTP-dependent Control of Cell Kinases and Apoptosis with PhSeZnCl: A New Seleno-organic Drug. Free Radical. Biol. Med. 2012, 53, S112–S113. [Google Scholar] [CrossRef]
- Sancineto, L.; Mariotti, A.; Bagnoli, L.; Marini, F.; Desantis, J.; Iraci, N.; Santi, C.; Pannecouque, C.; Tabarrini, O. Design and Synthesis of DiselenoBisBenzamides (DISeBAs) as Nucleocapsid Protein 7 (NCp7) Inhibitors with anti-HIV Activity. J. Med. Chem. 2015, 58, 9601–9614. [Google Scholar] [CrossRef]
- Krief, A.; Havesi, L. Organoselenium Chemistry; Springer: Berlin/Heidelberg, Germany, 1988. [Google Scholar]
- Krief, A. Alkylations of Sulfur- and Selenium-containing Carbanions. In Comprehensive Organometallic Chemistry; Trost, B.M., Fleming, I., Pattenden, G., Eds.; Elsevier: Amsterdam, The Netherlands, 1991; pp. 85–191. [Google Scholar]
- Wirth, T. Selenium. In Comprehensive Organometallic Chemistry III; Crabtree, R.H., Mingos, D.M.P., Eds.; Elsevier: Amsterdam, The Netherlands, 2006; pp. 457–500. [Google Scholar]
- Zhang, Y.; Bao, W.; Zheng, Y. Synthesis of Allyl-Type Selenides and α-Selenoesters in Aqueous Media Promoted by Cadmium. Synth. Commun. 2000, 30, 1731–1736. [Google Scholar] [CrossRef]
- Sonoda, N.; Okada, M.; Kuroki, T.; Watanabe, T.; Nishiyama, Y.; Nishino, T. One-Pot Synthetic Method of Unsymmetrical Diorganyl Selenides: Reaction of Diphenyl Diselenide with Alkyl Halides in the Presence of Lanthanum Metal. J. Org. Chem. 2002, 67, 8696–8698. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhang, Y.; Zhou, J.; Bao, W. A Novel Synthesis of Allyl and Propargyl Selenides in Aqueous Media Promoted by Indium. Tetrahedron Lett. 1996, 37, 9333–9334. [Google Scholar] [CrossRef]
- Mandal, T.; Samanta, S.; Ranu, B.C. Indium(I) Iodide-Mediated Cleavage of Diphenyl Diselenide. An Efficient One-Pot Procedure for the Synthesis of Unsymmetrical Diorganyl Selenides. Org. Lett. 2003, 5, 1439–1441. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhang, Y.; Guo, H. Convenient One-Pot Synthesis of α-Selenonitriles, α-Selenoesters and Asymmetrical Selenides by a Sm/ZnCl2 system in DMF-H2O. J. Chem. Res. 2001, 160–161. [Google Scholar]
- Guo, H.; Zhang, Y.; Zheng, Y. Synthesis of Allyl Selenides Promoted by an Sm/ZnCl2 Bimetal System in the Presence of Water. Chin. J. Chem. 2001, 19, 530–532. [Google Scholar] [CrossRef]
- Fujinami, T.; Sakai, S.; Fukuzawa, S. Samarium(II) Di-iodide Induced Synthesis of Allylic Phenyl Selenides from Allylic Acetates and Diphenyl Diselenide in the Presence of Palladium Catalyst. Chem. Lett. 1990, 927–930. [Google Scholar] [CrossRef]
- Bao, W.; Zhang, Y.; Liao, P. A Novel Synthesis of Allyl and Prop-2-ynyl Selenides Promoted by Tin in the Presence of Water. J. Chem. Res. (S) 1998, 150–151. [Google Scholar] [CrossRef]
