
Recent Advances in the Chemical Synthesis and Evaluation of Anticancer Nucleoside Analogues


1
Lennard-Jones Laboratory, School of Chemical and Physical Sciences, Keele University, Keele, Staffordshire ST5 5BG, UK
2
Medicinal Chemistry Knowledge Center, Stanford ChEM-H, 290 Jane Stanford Way, Stanford, CA 94305, USA
*
Author to whom correspondence should be addressed.
Molecules 2020, 25(9), 2050; https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25092050
Submission received: 2 April 2020
/
Revised: 20 April 2020
/
Accepted: 25 April 2020
/
Published: 28 April 2020
(This article belongs to the Special Issue Advances in Anticancer Drug Discovery)
Abstract
:Nucleoside analogues have proven to be highly successful chemotherapeutic agents in the treatment of a wide variety of cancers. Several such compounds, including gemcitabine and cytarabine, are the go-to option in first-line treatments. However, these materials do have limitations and the development of next generation compounds remains a topic of significant interest and necessity. Herein, we discuss recent advances in the chemical synthesis and biological evaluation of nucleoside analogues as potential anticancer agents. Focus is paid to 4′-heteroatom substitution of the furanose oxygen, 2′-, 3′-, 4′- and 5′-position ring modifications and the development of new prodrug strategies for these materials.
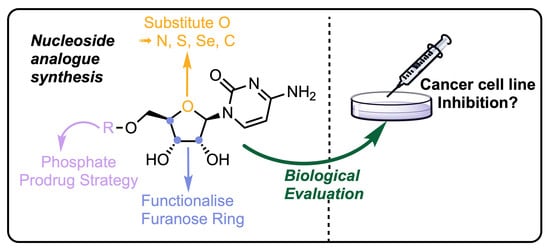
1. Introduction
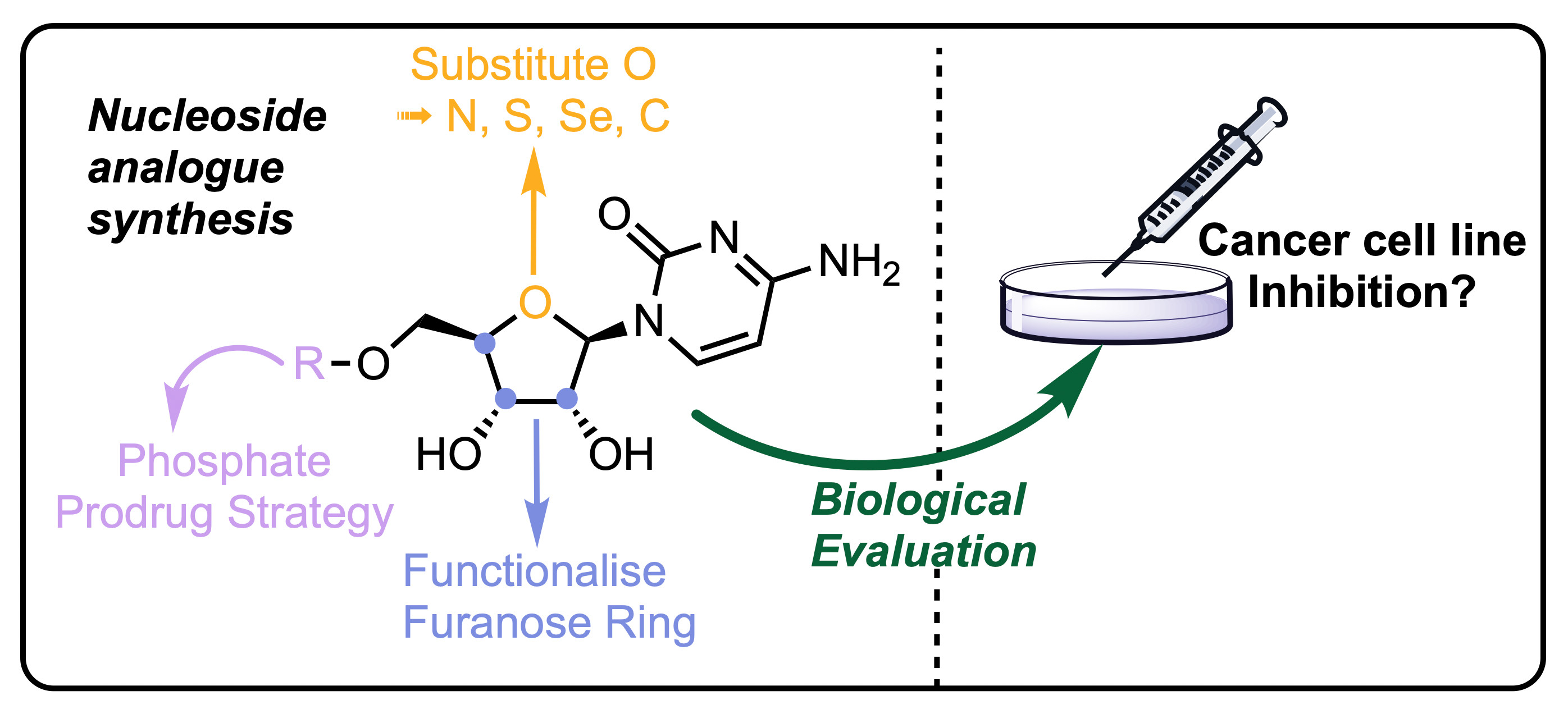
A significant proportion of current chemotherapeutic treatments for cancer involve the use of anti-metabolites, particularly modified nucleoside analogues that possess a capability to mimic native purine or pyrimidine nucleosides which can disrupt metabolic and regulatory pathways [1]. These molecules can be taken up by nucleoside transporters and then phosphorylated to their mono-, di- and triphosphate forms where they are able to interfere with DNA/RNA synthesis and repair; for example, by acting as chain terminators [2] or ribonucleotide reductase inhibitors [3]. Other notable modes of action include epigenetic regulation, through inhibition of DNA regulatory proteins, such as DNA methyltransferase [4]. Selected current examples of anticancer nucleoside analogues approved for chemotherapeutic treatment regimens include capecitabine, gemcitabine 1, clofarabine 2 and cytarabine (Ara-C) 3 (Figure 1).
Therapeutic intervention using nucleoside analogues is not without its problems and their use is often limited by poor cellular uptake, low conversion to the active triphosphate metabolite, rapid degradation or clearance and development of resistance profiles in certain cell types [5,6]. Consequently, research continues to develop next generations of nucleoside analogues that overcome some of these limitations and provide new therapeutic options.
This class of antimetabolite also possess a proud history, and current frontline treatment, as antiviral [7,8,9,10,11,12] and, more recently, antibacterial agents [13]. Indeed, the development of nucleoside analogues has a symbiotic relationship between compound class and final therapeutic treatment. For example, gemcitabine 1 was developed as an antiviral, but was subsequently shown to be very toxic to leukaemia cells.
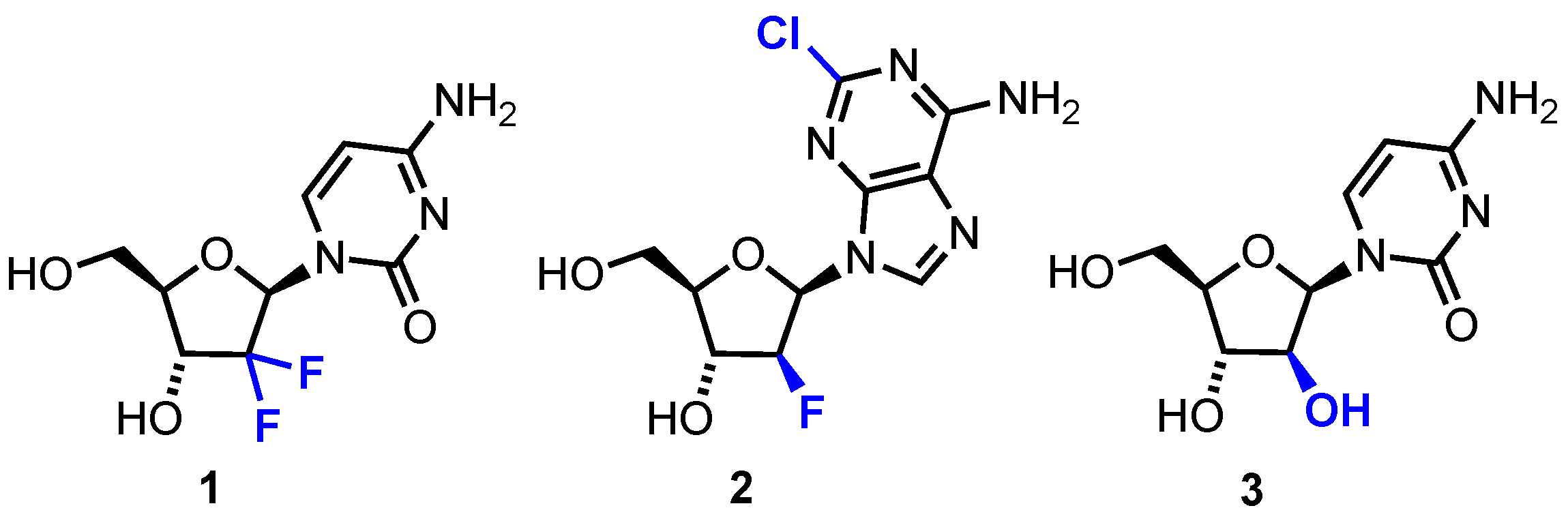
In this review, we survey developments from 2010 onwards for the chemical synthesis and evaluation of modified nucleoside analogues for anticancer research. Specifically, focusing on alterations to the native furanose ring (Figure 2) and generally retaining native purine or pyrimidine nucleobases. Comprehensive reviews concerning hetero-base modifications and general trends in nucleotide synthesis have been covered elsewhere [14,15]. The review is divided into sections that systematically consider: i) furanose 4’-oxygen atom replacements with N, S, Se and C ii) 2′-, 3′-, 4′- or 5′-position furanose ring modifications and iii) new prodrug approaches to deliver nucleoside analogues.
2. Furanose Oxygen Atom Replacements
2.1. Azanucleosides
Azanucleosides were originally defined as nucleoside analogues where the furanosyl oxygen is replaced by nitrogen, however this group of analogues has been extended to include nucleosides where the resultant pyrrolidine core has been replaced by other nitrogen containing rings, including heterocycles, heterotricycles and acyclic nitrogen-containing nucleosides [16,17]. This class of compound have proven successful in the treatment of cancer [18] and have also been established as having antiviral and antibacterial properties [19].
Development of Forodesine
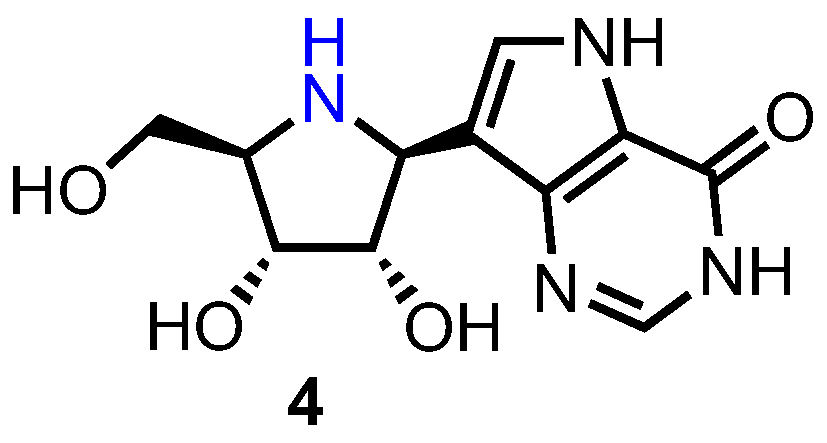
Purine nucleoside phosphorylases (PNPs) are responsible for the phosphorolytic metabolism of purine nucleosides to ribose/deoxyribose phosphate and the corresponding nucleobase. Patients with abnormally low levels of PNP possess little T-cell immunity due to a severely reduced degradation of deoxyguanosine, which results in the accumulation of the corresponding triphosphate (dGTP). This then reduces the activity of ribonucleotide reductase and induces apoptosis. As such, human PNP inhibitors are potential treatments for T-cell lymphomas [17]. Immucillin H (forodesine) 4 (Figure 3) is a highly potent PNP inhibitor (IC50 = 0.48–1.57 nM) which is effective against T-cell malignancies and was found to have excellent oral bioavailability in mice (63%) [20].
Forodesine is a gradual onset drug which binds tightly to PNP with a high affinity (Ki = 0.023 nM) [21]. Whilst clinical development of 4 was discontinued in the US and Europe, it was recently approved for use in the treatment of relapsed/refractory peripheral T-cell lymphoma (PCTL) in Japan (April 2017) [22].
Forodesine is a guanosine analogue and a transition state inhibitor of PNP with 100–1000-fold higher potency than previously identified inhibitors [23]. Due to the combined substitution of the furanosyl oxygen with nitrogen and the C-glycosidic bond, 4 is not incorporated into DNA, acting only as a highly selective PNP inhibitor [22]. Also noteworthy is an adenosine mimetic of 4 which is currently under development as a broad-spectrum antiviral [24].
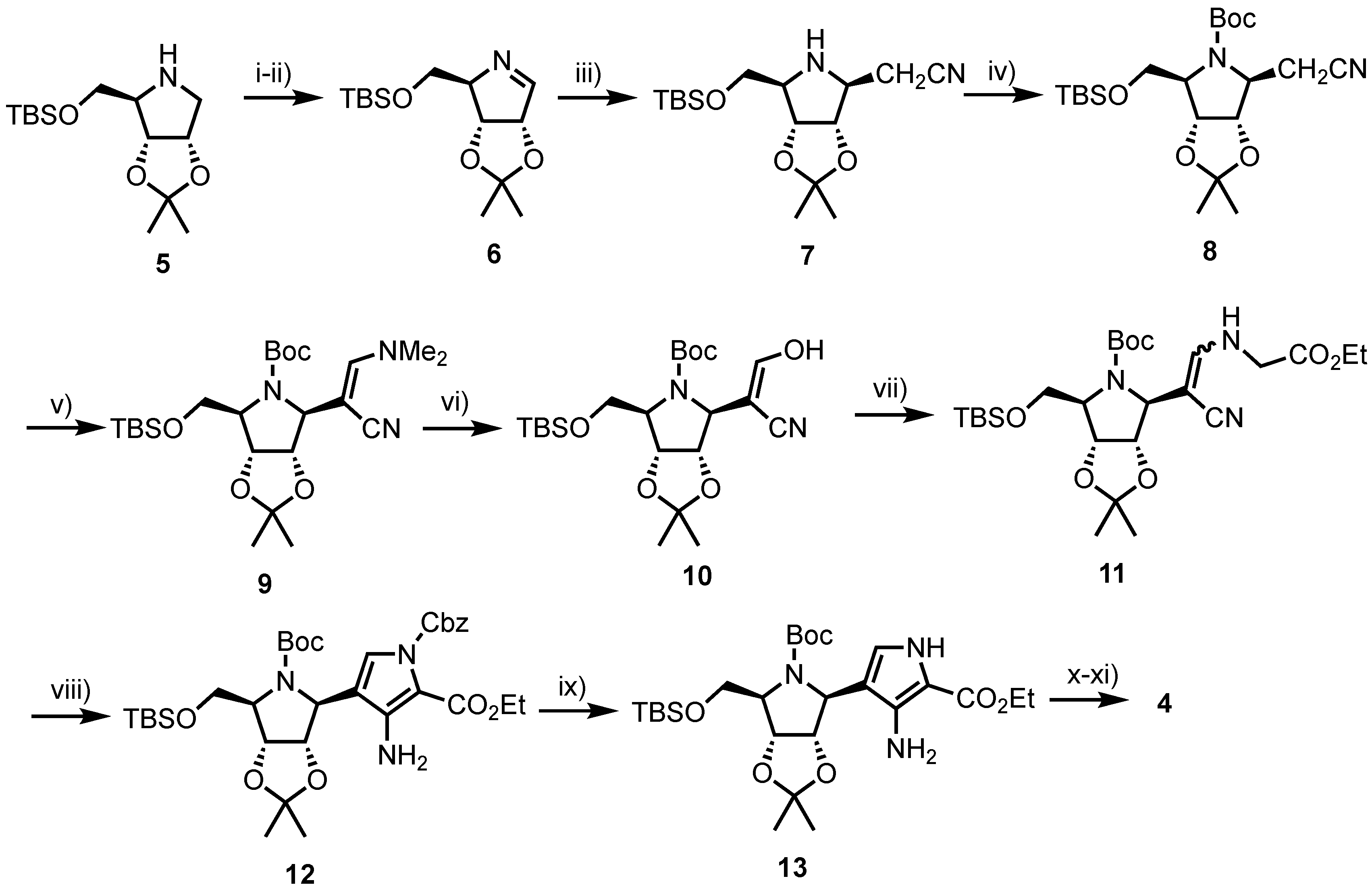
In 2000, Tyler et al. described a linear synthesis of 4 in a satisfactory 39% yield over 10 steps (Scheme 1) [25]. Starting from 5, synthesised in nine steps by known methods from d-gulonolactone [26], the pyrrolidine was treated with N-chlorosuccinimide (NCS), obtaining the 1-chloro anomeric glycoside which subsequently underwent elimination using lithium tetramethylpiperidine (LiTMP) to afford imine 6. The nucleobase was next assembled via addition of lithiated acetonitrile to afford 7, followed by protection of the furanosyl nitrogen giving 8 and treatment with Bredereck’s reagent to afford enamine 9. Acid-catalysed hydrolysis of 9 delivered enol 10 which was reacted with ethyl glycinate to obtain enamine 11. Treatment of 11 with excess benzyl chloroformate and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) revealed 12 and subsequent hydrogenolysis of the N-Cbz group provided pyrrole 13. Completion of the carbocyclic nucleobase was achieved via treatment of 13 with formamidine acetate and acidic removal of the silicon, nitrogen and isopropylidene protecting groups, to afford 4.
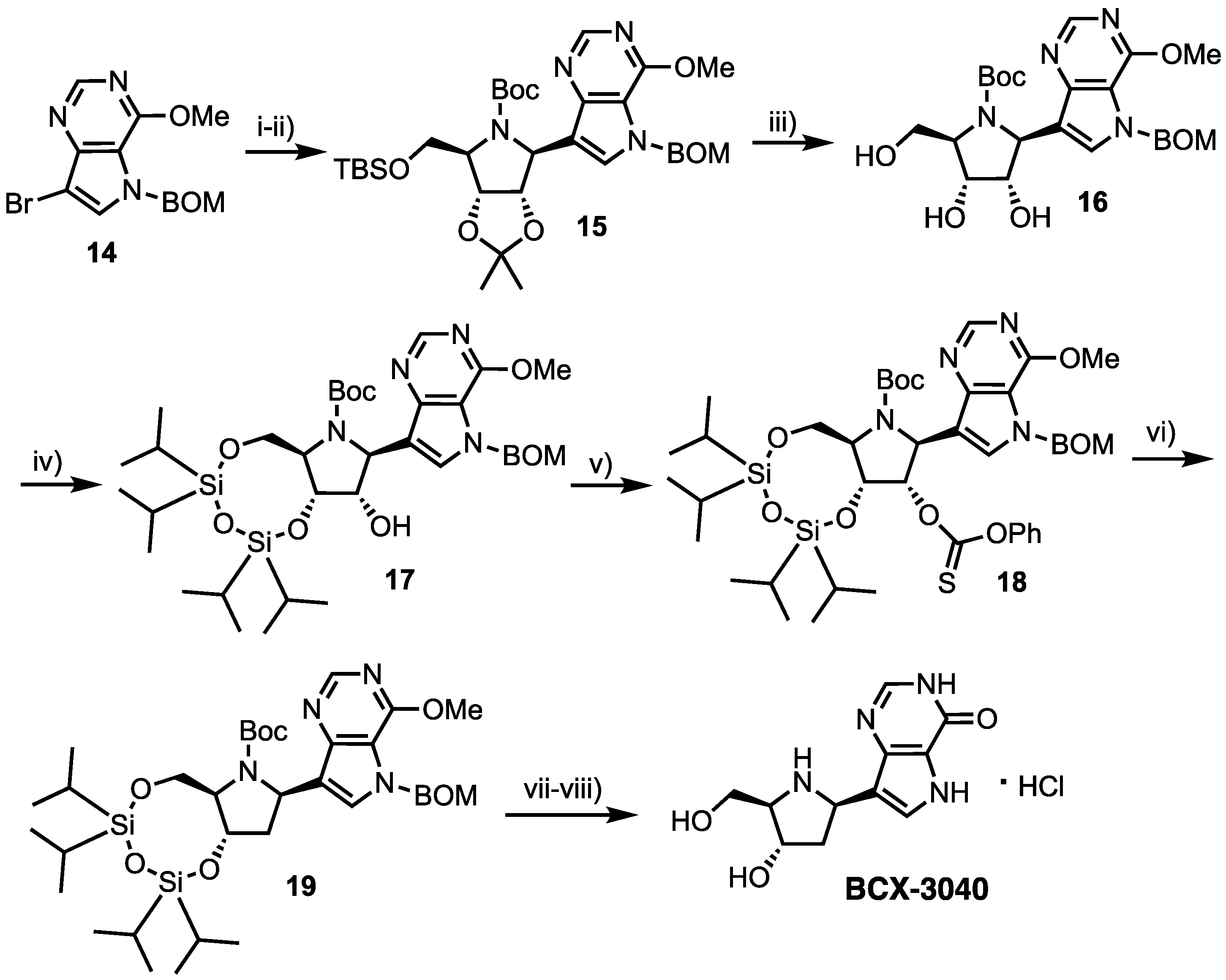
Forodesine was found to have low oral bioavailability (<11%) in primates, contrary to the case in mice (63% [20]) and was thus originally developed as an intravenous formulation [21]. In 2005, Morris, Jr et al. synthesised BCX-3040, the 2′-deoxy analogue of 4, and comparatively evaluated its oral pharmacokinetic and pharmacodynamic properties in an effort to maintain potency and deliver oral bioavailability [27]. This was hypothesised from 2′-deoxyguanosine exhibiting tight binding to PNP [28] and therefore a possible redundancy for the 2′-OH. Starting from vinyl bromide 14 (Scheme 2), the 9-deazapurine underwent bromine-lithium exchange and addition to imine 6, obtaining nucleoside 15 in 85% yield. Subsequent removal of 5′-O-TBS and 2′,3′-O-isopropylidene groups gave 16, followed by 3’,5’-OTIPDS protection to afford 17. The free 2′-hydroxyl group was then converted to thiocarbonate 18 and deoxygenated via treatment with 1,1′-azobis(cyclohexane-1-carbonitrile) in excellent yield (91%) to give 19. Deprotection was completed in two steps, first cleaving the CH2OCH2Ph (BOM) group, followed by acidic hydrolysis and hydrochloride salt formation to obtain BCX-3040 in 83% yield (from 19).
In vitro biological evaluation of BCX-3040 confirmed it to be a potent PNP inhibitor, with near identical IC50 values to 4 (BCX-3040 IC50 = 3.1 ± 0.50 nM and 4 IC50 = 1.2 ± 0.21 nM). Administration of 5.0 mg of 4 had a 12.6-fold greater 2′-deoxyguanosine response than administration of 10.0 mg of BCX-3040, indicating a reduced bioavailability for BCX-3040. Furthermore, following IV administration of 5 mg/kg of BCX-3040, the plasma concentration of BCX-3040 dropped rapidly to 3.0 ± 0.31 µg/mL. Overall, due to the observed rapid clearance and reduced bioavailability of BCX-3040, it was concluded to be a poorer PNP inhibitor candidate in comparison to 4.
2.2. Thionucleosides
4′-Thiofuranoses are of known importance in biological systems, for example as chemical biology or biomedical tools [29], and possess chemotherapeutic activity [30,31]. Furthermore, the thioaminal moiety within 4′-thiofuranosyl nucleosides has been proven to be resistant to metabolic hydrolysis in comparison to native 4′-oxa analogues [32]. In the early 1990s, Secrist, Montgomery and co-workers stimulated interest in this class of molecule with their synthesis and biological evaluation of 2′-deoxy-4′-thiopyrimidine nucleosides [30]. Since then, there has been a resurgence of interest in the synthesis and evaluation of these compounds as potential antiviral and chemotherapeutic agents.
2.2.1. 2′-Modfied Thionucleosides
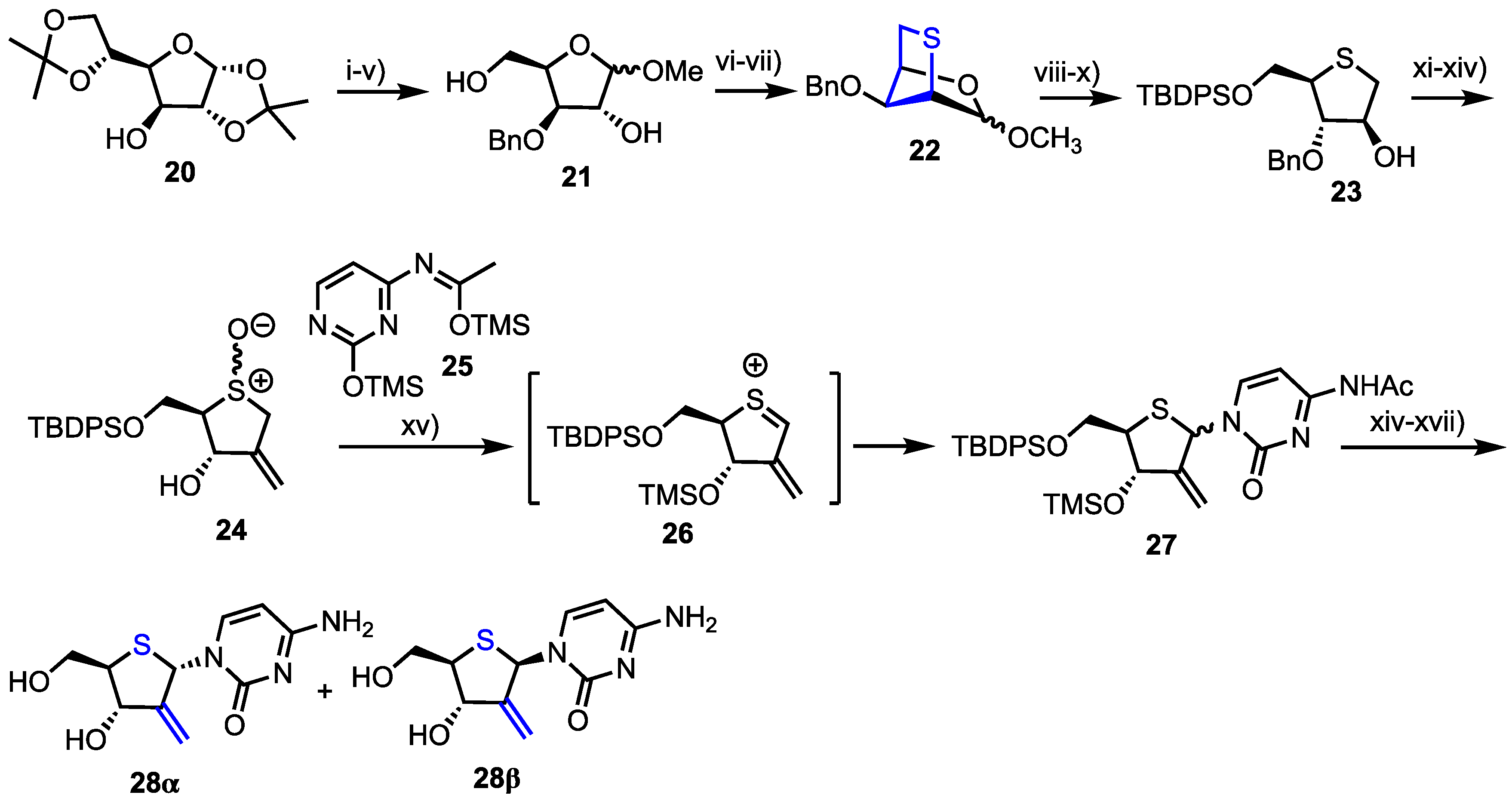
Yoshimura et al. reported the synthesis and biological evaluation of 4’-thia-1-(2-deoxy-2-C-methylene-β-d-erythro-pentofuranosyl)cytosine (4’-thio-DMDC) 28β and 2’-deoxy-2’-fluoro-arabino-4’-thiacytidine 33β as potential antitumour agents [33,34,35]. Their synthesis of 28 started from 1,2,5,6-diisopropylidene-d-glucose 20 and a series of protecting group manipulations delivered 3-O-benzyl d-xylose methyl glycoside 21 (Scheme 3). Mesylation of the 2- and 5-hydroxyl groups enabled reaction with sodium sulfide to afford 2,5-bicyclic intermediate 22 as a mixture of anomers. Following conversion of 22 to 4-thioarabinofuranose 23, the 2-position hydroxyl group was oxidised and the ketone homologated using a Wittig reagent to install the 2-methylene component, with this material then oxidised to sulfoxide 24.
The cytidine nucleobase was installed using a Pummerer-type thioglycosylation, via sulphenium ion 26, which afforded 27 as a mixture of anomers. These were fully deprotected to afford 28α and 28β and the desired 28β isolated using HPLC separation.
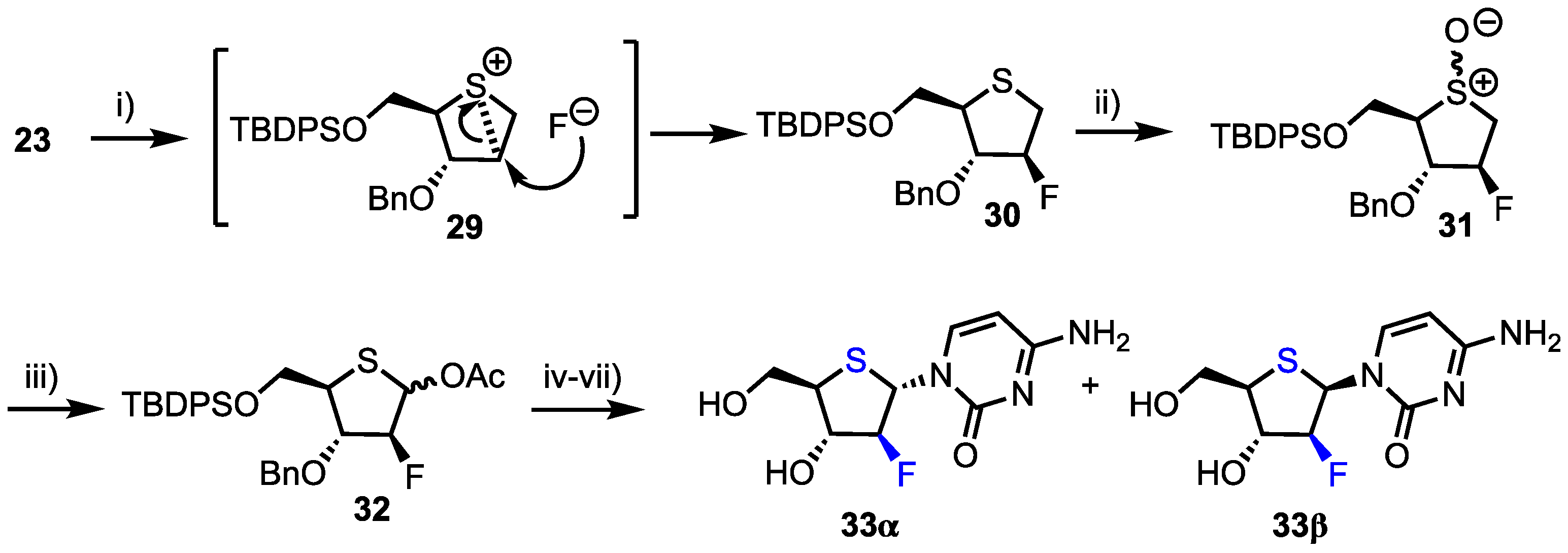
Additionally, 2′-deoxy-2′-fluoro-arabino-4′-thiacytidine 33 was prepared from intermediate 23 (Scheme 4). Stereospecific DAST fluorination of 23 proceeded through epi-sulphonium intermediate 29 which delivered the 2-deoxy-2-fluoroarabino intermediate 30 in 68% yield. m-CPBA oxidation to the sulfoxide 31 and subsequent Pummerer rearrangement formed anomeric acetate 32 in 77% yield from 30. Finally, thioglycosylation was successfully employed to access the corresponding 4’-thionucleoside mixture 33 (54% yield). Comparatively, the group found that using 32 as a donor and employing silyl-Hilbert-Johnson glycosylation conditions yielded 33α/β in higher yield (93%), but still as a mixture of anomers (2.9:1 α/β).
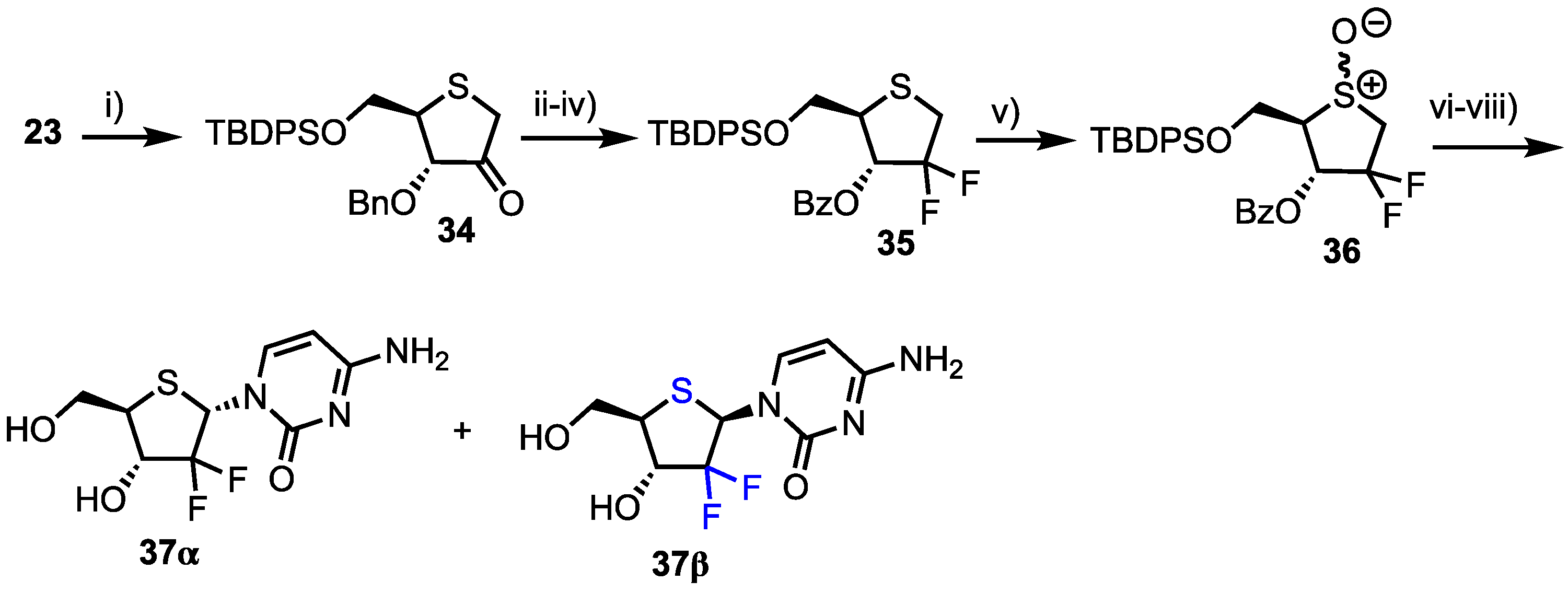
Finally, the group synthesised 2’-deoxy-2’-difluoro-1’(4’-thia-d-ribofuranose)cytosine 37, a thionucleoside analogue of the potent chemotherapeutic agent gemcitabine 1 (Scheme 5). Intermediate 23 was again utilised and oxidised using Albright-Goldman conditions to obtain ketone 34 which was difluorinated at C2 using DAST. The 3-O-benzyl group was next removed and replaced with benzoate to afford 35 in 79% overall yield from 34. Oxidation to the sulfoxide afforded 36 which subsequently underwent Pummerer rearrangement-glycosylation, before the remaining protecting groups were removed to afford 37α/β (α/β = 2.4/1) in a moderate yield of 51% from 36.
The antineoplastic activities of 28, 33 and 37 were evaluated and compared to arabinocytidine 3 (Ara-C) and 1-(2-deoxy-2-C-methylene-β-d-erythro-pentofuranosyl)cytosine (DMDC) (Table 1). As expected, all the α-anomer forms were found to be inactive against T-cell leukemia CCRF-HSB-2 cells. However, the β-anomers showed considerable cytotoxic activity against the same cell line. Notably, 28β and 33β were highly potent against both T-cell leukemia CCRF-HSB-2 cells and human solid tumour KB cells, with IC50 values of 0.01 µg/mL (CCRF-HSB-2) and 0.05 µg/mL (CCRF-HSB-2) for 28β and 33β, respectively. Indeed, the activity of 28β was significantly higher than that of its native counterpart, DMDC, against both cell lines, with an IC50 value 2.4 times lower in CCRF-HSB-2 cells and 3.7 times lower in KB cells. Interestingly, 4′-thiogemcitabine analogue 37β had poorer antineoplastic activity compared to 1, which the authors suggest may be due to a reduction in the phosphorylation efficacy of 37β by deoxycytidine kinase, a key enzyme which converts 2′-deoxycytidine analogues to their corresponding monophosphates.
2.2.2. 4′-Modfied-2′-deoxythionucleosides
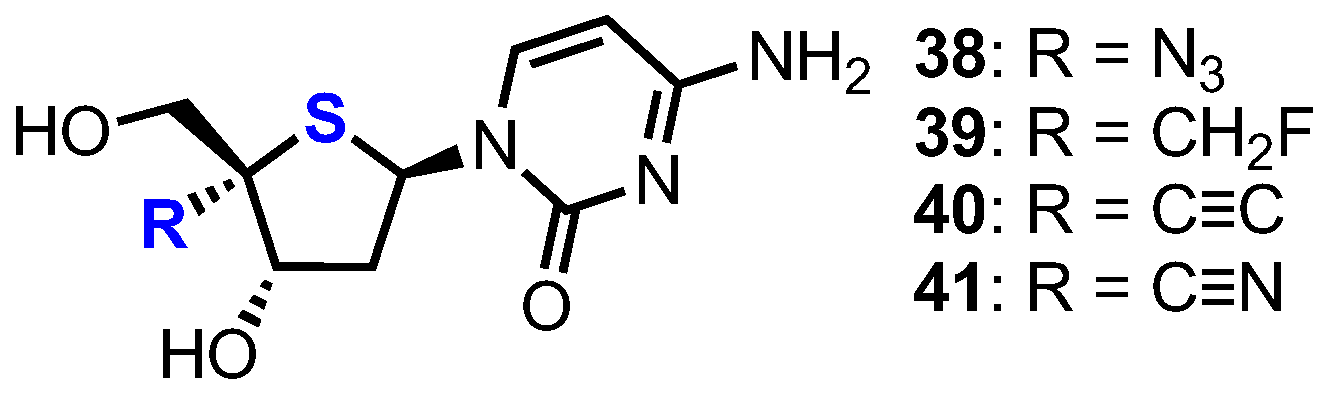
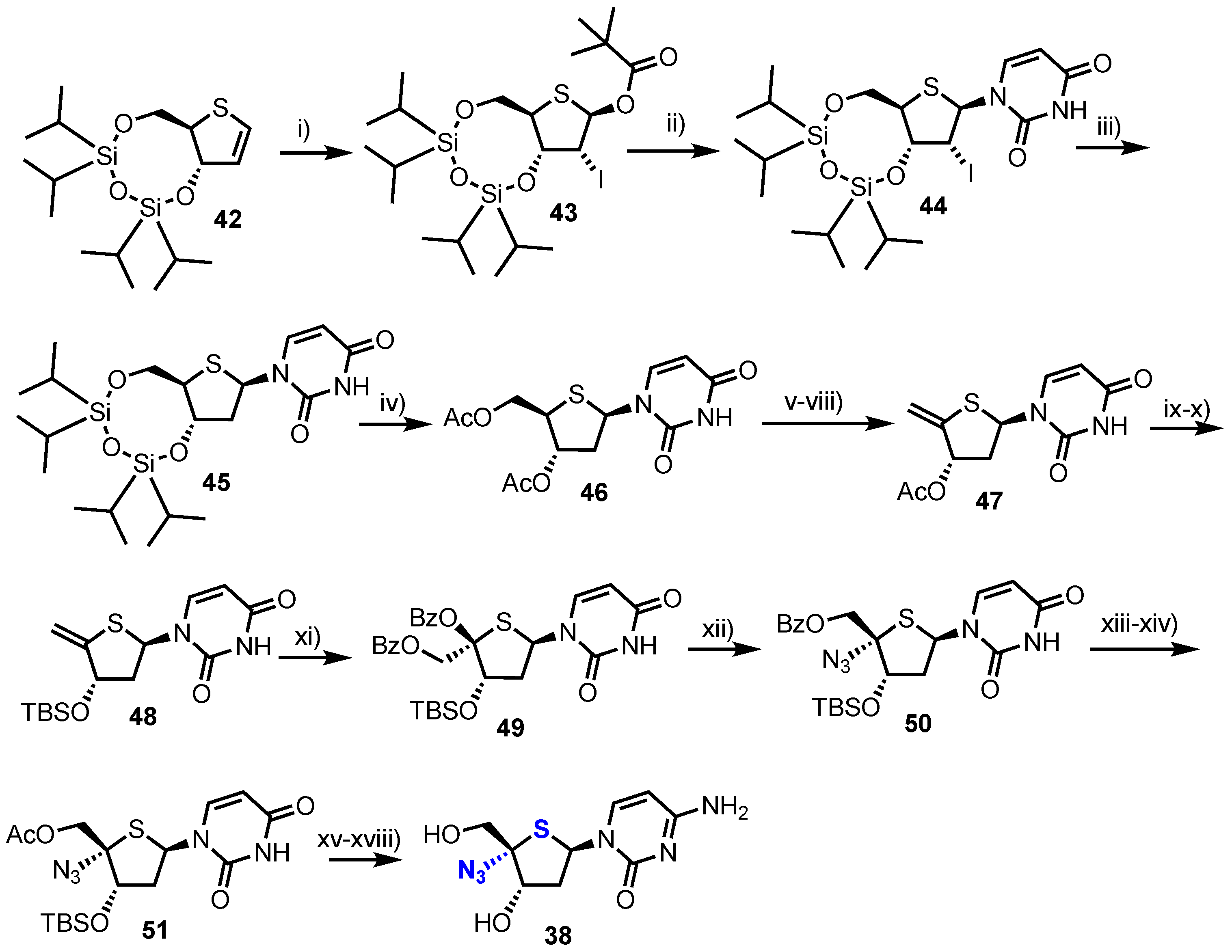
Following earlier work by Parker and colleagues, who identified 4′-thia-2′-deoxycytidine (T-dCyd) as being able to inhibit tumour growth [37], Haraguchi and colleagues reported the synthesis and evaluation of a small library of 4′-position modified 4′-thia-2′-deoxycytidine nucleosides 38–41 for their antineoplastic and antiviral activity (Figure 4) [38].
Towards analogue 38 the group started from thioglycal 42 (Scheme 6), obtained using established procedures from 2-deoxy-d-ribose in 12 steps [39]. This material was transformed into a glycosyl donor through treatment with N-iodosuccinamide (NIS) and pivalic acid, giving one diastereoisomer of 2-iodo derivative 43. Silylated uracil was then glycosylated with 43 using Vorbrüggen-type conditions to give β-4’-thiouridine 44 which was 2’-deoxygneated via a Bu3SnH mediated radical reduction to give 45. Subsequent TIPDS deprotection, followed by C3’ and C5’-O-acetylation delivered 46 to enable a four-step procedure to deliver 4’,5’-unsaturated-4’-thiouridine derivative 47. Exo-thioglycal 47 was next converted to silyl-protected 4’-thionucleoside 48, which when treated with Pb(OBz)4 yielded dibenzoate 49. SN2 inversion back to the native 4’-d-ribo configuration and 4’-azide installation was achieved by reaction with MeSiN3 in the presence of SnCl4 and afforded the desired 4’-α-azido derivative 50 as the major product. Finally, the nucleoside was converted to the cytidine form via intermediate 51 to give 38 in 7% yield over 18 steps.
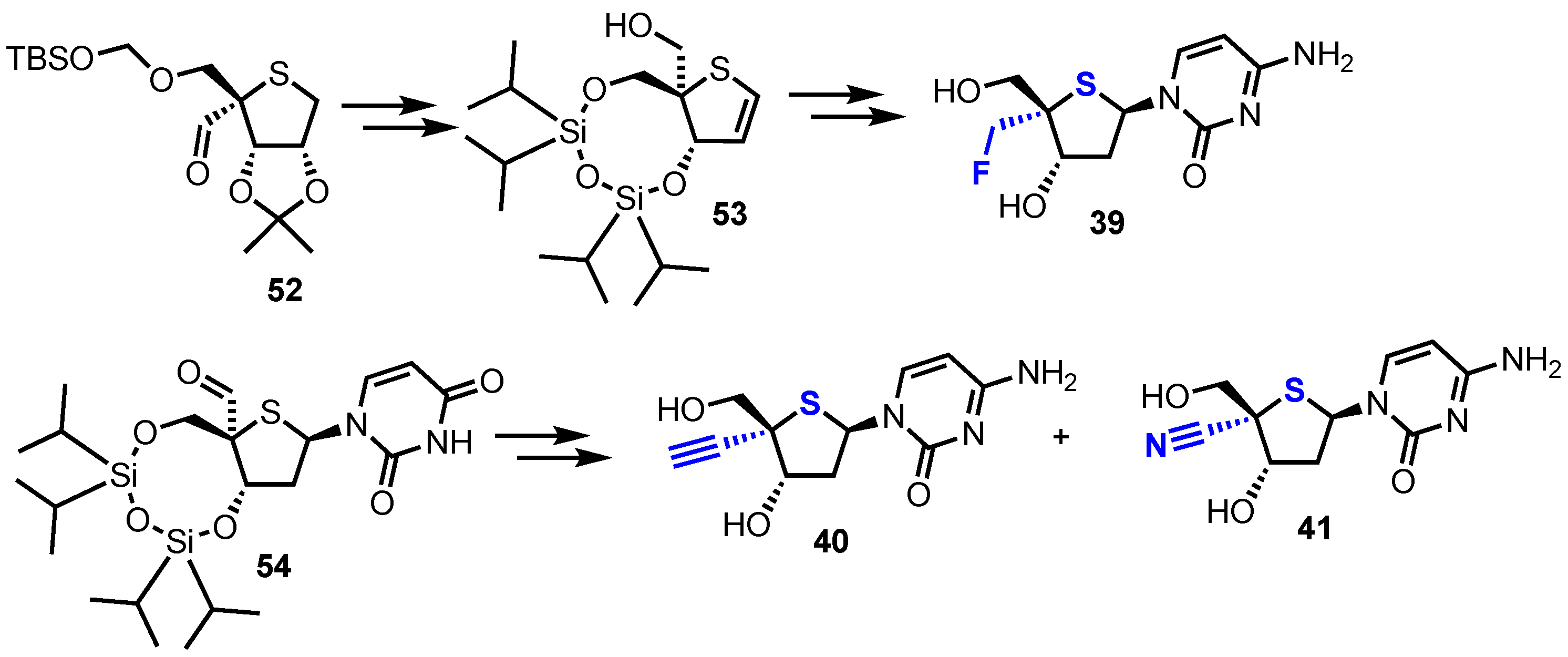
In order to access 2′-deoxy-4’-C-fluoromethyl-4’-thiacytidine 39, alcohol 53 was prepared from known aldehyde 52 [40,41]. Treatment of 53 with DAST successfully installed the key 4-α-fluoromethyl group which was then elaborated to 39 (Scheme 7). 4-α-alkynyl and nitrile analogues 40 and 41 were synthesised from aldehyde 54 using a late-stage insertion of the key functional group at the 4’-position in the presence of the nucleobase. Of the four 2’-deoxy-4’-modified thionucleosides 38-41, two analogues, 38 and 39, showed moderate cytotoxicity against human B-cell leukaemia (CCRF-SB; IC50 = 7.14 μM and 3.19 μM for 38 and 39 respectively) and human T-cell leukaemia cell lines (Molt-4; IC50 = 2.72 μM and 2.24 μM for 38 and 39 respectively).
