
Nitrosative Stress and Its Association with Cardiometabolic Disorders

Abstract
:
1. Introduction
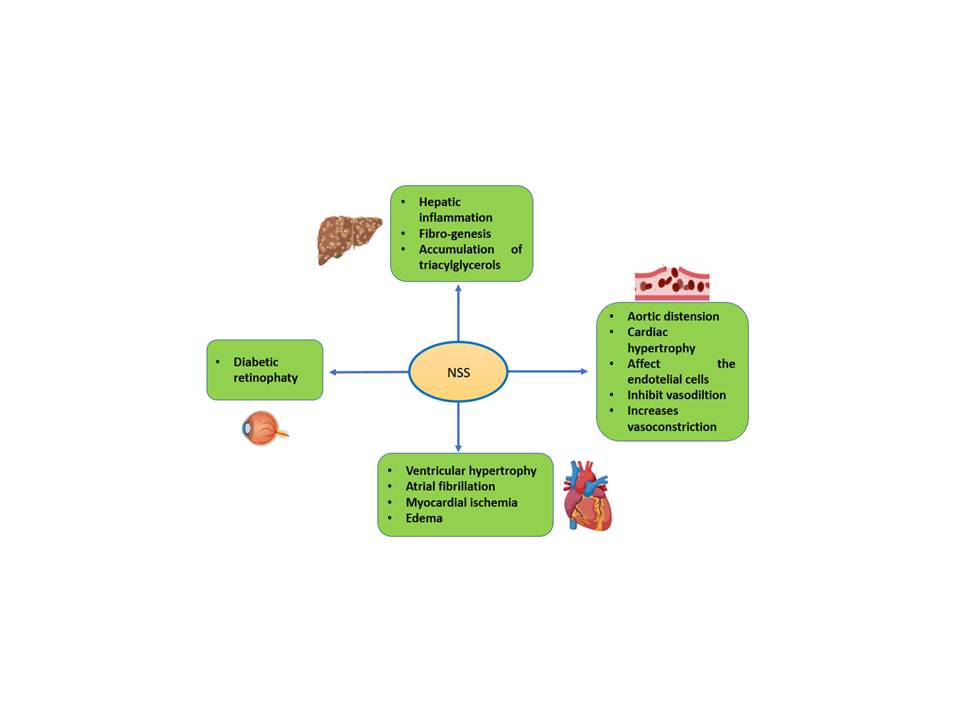
2. NO, Its Synthesis and the Generation of RNS
3. ONOO− and NO
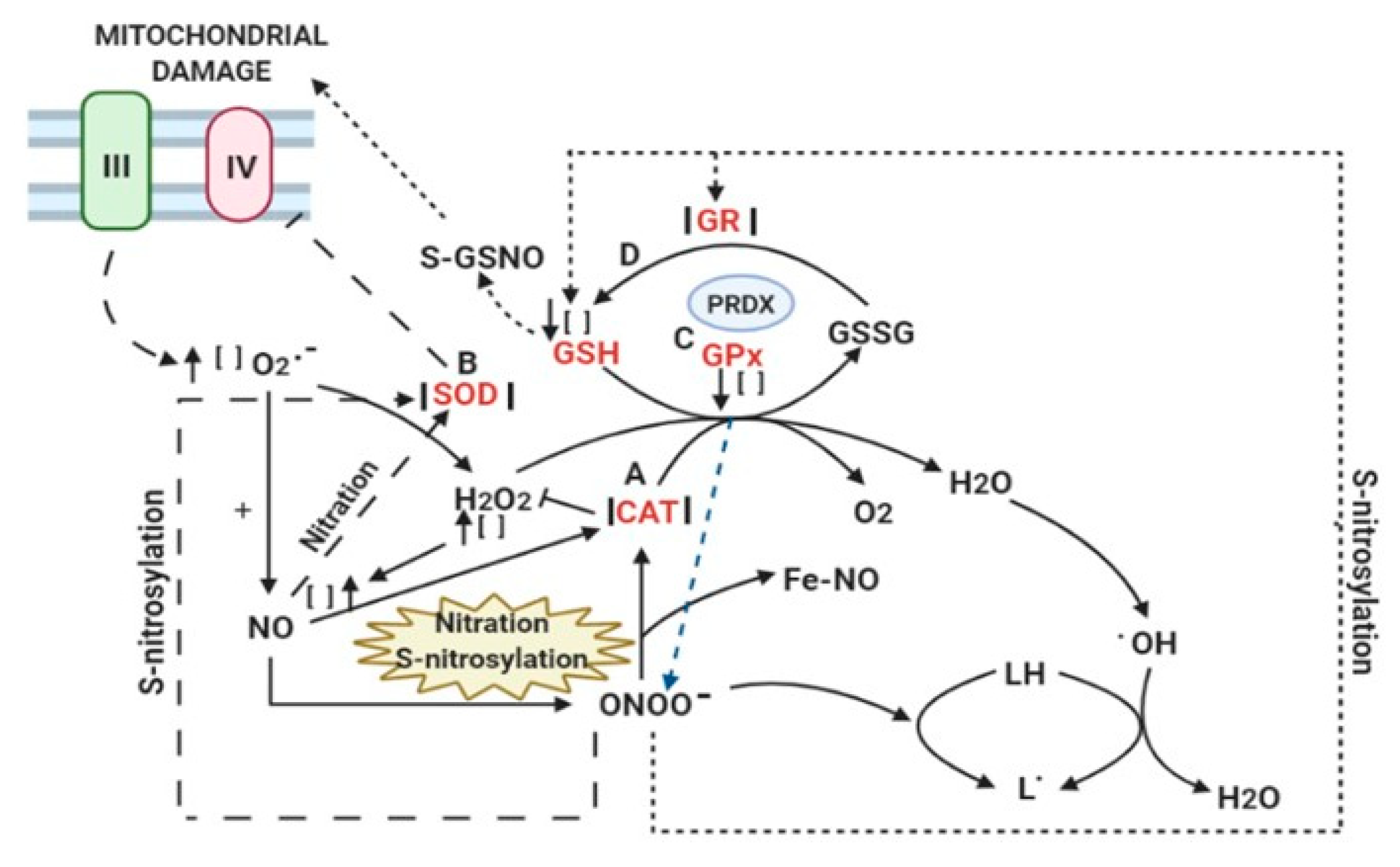
4. Mechanisms of Damage by RNS
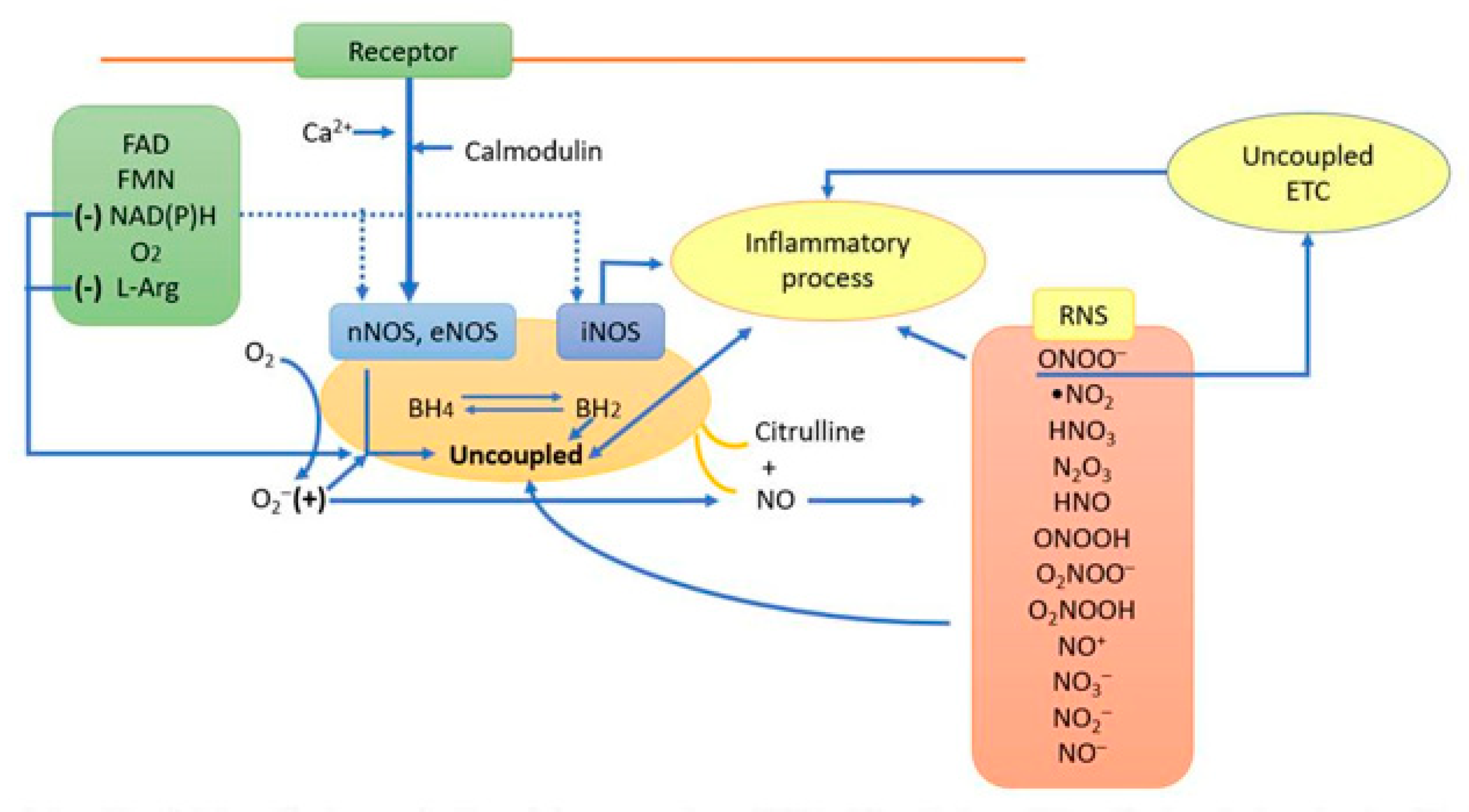
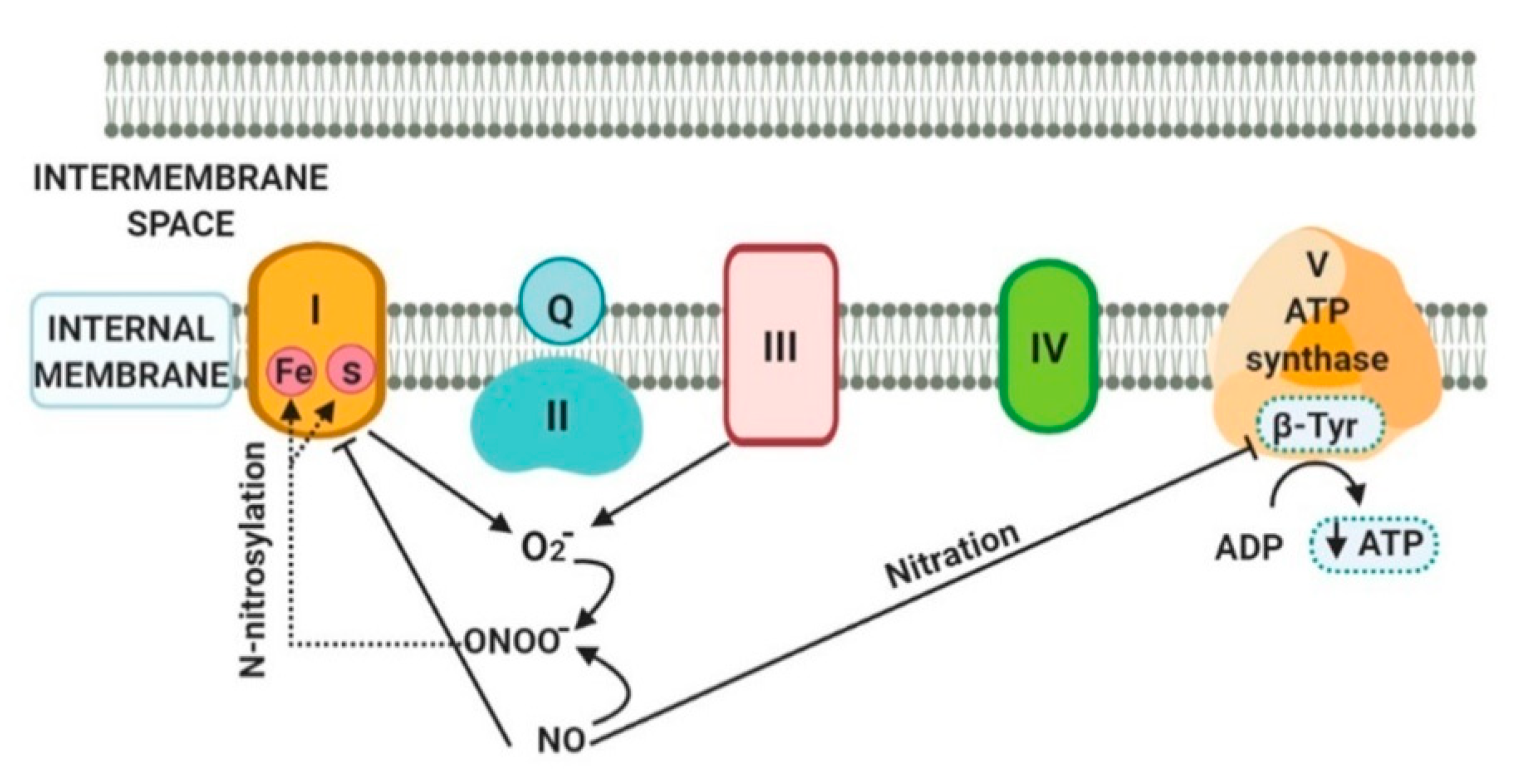
5. Mitochondrial Proteins and DNA
6. Connections with Antioxidant Enzymes
7. Inflammatory Process
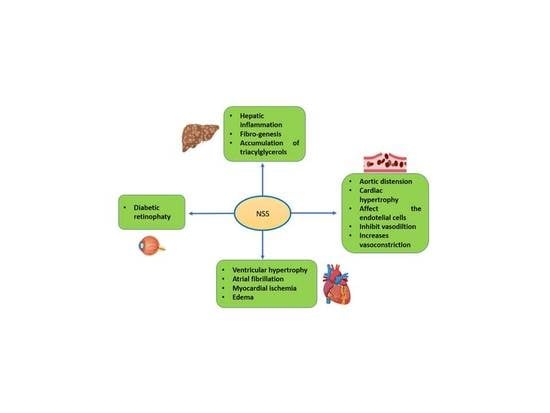
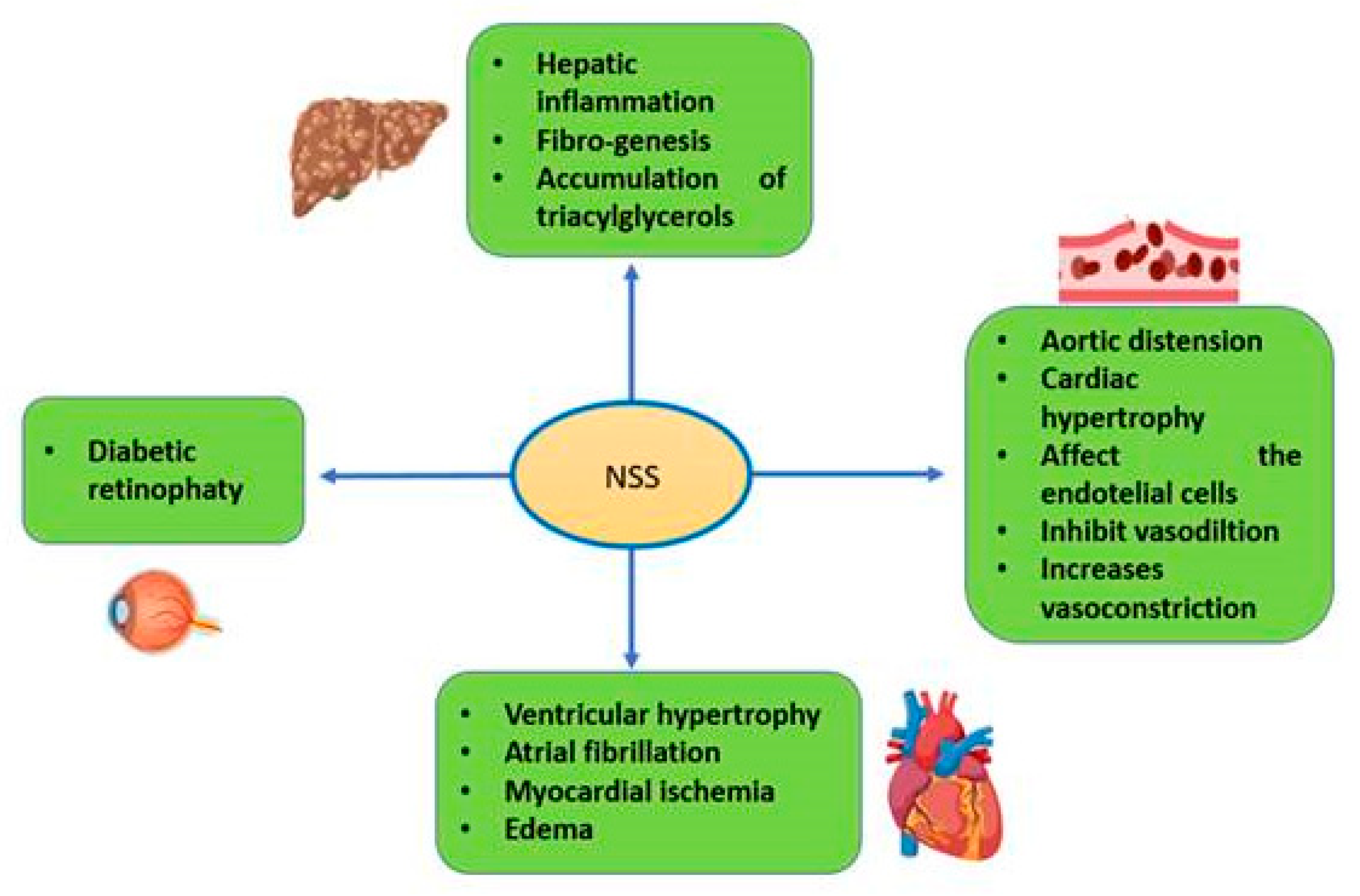
8. Metabolic Disorders
8.1. Diabetes Mellitus
8.2. Diabetic Retinopathy
8.3. Obesity
8.4. Fatty Liver Disease
9. Cardiovascular Disorders
9.1. Endothelial Dysfunction
9.2. Essential Arterial Hypertension and Metabolic Syndrome-Related Hypertension
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pérez-Torres, I.; Guarner-Lans, V.; Rubio-Ruiz, M.E. Reductive Stress in Inflammation-Associated Diseases and the Pro-Oxidant Effect of Antioxidant Agents. Int. J. Mol. Sci. 2017, 18, 2098. [Google Scholar] [CrossRef] [PubMed]
- Alp, N.J.; Channon, K.M. Regulation of Endothelial Nitric Oxide Synthase by Tetrahydrobiopterin in Vascular Disease. Arter. Thromb. Vasc. Biol. 2004, 24, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, J.R. Nitroxidative, Nitrosative, and Nitrative Stress: Kinetic Predictions of Reactive Nitrogen Species Chemistry Under Biological Conditions. Chem. Res. Toxicol. 2006, 19, 1160–1174. [Google Scholar] [CrossRef] [PubMed]
- Touyz, R.M. Reactive Oxygen Species, Vascular Oxidative Stress, and Redox Signaling in Hypertension. Hypertension 2004, 44, 248–252. [Google Scholar] [CrossRef] [Green Version]
- Griendling, K.K.; FitzGerald, G.A. Oxidative stress and cardiovascular injury. Part I: Basic Mechanisms and In Vivo Monitoring of ROS. Circulation 2003, 108, 1912–1916. [Google Scholar] [CrossRef] [Green Version]
- Kurutaş, E.B. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: Current state. Nutr. J. 2016, 15, 71. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, R.; Bredt, D.S.; Snyder, S.H.; Stone, R.A. The localization of nitric oxide synthase in the rat eye and related cranial ganglia. Neuroscience 1993, 54, 189–200. [Google Scholar] [CrossRef]
- Ferdinandy, P.; Danial, H.; Ambrus, I.; Rothery, R.; Schulz, R. Peroxynitrite is a major contributor to cytokine-induced myocardial contractile failure. Circ. Res. 2000, 87, 241–247. [Google Scholar] [CrossRef] [Green Version]
- Tegeder, I. Nitric oxide mediated redox regulation of protein homeostasis. Cell. Signal. 2019, 53, 348–356. [Google Scholar] [CrossRef]
- Moncada, S.; Palmer, R.M.J. Endothelium-Dependent Relaxation and the Discovery of the l-Arginine: NO Pathway. In Endothelial Regulation of Vascular Tone; Ryan, U.S., Rubanyi, G.M., Eds.; Marcel Dekker, Inc.: New York, NY, USA, 1992; pp. 21–36. [Google Scholar]
- Arral, M.L.; Halpern, J. Electrochemical Detection of NG-Hydroxy-l-arginine. ECS Trans. 2018, 85, 1163–1169. [Google Scholar] [CrossRef]
- Chakravarti, B.C.A.D.N. Protein Tyrosine Nitration: Role in Aging. Curr. Aging Sci. 2017, 10, 1. [Google Scholar] [CrossRef] [PubMed]
- Greenacre, S.A.; Ischiropoulos, H. Tyrosine nitration: Localisation, quantification, consequences for protein function and signal transduction. Free. Radic. Res. 2001, 34, 541–581. [Google Scholar] [CrossRef] [PubMed]
- Zielonka, J.; Sikora, A.; Joseph, J.; Kalyanaraman, B. Peroxynitrite Is the Major Species Formed from Different Flux Ratios of Co-generated Nitric Oxide and Superoxide. J. Biol. Chem. 2010, 285, 14210–14216. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Tocchetti, C.G.; Krieg, T.; Moens, A.L. Oxidative and nitrosative stress in the maintenance of myocardial function. Free. Radic. Biol. Med. 2012, 53, 1531–1540. [Google Scholar] [CrossRef]