- Zhang, Y.; Zheng, Y.; Li, Y.; Su, W. Facile Formation of Ytterbium Diiodide and Its Use in the Synthesis of Allyl Selenides. Chin. J. Chem. 2002, 20, 174–177. [Google Scholar] [CrossRef]
- Roy, S.; Kundu, A. Copper(II)/Tin(II) Reagent for Allylation, Propargylation, Alkynylation, and Benzylation of Diselenides: A Novel Bimetallic Reactivity. Organometallics 2000, 19, 105–107. [Google Scholar] [CrossRef]
- Reddy, K.M.; Mugesh, G. Application of Dehydroalanine as a Building Block for the Synthesis of Selenocysteine-containing Peptides. RSC Adv. 2019, 9, 34–43. [Google Scholar] [CrossRef] [Green Version]
- Krief, A.; Cravador, A. Nouvelles Methods de Synthèse des Méthylsélénoacétals et Cétals. C. R. Acad. Sci. Paris 1979, 289c, 267–269. [Google Scholar]
- Katsumata, K.; Watanabe, Y.; Toru, T.; Veno, Y.; Sakakakibara, M. A Convenient Procedure for the Preparation of Organic Selenides. Synthesis 1992, 377–379. [Google Scholar] [CrossRef]
- Miyazaki, T.; Ando, M.; Matsumura, Y.; Sakane, S.; Hattori, K.; Yamamoto, H.; Maruoka, K. Organoaluminum-promoted Beckmann Rearrangement of Oxime Sulfonates. J. Am. Chem. Soc. 1983, 105, 2831–2843. [Google Scholar] [CrossRef]
- Berkowitz, D.B.; Pedersen, M.L. Formal α-vinylation of Amino Acids. Use of a New Benzeneselenolate Equivalent. J. Org. Chem. 1993, 58, 6966–6975. [Google Scholar] [CrossRef]
- Block, H.D.; Schmidt, M. Notiz zur Vereinfachten Darstellung von Alkanselenolen. Chem. Ber. 1970, 103, 3348–3349. [Google Scholar] [CrossRef]
- Mlochowsky, J.; Syper, L. The Convenient Syntheses of Organoselenium Reagents. Synthesis 1984, 439–442. [Google Scholar] [CrossRef]
- Dan, W.; Deng, H.; Chen, J.; Liu, M.; Ding, M.; Wu, H. A New Odorless One-pot Synthesis of Thioesters and Selenoesters Promoted by Rongalite. Tetrahedron 2010, 66, 7384–7388. [Google Scholar] [CrossRef]
- Lakouraj, M.M.; Movassagh, B.; Fadaei, Z. Convenient Synthesis of Thiol Esters from Acyl Chlorides and Disulfides Using Zn/AlCl3. Monatsh. Chem. 2002, 133, 1085–1088. [Google Scholar] [CrossRef]
- Movassagh, B.; Lakouraj, M.M.; Fadaei, Z. A Convenient One-pot Synthesis of Thiol Esters from Disulfides Using a Zn/AlCl3 System. J. Chem. Res. (S) 2001, 22–23. [Google Scholar] [CrossRef]
- Tian, F.S.; Zhu, Y.M.; Zhang, S.L.; Wang, Y.L. A Novel Method for the Synthesis of Unsymmetrical Sulfides, Thioesters and β-thioesters. J. Chem. Res. (S) 2002, 11, 582–583. [Google Scholar] [CrossRef]
- Movassagh, B.; Tatar, A. Zn/RuCl3-Promoted Cleavage of Diselenides: An Efficient Michael Addition of Zinc Selenolates to Conjugated Alkenes in Aqueous Media. Synlett 2007, 12, 1954–1956. [Google Scholar] [CrossRef]
- Zhou, L.-H.; Zhang, Y.-M. An Effective Synthesis of β- Selenium and β-Tellurium Carbonyl Compounds Via Reaction of Diaryldiselenides Or Diarylditellurides with α,β-Unsaturated Carbonyl Compounds Induced by Low-Valent Titanium. Synth. Commun. 1999, 29, 533–540. [Google Scholar] [CrossRef]