2.3. Selenonucleosides
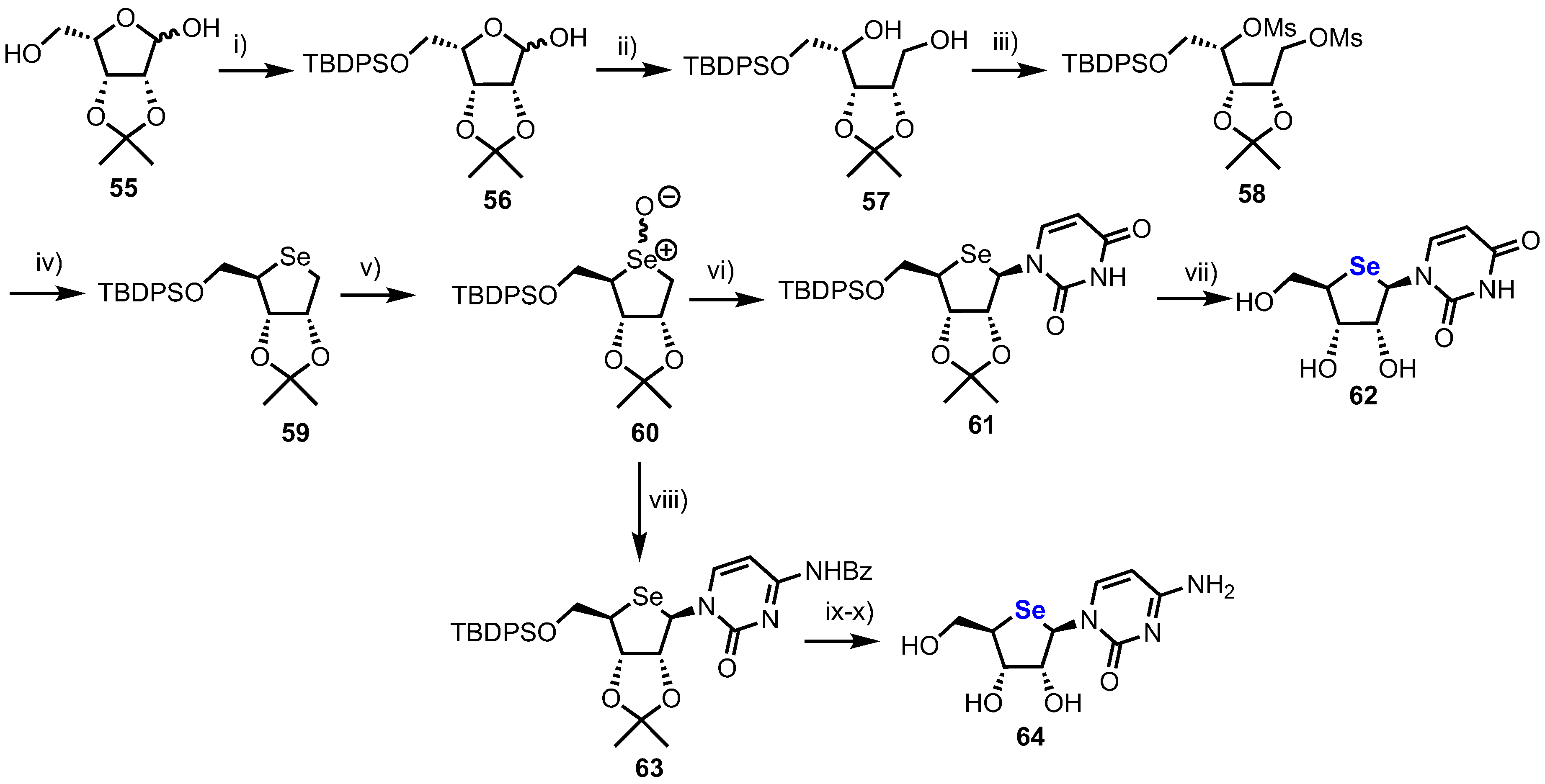
A first synthesis of pyrimidine 4’-selenonucleosides was reported by Jeong and colleagues in 2008 (Scheme 8), [42] starting from lyxose derivative 55, synthesised from d-gulonic-γ-lactone in four steps using established procedures [43].
Selective protection of the primary alcohol in 55 was achieved by reaction with TBDPSCl, giving hemiacetal 56 which was reduced with NaBH4, furnishing diol 57. Following double mesylation of 57 to give 58, cyclisation to give 4-selenosugar 59 was achieved by treatment with selenium in the presence of NaBH4. Oxidation of 59 to a diastereomeric selenoxide mixture 60 then enabled either a uracil or cytosine nucleobase to be installed via a Pummerer-type glycosylation, furnishing the β-anomers 61 or 63. 4’-selenouridine 62 was obtained after global deprotection of 61 with 50% aqueous TFA in an overall yield of 12% over 11 steps. 4’-selenocytidine 64 was similarly obtained in an overall yield of 9% over 11 steps. The crystal structure of 62 revealed the non-native ring adopted an unusual C2’-endo/C3’-exo twist (Southern confirmation), contrary to uridine, which shows a C2’-exo/C3’-endo twist (Northern conformation). This difference was explained by the introduction of the bulky selenium, whereby stereoelectronic effects observed in 4’-oxanucleosides are outweighed by the size of the heteroatom.
2’-Substituted-4’-selenoribofuranosyl Pyrimidines
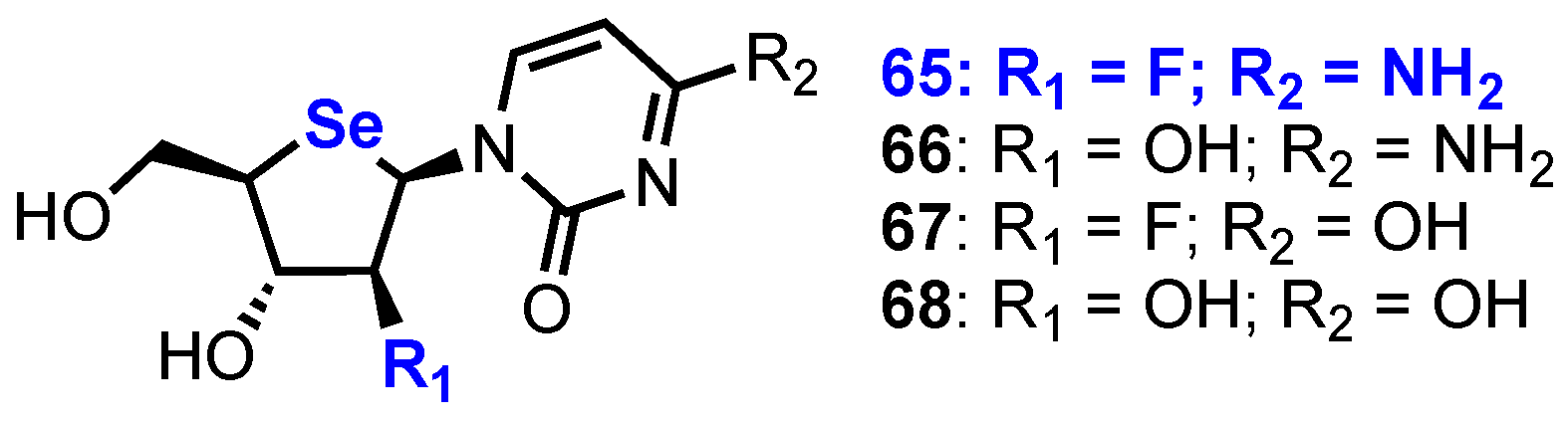
Building on their work in this area, Jeong and co-workers reported the synthesis and biological evaluation of a small library of 2’-substituted 4’-selenoarabinofuranosyl pyrimidine analogues 65–68 (Figure 5) [44,45].
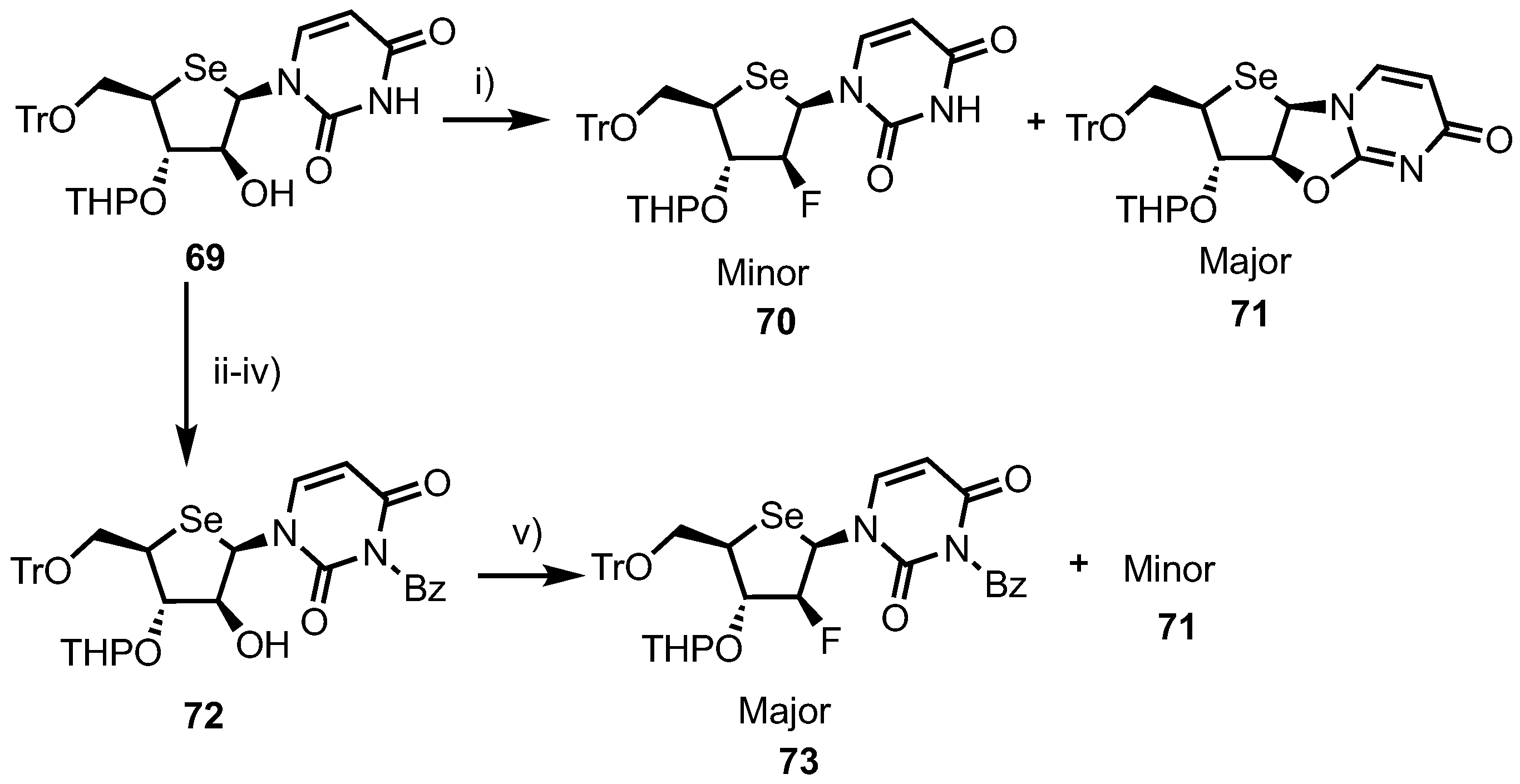
A 2’-fluoroarabino analogue was obtained via a 2’-position DAST fluorination of intermediate 69, synthesised from 4’-selenouridine 61 using a four-step process [44]. At first attempt, DAST fluorination of 69 led to the formation of the desired 2’-fluoro product 70 as the minor product in 23% yield, along with a major product, 2,2’-O-anhydro nucleoside 71, in 60% yield (Scheme 9). This ratio was subsequently improved by instead fluorinating an N3-benzoyl derivative 72, giving 73 in 45% yield, now as the major product, and 71 in reduced 30% yield.
Following global deprotection and uracil to cytosine conversion, four nucleoside analogues 65–68 were evaluated against several human cancer cell lines (HCT116, A549, SU638, T47D, PC-3 and K562) and compared to the established anticancer nucleosides 1 and 3 (Table 2). From this study, it was found that 2’-fluoro-4’-selenoarabinocytidine 65 was the most potent analogue, showing even greater potency than 3, in the majority of cell lines tested.
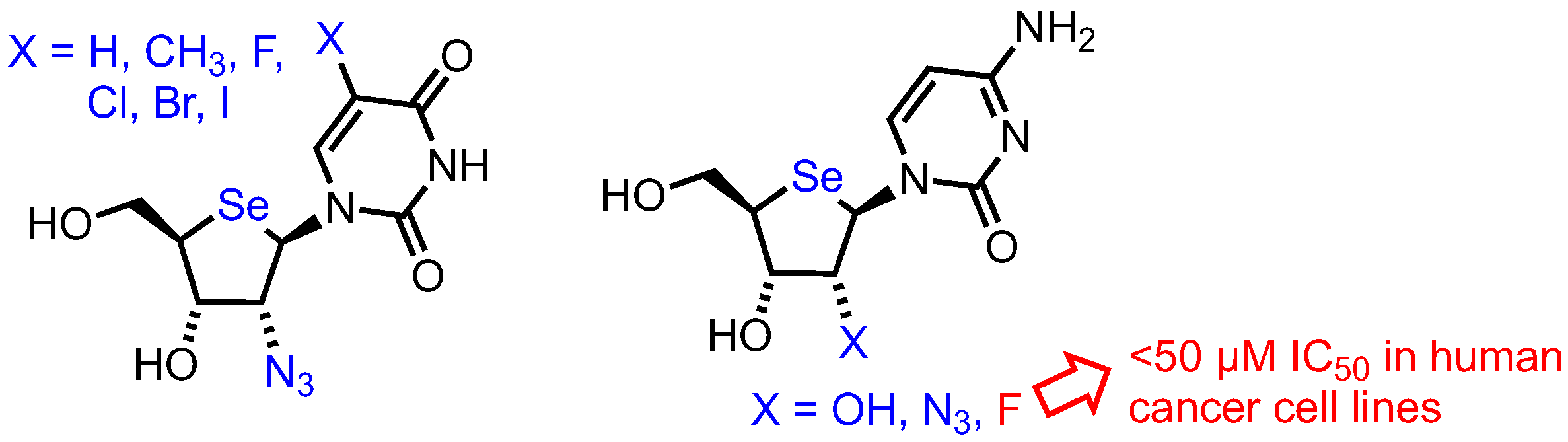
The group have also reported further 2’-substituted-4’-selenoribofuranosides [46], synthesising a series of pyrimidine 2’-substituted analogues and utilising a 2,2’-O-anhydro intermediate to enable regioselective nucleophilic ring opening to the desired ribo-configured products (Figure 6). Only a 2’-fluoro riboselenocytosine derivative showed significant activity (uracil, thymine and 5-halo uracil derivatives showed no activity up to 100 µM), but this was less potent than the arabino-configured analogue previously described [45].
2.4. Carbocyclic Nucleosides
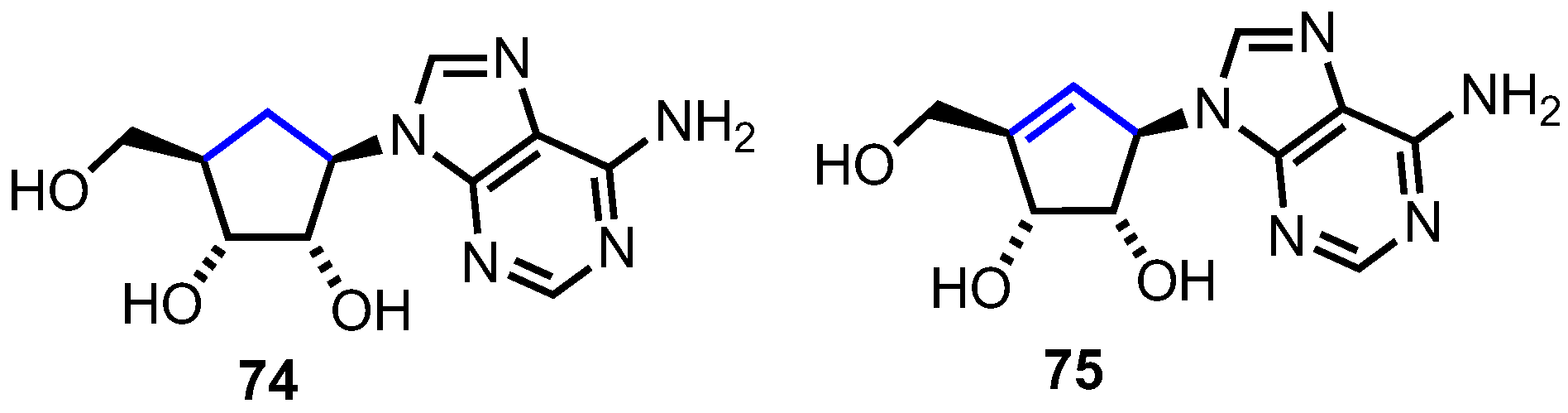
In carbocyclic nucleosides the furanosyl oxygen is replaced by CH2, forming a cyclopentane ring. The lack of a hemiaminal linkage between the nucleobase and the sugar leads to an increased chemical stability. Furthermore, due to the lack of a glycosidic bond, these nucleosides show an enhanced resistance towards phosphorylases [47]. Although considered a second generation of nucleoside analogues, there are two naturally occurring carbocyclic nucleosides, aristeromycin 74 and neplanocin A 75 (Figure 7), both of which exhibit substantial biological activity as antitumor agents [47].
2.4.1. Fluorinated Derivatives of Neplanocin A
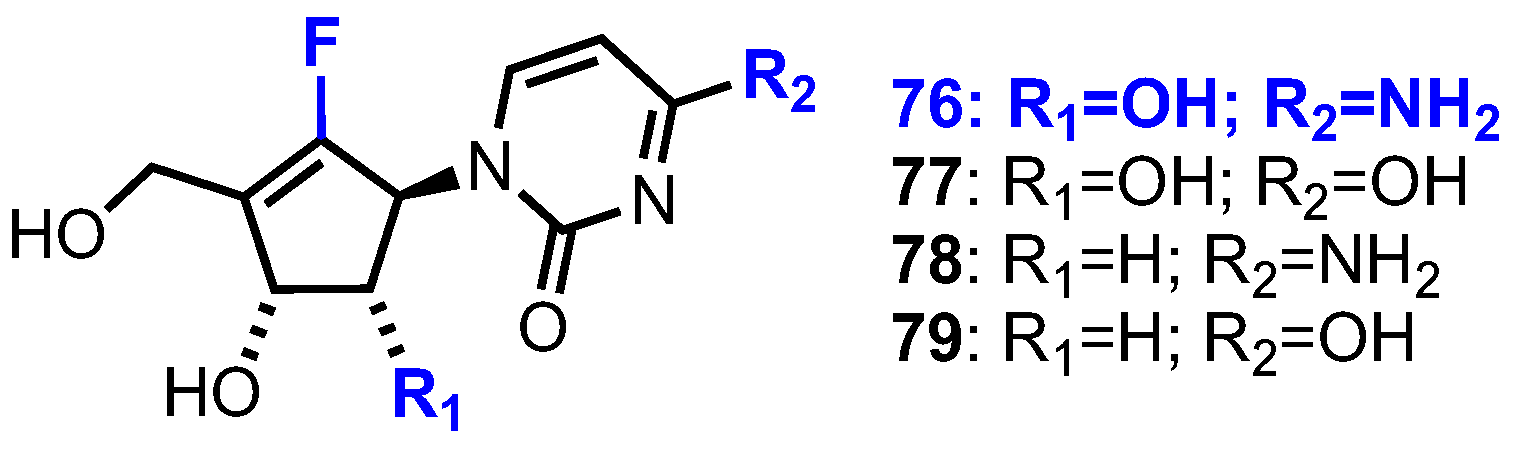
In 1988, Driscoll et al. reported that a cytosine analogue (CPE-C) of 75 showed substantial antitumour and antiviral activity [48]. On the basis of these findings, and the continuing clinical success of fluorine containing nucleoside chemotherapeutics [49,50], Jeong et al. subsequently reported the synthesis and biological evaluation of a small library of fluorocyclcopentenyl pyrimidine nucleosides 76–79 (Figure 8) [51].
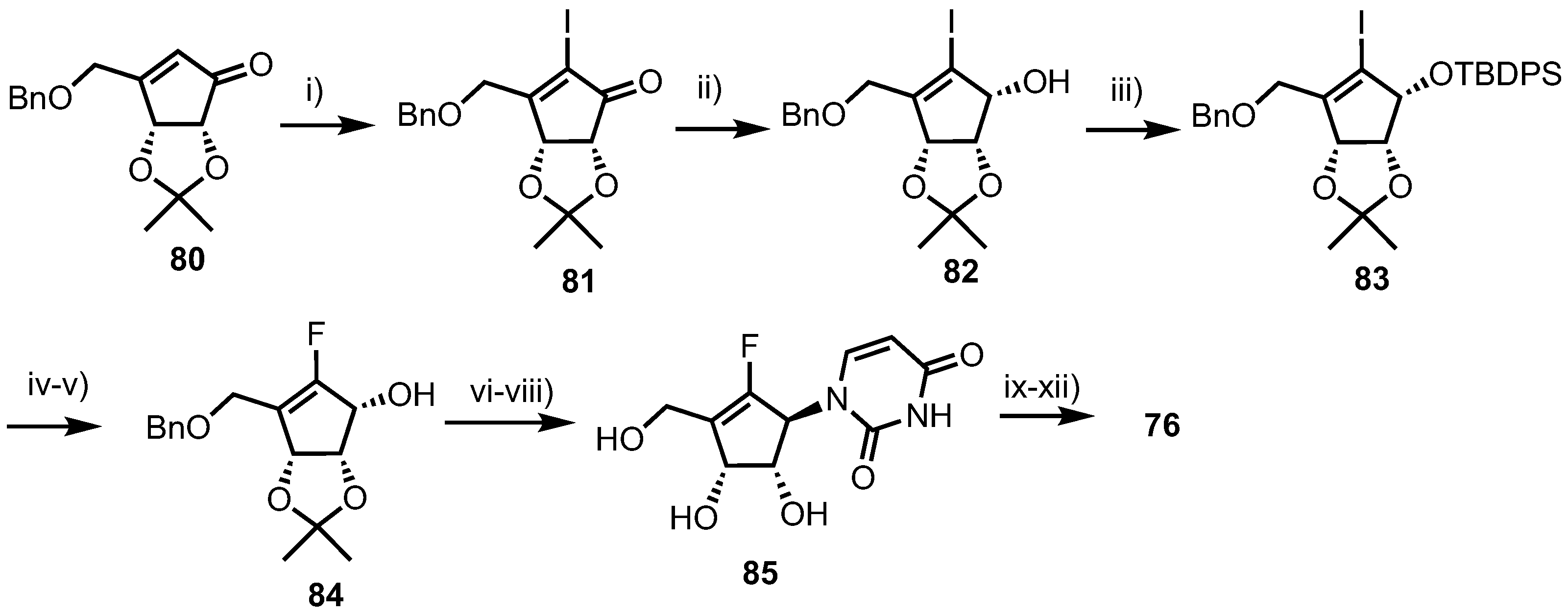
Fluorocyclopentenyl cytosine 76 was synthesised from cyclopentenone 80 (Scheme 10), which was obtained from d-ribose in nine steps using established procedures [52,53]. Iodination of 80 was accomplished by treatment with I2 and pyridine in THF, to give 81. Stereo- and regioselective reduction of 76 was achieved using Luche conditions to give 82, followed by TBDPS protection of the resulting alcohol to deliver 83. Following lithium-halogen exchange of 83 using n-BuLi, electrophilic fluorination was achieved by treatment with N-fluorobenzenesulfonimide (NFSI). Anomeric desilylation with TBAF then gave 84 to which a uracil nucleobase was installed via condensation with N3-benzoyluracil under Mitsunobu conditions. Finally, global deprotection was achieved by sequential treatment with methanolic ammonia and BBr3, providing uracil derivate 85 which was converted to 76 via a standard four-step process.
Of the four nucleoside analogues evaluated (Figure 8), derivative 76 showed significant potency against several human cancer cell lines (Table 3) [51,54,55]. Furthermore, Jeong reported that 76 also showed significant antitumour activity in a nude mouse xenograft model implanted with A549 human lung cancer cells, wherein after 38 days the inhibition of tumour growth was 32% and 58% at 3 and 10 mg/kg doses, respectively [54].
Carbocyclic nucleoside 76 has now been evaluated in more than 100 different cell lines, as well as several xenograft models, showing high potencies against numerous types of cancer, including gemcitabine resistant cell lines [56,57]. Pharmacokinetics and oral bioavailability for 76 were investigated in phase 0 clinical trials, wherein a small cohort of patients were administered a single oral dose (50 mg or 100 mg) of 76, or a single intravenous dose (20 mg, Table 4) [58]. This study found that the absolute bioavailability for 76 was 56% and 33% for 50 and 100 mg doses, respectively, suggesting it not to be dose-proportional. However, t1/2 was found to be 14 h and 21 h for 50 and 100 mg doses, respectively. This may suggest that 76 does exhibit some dose proportionality in some parameters, but not in others and it was noted that this result may be due to the small patient sample size. Analogue 76 is currently in phase II clinical trials for metastatic pancreatic cancer and advanced bladder cancer.
2.4.2. Norbornane-Derived (C2’,C4’-bridged) Carbocyclic Nucleosides
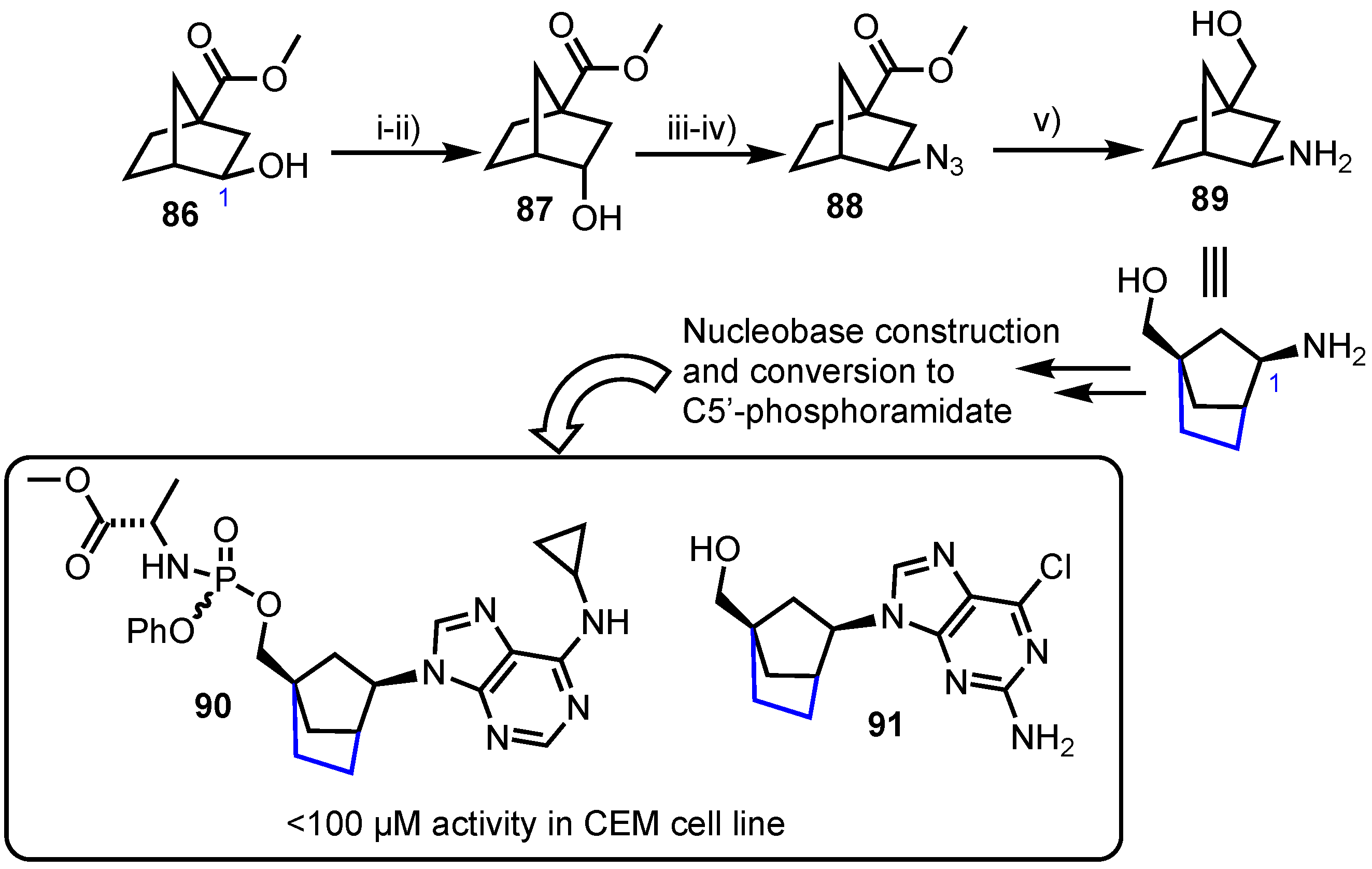
Nencka and colleagues reported the synthesis of norborane, C2’,C4’-bridged carbocyclic nucleosides [59], with a hypothesis of locking the nucleoside analogue in the 2’-exo or North conformation which has garnered attention in the field of carbocyclic antiviral nucleoside analogue development [60]. To access these compounds their synthesis started from known hydroxy ester 86 which was stereochemically inverted at the C1 position via an oxidation-reduction sequence to deliver 87 (Scheme 11). This enabled the desired stereochemistry to be attained at C1 when subsequently inserting an azide by nucleophilic displacement of a mesylate to afford 88 which was reduced to give 89. With scaffold 89 in hand, the group then elaborated the amine at C1 to several purine and pyrimidine nucleobase forms (including C6 purine analogues) and evaluated their cytotoxic potential. From this series, derivatives 90 and 91 showed IC50 activities below 100 µm in human T-lymphocyte (CEM) cells (IC50 = 88 µM for 90 and IC50 = 78 µM for 91), but no significant activity was observed in murine leukemia (L1210) or HeLa cell lines.
2.4.3. C3’,C5’-Bridged Carbocyclic L-Nucleosides
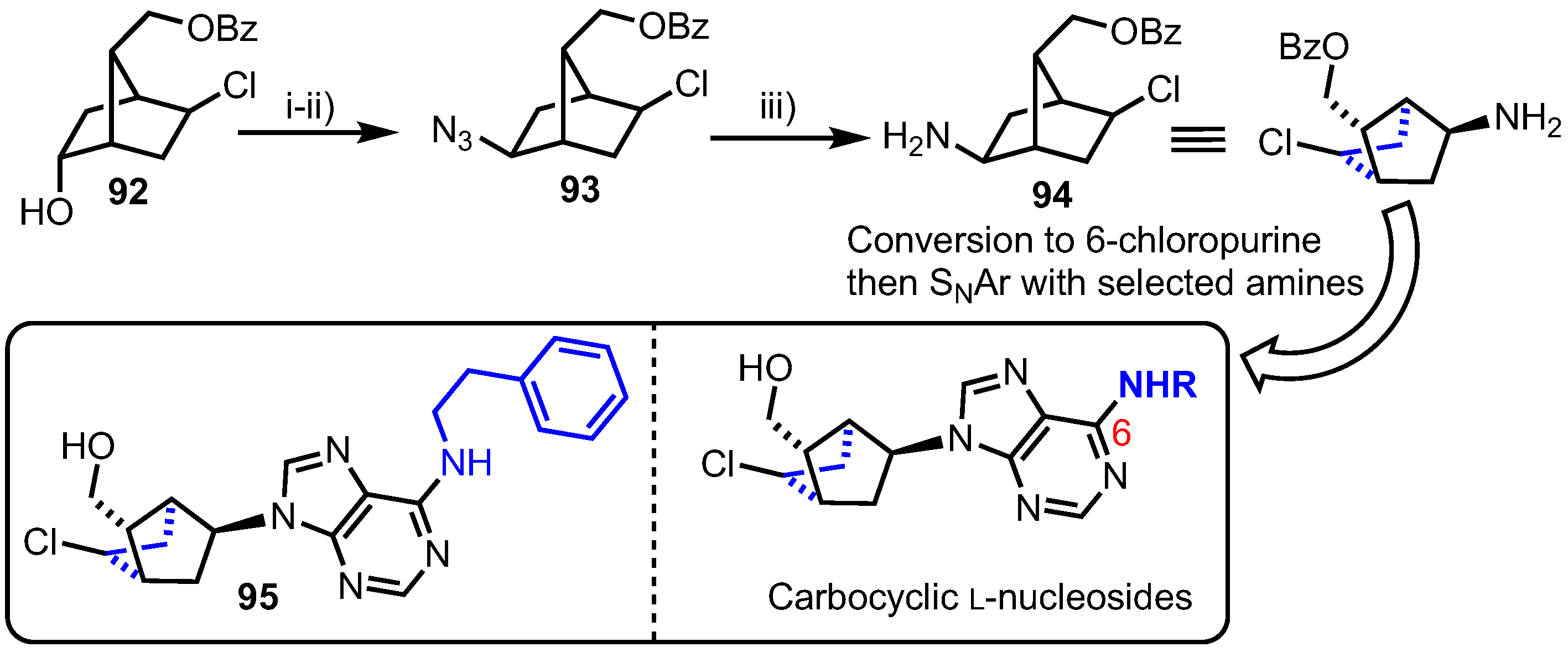
Tănase and colleagues reported the synthesis of an alternative carbocyclic system based on a similar bicyclo[2.2.1]heptane fragment, accessing a small series of 3’-5’-linked L-nucleoside analogues containing C6-amino modifications (Scheme 12) [61]. Their synthesis started from known alcohol 92 and proceeded through azide incorporation (to give 93) and reduction steps in good yields to deliver amine 94. The 6-chloropurine ring was then introduced using standard methods and elaborated at C6 with a series of amines via nucleophilic aromatic substitution. This small library was then screened in vitro at a single high dose (10−5 M) in the full NCI 58 human tumor cell screen panel. Phenethylamine derivative 95 exhibited growth inhibition of 74% on SK-MEL-5 melanoma and UO-31 renal cancer cell lines, but was not deemed sufficiently cytotoxic for studies to proceed further. The group followed up this report with further synthesis of 6-position carbocyclic analogues [62], derived from 94, noting that a 6-(4-methoxy-phenethyl)amino group was active, but again the analogue was not progressed further.
3. 2’-, 3’- and 5’-Furanose Ring Modifications
3.1. 2’-Furanose Modifications
Clofarabine 2 (Figure 1) is a purine nucleoside analogue which, in its corresponding 5’-O-triphosphate form, inhibits both ribonucleotide reductase and DNA polymerases and prevents effective DNA synthesis. Ultimately, this leads to cell apoptosis, particularly in rapidly proliferating and dormant cancer cells. The nucleoside analogue exhibits excellent cytotoxic activity in vitro, with an IC50 range of 0.028–0.29 μM across a variety of solid tumour and leukaemia cell lines [63], alongside substantial tissue distribution and a half-life of at least 24 h for the active triphosphate metabolite [64]. The use of 2 for treatment of paediatric patients with relapsed or refractory acute lymphoblastic leukaemia was approved by the food and drug administration (FDA) in 2004, the first nucleoside analogue of its kind to be approved in over a decade [65].
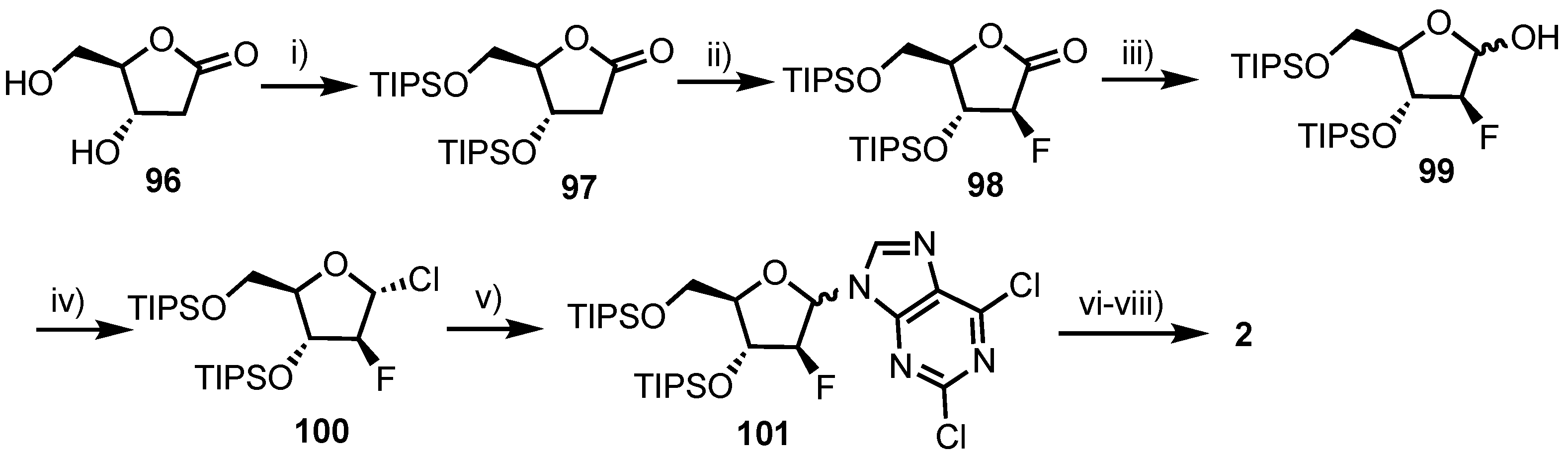
In 2010, Sauve and colleagues reported an improved, stereoselective synthesis of 2 [66], in an overall yield of 38% (Scheme 13). This compared favourably to a prior report by ILEX Products Inc., which detailed an overall yield of 14% in six steps, starting from a fully protected ribose derivative. Sauve’s work began from commercially available lactone 96 with 3,5-O protection using TIPS affording 97, which was then diastereoselectively fluorinated at the 2-position, obtaining the arabino-configured 98 exclusively in 72% yield. Conversion to the anomeric chloride (via hemi-acetal 99) delivered 100 which was condensed with 2,6-dichloropurine to afforded 101 in 66% yield over two steps and a β/α ratio of 3.5:1. Access to 2 was gained following diastereomeric separation of 101, 4-position ammonolysis and deprotection.
3.2. 2’-O,4-C’-Bridged Nucleosides
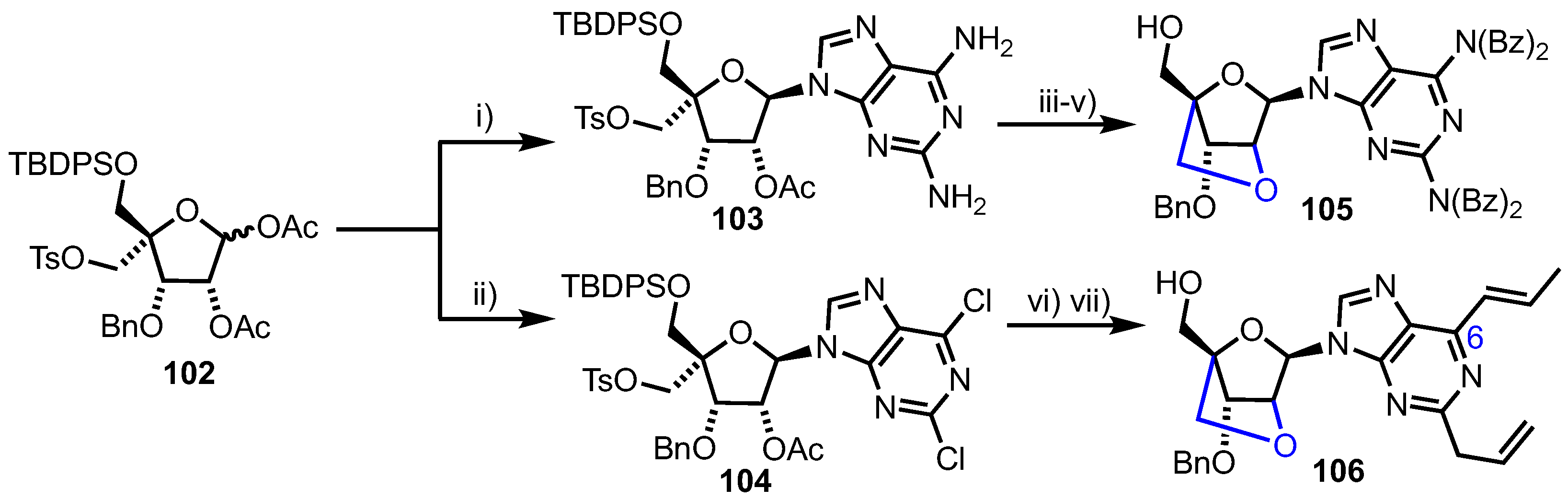
In 2011, Nicolaou and colleagues reported the synthesis and evaluation of a small library of 2’,4’- and 3’,4’-bridged nucleoside analogues, presenting conformationally restrained 3’-endo (North) and 2’-endo (South) systems [67]. These compounds were evaluated for their antiviral, antitumour and antibacterial properties, with 2’,4’-bridged purines 105 and 106 showing µM inhibitory activity against CEM (IC50 = 0.36 µM for 105 and 7.6 µM for 106) and Raji (IC50 = 0.25 µM for 105 and 5.8 µM for 106) cancer cell lines.
The synthesis of 105 and 106 derived from a common 4-disubstituted acetate 102, available using established chemistry from diacetone-d-glucose (Scheme 14) [68,69]. The group used Vorbrüggen glycosylation of either 2,6-diaminopurine or 2,6-dicholoro-9H-purine to afford 103 or 104 respectively, with the substitution proceeding in good yields (100% for 103 and 64% for 104), but noting a requirement to control the amount of N,O-bis(trimethylsilyl)acetamide (BSA) used to silylate the purine to 2.5 equivalents. 2’,4’-bridged compound 105 was constructed via base mediated cyclisation between C2’ and C4’, followed by per-benzoylation of purine nitrogen and silicon protecting group removal.
To access allyl substituted 2’,4’-system, Stille couplings were completed at positions 2 and 6 of the purine, followed by a similar base-mediated C2’ deacetylation and intramolecular cyclisation to afford, after C5’-protecting group removal, 106. Isomerisation of the allyl group at C6 of the purine ring was observed during the palladium and base-mediated cyclisation steps. The antitumour activities of 105 and 106 were considerably lower than for the known anticancer nucleoside cladribine (CEM IC50 = 0.5 nM and Raji IC50 = 9.0 nM), as was the activity (IC50 >10 µM) of the 2’4’-bridged analogue of cladribine, suggesting that the inclusion of this conformational restraint or the addition of an extra CH2O unit was enough to remove antitumour properties.
3.3. 3’-Modified Nucleosides
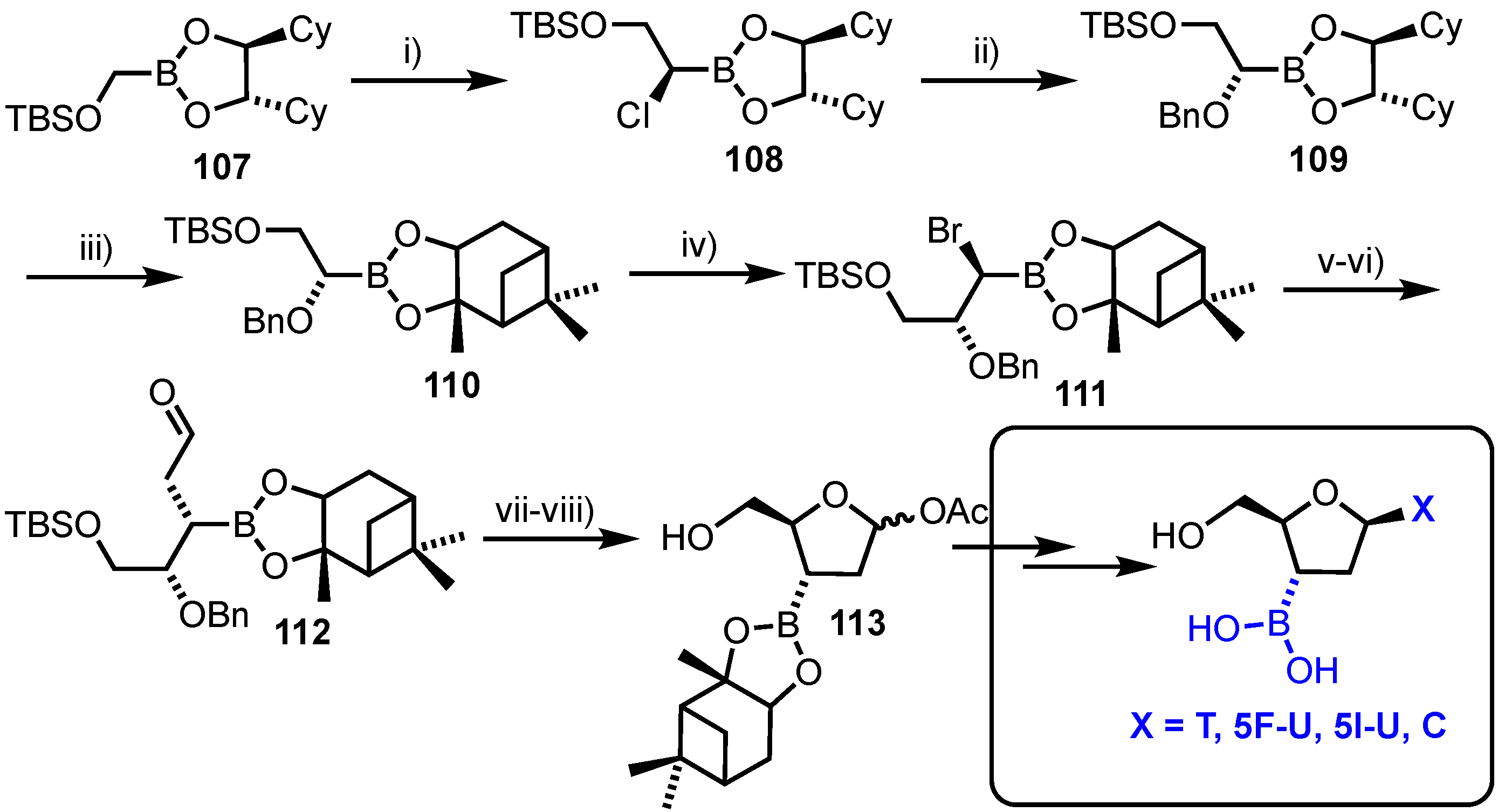
Cheng and co-workers reported an asymmetric synthesis of 2’,3’-dideoxy-3’-boronic acid pyrimidine nucleosides [70]. The group used two highly diastereoselective reactions of chiral boronic esters with (dihalomethyl)lithium reagents to install the ultimate stereochemistry required at C3’ and C4’ (Scheme 15). Starting from (S,S)-1,2-dicyclohexyl-1,2-ethanediol (DICHED) derived boronate ester 107, homologation was completed with (dichloromethyl)lithium to afford 108, the newly formed stereochemistry of which was inverted to give boronate ester 109. The chiral auxiliary component was next switched from DICHED to a pinanediol (derived from (+)-pinene), delivering 110. A second diastereoselective homologation was completed with (dibromomethyl)lithium, affording bromo boronic ester 111. Nucleophilic displacement and inversion of this bromide with allylmagnesium bromide followed by oxidative cleavage of the alkene afforded aldehyde 112. Hydrogenolysis of the C4-O-Bn group in 112, concomitant cyclisation and anomeric acetylation delivered the final 2-deoxy-ribo configured scaffold 113. This was divergently converted to a series of pyrimidine containing nucleoside analogues containing a unique C3’-boronic acid. Unfortunately, biological evaluation of these compounds demonstrated no significant cytotoxicity towards the HepG2 cell line and all derivatives were observed to undergo gradual hydrolytic cleavage under biological conditions.
More recently, Borbas and colleagues reported a small library of 3’-deoxy-3’-thio substituted xylofuranosyl pyrimidines [71]. Utilising a photoinduced thiol-ene reaction the workers were able to effect addition of several different thiols to an appropriately protected 3’-exomethylene ribopyrimidine system. This afforded d-xylo configured products in high diastereomeric excess which were shown to have cytostatic activity in the low micromolar range.
3.4. C5′-N-Cyclopropylcarboxamido-C6-amino-C2-alkynylated Analogues
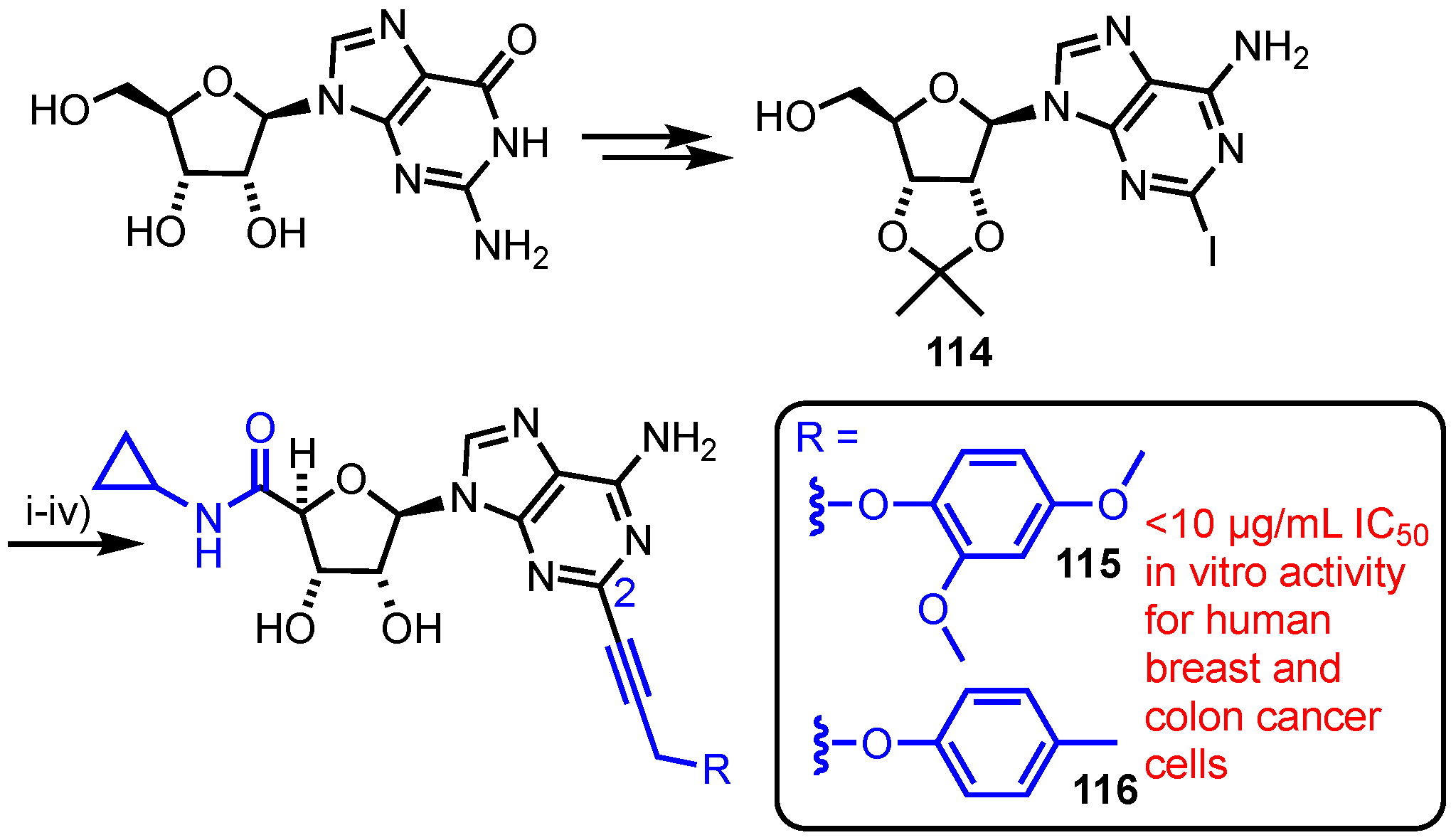
In 2017, Mohan et al. synthesised and evaluated a series of C5′-N-cyclopropylcarboxamido-C6-amino-C2-alkynylated purine nucleoside analogues (Scheme 16) [72]. Starting from guanosine, the group accessed C2-aryl iodide derivative 114 which was oxidised at C5’ using KMnO4, followed by amide coupling to install the C5′-N-cyclopropylcarboxamido group. Following 2’,3’-O-acetonide removal, the C2-iodide underwent a series of divergent Sonogoshira couplings to deliver a library of seven purine analogues. From this library, compounds 115 and 116 showed in vitro cytotoxic effects comparable to doxorubicin against human breast (MDA-MB-2312) and human colon (Caco2) cell lines.