- Liu, F.; Li, L.; Zhang, B.; Fan, W.; Zhang, R.; Liu, G.; Liu, X. A novel electrochemical sensor based on microporous polymeric nanospheres for measuring peroxynitrite anion released by living cells and studying the synergistic effect of antioxidants. Analytical 2019, 144, 6905–6913. [Google Scholar] [CrossRef]
- Hodges, G.R.; Marwaha, J.; Paul, T.; Ingold, K.U. A novel procedure for generating both nitric oxide and superoxide in situ from chemical sources at any chosen mole ratio. First application: Tyrosine oxidation and a comparison with preformed peroxynitrite. Chem. Res. Toxicol. 2000, 13, 1287–1293. [Google Scholar] [CrossRef]
- Minko, I.G.; Kozekov, I.D.; Harris, T.M.; Rizzo, C.J.; Lloyd, R.S.; Stone, M.P. Chemistry and Biology of DNA Containing 1,N2-Deoxyguanosine Adducts of the α,β-Unsaturated Aldehydes Acrolein, Crotonaldehyde, and 4-Hydroxynonenal. Chem. Res. Toxicol. 2009, 22, 759–778. [Google Scholar] [CrossRef] [Green Version]
- Yamakura, F.; Taka, H.; Fujimura, T.; Murayama, K. Inactivation of human manganese-superoxide dismutase by peroxynitrite is caused by exclusive nitration of tyrosine 34 to 3-nitrotyrosine. J. Biol. Chem. 1998, 273, 14085–14089. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, D.; Erwin, P.A.; Michel, T.; Marletta, M.A. S-Nitrosation and Regulation of Inducible Nitric Oxide Synthase. Biochemistry 2005, 44, 4636–4647. [Google Scholar] [CrossRef]
- Münzel, T.; Daiber, A.; Ullrich, V.; Mülsch, A. Vascular Consequences of Endothelial Nitric Oxide Synthase Uncoupling for the Activity and Expression of the Soluble Guanylyl Cyclase and the cGMP-Dependent Protein Kinase. Arter. Thromb. Vasc. Biol. 2005, 25, 1551–1557. [Google Scholar] [CrossRef]
- Paolocci, N.; Ekelund, U.E.G.; Isoda, T.; Ozaki, M.; Vandegaer, K.; Georgakopoulos, D.; Harrison, R.W.; Kass, D.A.; Hare, J.M. cGMP-independent inotropic effects of nitric oxide and peroxynitrite donors: Potential role for nitrosylation. Am. J. Physiol. Circ. Physiol. 2000, 279, H1982–H1988. [Google Scholar] [CrossRef] [PubMed]
- Gaut, J.P.; Byun, J.; Tran, H.D.; Lauber, W.M.; Carroll, J.A.; Hotchkiss, R.S.; Belaaouaj, A.; Heinecke, J.W. Myeloperoxidase produces nitrating oxidants in vivo. J. Clin. Investig. 2002, 109, 1311–1319. [Google Scholar] [CrossRef] [PubMed]
- Broniowska, K.; Hogg, N. The Chemical Biology of S-Nitrosothiols. Antioxid. Redox Signal. 2012, 17, 969–980. [Google Scholar] [CrossRef] [PubMed]
- Heidrich, F.M.; Jercke, M.C.; Ritzkat, A.; Ebner, A.; Poitz, D.M.; Pfluecke, C.; Quick, S.; Speiser, U.; Simonis, G.; Wäßnig, N.K.; et al. The endothelial nitric oxide synthase cofactor tetrahydrobiopterin shields the remote myocardium from apoptosis after experimental myocardial infarction in vivo. Kardiol. Pol. 2017, 75, 1339–1350. [Google Scholar] [CrossRef] [Green Version]
- Kuzkaya, N.; Weissmann, N.; Harrison, D.G.; Dikalov, S. Interactions of Peroxynitrite, Tetrahydrobiopterin, Ascorbic Acid, and Thiols: Implications for Uncoupling Endothelial Nitric-Oxide Synthase. J. Biol. Chem. 2003, 278, 22546–22554. [Google Scholar] [CrossRef] [Green Version]
- Vasquez-Vivar, J.; Martasek, P.; Whitsett, J.; Joseph, J.; Kalyanaraman, B. The ratio between tetrahydrobiopterin and oxidized tetrahydrobiopterin analogues controls superoxide release from endothelial nitric oxide synthase: An EPR spin trapping study. Biochem. J. 2002, 362, 733–739. [Google Scholar] [CrossRef]
- Durante, W.; Johnson, F.K.; Johnson, R.A. Arginase: A Critical Regulator of Nitric Oxide Synthesis and Vascular Function. Clin. Exp. Pharmacol. Physiol. 2007, 34, 906–911. [Google Scholar] [CrossRef] [Green Version]
- Bevers, L.M.; Braam, B.; Post, J.A.; Van Zonneveld, A.J.; Rabelink, T.J.; Koomans, H.A.; Verhaar, M.C.; Joles, J. Tetrahydrobiopterin, but Not l-Arginine, Decreases NO Synthase Uncoupling in Cells Expressing High Levels of Endothelial NO Synthase. Hypertension 2006, 47, 87–94. [Google Scholar] [CrossRef] [Green Version]