- Nunes, V.L.; de Oliveira, I.C.; do Rego Barros, O.S. Organylzinc Chalcogenolate Promoted Michael-Type Addition of α,β-Unsaturated Carbonyl Compounds. Eur. J. Org. Chem. 2014, 1525–1530. [Google Scholar] [CrossRef]
- Sancineto, L.; Vargas, J.P.; Monti, B.; Arca, M.; Lippolis, V.; Perin, G.; Lenardão, E.J.; Santi, C. Atom Efficient Preparation of Zinc Selenates for the Synthesis of Selenol Esters under “On Water” Conditions. Molecules 2017, 22, 953. [Google Scholar] [CrossRef] [Green Version]
- Santi, C.; Santoro, S.; Battistelli, B.; Testaferri, L.; Tiecco, M. Preparation of the First Bench-Stable Phenyl Selenolate: An Interesting “On Water” Nucleophilic Reagent. Eur. J. Org. Chem. 2008, 5387–5390. [Google Scholar] [CrossRef]
- Santoro, S.; Battistelli, B.; Testaferri, L.; Tiecco, M.; Santi, C. Vinylic Substitutions Promoted by PhSeZnCl: Synthetic and Theoretical Investigations. Eur. J. Org. Chem. 2009, 4921–4925. [Google Scholar] [CrossRef]
- Santi, C.; Battistelli, B.; Testaferri, L.; Tiecco, M. On Water Preparation of Phenylselenoesters. Green Chem. 2012, 14, 1277–1280. [Google Scholar] [CrossRef]
- Battistelli, B.; Testaferri, L.; Tiecco, M.; Santi, C. “On-Water” Michael-Type Addition Reactions Promoted by PhSeZnCl. Eur. J. Org. Chem. 2011, 1848–1851. [Google Scholar] [CrossRef]
- Salman, S.; Schwab, R.; Alberto, E.E.; Vargas, J.; Dornelles, L.; Rodrigues, O.E.D.; Braga, A.L. Efficient Ring Opening of Protected and Unprotected Aziridines Promoted by Stable Zinc Selenolate in Ionic Liquid. Synlett 2011, 69–72. [Google Scholar] [CrossRef]
- Nagasawa, T.; Shimada, N.; Torihata, M.; Kuwahara, S. Enantioselective Total Synthesis of Idesolide via NaHCO3-promoted Dimerization. Tetrahedron 2010, 66, 4965–4969. [Google Scholar] [CrossRef]
- Jiang, H.; Pan, X.; Li, N.; Zhang, Z.; Zhu, J.; Zhu, X. Selenide-containing High Refractive Index Polymer Material with Adjustable Refractive Index and Abbe’s Number. React. Funct. Polym. 2017, 111, 1–6. [Google Scholar] [CrossRef]
- Kim, Y.; Mulay, S.V.; Choi, M.; Yu, S.B.; Jon, S.; Churchill, D.G. Exceptional Time Response, Stability and Selectivity in Doubly-activated Phenyl Selenium-based Glutathione-selective Platform. Chem. Sci. 2015, 6, 5435–5439. [Google Scholar] [CrossRef] [Green Version]
- Jardim, G.A.M.; Bozzi, I.A.O.; Oliveira, W.X.C.; Mesquita-Rodrigues, C.; Menna-Barreto, R.F.S.; Kumar, R.A.; Gravel, E.; Doris, E.; Braga, A.L.; da Silva Júnior, E.N. Copper Complexes and Carbon nanotube–copper Ferrite-catalyzed Benzenoid A-ring Selenation of Quinones: An Efficient Method for the Synthesis of Trypanocidal agents. New J. Chem. 2019, 43, 13751–13763. [Google Scholar] [CrossRef]