3.5. 5′-β-Hydroxyphosphonate Analogues
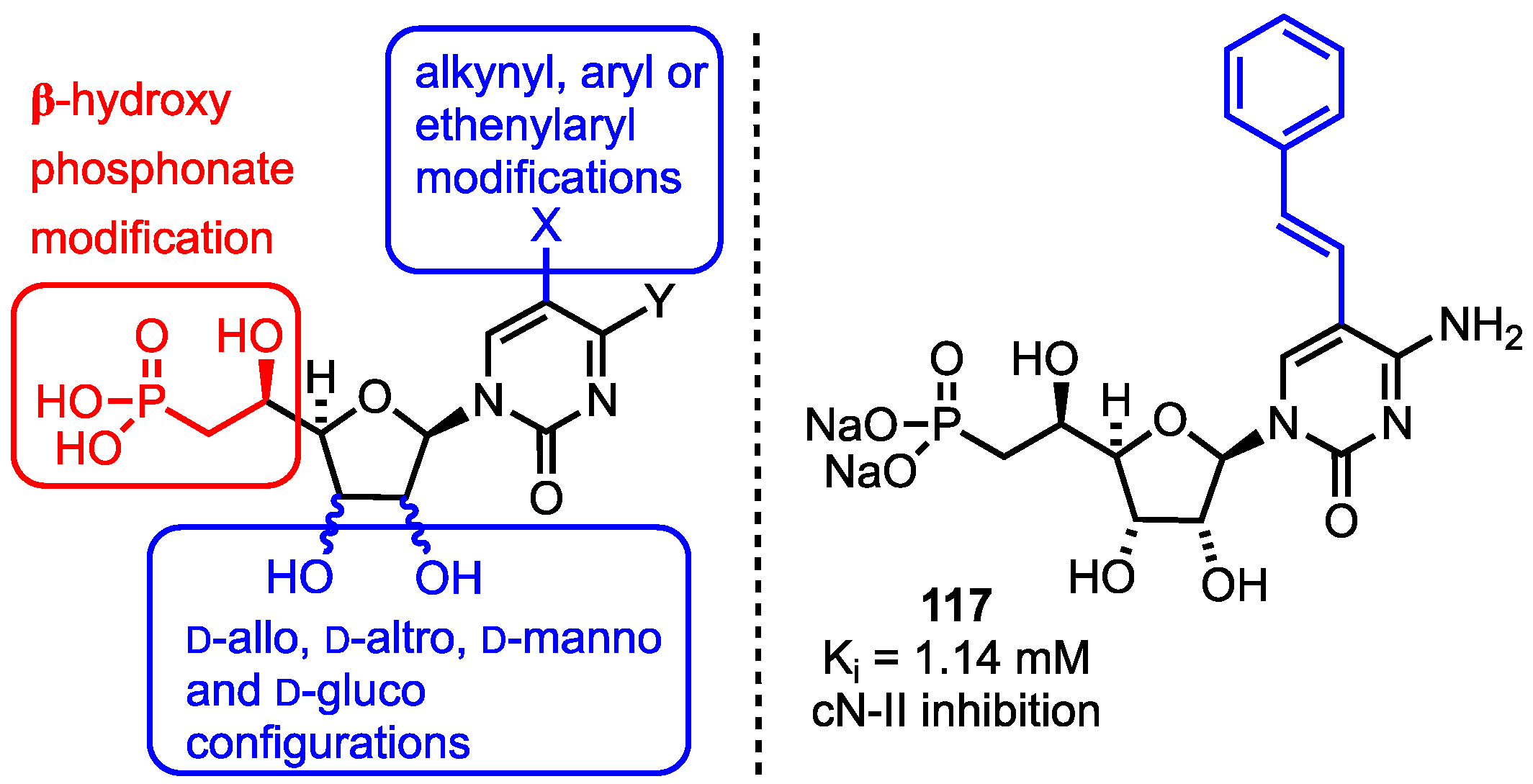
In 2014, Peyrottes and co-workers reported their synthesis of a series of β-hydroxyphosphonate nucleosides, targeting 2’ and 3’ hydroxyl group stereochemistry changes and the introduction of 5-position substituents to the nucleobase (Figure 9) [73]. This report built upon the group’s earlier work developing this class of compound as 5’-nucleotidase inhibitors (specifically the cytosolic cN-II 5’-nucleotidase II) [74]. These enzymes catabolise nucleoside 5’-monophosphates and their expression level is of crucial interest for patients undergoing nucleoside analogue chemotherapy, with a higher expression level often associated with a worsened clinical outcome [75].
The group completed the synthesis of a library of 32 β-hydroxyphosphonate analogues which included d-allo, d-altro, d-manno and d-gluco configurations of the furanose 2’- and 3’-positions along with C2’-C3’ ring opened derivatives. They also installed alkynyl, aryl or ethenylaryl groups at C5 of the cytidine or uridine nucleobase using transition-metal cross-couplings of the corresponding C5-iodide. Biological activity data was obtained using recombinant, purified cN-II with inosine monophosphate as substrate. From the resultant SAR study, cytosine-based analogues were generally more active than their uracil counterparts, but the configurational isomers at C2’ or C3’ were generally less active than the parent compounds (allofuranose). Modification of the nucleobase was generally well tolerated with an observed Ki value of 1.14 mM for phenylethenyl substituted derivative 117 (Figure 9), noting that high substrate concentrations (in the mM range) are required for activity with cN-II.
3.6. Ferronucleosides
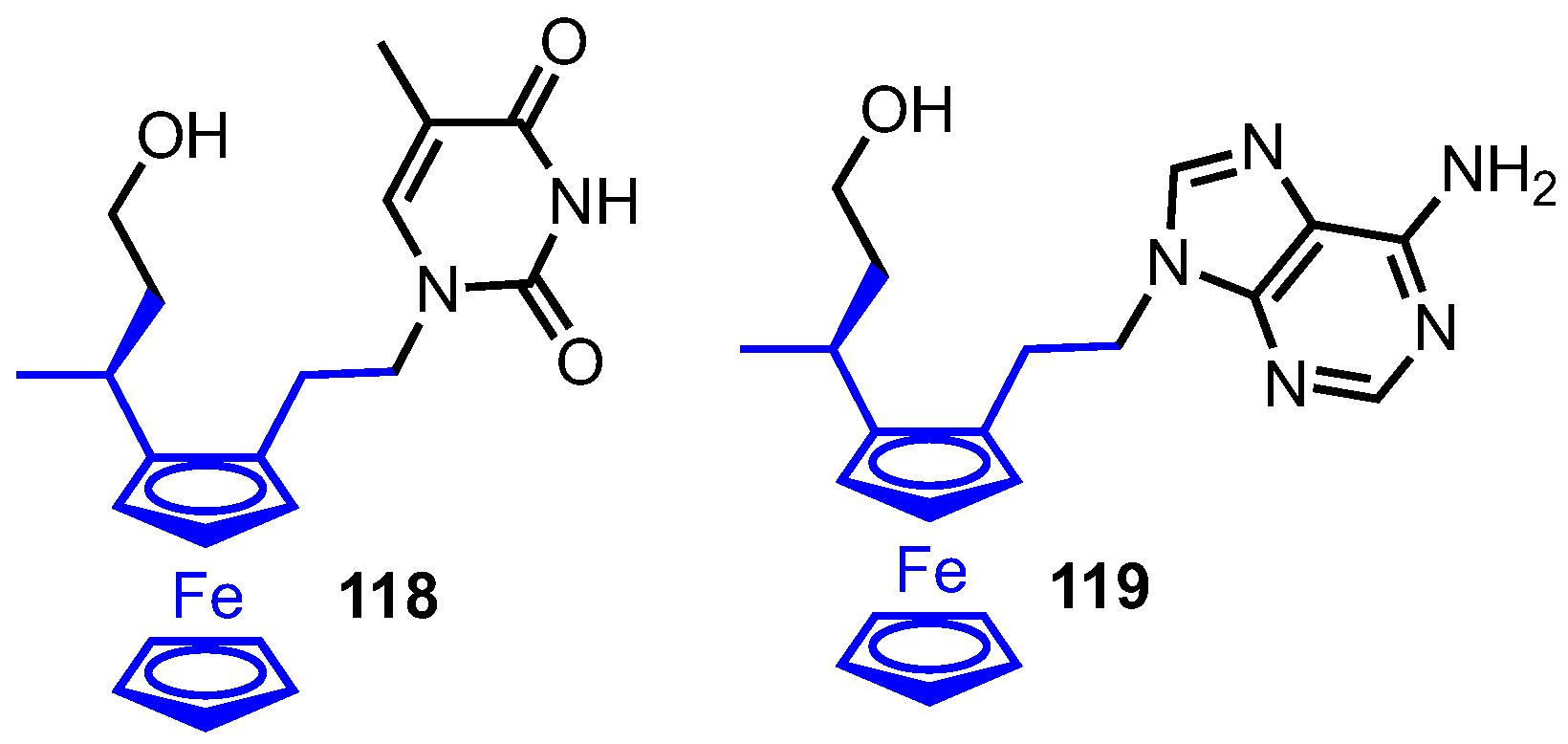
Tucker and colleagues recently described their synthesis of ferronucleosides, an important new development in the field of medicinal bioorganometallic chemistry [76]. Here the furanose ring was exchanged for the five-membered cyclopentadienyl ring of a ferrocene unit, but retained the nucleobase and hydroxymethyl group as key components appended in a 2,3 relationship to the ferrocene core (compounds 118 and 119, Figure 10). Using thymine or adenine as the nucleobase, the compounds were tested with a control series (where the hydroxymethyl or base component were absent) for their cytostatic activity and compared to established chemotherapeutic agents 5-fluorouracil (5F-U) and cisplatin. In a proliferation activity assay on three tumour cell lines (L1210, HeLa and CEM), both 118 and 119 had activities in the low µM range, with 118 and 119 20 to 50-fold more active than 5F-U in CEM cell cultures (0.9 µM for 118, 0.35 µM for 119 versus 18 µM for 5F-U). The compounds also performed promisingly in cell growth inhibition (oesophageal cancer cell line) and cellular viability (MTT assay) studies, with the data indicating that both functional groups appended to the ferrocene component were required for optimum cytostatic activity.
4. Nucleoside Analogue Prodrugs
4.1. Phosphorodiamidate Prodrugs
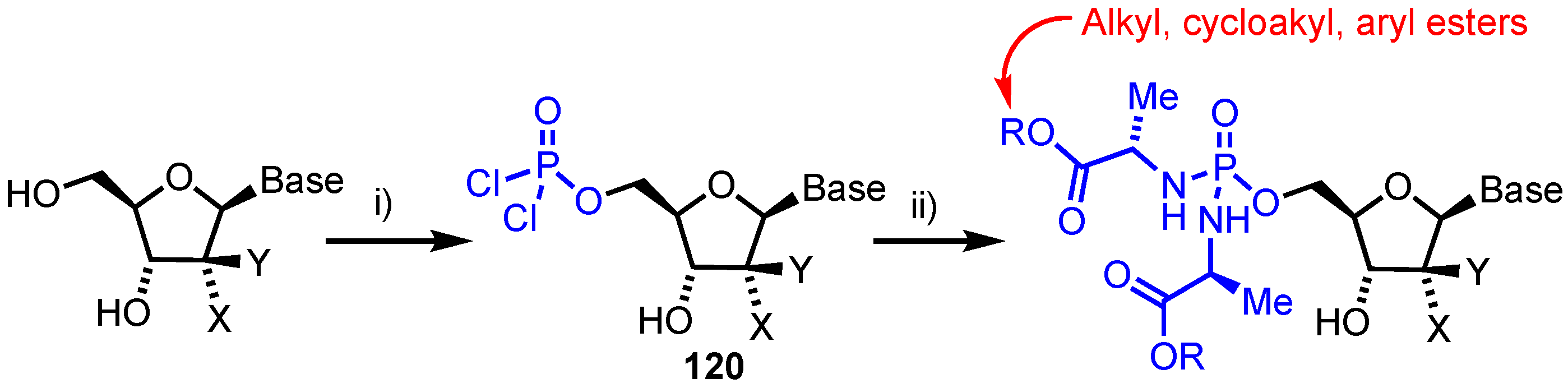
In 2018, Slusarczyk’s team reported the synthesis of phosphorodiamidates for a series of established anticancer nucleoside analogues [77]. This was envisioned following the successful ProTide technology developed by McGuigan [78], to deliver nucleoside monophosphate analogues into cells and recent reports of phosphorothioamidate systems [79]. A lack of chirality at phosphorous in a phosphorodiamidate was envisioned to confer advantages in not having to resolve diastereomeric mixtures, often a requirement in earlier generations of ProTides, as SP and RP diastereoisomers exhibited markedly different biological activities [80]. Accordingly, seven different anticancer nucleosides (1, FUdR, 8-chloroadenosine, fludarabine, AraG, thioinosine and thioguanosine) were converted to their phosphorodiamidate form using a one-pot, two-stage strategy (Scheme 17). First the 5’-OH was converted to a phosphorodichloridate intermediate 120, followed by double phosphoramidation using an appropriate amino acid ester salt. The panel of analogues were then evaluated in vitro against a wide range of solid tumour and haematological cell lines with the potential for this approach confirmed for FUdR and 8-chloroadenosine, where similar or improved inhibitory activities compared to the parent nucleosides were observed. FUdR and its phosphorodiamidate prodrug showed activity in the sub-micromolar range with IC50 values of 0.0046–0.073 µM for FUdR and 0.01–0.40 µM for the FUdR phosphorodiamidates against the wild-type cell lines. Also of note was the inactivity of arabino configured analogues, suggesting conversion to the monophosphate might be prevented through their being poor substrates for phosphoramidase activity [81]. Enzymatic studies were undertaken to investigate the bioactivation pathway of this class of nucleoside prodrug with a carboxypeptidase Y assay and 31P-NMR confirming the hydrolysis of both esters followed by loss of one phosphoramidate group.
4.2. Vitamin E Phosphate Prodrugs
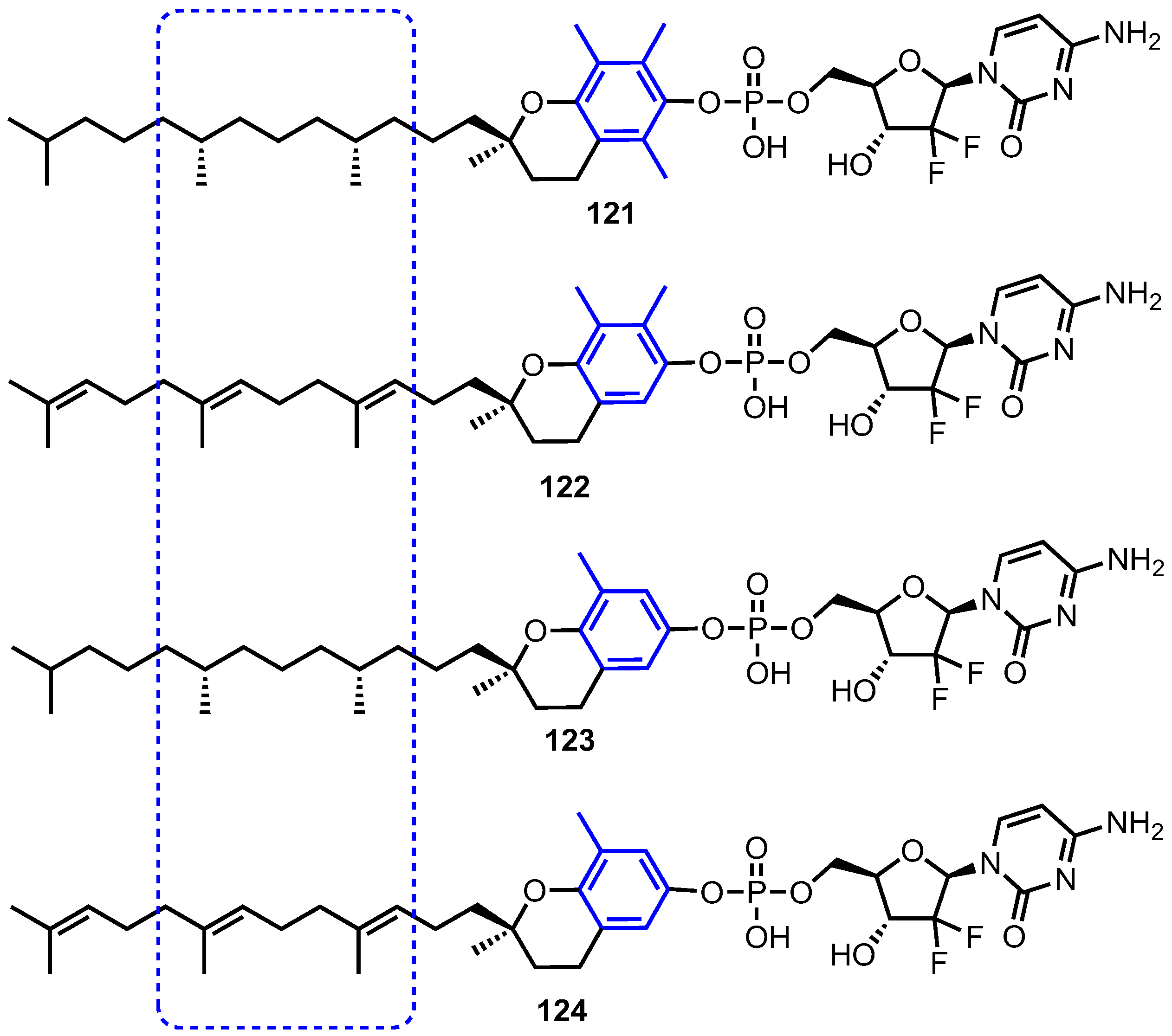
Nucleoside analogue therapy can suffer from inducive and constitutive resistance, which limits the efficacy of the treatment. Isoforms of vitamin E, in particular δ-tocopherol and tocotrienols have displayed anticancer activity. As such, vitamin E phosphate nucleoside analogue prodrugs were envisaged to combat two mechanisms of resistance: i) downregulation of metabolic kinases (deoxycytidine kinase, dCK, in the case of 1) and ii) nucleoside transport [82,83]. Accordingly, Daifuku and co-workers synthesised and evaluated four isoforms of vitamin E conjugated to 1 (Figure 11, compounds 121–124) [84].
The in vitro GI50 of 1, vitamin E phosphate (VEP) isoforms and compounds 121–124 showed these VEP-gemcitabine prodrugs to exhibit anticancer activity, consistent with their catabolism to vitamin E and gemcitabine monophosphate. Conjugate 122 displayed the best activity with GI50 values <5 μM in breast MDA (MB-231), non-small cell lung (NCI-H460) and colon (HCT-116) cell lines. The authors suggested this was due to steric hindrance, from the methyl groups proximal to the vitamin E-phosphate bond, reducing the rate of enzymatic cleavage to the monophosphate counterpart.
Prodrugs 122 and 123 were then evaluated in the presence of an inhibitor of nucleoside transport, dipyridamole (DP, Table 5) with the data indicating that both were largely unaffected by the presence of DP in comparison to 1. This suggested that these prodrugs bypass nucleoside membrane transporters and may thus be beneficial in the treatment of patients with gemcitabine resistant cells.
In particular, as conjugate 123 displayed significant potency against the three DP(-) cell lines and moderate potency against the DP-dosed cell lines (Table 5), it was selected to further compare activity against 1 in in vitro wild-type leukemic CEM cells and CEM cells deficient in dCK. In dCK(−) cells 1 is not phosphorylated, with GI50 values increasing from 0.002 μM in wild-type to 124.5 μM in dCK(−). For 123 the GI50 increase was significantly lower than that of 1 (from 0.59 μM to 19.2 μM). Furthermore, the half-life of 123 in mice was shown to be 4 h, a 13.9-fold increase to that of 1 (0.3 h [85]). Overall, this study showed an interesting proof of concept for VEP-1 prodrugs against resistant cancer cell lines. However, further optimisation will be required to obtain promising candidate compounds with increased potencies.
5. Conclusions
Nucleoside analogues are an historically accomplished class of drugs with highly diversifiable scaffolds and proven potential to treat a wide range of cancer cell types. Recent synthetic trends have focused on furanose oxygen substitution with heteroatoms or carbon and fluorocyclopentenyl cytosine is proving a promising new clinical candidate in this regard. Alongside this, examples of templating such modifications onto established nucleoside analogue scaffolds, such as Ara-C and forodesine, are emerging. As further pharmacokinetic and pharmacodynamic parameters are evaluated for these architectures, their pharmaceutical utility will be established. Finally, the continued advancement of prodrug strategies to deliver these compounds more effectively and provide options to treat drug-resistant cancer cell types sets an exciting future for nucleoside analogue chemotherapeutics and the underpinning requirement of chemical synthesis in realising this.
Author Contributions
G.J.M., M.G. and C.B. conceptualised and wrote the manuscript. M.G., C.B., M.S., G.J.M. reviewed the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
Riboscience LLC and Keele University are thanked for PhD scholarship funding to M.G and C.B.
Conflicts of Interest
M.S. is a Founder and owns equity in Riboscience LLC.
References
- Jordheim, L.P.; Durantel, D.; Zoulim, F.; Dumontet, C. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat. Rev. Drug. Discov. 2013, 12, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Clercq, E.D.; Neyts, J. Handbook of Experimental Pharmacology. Handb. Exp. Pharmacol. 2009, 53–84. [Google Scholar]
- Chapman, T.R.; Kinsella, T.J. Ribonucleotide reductase inhibitors: A new look at an old target for radiosensitization. Front. Oncol. 2012, 1, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flotho, C.; Claus, R.; Batz, C.; Schneider, M.; Sandrock, I.; Ihde, S.; Plass, C.; Niemeyer, C.M.; Lübbert, M. The DNA methyltransferase inhibitors azacitidine, decitabine and zebularine exert differential effects on cancer gene expression in acute myeloid leukemia cells. Leukemia 2009, 23, 1019–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galmarini, C.; Mackey, J.; Dumontet, C. Nucleoside analogues: Mechanisms of drug resistance and reversal strategies. Leukemia 2001, 15, 875–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordheim, L.; Dumontet, C. Review of recent studies on resistance to cytotoxic deoxynucleoside analogues. Biochim. Biophys. Acta 2007, 1776, 138–159. [Google Scholar] [CrossRef]
- Yates, M.K.; Seley-Radtke, K.L. The evolution of antiviral nucleoside analogues: A review for chemists and non-chemists. Part II: Complex modifications to the nucleoside scaffold. Antivir. Res. 2018, 162, 5–21. [Google Scholar] [CrossRef]
- Seley-Radtke, K.L.; Yates, M.K. The evolution of nucleoside analogue antivirals: A review for chemists and non-chemists. Part 1: Early structural modifications to the nucleoside scaffold. Antivir. Res. 2018, 154, 66–86. [Google Scholar] [CrossRef]
- Siegel, D.; Hui, H.C.; Doerffler, E.; Clarke, M.O.; Chun, K.; Zhang, L.; Neville, S.; Carra, E.; Lew, W.; Ross, B.; et al. Discovery and Synthesis of a Phosphoramidate Prodrug of a Pyrrolo[2,1-f][triazin-4-amino] Adenine C-Nucleoside (GS-5734) for the Treatment of Ebola and Emerging Viruses. J. Med. Chem. 2017, 60, 1648–1661. [Google Scholar] [CrossRef] [Green Version]
- Warren, T.K.; Jordan, R.; Lo, M.K.; Ray, A.S.; Mackman, R.L.; Soloveva, V.; Siegel, D.; Perron, M.; Bannister, R.; Hui, H.C.; et al. Therapeutic efficacy of the small molecule GS-5734 against Ebola virus in rhesus monkeys. Nature 2016, 531, 381–385. [Google Scholar] [CrossRef]
- Sheahan, T.P.; Sims, A.C.; Leist, S.R.; Schäfer, A.; Won, J.; Brown, A.J.; Montgomery, S.A.; Hogg, A.; Babusis, D.; Clarke, M.O.; et al. Comparative therapeutic efficacy of remdesivir and combination lopinavir, ritonavir, and interferon beta against MERS-CoV. Nat. Commun. 2020, 11, 222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, A.; Saunders, O.L.; Butler, T.; Zhang, L.; Xu, J.; Vela, J.E.; Feng, J.Y.; Ray, A.S.; Kim, C.U. Synthesis and antiviral activity of a series of 1′-substituted 4-aza-7,9-dideazaadenosine C-nucleosides. Bioorg. Med. Chem. Lett. 2012, 22, 2705–2707. [Google Scholar] [CrossRef] [PubMed]
- Serpi, M.; Ferrari, V.; Pertusati, F. Nucleoside Derived Antibiotics to Fight Microbial Drug Resistance: New Utilities for an Established Class of Drugs? J. Med. Chem. 2016, 59, 10343–10382. [Google Scholar] [CrossRef] [PubMed]
- Romeo, G.; Chiacchio, U.; Corsaro, A.; Merino, P. Chemical Synthesis of Heterocyclic−Sugar Nucleoside Analogues. Chem. Rev. 2010, 110, 3337–3370. [Google Scholar] [CrossRef] [PubMed]
- Roy, B.; Depaix, A.; Peérigaud, C.; Peyrottes, S. Recent Trends in Nucleotide Synthesis. Chem. Rev. 2016, 116, 7854–7897. [Google Scholar] [CrossRef]
- Kicsák, M.; Mándi, A.; Varga, S.; Herczeg, M.; Batta, G.; Bényei, A.; Borbás, A.; Herczegh, P. Tricyclanos: Conformationally constrained nucleoside analogues with a new heterotricycle obtained from a d-ribofuranose unit. Org. Biomol. Chem. 2018, 16, 393–401. [Google Scholar] [CrossRef] [Green Version]
- Hernández, D.; Boto, A. Nucleoside Analogues: Synthesis and Biological Properties of Azanucleoside Derivatives. Eur. J. Org. Chem. 2014, 2014, 2201–2220. [Google Scholar] [CrossRef]
- Quintás-Cardama, A.; Santos, F.P.S.; Garcia-Manero, G. Therapy with azanucleosides for myelodysplastic syndromes. Nat. Rev. Clin. Oncol. 2010, 7, 433–444. [Google Scholar] [CrossRef]
- Bouton, J.; Hecke, K.V.; Calenbergh, S.V. Efficient diastereoselective synthesis of a new class of azanucleosides: 2′-homoazanucleosides. Tetrahedron 2017, 73, 4307–4316. [Google Scholar] [CrossRef] [Green Version]
- Bantia, S.; Miller, P.J.; Parker, C.D.; Ananth, S.L.; Horn, L.L.; Kilpatrick, J.M.; Morris, P.E.; Hutchison, T.L.; Montgomery, J.A.; Sandhu, J.S. Purine nucleoside phosphorylase inhibitor BCX-1777 (Immucillin-H)—A novel potent and orally active immunosuppressive agent. Int. Immunopharmacol. 2001, 1, 1199–1210. [Google Scholar] [CrossRef]
- Miles, R.W.; Tyler, P.C.; Furneaux, R.H.; Bagdassarian, C.K.; Schramm, V.L. One-third-the-sites transition-state inhibitors for purine nucleoside phosphorylase. Biochemistry 1998, 37, 8615–8621. [Google Scholar] [CrossRef] [PubMed]
- Makita, S.; Maeshima, A.M.; Maruyama, D.; Izutsu, K.; Tobinai, K. Forodesine in the treatment of relapsed/refractory peripheral T-cell lymphoma: An evidence-based review. Oncotargets Ther. 2018, 11, 2287–2293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Kali, A.; Gandhi, V.; Ayoubi, M.; Keating, M.; Ravindi, F. Forodesine: Review of preclinical and clinical data. Future Oncol. 2010, 6, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Warren, T.K.; Wells, J.; Panchal, R.G.; Stuthman, K.S.; Garza, N.L.; Tongeren, S.A.V.; Dong, L.; Retterer, C.J.; Eaton, B.P.; Pegoraro, G.; et al. Protection against filovirus diseases by a novel broad-spectrum nucleoside analogue BCX4430. Nature 2014, 508, 402–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, G.B.; Furneaux, R.H.; Gainsford, G.J.; Schramm, V.L.; Tyler, P.C. Synthesis of Transition State Analogue Inhibitors for Purine Nucleoside Phosphorylase and N-Riboside Hydrolases. Tetrahedron 2000, 56, 3053–3062. [Google Scholar] [CrossRef]
- Fleet, G.W.J.; Son, J.C. Polyhydroxylated pyrrolidines from sugar lactomes: Synthesis of 1,4-dideoxy-1,4-imino-d-glucitol from d-galactonolactone and syntheses of 1,4-dideoxy-1,4-imino-d-allitol, 1,4-dideoxy-1,4-imino-d-ribitol, and (2s,3r,4s)-3,4-dihydroxyproline from d-gulonolactone. Tetrahedron 1988, 44, 2637–2647. [Google Scholar]
- Kezar, H.S.; Kilpatrick, J.M.; Phillips, D.; Kellogg, D.; Zhang, J.; Morris, P.E. Synthesis and Pharmacokinetic and Pharmacodynamic Evaluation of the Forodesine HCl Analog BCX-3040. Nucleosides Nucleotides Nucleic Acids 2005, 24, 1817–1830. [Google Scholar] [CrossRef]