- Crijns, H.J.; Schotten, U.; Moens, A.L. Is NOS uncoupling the missing link between atrial fibrillation and chronic non-ischaemic cardiomyopathy? Cardiovasc. Res. 2011, 91, 556–558. [Google Scholar] [CrossRef]
- Lim, G.; Venetucci, L.; Eisner, D.; Casadei, B. Does nitric oxide modulate cardiac ryanodine receptor function? Implications for excitation-contraction coupling. Cardiovasc. Res. 2007, 77, 256–264. [Google Scholar] [CrossRef]
- Tocchetti, C.G.; Stanley, B.A.; Murray, C.I.; Sivakumaran, V.; Donzelli, S.; Mancardi, D.; Pagliaro, P.; Gao, W.D.; Van Eyk, J.; Kass, D.A.; et al. Playing with cardiac ‘‘redox switches’’: The ‘‘HNO way’’ to modulate cardiac function. Antioxid. Redox. Signal. 2011, 14, 1687–1698. [Google Scholar] [CrossRef] [Green Version]
- Kohr, M.J.; Kaludercic, N.; Tocchetti, C.G.; Dong, W.; Kass, D.A.; Janssen, P.M.; Paolocci, N.; Ziolo, M.T. Nitroxyl enhances myocyte Ca2þ transients by exclusively targeting SR Ca2þ-cycling. Front. Biosci. 2010, 2, 614–626. [Google Scholar]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2011, 33, 829–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gliozzi, M.; Scicchitano, M.; Bosco, F.; Musolino, V.; Carresi, C.; Scarano, F.; Maiuolo, J.; Nucera, S.; Maretta, A.; Paone, S.; et al. Modulation of Nitric Oxide Synthases by Oxidized LDLs: Role in Vascular Inflammation and Atherosclerosis Development. Int. J. Mol. Sci. 2019, 20, 3294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Torres, I.; Guarner, V.; El Hafidi, M.; Baños, G. Sex hormones, metabolic syndrome and kidney. Curr. Top. Med. Chem. 2011, 11, 1694–1705. [Google Scholar] [CrossRef]
- Ischiropoulos, H. Biological selectivity and functional aspects of protein tyrosine nitration. Biochem. Biophys. Res. Commun. 2003, 305, 776–783. [Google Scholar] [CrossRef]
- Radi, R. Nitric oxide, oxidants, and protein tyrosine nitration. Proc. Natl. Acad. Sci. USA 2004, 101, 4003–4008. [Google Scholar] [CrossRef] [Green Version]
- Wolin, M.S. Interactions of oxidants with vascular signaling systems. Arter. Thromb. Vasc. Biol. 2000, 20, 1430–1442. [Google Scholar] [CrossRef] [Green Version]
- Miseta, A.; Csutora, P. Relationship between the occurrence of cysteine in proteins and the complexity of organisms. Mol. Biol. Evol. 2000, 17, 1232–1239. [Google Scholar] [CrossRef] [Green Version]
- Landar, A.; Oh, J.-Y.; Giles, N.M.; Isom, A.; Kirk, M.; Barnes, S.; Darley-Usmar, V. A sensitive method for the quantitative measurement of protein thiol modification in response to oxidative stress. Free Radic. Biol. Med. 2006, 40, 459–468. [Google Scholar] [CrossRef]
- Trachootham, D.; Lu, W.; Ogasawara, M.A.; Valle, N.R.-D.; Huang, P. Redox Regulation of Cell Survival. Antioxid. Redox Signal. 2008, 10, 1343–1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benhar, M.; Forrester, M.T.; Stamler, J.S. Protein denitrosylation: Enzymatic mechanisms and cellular functions. Nat. Rev. Mol. Cell. Biol. 2009, 10, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Nakato, R.; Ohkubo, Y.; Konishi, A.; Shibata, M.; Kaneko, Y.; Iwawaki, T.; Nakamura, T.; Lipton, S.A.; Uehara, T. Regulation of the unfolded protein response via S-nitrosylation of sensors of endoplasmic reticulum stress. Sci. Rep. 2015, 5, 14812. [Google Scholar] [CrossRef] [PubMed]
- Beigi, F.; Gonzalez, D.R.; Minhas, K.M.; Sun, Q.-A.; Foster, M.W.; Khan, S.A.; Treuer, A.V.; Dulce, R.A.; Harrison, R.W.; Saraiva, R.M.; et al. Dynamic denitrosylation via S-nitrosoglutathione reductase regulates cardiovascular function. Proc. Natl. Acad. Sci. USA 2012, 109, 4314–4319. [Google Scholar] [CrossRef] [Green Version]