- Santi, C.; Santoro, S.; Testaferri, L.; Tiecco, M. A Simple Zinc-Mediated Preparation of Selenols. Synlett 2008, 1471–1474. [Google Scholar] [CrossRef]
- Braga, A.L.; Schwab, R.S.; Alberto, E.E.; Salman, S.M.; Vargas, J.; Azeredo, J.B. Ring Opening of Unprotected Aziridines by Zinc Selenolates in a Biphasic System. Tetrahedron Lett. 2009, 50, 2309–2311. [Google Scholar] [CrossRef]
- Bellino, G.; Scisciani, M.; Vargas, J.P.; Sancineto, L.; Bagnoli, L.; Marini, L.; Lüdtke, D.S.; Lenardão, E.J.; Santi, C. Reaction of Acyl Chlorides with In Situ Formed Zinc Selenolates: Synthesis of Selenoesters versus Ring-Opening Reaction of Tetrahydrofuran. J. Chem. 2016, 1–8. [Google Scholar] [CrossRef]
- Flemer, S., Jr. A Comprehensive One-Pot Synthesis of Protected Cysteine and Selenocysteine SPPS Derivatives. Prot. Pept. Lett. 2014, 21, 1257–1264. [Google Scholar]
- Flemer, S., Jr. Fmoc-Sec(Xan)-OH: Synthesis and Utility of Fmoc Selenocysteine SPPS Derivatives with Acid-Labile Sidechain Protection. J. Pept. Sci. 2015, 21, 53–59. [Google Scholar] [CrossRef] [Green Version]
- Tidei, C.; Sancineto, L.; Bagnoli, L.; Battistelli, B.; Marini, F.; Santi, C. A Recyclable Biphasic System for Stereoselective and Easily Handled Hydrochalcogenations. Eur. J. Org. Chem. 2014, 5968–5975. [Google Scholar] [CrossRef]
- Santi, C.; Jacob, R.G.; Monti, B.; Bagnoli, L.; Sancineto, L.; Lenardão, E.J. Water and Aqueous Mixtures as Convenient Alternative Media for Organoselenium Chemistry. Molecules 2016, 21, 1482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, S.-C.; Chen, F.; Hou, D.; Kim-Meade, A.; Bernard, C.; Liu, J.; Levy, S.; Wu, G.G. A Novel Enantioselective Alkylation and Its Application to the Synthesis of an Anticancer Agent. J. Org. Chem. 2013, 68, 4984–4987. [Google Scholar] [CrossRef] [PubMed]
- Prat, D.; Wells, A.; Hayler, J.; Sneddon, H.; McElroy, C.R.; Abou-Shehada, S.; Dunn, P.J. CHEM21 Selection Guide of Classical- and Less Classical-solvents. Green Chem. 2016, 18, 288–296. [Google Scholar] [CrossRef] [Green Version]
- Bickley, J.F.; Evans, P.; Meek, A.; Morgan, B.S.; Roberts, S.M. Novel Preparation of (−)-4-Hydroxycyclohex-2-enone: Reaction of 4-hydroxycyclohex-2-enone and 4-hydroxycyclopent-2-enone with some Thiols. Tetrahedron: Asymmetry 2006, 17, 355–362. [Google Scholar] [CrossRef]
- O’Byrne, A.; Murray, C.; Keegan, D.; Palacio, C.; Evans, P.; Morgan, B.S. The Thio-adduct Facilitated, Enzymatic Kinetic Resolution of 4-hydroxycyclopentenone and 4-hydroxycyclohexenone. Org. Biomol. Chem. 2010, 8, 539–545. [Google Scholar] [CrossRef]
- Johnson, C.R.; Braun, M.P. A Two-Step, Three-Component Synthesis of PGE1: Utilization of α-Iodoenones in Pd(0)-Catalyzed Cross-Couplings of Organoboranes. J. Am. Chem. Soc. 1993, 115, 11014–11015. [Google Scholar] [CrossRef]