- Wortmann, R.L.; Andres, C.; Kaminska, J.; Mejias, E.; Gelf, E.; Arnold, W.; Rich, K.; Fox, I.H. Purine Nucleoside Phosphorylase Deficiency. Arthritis Rheum. 1979, 22, 524–531. [Google Scholar] [CrossRef]
- Witczak, Z.J.; Culhane, J.M. Thiosugars: New perspectives regarding availability and potential biochemical and medicinal applications. Appl. Microbiol. Biotechnol. 2005, 69, 237–244. [Google Scholar] [CrossRef]
- Secrist, J.A.; Tiwari, K.N.; Riordan, J.M.; Montgomery, J.A. Synthesis and biological activity of 2’-deoxy-4’-thio pyrimidine nucleosides. J. Med. Chem. 1991, 34, 2361–2366. [Google Scholar] [CrossRef]
- Pejanović, V.; Stokić, Z.; Stojanović, B.; Piperski, V.; Popsavin, M.; Popsavin, V. Synthesis and biological evaluation of some novel 4′-Thio-L-ribonucleosides with modified nucleobase moieties. Bioorg. Med. Chem. Lett. 2003, 13, 1849–1852. [Google Scholar] [CrossRef]
- Zheng, F.; Zhang, X.-H.; Qiu, X.-L.; Zhang, X.; Qing, F.-L. Synthesis of l-β-3‘-Deoxy-3‘,3‘-difluoro-4‘-thionucleosides. Org. Lett. 2006, 8, 6083–6086. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, Y.; Kitano, K.; Yamada, K.; Satoh, H.; Watanabe, M.; Miura, S.; Sakata, S.; Sasaki, T.; Matsuda, A. A Novel Synthesis of 2′-Modified 2′-Deoxy-4′-thiocytidines from D-Glucose. J. Org. Chem. 1997, 62, 3140–3152. [Google Scholar] [CrossRef]
- Yoshimura, Y.; Kitano, K.; Satoh, H.; Watanabe, M.; Miura, S.; Sakata, S.; Sasaki, T.; Matsuda, A. A Novel Synthesis of New Antineoplastic 2‘-Deoxy-2‘-substituted-4‘-thiocytidines. J. Org. Chem. 1996, 61, 822–823. [Google Scholar] [CrossRef]
- Yoshimura, Y.; Saito, Y.; Natori, Y.; Wakamatsu, H. Synthesis of 4′-Thionucleosides as Antitumor and Antiviral Agents. Chem. Pharm. Bull. 2018, 66, 139–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshimura, Y.; Kano, F.; Miyazaki, S.; Ashida, N.; Sakata, S.; Haraguchi, K.; Itoh, Y.; Tanaka, H.; Miyasaka, T. Synthesis and biological evaluation of 1′-C-cyano-pyrimidine nucleosides. Nucleosides Nucleotides 1996, 15, 305–324. [Google Scholar] [CrossRef]
- Thottassery, J.V.; Sambandam, V.; Allan, P.W.; Maddry, J.A.; Maxuitenko, Y.Y.; Tiwari, K.; Hollingshead, M.; Parker, W.B. Novel DNA methyltransferase-1 (DNMT1) depleting anticancer nucleosides, 4′-thio-2′-deoxycytidine and 5-aza-4′-thio-2′-deoxycytidine. Cancer Chemother. Pharmacol. 2014, 74, 291–302. [Google Scholar] [CrossRef] [Green Version]
- Haraguchi, K.; Kumamoto, H.; Konno, K.; Yagi, H.; Tatano, Y.; Odanaka, Y.; Matsubayashi, S.S.; Snoeck, R.; Andrei, G. Synthesis of 4′-substituted 2′-deoxy-4′-thiocytidines and its evaluation for antineoplastic and antiviral activities. Tetrahedron 2019, 75, 4542–4555. [Google Scholar] [CrossRef]
- Haraguchi, K.; Takahashi, H.; Shiina, N.; Horii, C.; Yoshimura, Y.; Nishikawa, A.; Sasakura, E.; Nakamura, K.T.; Tanaka, H. Stereoselective Synthesis of the β-Anomer of 4‘-Thionucleosides Based on Electrophilic Glycosidation to 4-Thiofuranoid Glycals. J. Org. Chem. 2002, 67, 5919–5927. [Google Scholar] [CrossRef]
- Jayakanthan, K.; Johnston, B.D.; Pinto, B.M. Stereoselective synthesis of 4′-selenonucleosides using the Pummerer glycosylation reaction. Carbohydr. Res. 2008, 343, 1790–1800. [Google Scholar] [CrossRef]
- Haraguchi, K.; Shimada, H.; Kimura, K.; Akutsu, G.; Tanaka, H.; Abe, H.; Hamasaki, T.; Baba, M.; Gullen, E.A.; Dutschman, G.E.; et al. Synthesis of 4′-Ethynyl-2′-deoxy-4′-thioribonucleosides and Discovery of a Highly Potent and Less Toxic NRTI. ACS. Med. Chem. Lett. 2011, 2, 692–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, L.S.; Tosh, D.K.; Kim, H.O.; Wang, T.; Hou, X.; Yun, H.S.; Kwon, Y.; Lee, S.K.; Choi, J.; Zhao, L.X. First Synthesis of 4‘-Selenonucleosides Showing Unusual Southern Conformation. Org. Lett. 2008, 10, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Varela, O.; Zunszain, P.A. First synthesis of aldopentono-1,4-thiolactones. J. Org. Chem. 1993, 58, 7860–7864. [Google Scholar] [CrossRef]
- Jeong, L.S.; Tosh, D.K.; Choi, W.J.; Lee, S.K.; Kang, Y.-J.; Choi, S.; Lee, J.H.; Lee, H.; Lee, H.W.; Kim, H.O. Discovery of a New Template for Anticancer Agents: 2′-deoxy-2′-fluoro-4′-selenoarabinofuranosyl-cytosine (2′-F-4′-Seleno-ara-C). J. Med. Chem. 2009, 52, 5303–5306. [Google Scholar] [CrossRef]
- Kim, J.-H.; Yu, J.; Alexander, V.; Choi, J.H.; Song, J.; Lee, H.W.; Kim, H.O.; Choi, J.; Lee, S.K.; Jeong, L.S. Structure–activity relationships of 2′-modified-4′-selenoarabinofuranosyl-pyrimidines as anticancer agents. Eur. J. Med. Chem. 2014, 83, 208–225. [Google Scholar] [CrossRef] [PubMed]
- Alexander, V.; Song, J.; Yu, J.; Choi, J.H.; Kim, J.-H.; Lee, S.K.; Choi, W.J.; Jeong, L.S. Synthesis and biological evaluation of 2’-substituted-4’-selenoribofuranosyl pyrimidines as antitumor agents. Arch. Pharm. Sci. Res. 2014, 38, 966–972. [Google Scholar] [CrossRef]
- Marquez, V.E. Advances in Antiviral Drug Design. In Advances in Antiviral Drug Design; Clercq, E.D., Ed.; Elsevier: Amsterdam, The Netherlands, 1996; pp. 89–146. [Google Scholar]
- Marquez, V.E.; Lim, M.I.; Treanor, S.P.; Plowman, J.; Priest, M.A.; Markovac, A.; Khan, M.S.; Kaskar, B.; Driscoll, J.S. Cyclopentenylcytosine. A carbocyclic nucleoside with antitumor and antiviral properties. J. Med. Chem. 1988, 31, 1687–1694. [Google Scholar] [CrossRef]
- Cavaliere, A.; Probst, K.C.; Westwell, A.D.; Slusarczyk, M. Fluorinated nucleosides as an important class of anticancer and antiviral agents. Future Med. Chem. 2017, 9, 1809–1833. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, X.; Wang, Q.; Zhang, Y.; Jiang, J.; Guo, X.; Fan, Q.; Zheng, L.; Yu, X.; Wang, N.; et al. FNC, a novel nucleoside analogue inhibits cell proliferation and tumor growth in a variety of human cancer cells. Biochem. Pharmacol. 2011, 81, 848–855. [Google Scholar] [CrossRef]
- Jeong, L.S.; Zhao, L.X.; Choi, W.J.; Pal, S.; Park, Y.H.; Lee, S.K.; Chun, M.W.; Lee, Y.B.; Ahn, C.H.; Moon, H.R. Synthesis and Antitumor Activity of Fluorocyclopentenyl-Pyrimidines. Nucleosides Nucleotides Nucleic Acids 2007, 26, 713–716. [Google Scholar] [CrossRef]
- Moon, H.R.; Choi, W.J.; Kim, H.O.; Jeong, L.S. Improved and alternative synthesis of d- and l-cyclopentenone derivatives, the versatile intermediates for the synthesis of carbocyclic nucleosides. Tet. Asymm. 2002, 13, 1189–1193. [Google Scholar] [CrossRef]
- Choi, W.J.; Moon, H.R.; Kim, H.O.; Yoo, B.N.; Lee, J.A.; Shin, D.H.; Jeong, L.S. Preparative and Stereoselective Synthesis of the Versatile Intermediate for Carbocyclic Nucleosides: Effects of the Bulky Protecting Groups to Enforce Facial Selectivity. J. Org. Chem. 2004, 69, 2634–2636. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.J.; Chung, H.-J.; Chandra, G.; Alexander, V.; Zhao, L.X.; Lee, H.W.; Nayak, A.; Majik, M.S.; Kim, H.O.; Kim, J.-H.; et al. Fluorocyclopentenyl-cytosine with Broad Spectrum and Potent Antitumor Activity. J. Med. Chem. 2012, 55, 4521–4525. [Google Scholar] [CrossRef] [PubMed]
- Balboni, B.; Hassouni, B.E.; Honeywell, R.J.; Sarkisjan, D.; Giovannetti, E.; Poore, J.; Heaton, C.; Peterson, C.; Benaim, E.; Lee, Y.B.; et al. RX-3117 (fluorocyclopentenyl cytosine): A novel specific antimetabolite for selective cancer treatment. Expert. Opin. Inv. Drug. 2019, 28, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Peters, G.J.; Smid, K.; Vecchi, L.; Kathmann, I.; Sarkisjan, D.; Honeywell, R.J.; Losekoot, N.; Ohne, O.; Orbach, A.; Blaugrund, E.; et al. Metabolism, mechanism of action and sensitivity profile of fluorocyclopentenylcytosine (RX-3117; TV-1360). Investig. New Drugs 2013, 31, 1444–1457. [Google Scholar] [CrossRef]
- Yang, M.Y.; Lee, Y.B.; Ahn, C.-H.; Kaye, J.; Fine, T.; Kashi, R.; Ohne, O.; Smid, K.; Peters, G.J.; Kim, D.J. A novel cytidine analog, RX-3117, shows potent efficacy in xenograft models, even in tumors that are resistant to gemcitabine. Anticancer Res. 2014, 34, 6951–6959. [Google Scholar]
- Udvaros, I.; Rethy, A.; Lang, I.; Hitre, E.; Peterson, C. A phase 1 exploratory study of RX-3117 to determine oral bioavailability in cancer subjects with solid tumors. J. Clin. Oncol. 2015, 33, e13545. [Google Scholar] [CrossRef]
- Dejmek, M.; Šála, M.; Hřebabecký, H.; Dračínský, M.; Procházková, E.; Chalupská, D.; Klíma, M.; Plačková, P.; Hájek, M.; Andrei, G.; et al. Norbornane-based nucleoside and nucleotide analogues locked in North conformation. Bioorg. Med. Chem. 2015, 23, 184–191. [Google Scholar] [CrossRef]
- Marquez, V.E.; Siddiqui, M.A.; Ezzitouni, A.; Russ, P.; Wang, J.; Wagner, R.W.; Matteucci, M.D. Nucleosides with a Twist. Can Fixed Forms of Sugar Ring Pucker Influence Biological Activity in Nucleosides and Oligonucleotides? J. Med. Chem. 1996, 39, 3739–3747. [Google Scholar] [CrossRef]
- Tănase, C.I.; Drăghici, C.; Căproiu, M.T.; Shova, S.; Mathe, C.; Cocu, F.G.; Enache, C.; Maganu, M. New carbocyclic nucleoside analogues with a bicyclo[2.2.1]heptane fragment as sugar moiety; Synthesis, X-ray crystallography and anticancer activity. Bioorg. Med. Chem. 2014, 22, 513–522. [Google Scholar] [CrossRef]
- Tănase, C.I.; Drăghici, C.; Cojocaru, A.; Galochkina, A.V.; Orshanskaya, J.R.; Zarubaev, V.V.; Shova, S.; Enache, C.; Maganu, M. New carbocyclic N6-substituted adenine and pyrimidine nucleoside analogues with a bicyclo[2.2.1]heptane fragment as sugar moiety; synthesis, antiviral, anticancer activity and X-ray crystallography. Bioorg. Med. Chem. 2015, 23, 6346–6354. [Google Scholar] [CrossRef] [PubMed]
- Waud, W.R.; Schmid, S.M.; Montgomery, J.A.; Secrist, J.A. Preclinical Antitumor Activity of 2-Chloro-9-(2-deoxy-2-fluoro-β-D-arabinofuranosyl)adenine (C1-F-Ara-A). Nucleosides Nucleotides Nucleic Acids 2000, 19, 447–460. [Google Scholar] [CrossRef] [PubMed]
- Albertioni, F.; Lindemalm, S.; Reichelova, V.; Pettersson, B.; Eriksson, S.; Juliusson, G.; Liliemark, J. Pharmacokinetics of cladribine in plasma and its 5’-monophosphate and 5’-triphosphate in leukemic cells of patients with chronic lymphocytic leukemia. Clin. Cancer Res. 1998, 4, 653–658. [Google Scholar] [PubMed]
- Pui, C.H.; Jeda, S.; Kirkpatrick, P. Clofarabine. Nat. Rev. Drug. Discov. 2005, 4, 369–370. [Google Scholar] [CrossRef] [PubMed]
- Cen, Y.; Sauve, A.A. Efficient syntheses of clofarabine and gemcitabine from 2-deoxyribonolactone. Nucleosides Nucleotides Nucleic Acids 2010, 29, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, K.C.; Ellery, S.P.; Rivas, F.; Saye, K.; Rogers, E.; Workinger, T.J.; Schallenberger, M.; Tawatao, R.; Montero, A.; Hessell, A.; et al. Synthesis and biological evaluation of 2′,4′- and 3′,4′-bridged nucleoside analogues. Bioorg. Med. Chem. 2011, 19, 5648–5669. [Google Scholar] [CrossRef] [Green Version]
- Obika, S.; Morio, K.; Nanbu, D.; Hari, Y.; Itoh, H.; Imanishi, T. Synthesis and conformation of 3′,4′-BNA monomers, 3′-O,4′-C-methyleneribonucleosides. Tetrahedron 2002, 58, 3039–3049. [Google Scholar] [CrossRef]
- Youssefyeh, R.D.; Verheyden, J.P.H.; Moffatt, J.G. 4′-Substituted nucleosides. 4. Synthesis of some 4′-hydroxymethyl nucleosides. J. Org. Chem. 1979, 44, 1301–1309. [Google Scholar] [CrossRef]
- Kim, B.J.; Zhang, J.; Tan, S.; Matteson, D.S.; Prusoff, W.H.; Cheng, Y.-C. Synthesis and properties of 1-(3′-dihydroxyboryl-2′,3′-dideoxyribosyl)pyrimidines. Org. Biomol. Chem. 2012, 10, 9349. [Google Scholar] [CrossRef]
- Bege, M.; Kiss, A.; Kicsák, M.; Bereczki, I.; Baksa, V.; Király, G.; Szemán-Nagy, G.; Szigeti, M.Z.; Herczegh, P.; Borbás, A. Synthesis and Cytostatic Effect of 3’-deoxy-3’-C-Sulfanylmethyl Nucleoside Derivatives with d-xylo Configuration. Molecules 2019, 24, 2173. [Google Scholar] [CrossRef] [Green Version]
- Mohan, A.A.; Sharma, G.V.R.; Vidavalur, S. Synthesis, characterization and biological evaluation of C5’-N-cyclopropylcarboxamido-C6-amino-C2-alkynylated purine nucleoside analogues. Nucleosides Nucleotides Nucleic Acids 2017, 36, 1–15. [Google Scholar]
- Meurillon, M.; Marton, Z.; Hospital, A.; Jordheim, L.P.; Béjaud, J.; Lionne, C.; Dumontet, C.; Périgaud, C.; Chaloin, L.; Peyrottes, S. Structure–activity relationships of β-hydroxyphosphonate nucleoside analogues as cytosolic 5′-nucleotidase II potential inhibitors: Synthesis, in vitro evaluation and molecular modeling studies. Eur. J. Med. Chem. 2014, 77, 18–37. [Google Scholar] [CrossRef] [PubMed]
- Gallier, F.; Lallemand, P.; Meurillon, M.; Jordheim, L.P.; Dumontet, C.; Périgaud, C.; Lionne, C.; Peyrottes, S.; Chaloin, L. Structural Insights into the Inhibition of Cytosolic 5′-Nucleotidase II (cN-II) by Ribonucleoside 5′-Monophosphate Analogues. PLoS Comput. Biol. 2011, 7, e1002295. [Google Scholar] [CrossRef] [PubMed]
- Galmarini, C.M.; Thomas, X.; Graham, K.; Jafaari, A.E.; Cros, E.; Jordheim, L.; Mackey, J.R.; Dumontet, C. Deoxycytidine kinase and cN-II nucleotidase expression in blast cells predict survival in acute myeloid leukaemia patients treated with cytarabine. Br. J. Haematol. 2003, 122, 53–60. [Google Scholar] [CrossRef]
- Nguyen, H.V.; Sallustrau, A.; Balzarini, J.; Bedford, M.R.; Eden, J.C.; Georgousi, N.; Hodges, N.J.; Kedge, J.; Mehellou, Y.; Tselepis, C.; et al. Organometallic nucleoside analogues with ferrocenyl linker groups: Synthesis and cancer cell line studies. J. Med. Chem. 2014, 57, 5817–5822. [Google Scholar] [CrossRef] [Green Version]
- Slusarczyk, M.; Ferrari, V.; Serpi, M.; Gönczy, B.; Balzarini, J.; McGuigan, C. Symmetrical Diamidates as a Class of Phosphate Prodrugs to Deliver the 5′-Monophosphate Forms of Anticancer Nucleoside Analogues. ChemMedChem 2018, 13, 2305–2316. [Google Scholar] [CrossRef]
- Cahard, D.; McGuigan, C.; Balzarini, J. Aryloxy phosphoramidate triesters as pro-tides. Mini-Rev. Med. Chem. 2004, 4, 371–381. [Google Scholar] [CrossRef]
- Liu, W.; Zhang, L.; Zhou, H.; Yang, C.; Miao, Z.; Zhao, Y. Synthesis of Novel Nucleoside Analogue Phosphorothioamidate Prodrugs and in vitro Anticancer Evaluation Against RKO Human Colon Carcinoma Cells. Nucleosides Nucleotides Nucleic Acids 2013, 32, 161–173. [Google Scholar] [CrossRef]
- Sofia, M.J.; Bao, D.; Chang, W.; Du, J.; Nagarathnam, D.; Rachakonda, S.; Reddy, P.G.; Ross, B.S.; Wang, P.; Zhang, H.-R.; et al. Discovery of a β- d -2′-Deoxy-2′-α-fluoro-2′-β- C -methyluridine Nucleotide Prodrug (PSI-7977) for the Treatment of Hepatitis C Virus. J. Med. Chem. 2010, 53, 7202–7218. [Google Scholar] [CrossRef]
- Mehellou, Y.; Valente, R.; Mottram, H.; Walsby, E.; Mills, K.I.; Balzarini, J.; McGuigan, C. Phosphoramidates of 2’-beta-D-arabinouridine (AraU) as phosphate prodrugs; design, synthesis, in vitro activity and metabolism. Bioorg. Med. Chem. 2010, 18, 2439–2446. [Google Scholar] [CrossRef]
- Jordheim, L.P.; Galmarini, C.M.; Dumontet, C. Gemcitabine resistance due to deoxycytidine kinase deficiency can be reverted by fruitfly deoxynucleoside kinase, DmdNK, in human uterine sarcoma cells. Cancer Chemother. Pharmacol. 2006, 58, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Damaraju, V.L.; Damaraju, S.; Young, J.D.; Baldwin, S.A.; Mackey, J.; Sawyer, M.B.; Cass, C.E. Nucleoside anticancer drugs: The role of nucleoside transporters in resistance to cancer chemotherapy. Oncogene 2003, 22, 7524–7536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daifuku, R.; Koratich, M.; Stackhouse, M. Vitamin E Phosphate Nucleoside Prodrugs: A Platform for Intracellular Delivery of Monophosphorylated Nucleosides. Pharmaceuticals 2018, 11, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shipley, L.A.; Brown, T.J.; Cornpropst, J.D.; Hamilton, M.; Daniels, W.D.; Culp, H.W. Metabolism and disposition of gemcitabine, an oncolytic deoxycytidine analog, in mice, rats, and dogs. Drug Metab. Dispos. 1992, 20, 849–855. [Google Scholar] [PubMed]
Figure 1.
Gemcitabine 1, clofarabine 2 and Ara-C 3. Modifications compared to native d-ribo-configured purine or pyrimidine nucleosides are shown in blue.
Figure 1.
Gemcitabine 1, clofarabine 2 and Ara-C 3. Modifications compared to native d-ribo-configured purine or pyrimidine nucleosides are shown in blue.