- Diers, A.R.; Broniowska, K.; Hogg, N. Nitrosative stress and redox-cycling agents synergize to cause mitochondrial dysfunction and cell death in endothelial cells. Redox Biol. 2013, 1, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Bedard, K.; Krause, K.-H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Park, J.-W.; Yang, E.S.; Park, J.-W. Inactivation of NADP+-dependent Isocitrate Dehydrogenase by Peroxynitrite. J. Biol. Chem. 2003, 278, 51360–51371. [Google Scholar] [CrossRef] [Green Version]
- Mailloux, R.J. Mitochondrial Antioxidants and the Maintenance of Cellular Hydrogen Peroxide Levels. Oxid. Med. Cell. Longev. 2018, 2018, 1–10. [Google Scholar] [CrossRef]
- Moon, K.-H.; Hood, B.L.; Kim, B.-J.; Hardwick, J.P.; Conrads, T.P.; Veenstra, T.D.; Song, B.J. Inactivation of oxidized andS-nitrosylated mitochondrial proteins in alcoholic fatty liver of rats. Hepatology 2006, 44, 1218–1230. [Google Scholar] [CrossRef]
- Wallace, K. Mitochondrial off targets of drug therapy. Trends Pharmacol. Sci. 2008, 29, 361–366. [Google Scholar] [CrossRef]
- Lu, H.; Koshkin, V.; Allister, E.M.; Gyulkhandanyan, A.V.; Wheeler, M.B. Molecular and Metabolic Evidence for Mitochondrial Defects Associated With β-Cell Dysfunction in a Mouse Model of Type 2 Diabetes. Diabetes 2009, 59, 448–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric Oxide and Peroxynitrite in Health and Disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riobó, N.; Clementi, E.; Melani, M.; Boveris, A.; Cadenas, E.; Moncada, S.; Poderoso, J.J. Nitric oxide inhibits mitochondrial NADH: Ubiquinone reductase activity through peroxynitrite formation. Biochem. J. 2001, 359, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Apostolova, N.; Garcia-Bou, R.; Hernandez-Mijares, A.; Herance, R.; Rocha, M.; Victor, V.M.; Herance, J.R. Mitochondrial Antioxidants Alleviate Oxidative and Nitrosative Stress in a Cellular Model of Sepsis. Pharm. Res. 2011, 28, 2910–2919. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, P.; Moinuddin; Dixit, K.; Mir, A.R.; Habib, S.; Alam, K.; Ali, A. Peroxynitrite modified DNA presents better epitopes for anti-DNA autoantibodies in diabetes type 1 patients. Cell. Immunol. 2014, 290, 30–38. [Google Scholar] [CrossRef]
- Lange, M.; Connelly, R.; Traber, D.L.; Hamahata, A.; Nakano, Y.; Esechie, A.; Jonkam, C.; Von Borzyskowski, S.; Traber, L.D.; Schmalstieg, F.C.; et al. Time course of nitric oxide synthases, nitrosative stress, and poly(ADP ribosylation) in an ovine sepsis model. Crit. Care 2010, 14, R129. [Google Scholar] [CrossRef] [Green Version]
- Jagtap, P.; Szabo, C. Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat. Rev. Drug Discov. 2005, 4, 421–440. [Google Scholar] [CrossRef]
- Mathews, M.T.; Berk, B.C. PARP-1 Inhibition Prevents Oxidative and Nitrosative Stress–Induced Endothelial Cell Death via Transactivation of the VEGF Receptor 2. Arter. Thromb. Vasc. Biol. 2008, 28, 711–717. [Google Scholar] [CrossRef] [Green Version]
- Radovits, T.; Lin, L.-N.; Zotkina, J.; Gero, D.; Szabo, C.; Karck, M.; Szabo, G. Poly(ADP-ribose) polymerase inhibition improves endothelial dysfunction induced by reactive oxidant hydrogen peroxide in vitro. Eur. J. Pharmacol. 2007, 564, 158–166. [Google Scholar] [CrossRef]
- Szabo, C. Role of nitrosative stress in the pathogenesis of diabetic vascular dysfunction. Br. J. Pharmacol. 2009, 156, 713–727. [Google Scholar] [CrossRef] [Green Version]
- Purwar, N.; McGarry, J.M.; Kostera, J.; Pacheco, A.A.; Schmidt, M. Interaction of Nitric Oxide with Catalase: Structural and Kinetic Analysis. Biochemistry 2011, 50, 4491–4503. [Google Scholar] [CrossRef] [PubMed]
- Chakravarti, R.; Gupta, K.; Majors, A.; Ruple, L.; Aronica, M.; Stuehr, D.J. Novel insights in mammalian catalase heme maturation: Effect of NO and thioredoxin-1. Free Radic. Biol. Med. 2015, 82, 105–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Díaz-Vilchis, A.; Loewen, P.C.; Fita, I.; Carpena, X. Thirty years of heme catalases structural biology. Arch. Biochem. Biophys. 2012, 525, 102–110. [Google Scholar] [CrossRef]