- Dauvergne, J.; Happe, A.M.; Jadhav, V.; Justice, D.; Matos, M.-C.; McCormack, P.J.; Pitts, M.R.; Roberts, S.M.; Singh, S.K.; Snape, T.J.; et al. Synthesis of 4-azacyclopent-2-enones and 5,5-diakyl-4-azacyclopent-2-enones. Tetrahedron 2004, 60, 2559–2567. [Google Scholar] [CrossRef]
- Zhao, X.; Yu, Z.; Zeng, F.; Chen, J.; Wu, X.; Wu, S.; Xiao, W.-J.; Zheng, Z. Highly Efficient Route to Diselenides from the Reactions of Imines and Selenium in the Presence of Carbon Monoxide and Water. Adv. Synth. Catal. 2005, 347, 877–882. [Google Scholar] [CrossRef]
- Liu, J.; Zheng, F.; Cheng, R.; Li, S.; Rozovsky, S.; Wang, Q.; Wang, L. Site-Specific Incorporation of Selenocysteine Using an Expanded Genetic Code and Palladium-Mediated Chemical Deprotection. J. Am. Chem. Soc. 2018, 140, 8807–8816. [Google Scholar] [CrossRef]
- Chu, C.-M.; Gao, S.; Sastry, M.N.V.; Kuo, C.-W.; Lu, C.; Liu, J.-T.; Yao, C.-F. Ceric Ammonium Nitrate (CAN) as a Green and Highly Efficient Promoter for the 1,4-addition of Thiols and Benzeneselenol to α,β-Unsaturated Ketones. Tetrahedron 2007, 63, 1863–1871. [Google Scholar] [CrossRef]
- Pedrosa, R.; Andrés, C.; Duque-Soladana, J.P.; Rosón, C.D. Regio- and Stereoselective 6-exo-trig Radical Cyclisations onto Chiral Perhydro-1,3-benzoxazines: Synthesis of Enantiopure 3-alkylpiperidines. Tetrahedron 2000, 11, 2809–2821. [Google Scholar] [CrossRef]
- Sanz, X.; Vogels, C.M.; Decken, A.; Bo, C.; Westcott, T.A.; Fernandez, E. Face to Face Activation of a Phenylselenium Borane with α,β-Unsaturated Carbonyl Substrates: Facile Synthesis of C–Se Bonds. Chem. Comm. 2014, 50, 8420–8423. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, Y.; Asano, T.; Kishimoto, Y.; Itoh, K.; Ishii, Y. Selective Synthesis of 1-Alkoxy-3-phenylseleno-1-alkenes and 3-Phenylselenoalkanals by the Reaction of Diisobutylaluminum Phenylselenolate with α,β-Unsaturated Acetals. Tetrahedron Lett. 1998, 39, 8685–8686. [Google Scholar] [CrossRef]
- Bhalla, A.; Sharma, S.; Bhasin, K.K.; Bari, S.S. Convenient Preparation of Benzylseleno- and Phenylselenoalkanoic Acids: Reagents for Synthesis of Organoselenium Compounds. Synth. Commun. 2007, 37, 783–793. [Google Scholar] [CrossRef]
- Miyashita, M.; Toshikoshi, A. Facile and Highly Efficient Conjugate Addition of Benzeneselenol to α,β-Unsaturated Carbonyl Compounds. Synthesis 1980, 8, 664–666. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |
Scheme 1.
One-pot zinc-mediated biphasic diselenide reduction-selenylation reaction. 1st step: diselenide reduction evidenced by discoloration; 2nd step selenylation (substitution and addition reactions).
Scheme 1.
One-pot zinc-mediated biphasic diselenide reduction-selenylation reaction. 1st step: diselenide reduction evidenced by discoloration; 2nd step selenylation (substitution and addition reactions).