Figure 2.
General scope for nucleoside analogues covered in this review. Base = purine or pyrimidine (i.e., C, U, T, A, G or close derivative thereof). X = heteroatom or carbon and Y and Z = ring functional group or modification of native d-ribo stereochemistry.
Figure 2.
General scope for nucleoside analogues covered in this review. Base = purine or pyrimidine (i.e., C, U, T, A, G or close derivative thereof). X = heteroatom or carbon and Y and Z = ring functional group or modification of native d-ribo stereochemistry.

Figure 3.
Structure of forodesine 4.

Scheme 1.
Reagents and conditions: (i) NCS, pentane; (ii) LiTMP, −78 °C, 36% over two steps; (iii) nBuLi, MeCN, THF, −78 °C then tetramethylpiperidine −78 °C, 100%; (iv) (Boc)2O, CH2Cl2; (v) tBuOCH(NMe2)2, DMF, 70 °C; (vi) THF, AcOH, H2O, 72% from 7; (vii) H2NCH2CO2Et·HCl, NaOAc, MeOH (viii) ClCO2Bn, DBU, CH2Cl2, reflux, 67% from 10; (ix) H2, Pd/C, EtOH; (x) H2NCH=NH·AcOH, EtOH, reflux, 91% from 12 and (xi) TFA, 81%.
Scheme 1.
Reagents and conditions: (i) NCS, pentane; (ii) LiTMP, −78 °C, 36% over two steps; (iii) nBuLi, MeCN, THF, −78 °C then tetramethylpiperidine −78 °C, 100%; (iv) (Boc)2O, CH2Cl2; (v) tBuOCH(NMe2)2, DMF, 70 °C; (vi) THF, AcOH, H2O, 72% from 7; (vii) H2NCH2CO2Et·HCl, NaOAc, MeOH (viii) ClCO2Bn, DBU, CH2Cl2, reflux, 67% from 10; (ix) H2, Pd/C, EtOH; (x) H2NCH=NH·AcOH, EtOH, reflux, 91% from 12 and (xi) TFA, 81%.