- Davis, K.L.; Martin, E.; Turko, I.V.; Murad, F. Novel effects of nitric oxide. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 203–236. [Google Scholar] [CrossRef]
- Brunelli, L.; Yermilov, V.; Beckman, J.S. Modulation of catalase peroxidatic and catalatic activity by nitric oxide. Free Radic. Biol. Med. 2001, 30, 709–714. [Google Scholar] [CrossRef]
- Pérez-Torres, I.; Torres-Narváez, J.C.; Guarner-Lans, V.; Díaz-Díaz, E.; Perezpeña-Diazconti, M.; Palacios, A.R.; Manzano-Pech, L. Myocardial Protection from Ischemia-Reperfusion Damage by the Antioxidant Effect of Hibiscus sabdariffa Linnaeus on Metabolic Syndrome Rats. Oxid. Med. Cell. Longev. 2019, 2019, 1724194. [Google Scholar] [CrossRef] [Green Version]
- Soto, M.E.; Zuñiga-Muñoz, A.; Guarner-Lans, V.; Duran-Hernández, E.J.; Pérez-Torres, I. Infusion ofHibiscus sabdariffa L.Modulates Oxidative Stress in Patients with Marfan Syndrome. Mediat. Inflamm. 2016, 2016, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.; Berk, M.; Klein, H.; Walder, K.; Galecki, P.; Maes, M. Nitrosative Stress, Hypernitrosylation, and Autoimmune Responses to Nitrosylated Proteins: New Pathways in Neuroprogressive Disorders Including Depression and Chronic Fatigue Syndrome. Mol. Neurobiol. 2016, 54, 4271–4291. [Google Scholar] [CrossRef]
- Quijano, C.; Hernandez-Saavedra, D.; Castro, L.; McCord, J.M.; Freeman, B.A.; Radi, R. Reaction of peroxynitrite with Mn-superoxide dismutase. Role of the metal center in decomposition kinetics and nitration. J. Biol. Chem. 2001, 276, 11631–11638. [Google Scholar] [CrossRef] [Green Version]
- Schiffrin, E.L. Oxidative stress, nitric oxide synthase, and superoxide dismutase: A matter of imbalance underlies endothelial dysfunction in the human coronary circulation. Hypertension 2007, 51, 31–32. [Google Scholar] [CrossRef] [Green Version]
- Yamakura, F.; Kawasaki, H. Post-translational modifications of superoxide dismutase. Biochim. Biophys. Acta BBA Proteins Proteom. 2010, 1804, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Franco, M.C.; Estevez, A.G. Tyrosine nitration as mediator of cell death. Cell. Mol. Life Sci. 2014, 71, 3939–3950. [Google Scholar] [CrossRef] [PubMed]
- Arora, D.; Jain, P.; Singh, N.; Kaur, H.; Bhatla, S.C. Mechanisms of nitric oxide crosstalk with reactive oxygen species scavenging enzymes during abiotic stress tolerance in plants. Free Radic. Res. 2016, 50, 291–303. [Google Scholar] [CrossRef] [PubMed]
- Romero-Puertas, M.C.; Laxa, M.; Mattè, A.; Zaninotto, F.; Finkemeier, I.; Jones, A.M.; Perazzolli, M.; Vandelle, E.G.G.; Dietz, K.-J.; Delledonne, M. S-Nitrosylation of Peroxiredoxin II E Promotes Peroxynitrite-Mediated Tyrosine Nitration. Plant Cell 2007, 19, 4120–4130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pech, L.G.M.; Caballero-Chacón, S.D.C.; Guarner-Lans, V.; Díaz-Díaz, E.; Gómez, A.M.; Pérez-Torres, I. Effect of oophorosalpingo-hysterectomy on serum antioxidant enzymes in female dogs. Sci. Rep. 2019, 9, 9674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Presnell, C.E.; Bhatti, G.; Numan, L.S.; Lerche, M.; Alkhateeb, S.K.; Ghalib, M.; Shammaa, M.; Kavdia, M. Computational insights into the role of glutathione in oxidative stress. Curr. Neurovasc. Res. 2013, 10, 185–194. [Google Scholar] [CrossRef]
- Yamada, T.; Fedotovskaya, O.; Cheng, A.J.; Cornachione, A.S.; Minozzo, F.C.; Aulin, C.; Fridén, C.; Turesson, C.; Andersson, D.; Glenmark, B.; et al. Nitrosative modifications of the Ca2+release complex and actin underlie arthritis-induced muscle weakness. Ann. Rheum. Dis. 2014, 74, 1907–1914. [Google Scholar] [CrossRef] [Green Version]