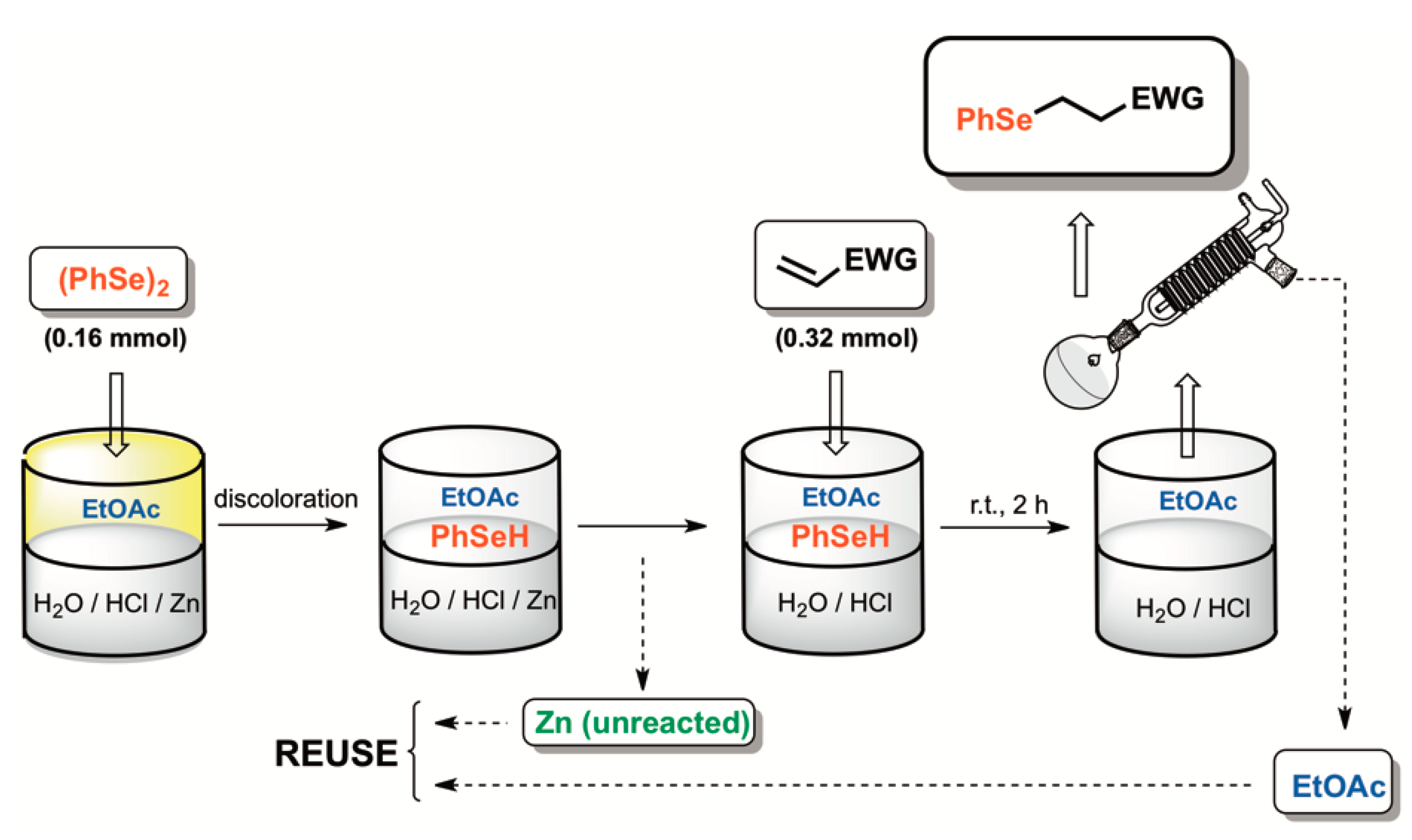
Scheme 2.
One-pot zinc-mediated biphasic diselenide reduction-Se-conjugate addition reaction.

Scheme 3.
Reuse of the aqueous phase in the Se-conjugate addition of diphenyl diselenide 1 to cyclohexenone 21.
Scheme 3.
Reuse of the aqueous phase in the Se-conjugate addition of diphenyl diselenide 1 to cyclohexenone 21.

Figure 1.
NMR conversion in the first seven reuses of the aqueous phase in the Se-conjugate addition of diphenyl diselenide 1 to cyclohexenone 21.
Figure 1.