Scheme 2.
Reagents and conditions: (i) 6, nBuLi, anisole, ether, −70 °C; (ii) (Boc)2O, CH2Cl2, 85% over two steps; (iii) 1M HCl, MeOH, 30 °C, 96%; (iv) 1,3-Dichloro-1,1-3,3-tetraisopropyldisiloxane, pyridine, 81%; (v) O-Phenyl chlorothionoformate, MeCN, 90%; (vi) 1,1′-Azobis(cyclohexane-1-carbonitrile), toluene reflux, 91%; (vii) Pd(OH)2, H2, EtOH, conc. NH4OH, 90% and (viii) conc. HCl, MeOH, reflux, 83%.
Scheme 2.
Reagents and conditions: (i) 6, nBuLi, anisole, ether, −70 °C; (ii) (Boc)2O, CH2Cl2, 85% over two steps; (iii) 1M HCl, MeOH, 30 °C, 96%; (iv) 1,3-Dichloro-1,1-3,3-tetraisopropyldisiloxane, pyridine, 81%; (v) O-Phenyl chlorothionoformate, MeCN, 90%; (vi) 1,1′-Azobis(cyclohexane-1-carbonitrile), toluene reflux, 91%; (vii) Pd(OH)2, H2, EtOH, conc. NH4OH, 90% and (viii) conc. HCl, MeOH, reflux, 83%.