- Laroux, F.S.; Lefer, D.J.; Kawachi, S.; Scalia, R.; Cockrell, A.S.; Gray, L.; Van Der Heyde, H.; Hoffman, J.M.; Grisham, M.B. Role of Nitric Oxide in the Regulation of Acute and Chronic Inflammation. Antioxid. Redox Signal. 2000, 2, 391–396. [Google Scholar] [CrossRef]
- Van Der Goes, A.; Wouters, D.; Pol, S.M.A.; Huizinga, R.; Ronken, E.; Adamson, P.; Greenwood, J.; Dijkstra, C.D.; Vries, H.E. Reactive oxygen species enhance the migration of monocytes across the blood-brain barrier in vitro. FASEB J. 2001, 15, 1852–1854. [Google Scholar] [CrossRef]
- Ceriello, A.; Quagliaro, L.; Catone, B.; Pascon, R.C.; Piazzola, M.; Bais, B.; Marra, G.; Tonutti, L.; Taboga, C.; Motz, E. Role of hyperglycemia in nitrotyrosine postprandial generation. Diabetes Care 2002, 25, 1439–1443. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Lee, K.-S.; Choi, S.; Kim, J.; Lee, D.-K.; Park, M.; Park, W.; Kim, T.-H.; Hwang, J.Y.; Won, M.H.; et al. NF-κB–responsive miRNA-31-5p elicits endothelial dysfunction associated with preeclampsia via down-regulation of endothelial nitric-oxide synthase. J. Biol. Chem. 2018, 293, 18989–19000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Veremeyko, T.; Wong, A.H.K.; El Fatimy, R.; Wei, Z.; Cai, W.; Krichevsky, A.M. Downregulation of miR-132/212 impairs S-nitrosylation balance and induces tau phosphorylation in Alzheimer’s disease. Neurobiol. Aging. 2017, 51, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, C.S.; Gutterman, D. Focus on oxidative stress in the cardiovascular and renal systems. Am. J. Physiol. Circ. Physiol. 2005, 288, H3–H6. [Google Scholar] [CrossRef] [PubMed]
- Nagareddy, P.R.; Xia, Z.; McNeill, J.H.; MacLeod, K.M. Increased expression of iNOS is associated with endothelial dysfunction and impaired pressor responsiveness in streptozotocin-induced diabetes. Am. J. Physiol. Circ. Physiol. 2005, 289, H2144–H2152. [Google Scholar] [CrossRef] [PubMed]
- Piconi, L.; Quagliaro, L.; Da Ros, R.; Assaloni, R.; Giugliano, D.; Esposito, K.; Szabó, C.; Ceriello, A. Intermittent high glucose enhances ICAM-1, VCAM-1, E-selectin and interleukin-6 expression in human umbilical endothelial cells in culture: The role of poly(ADP-ribose) polymerase. J. Thromb. Haemost. 2004, 2, 1453–1459. [Google Scholar] [CrossRef]
- Cheng, X.; Pang, C.C. Increased vasoconstriction to noradrenaline by 1400W, inhibitor of iNOS, in rats with streptozotocin-induced diabetes. Eur. J. Pharmacol. 2004, 484, 263–268. [Google Scholar] [CrossRef]
- Pop-Busui, R.; Oral, E.A.; Raffel, D.; Byun, J.; Bajirovic, V.; Vivekanandan-Giri, A.; Kellogg, A.; Pennathur, S.; Stevens, M.J. Impact of rosiglitazone and glyburide on nitrosative stress and myocardial blood flow regulation in type 2 diabetes mellitus. Metabolism 2009, 58, 989–994. [Google Scholar] [CrossRef]
- Guzik, T.J.; West, N.E.J.; Black, E.; McDonald, D.; Ratnatunga, C.; Pillai, R.; Channon, K.M. Vascular superoxide production by NAD(P)H oxidase: Association with endothelial dysfunction and clinical risk factors. Circ. Res. 2000, 86, 85. [Google Scholar] [CrossRef] [Green Version]
- Szabó, C.; Zanchi, A.; Komjáti, K.; Pacher, P.; Krolewski, A.S.; Quist, W.C.; LoGerfo, F.W.; Horton, E.S.; Veves, A. Poly(ADP-Ribose) Polymerase Is Activated in Subjects at Risk of Developing Type 2 Diabetes and Is Associated With Impaired Vascular Reactivity. Circulation 2002, 106, 2680–2686. [Google Scholar] [CrossRef] [Green Version]
- Du, Y.; Smith, M.A.; Miller, C.M.; Kern, T.S. Diabetes-induced nitrative stress in the retina, and correction by aminoguanidine. J. Neurochem. 2002, 80, 771–779. [Google Scholar] [CrossRef]
- Leal, E.C.; Aveleira, C.; Castilho, Á.F.; Serra, A.M.; Baptista, F.; Hosoya, K.-I.; Forrester, J.V.; Ambrósio, A.F. High glucose and oxidative/nitrosative stress conditions induce apoptosis in retinal endothelial cells by a caspase-independent pathway. Exp. Eye Res. 2009, 88, 983–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bereczki, D.; Balla, J. Heme Oxygenase-1: Clinical Relevance in Ischemic Stroke. Curr. Pharm. Des. 2018, 24, 2229–2235. [Google Scholar] [CrossRef] [PubMed]