NMR conversion in the first seven reuses of the aqueous phase in the Se-conjugate addition of diphenyl diselenide 1 to cyclohexenone 21.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Telescoped diselenide reduction-conjugate addition to α,β-unsaturated carbonyl derivatives 2–10.
Table 1.
Telescoped diselenide reduction-conjugate addition to α,β-unsaturated carbonyl derivatives 2–10.
 | |||
|---|---|---|---|
| Entry | Substrate a | Se-Adduct | Yield b |
| 1 |  2 |  11 | 91% |
| 2 |  3 |  12 | 44% |
| 3 |  4 |  13 | 70% |
| 4 |  5 |  14 | 60% |
| 5 |  6 |  15 | 22% |
| 6 |  7 |  16 | 79% |
| 7 |  8 |  17 | 60% |
| 8 |  9 |  18 | 25% |
| 9 |  10 |  19 | 95% c (62:38) |
a Phenyl vinyl sulfone, acrylonitrile, and chalcones did not afford the corresponding target compound. b Quoted for analytically pure material, obtained either directly, or following purification by flash column chromatography. c Obtained as an undetermined mixture of diastereomers (62:38) determined by the 1H-NMR of the crude integrating the doublets of the methyl group at 1.02 ppm for the major isomer and 0.97 for the minor.
Table 2.
Telescoped diselenide reduction-conjugate addition to cyclic enones 20–24.
 | |||
|---|---|---|---|
| Entry | Substrate | Se-Adduct | Yield a |
| 1 |  20 |  25 | 93% |
| 2 |  21 |  26 | 88% |
| 3 |  22 |  27 | 95% |
| 4 |  23 |  28 | 90% b (75:25) |
| 5 |  24 |  29 | 29% c |
a Quoted for analytically pure material, obtained either directly or following purification by flash column chromatography. b NMR conversion of the diastereomeric mixture from which the major isomer was isolated in 62% yield after flash chromatography. Even if the attempts to clarify the relative configuration by NOE experiments failed and we were not able to obtain crystals suitable for X-ray analysis, in accordance with similar sulphur derivatives reported by some of us, we can assume a trans-geometry between substituents in C-3 and C-4 [65,66]. c Obtained as a single undetermined diastereomer.
Table 3.
Reaction of 1,2-bis(3-phenylpropyl)diselenide 30 and protected selenocystine 31 with enones.
Table 3.
Reaction of 1,2-bis(3-phenylpropyl)diselenide 30 and protected selenocystine 31 with enones.
 | |||
|---|---|---|---|
| Entry | Substrate | Se-Adduct | Yield a |
| 1 |  21 |  33 | 65% |
| 2 |  32 |  34 | 90% b (89:11) |
| 3 |  2 |  35 | 30% |
| 4 |  21 |  36 | 53% c (66:34) |
| 5 |  23 |  37 | 30% c (55:45) |
a Quoted for analytically pure material, obtained either directly or following purification by flash column chromatography. b NMR conversion of the diastereomeric mixture from which the major isomer was isolated in 49% yield after flash chromatography. In analogy with 28, we can assume a trans-geometry between substituents in C-3 and C-4 [65,66]. c Obtained as an inseparable and undetermined mixture of diastereomers determined by the 1H-NMR of the crude.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Nacca, F.G.; Monti, B.; Lenardão, E.J.; Evans, P.; Santi, C. A Simple Zinc-Mediated Method for Selenium Addition to Michael Acceptors. Molecules 2020, 25, 2018. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25092018
AMA Style
Nacca FG, Monti B, Lenardão EJ, Evans P, Santi C. A Simple Zinc-Mediated Method for Selenium Addition to Michael Acceptors. Molecules. 2020; 25(9):2018. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25092018
Chicago/Turabian StyleNacca, Francesca Giulia, Bonifacio Monti, Eder João Lenardão, Paul Evans, and Claudio Santi. 2020. "A Simple Zinc-Mediated Method for Selenium Addition to Michael Acceptors" Molecules 25, no. 9: 2018. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25092018