Scheme 3.
Reagents and conditions: (i) BnBr, NaH, DMF, THF; (ii) 2M HCl, THF; (iii) NaIO4, H2O, MeOH; (iv) NaBH4, MeOH, 84% from 20; (v) 5% HCl/MeOH, 91%; (vi) MsCl, pyridine; (vii) Na2S, DMF, α-anomer 78% from 21, β-anomer 73% from 21; (viii) 4M HCl, THF; (ix) NaBH4, MeOH, 90% from 2; (x) TBDPSCl, imidazole, DMF, 87%; (xi) Ac2O, DMSO; (xii) Ph3PCH3Br, NaH, t-amyl alcohol, THF; (xiii) BCl3, CH2Cl2, −78 °C then MeOH, pyridine, 92%; (xiv) m-CPBA, CH2Cl2, −78 °C, 74% from 23; (xv) 25, TMSOTf, ClCH2CH2Cl, 0 °C, 29%; (xvi) TBAF, THF and (xvii) aqueous NH3, MeOH then HPLC separation.
Scheme 3.
Reagents and conditions: (i) BnBr, NaH, DMF, THF; (ii) 2M HCl, THF; (iii) NaIO4, H2O, MeOH; (iv) NaBH4, MeOH, 84% from 20; (v) 5% HCl/MeOH, 91%; (vi) MsCl, pyridine; (vii) Na2S, DMF, α-anomer 78% from 21, β-anomer 73% from 21; (viii) 4M HCl, THF; (ix) NaBH4, MeOH, 90% from 2; (x) TBDPSCl, imidazole, DMF, 87%; (xi) Ac2O, DMSO; (xii) Ph3PCH3Br, NaH, t-amyl alcohol, THF; (xiii) BCl3, CH2Cl2, −78 °C then MeOH, pyridine, 92%; (xiv) m-CPBA, CH2Cl2, −78 °C, 74% from 23; (xv) 25, TMSOTf, ClCH2CH2Cl, 0 °C, 29%; (xvi) TBAF, THF and (xvii) aqueous NH3, MeOH then HPLC separation.