- Castilho, Á.F.; Aveleira, C.; Leal, E.C.; Simões, N.F.; Fernandes, C.R.; Meirinhos, R.I.; Baptista, F.; Ambrósio, A.F. Heme Oxygenase-1 Protects Retinal Endothelial Cells against High Glucose- and Oxidative/Nitrosative Stress-Induced Toxicity. PLoS ONE 2012, 7, e42428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández-Ramírez, E.; Sánchez-Chávez, G.; Estrella-Salazar, L.A.; Salceda, R. Nitrosative Stress in the Rat Retina at the Onset of Streptozotocin-Induced Diabetes. Cell. Physiol. Biochem. 2017, 42, 2353–2363. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Saxena, S.; Srivastav, K.; Shukla, R.K.; Mishra, N.; Meyer, C.H.; Kruzliak, P.; Khanna, V.K. Nitric oxide and oxidative stress is associated with severity of diabetic retinopathy and retinal structural alterations. Clin. Exp. Ophthalmol. 2015, 43, 429–436. [Google Scholar] [CrossRef]
- Obrosova, I.G.; Kador, P.F. Aldose reductase/polyol inhibitors for diabetic retinopathy. Curr. Pharm. Biotechnol. 2011, 12, 373–385. [Google Scholar] [CrossRef]
- Rajesh, M.; Sulochana, K.N.; Punitham, R.; Biswas, J.; Lakshmi, S.; Ramakrishnan, S. Involvement of oxidative and nitrosative stress in promoting retinal vasculitis in patients with Eales’ disease. Clin. Biochem. 2003, 36, 377–385. [Google Scholar] [CrossRef]
- Morelli, N.R.; Scavuzzi, B.M.; Miglioranza, L.H.D.S.; Lozovoy, M.A.B.; Simão, A.N.C.; Dichi, I. Metabolic syndrome components are associated with oxidative stress in overweight and obese patients. Arch. Endocrinol. Metab. 2018, 62, 309–318. [Google Scholar] [CrossRef] [Green Version]
- Rubio-Ruiz, M.E.; Peredo-Escárcega, A.E.; Cano-Martínez, A.; Guarner-Lans, V. An Evolutionary Perspective of Nutrition and Inflammation as Mechanisms of Cardiovascular Disease. Int. J. Evol. Biol. 2015, 2015, 1–10. [Google Scholar] [CrossRef]
- Ovadia, H.; Haim, Y.; Nov, O.; Almog, O.; Kovsan, J.; Bashan, N.; Benhar, M.; Rudich, A. Increased Adipocyte S-Nitrosylation Targets Anti-lipolytic Action of Insulin. J. Biol. Chem. 2011, 286, 30433–30443. [Google Scholar] [CrossRef] [Green Version]
- Choi, M.S.; Jung, J.-Y.; Kim, H.-J.; Ham, M.R.; Lee, T.R.; Shin, N.W. S-nitrosylation of fatty acid synthase regulates its activity through dimerization. J. Lipid Res. 2016, 57, 607–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Torres, I.; Gutiérrez-Alvarez, Y.; Guarner-Lans, V.; Díaz-Díaz, E.; Pech, L.M.; Caballero-Chacón, S.D.C. Intra-Abdominal Fat Adipocyte Hypertrophy through a Progressive Alteration of Lipolysis and Lipogenesis in Metabolic Syndrome Rats. Nutrition 2019, 11, 1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikura, Y.; Ohsawa, M.; Suekane, T.; Fukushima, H.; Itabe, H.; Jomura, H.; Nishiguchi, S.; Inoue, T.; Naruko, T.; Ehara, S.; et al. Localization of oxidized phosphatidylcholine in nonalcoholic fatty liver disease: Impact on disease progression. Hepatology 2006, 43, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Botham, K.M.; Zheng, X.; Napoletano, M.; Avella, M.; Cavallari, C.; Rivabene, R.; Bravo, E. The effects of dietary n3 polyunsaturated fatty acids delivered in chylomicron renmants on the transcription of genes regulating synthesis and secretion of very-lowdensity lipoprotein by the liver: Modulation by cellular oxidative state. Exp. Biol. Med. 2003, 228, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Song, B.-J.; Abdelmegeed, M.A.; Henderson, L.E.; Yoo, S.-H.; Wan, J.; Purohit, V.; Hardwick, J.P.; Moon, K.-H. Increased Nitroxidative Stress Promotes Mitochondrial Dysfunction in Alcoholic and Nonalcoholic Fatty Liver Disease. Oxid. Med. Cell. Longev. 2013, 2013, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pennathur, S.; Heinecke, J.W. Mechanisms for Oxidative Stress in Diabetic Cardiovascular Disease. Antioxid. Redox Signal. 2007, 9, 955–969. [Google Scholar] [CrossRef] [PubMed]