Scheme 4.
Reagents and conditions: (i) DAST, CH2Cl2, −78 °C, 77%; (ii) m-CPBA, CH2Cl2, −78 °C; (iii) Ac2O, 100 °C, 77% from 23; (iv) 25, SnCl4, MeCN, 93%; (v) BBr3, MeOH; (vi) NH4F, MeOH, 60 °C and (vii) aqueous NH3, MeOH then HPLC separation, 43% (β-anomer) and 17% (α-anomer) from 32.
Scheme 4.
Reagents and conditions: (i) DAST, CH2Cl2, −78 °C, 77%; (ii) m-CPBA, CH2Cl2, −78 °C; (iii) Ac2O, 100 °C, 77% from 23; (iv) 25, SnCl4, MeCN, 93%; (v) BBr3, MeOH; (vi) NH4F, MeOH, 60 °C and (vii) aqueous NH3, MeOH then HPLC separation, 43% (β-anomer) and 17% (α-anomer) from 32.

Scheme 5.
Reagents and conditions: (i) Ac2O, DMSO; (ii) DAST, benzene, 0 °C-r.t., 48%; (iii) BCl3, CH2Cl2, −78 °C, then MeOH, pyridine; (iv) Bz2O, Et3N, DMAP, MeCN, 79% from 34; (v) m-CPBA, CH2Cl2, −78 °C; (vi) 25, TMSOTf, ClCH2CH2Cl, 0 °C, 57% from 35; (vii) TBAF, THF and (viii) aqueous NH3, MeOH, then HPLC separation, 36% (α-anomer) and 15% (β-anomer) from 36.
Scheme 5.
Reagents and conditions: (i) Ac2O, DMSO; (ii) DAST, benzene, 0 °C-r.t., 48%; (iii) BCl3, CH2Cl2, −78 °C, then MeOH, pyridine; (iv) Bz2O, Et3N, DMAP, MeCN, 79% from 34; (v) m-CPBA, CH2Cl2, −78 °C; (vi) 25, TMSOTf, ClCH2CH2Cl, 0 °C, 57% from 35; (vii) TBAF, THF and (viii) aqueous NH3, MeOH, then HPLC separation, 36% (α-anomer) and 15% (β-anomer) from 36.

Figure 4.
Structures of 2′-deoxy-4’-thiacytidine nucleosides 38–41.

Scheme 6.
Reagents and conditions: (i) NIS, pivalic acid, MeCN/CH2Cl2, 93%; (ii) Uracil, N,O-bis(trimethylsilyl)acetamide (BSA), TMSOTf, MeCN/CH2Cl2, 87%; (iii) Bu3SnH, Et3B, toluene, O2, −60 °C, 98%; (iv) TBAF, Ac2O, THF, 100%; (v) NH3/MeOH; (vi) I2, PPh3, pyridine, dioxane; (vii) Ac2O, DMAP, DIPEA, CH2Cl2; (viii) BDN, MeCN, 65% over four steps; (ix) NH3/MeOH; (x) TBDMSCl, imidazole, DMF, 61% over two steps; (xi) Pb(OBz)4, toluene, 63%; (xii) TMSN3, SnCl4, CH2Cl2, 61%; (xiii) NaOMe, MeOH; (xiv) Ac2O, DMAP, DIPEA, MeCN, 83% over two steps; (xv) TPSCl, K2O4, MeCN, 60 °C; (xvi) NH4OH, THF; (xvii) TBAF, Ac2O, THF and (xviii) NaOMe, MeOH, 66% over four steps.
Scheme 6.
Reagents and conditions: (i) NIS, pivalic acid, MeCN/CH2Cl2, 93%; (ii) Uracil, N,O-bis(trimethylsilyl)acetamide (BSA), TMSOTf, MeCN/CH2Cl2, 87%; (iii) Bu3SnH, Et3B, toluene, O2, −60 °C, 98%; (iv) TBAF, Ac2O, THF, 100%; (v) NH3/MeOH; (vi) I2, PPh3, pyridine, dioxane; (vii) Ac2O, DMAP, DIPEA, CH2Cl2; (viii) BDN, MeCN, 65% over four steps; (ix) NH3/MeOH; (x) TBDMSCl, imidazole, DMF, 61% over two steps; (xi) Pb(OBz)4, toluene, 63%; (xii) TMSN3, SnCl4, CH2Cl2, 61%; (xiii) NaOMe, MeOH; (xiv) Ac2O, DMAP, DIPEA, MeCN, 83% over two steps; (xv) TPSCl, K2O4, MeCN, 60 °C; (xvi) NH4OH, THF; (xvii) TBAF, Ac2O, THF and (xviii) NaOMe, MeOH, 66% over four steps.

Scheme 7.
Key intermediates to access 2’-deoxy-4’-thiocytidine nucleosides 39–41.

Scheme 8.
Reagents and conditions: (i) TBDPSCl, Et3N, DMAP, CH2Cl2, 92%; (ii) NaBH4, MeOH, 98%; (iii) MsCl, Et3N, DMAP, CH2Cl2, 97%; (iv) Se, NaBH4, EtOH, THF, 60 °C, 96%; (v) m-CPBA, CH2Cl2, –78 °C, 85%; (vi) Uracil, Et3N, TMSOTf, toluene, CH2Cl2, 53%; (vii) 50% aq. TFA, 81%; (viii) N3-benzoylcytosine, Et3N, TMSOTf, toluene, CH2Cl2, 35%; (ix) 50% Aq. TFA and (x) NH3, MeOH, 82% over two steps.
Scheme 8.
Reagents and conditions: (i) TBDPSCl, Et3N, DMAP, CH2Cl2, 92%; (ii) NaBH4, MeOH, 98%; (iii) MsCl, Et3N, DMAP, CH2Cl2, 97%; (iv) Se, NaBH4, EtOH, THF, 60 °C, 96%; (v) m-CPBA, CH2Cl2, –78 °C, 85%; (vi) Uracil, Et3N, TMSOTf, toluene, CH2Cl2, 53%; (vii) 50% aq. TFA, 81%; (viii) N3-benzoylcytosine, Et3N, TMSOTf, toluene, CH2Cl2, 35%; (ix) 50% Aq. TFA and (x) NH3, MeOH, 82% over two steps.

Figure 5.
Structures of 2’-substituted 4’-selenoarabinofuranosyl pyrimidine analogues 65–68.

Scheme 9.
Reagents and conditions: (i) DAST, CH2Cl2, −78 °C, 23%; (ii) TESCl, DMAP, CH2Cl2; (iii) BzCl, pyridine, DIPEA, 70 °C; (iv) TBAF, THF, 0 °C, 81% over three steps and (v) DAST, CH2Cl2, −78 °C, 45%.
Scheme 9.
Reagents and conditions: (i) DAST, CH2Cl2, −78 °C, 23%; (ii) TESCl, DMAP, CH2Cl2; (iii) BzCl, pyridine, DIPEA, 70 °C; (iv) TBAF, THF, 0 °C, 81% over three steps and (v) DAST, CH2Cl2, −78 °C, 45%.

Figure 6.
2’-Substituted-4’selenoribofuranosyl nucleosides reported by Jeong.

Figure 7.
Structures of aristeromycin, 74, and neplanocin A, 75.

Figure 8.
Structures of fluorocyclopentenyl nucleoside analogues 76–79.

Scheme 10.
Reagents and conditions: (i) I2, pyridine, THF, 55%; (ii) NaBH4, CeCl3, MeOH, 93%; (iii) TBDPSCl, imidazole, DMF, 97%; (iv) NFSI, nBuLi, THF, −78 °C, 80%; (v) TBAF, THF, 80%; (vi) N3-benzoyluracil, DEAD, PPh3, THF; (vii) NH3/MeOH; (viii) BBr3, DCM, −78 °C, 49% over three steps; (ix) Ac2O, pyridine; (x) POCl3, Et3N, 1,2,3-triazole; (xi) NH4OH, 1,4-dioxane and (xii) NH3/MeOH, 40% over four steps.
Scheme 10.
Reagents and conditions: (i) I2, pyridine, THF, 55%; (ii) NaBH4, CeCl3, MeOH, 93%; (iii) TBDPSCl, imidazole, DMF, 97%; (iv) NFSI, nBuLi, THF, −78 °C, 80%; (v) TBAF, THF, 80%; (vi) N3-benzoyluracil, DEAD, PPh3, THF; (vii) NH3/MeOH; (viii) BBr3, DCM, −78 °C, 49% over three steps; (ix) Ac2O, pyridine; (x) POCl3, Et3N, 1,2,3-triazole; (xi) NH4OH, 1,4-dioxane and (xii) NH3/MeOH, 40% over four steps.

Scheme 11.
Reagents and conditions: (i) PDC, CH2Cl2, 82%; (ii) NaBH4, MeOH, 88%; (iii) MsCl, pyridine, 99%; (iv) NaN3, DMF, 115 °C, 92% and (v) LiAlH4, THF, 59%.
Scheme 11.
Reagents and conditions: (i) PDC, CH2Cl2, 82%; (ii) NaBH4, MeOH, 88%; (iii) MsCl, pyridine, 99%; (iv) NaN3, DMF, 115 °C, 92% and (v) LiAlH4, THF, 59%.

Scheme 12.
Reagents and conditions: (i) MsCl, pyridine, CH2Cl2, 98%; (ii) NaN3, DMF, 110 °C, 91% and (iii) Pd(OH)2/C, MeOH, 87%.
Scheme 12.
Reagents and conditions: (i) MsCl, pyridine, CH2Cl2, 98%; (ii) NaN3, DMF, 110 °C, 91% and (iii) Pd(OH)2/C, MeOH, 87%.

Scheme 13.
Reagents and conditions: (i) TIPSCl, imidazole, DMF, 92%; (ii) NFSI, LiHMDS, THF, −78 °C, 72%; (iii) DIBAL-H, toluene, −78 °C, 91%; (iv) MsCl, Et3N, CH2Cl2, quant.; (v) 2,6-dichloropurine, dichloroethane, reflux; (vi) NH3/isopropanol, sealed tube, 105 °C, 66% over two steps and (vii) Me4NF, AcOH, DMF, 90%.
Scheme 13.
Reagents and conditions: (i) TIPSCl, imidazole, DMF, 92%; (ii) NFSI, LiHMDS, THF, −78 °C, 72%; (iii) DIBAL-H, toluene, −78 °C, 91%; (iv) MsCl, Et3N, CH2Cl2, quant.; (v) 2,6-dichloropurine, dichloroethane, reflux; (vi) NH3/isopropanol, sealed tube, 105 °C, 66% over two steps and (vii) Me4NF, AcOH, DMF, 90%.

Scheme 14.
Reagents and conditions: (i) 2,6-diaminopurine, BSA, TMSOTf, MeCN, 100%; (ii) 2,6-dicholoro-9H-purine, BSA, TMSOTf, 64%; (iii) NaOH, THF, 94%; (iv) BzCl, pyridine, 52%; (v) HF, pyridine, 50%; (vi) Allyl(tri-n-butyl)tin, PdCl2(PPh3)2, DMF then NaOH, THF, 42%, 2 steps and (vii) HF, pyridine, 87%.
Scheme 14.
Reagents and conditions: (i) 2,6-diaminopurine, BSA, TMSOTf, MeCN, 100%; (ii) 2,6-dicholoro-9H-purine, BSA, TMSOTf, 64%; (iii) NaOH, THF, 94%; (iv) BzCl, pyridine, 52%; (v) HF, pyridine, 50%; (vi) Allyl(tri-n-butyl)tin, PdCl2(PPh3)2, DMF then NaOH, THF, 42%, 2 steps and (vii) HF, pyridine, 87%.

Scheme 15.
Reagents and conditions: (i) LiCHCl2, THF then ZnCl2; (ii) PhCH2ONa, 97%; (iii) Pinanediol, Et2O, H2O, 96%; (iv) CH2Br2, LDA, THF then ZnCl2, 83%; (v) CH2CHCH2MgBr, 86%; (vi) NaIO4/K2OsO4, 2,6-lutidine, dioxane/H2O, 63%; (vii) H2 Pd/C, EtOAc, 81% and (viii) Ac2O,DMAP, CH2Cl2, 98%. Cy = cyclohexyl, T = thymine, 5F-U = 5-fluorouracil, C = cytosine, 5I-U = 5-iodouracil.
Scheme 15.
Reagents and conditions: (i) LiCHCl2, THF then ZnCl2; (ii) PhCH2ONa, 97%; (iii) Pinanediol, Et2O, H2O, 96%; (iv) CH2Br2, LDA, THF then ZnCl2, 83%; (v) CH2CHCH2MgBr, 86%; (vi) NaIO4/K2OsO4, 2,6-lutidine, dioxane/H2O, 63%; (vii) H2 Pd/C, EtOAc, 81% and (viii) Ac2O,DMAP, CH2Cl2, 98%. Cy = cyclohexyl, T = thymine, 5F-U = 5-fluorouracil, C = cytosine, 5I-U = 5-iodouracil.

Scheme 16.
Reagents and conditions: (i) KMnO4, KOH, H2O, 79%; (ii) EDC, HOBt, cyclopropylamine, Et3N, DMF, 70%; (iii) 50% Aq. HCO2H, 73% and (iv) Pd(PPh3)2Cl2, CuI, Et3N, DMF, MeCN, R = 115 = 86%, R = 116 = 85%.
Scheme 16.
Reagents and conditions: (i) KMnO4, KOH, H2O, 79%; (ii) EDC, HOBt, cyclopropylamine, Et3N, DMF, 70%; (iii) 50% Aq. HCO2H, 73% and (iv) Pd(PPh3)2Cl2, CuI, Et3N, DMF, MeCN, R = 115 = 86%, R = 116 = 85%.

Figure 9.
5’-β-Hydroxyphosphonate analogues developed by Peyrottes and active compound 117 from this series, Y = NH2 or OH.
Figure 9.
5’-β-Hydroxyphosphonate analogues developed by Peyrottes and active compound 117 from this series, Y = NH2 or OH.

Figure 10.
Ferronucleosides developed by Tucker.

Scheme 17.
Reagents and conditions: (i) (Me)3PO4, POCl3 and (ii) Amino acid ester salt, DIPEA. X and Y represent different functional groups for analogue classes e.g. X = Y = F, Base = C = gemcitabine phosphorodiamidate. See Reference [77] for full list of analogues synthesised and evaluated.
Scheme 17.
Reagents and conditions: (i) (Me)3PO4, POCl3 and (ii) Amino acid ester salt, DIPEA. X and Y represent different functional groups for analogue classes e.g. X = Y = F, Base = C = gemcitabine phosphorodiamidate. See Reference [77] for full list of analogues synthesised and evaluated.

Figure 11.
Structures of vitamin E-gemcitabine conjugates 121–124, highlighting changes to the tocopherol or tocotrienol component.
Figure 11.
Structures of vitamin E-gemcitabine conjugates 121–124, highlighting changes to the tocopherol or tocotrienol component.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Antineoplastic activities of 2′-modified-4′-thionucleosides.
| Antineoplastic Activities IC50 (µg/mL) | |||
|---|---|---|---|
| Compound (Anomer) | 2′-Substituent | CCRF-HSB-2 a | KB Cells b |
| 28α | CH2 | >10 | ND c |
| 28β | CH2 | 0.01 | 0.12 |
| 33α | F (arabino) | >10 | ND |
| 33β | F (arabino) | 0.05 | 0.02 |
| 37α | F2 | >10 | ND |
| 37β | F2 | 1.5 | 17 |
| Ara-C 3 | 0.05 | 0.26 | |
| DMDC | 0.02 | 0.44 | |
Table 2.
Anticancer activity of 2’-modified 4’-selenoarabino nucleosides 65–69 compared to 1 and 3 across several human cancer cell lines.
Table 2.
Anticancer activity of 2’-modified 4’-selenoarabino nucleosides 65–69 compared to 1 and 3 across several human cancer cell lines.
| IC50 (μM) | ||||||
|---|---|---|---|---|---|---|
| Compound | HCT116 a | A549 b | SNU638 c | T47D d | PC-3 e | K562 f |
| 65 | 1.1 | 0.47 | 0.14 | 0.79 | 0.58 | 0.63 |
| 66 | 7.13 | 8.83 | 4.72 | ND | ND | 86.6 |
| 67 | >100 | >100 | >100 | >100 | >100 | >100 |
| 68 | >100 | >100 | >100 | >100 | >100 | >100 |
| 3 | 5.30 | 1.90 | 0.15 | 2.70 | 55.9 | 0.05 |
| 1 | 0.01 | 0.09 | ND | ND | 0.04 | ND |
Human cancer cell tissue type a colon; b lung; c stomach; d breast; e prostate; f myelogenous leukemia.
Table 3.
Anticancer activity of 76 in human cancer cell lines.
| Cancer Cell Line | |||||||
|---|---|---|---|---|---|---|---|
| HTC-116 a | MDA-MB-231 b | PANC-1 c | MCF-7 d | A549 e | MKN45 f | U251 g | |
| IC50 (μM) | 0.39 | 0.18 | 0.62 | 0.34 | 0.34 | 0.50 | 0.83 |
Human cancer cell tissue type a colon; b breast; c pancreatic; d hormone-dependent breast; e lung; f stomach; g brain.
Table 4.
Pharmacokinetic data for 76 from phase 0 clinical studies.
| Dose (mg) | Tmax (h) | Cmax (ng/mL) | t1/2 (h) | Oral Bioavailability (%) |
|---|---|---|---|---|
| 20 * | 0.3 | 1144 | - | - |
| 50 | 2.2 | 303 | 14 | 56 |
| 100 | 2.5 | 311 | 21 | 33 |
* Intravenous.
Table 5.
GI50 values of 1, 122 and 123 in the presence or absence of dipyridamole.
| Cancer Cell Line | ||||||
|---|---|---|---|---|---|---|
| Breast MDA-MB-231 (μM) | Non-Small Cell Lung NCI-H460 (μM) | Colon HCT-116 (μM) | ||||
| Compound | DP (−) | DP (20 μM) | DP (−) | DP (20 μM) | DP (−) | DP (20 μM) |
| 1 | 3.08 | 56.8 | 0.02 | 0.82 | 0.03 | 2.39 |
| 122 | 30.3 | 27.8 | 7.16 | 16.0 | 5.55 | 12.6 |
| 123 | 17.2 | 23.3 | 2.14 | 1.47 | 3.07 | 6.74 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Guinan, M.; Benckendorff, C.; Smith, M.; Miller, G.J. Recent Advances in the Chemical Synthesis and Evaluation of Anticancer Nucleoside Analogues. Molecules 2020, 25, 2050. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25092050
AMA Style
Guinan M, Benckendorff C, Smith M, Miller GJ. Recent Advances in the Chemical Synthesis and Evaluation of Anticancer Nucleoside Analogues. Molecules. 2020; 25(9):2050. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25092050
Chicago/Turabian StyleGuinan, Mieke, Caecilie Benckendorff, Mark Smith, and Gavin J. Miller. 2020. "Recent Advances in the Chemical Synthesis and Evaluation of Anticancer Nucleoside Analogues" Molecules 25, no. 9: 2050. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25092050