- Keira, N.; Tatsumi, T.; Matoba, S.; Shiraishi, J.; Yamanaka, S.; Akashi, K.; Kobara, M.; Asayama, J.; Fushiki, S.; Fliss, H.; et al. Lethal Effect of Cytokine-induced Nitric Oxide and Peroxynitrite on Cultured Rat Cardiac Myocytes. J. Mol. Cell. Cardiol. 2002, 34, 583–596. [Google Scholar] [CrossRef]
- Cheng, X.; Kuo, K.-H.; Pang, C.C. Inhibition of iNOS augments cardiovascular action of noradrenaline in streptozotocin-induced diabetes. Cardiovasc. Res. 2004, 64, 298–307. [Google Scholar] [CrossRef]
- Baldelli, S.; Ciriolo, M.R. Altered S-nitrosylation of p53 is responsible for impaired antioxidant response in skeletal muscle during aging. Aging 2016, 8, 3450–3467. [Google Scholar] [CrossRef] [Green Version]
- Burwell, L.; Nadtochiy, S.M.; Tompkins, A.; Young, S.; Brookes, P.S. Direct evidence for S-nitrosation of mitochondrial complex I. Biochem. J. 2006, 394, 627–634. [Google Scholar] [CrossRef]
- Brown, G.C. Regulation of mitochondrial respiration by nitric oxide inhibition of cytochrome c oxidase. Biochim. Biophys. Acta BBA Bioenerg. 2001, 1504, 46–57. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.Y.; Griendling, K. Redox Signaling, Vascular Function, and Hypertension. Antioxid. Redox Signal. 2008, 10, 1045–1059. [Google Scholar] [CrossRef] [PubMed]
- Julius, U.; Drel, V.; Grässler, J.; Obrosova, I.G.; Gräßler, J. Nitrosylated proteins in monocytes as a new marker of oxidative-nitrosative stress in diabetic subjects with macroangiopathy. Exp. Clin. Endocrinol. Diabetes 2008, 117, 72–77. [Google Scholar] [CrossRef] [Green Version]
- Csányi, G.; Taylor, W.R.; Pagano, P.J. NOX and inflammation in the vascular adventitia. Free. Radic. Biol. Med. 2009, 47, 1254–1266. [Google Scholar] [CrossRef] [Green Version]
- Bertuglia, S.; Giusti, A. Microvascular oxygenation, oxidative stress, NO suppression and superoxide dismutase during postischemic reperfusion. Am. J. Physiol. Circ. Physiol. 2003, 285, H1064–H1071. [Google Scholar] [CrossRef] [Green Version]
- Hogg, N.; Broniowska, K.A.; Novalija, J.; Kettenhofen, N.J.; Novalija, E. Role of S-nitrosothiol transport in the cardioprotective effects of S-nitrosocysteine inrat hearts. Free Radic. Biol. Med. 2007, 43, 1086–1094. [Google Scholar] [CrossRef]
- Prime, T.A.; Blaikie, F.H.; Evans, C.; Nadtochiy, S.M.; James, A.M.; Dahm, C.C.; Vitturi, D.A.; Patel, R.P.; Hiley, C.R.; Abakumova, I.; et al. A mitochondria-targeted S-nitro-sothiol modulates respiration, nitrosates thiols, and protects againstischemia-reperfusion injury. Proc. Natl. Acad. Sci. USA 2009, 106, 10764–10769. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, P.; Rajesh, M.; Bátkai, S.; Kashiwaya, Y.; Haskó, G.; Liaudet, L.; Szabó, C.; Pacher, P. Role of superoxide, nitric oxide, and peroxynitrite in doxorubicin-induced cell death in vivo and in vitro. Am. J. Physiol. Circ. Physiol. 2009, 296, H1466–H1483. [Google Scholar] [CrossRef] [Green Version]
- Forstermann, U.; Munzel, T. Endothelial Nitric Oxide Synthase in Vascular Disease. Circulation 2006, 113, 1708–1714. [Google Scholar] [CrossRef] [Green Version]
- Wood, K.C.; Hebbel, R.P.; Lefer, D.J.; Granger, D.N. Critical role of endothelial cell-derived nitric oxide synthase in sickle cell disease-induced microvascular dysfunction. Free Radic. Biol. Med. 2006, 40, 1443–1453. [Google Scholar] [CrossRef]
- Tousoulis, D.; Kampoli, A.-M.; Papageorgiou, C.T.N.; Stefanadis, C. The Role of Nitric Oxide on Endothelial Function. Curr. Vasc. Pharmacol. 2012, 10, 4–18. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, M.J.; Smith, C.L.; Lam, G.; Goligorsky, M.S.; Gross, S. Ratio of 5,6,7,8-tetrahydrobiopterin to 7,8-dihydrobiopterin in endothelial cells determines glucose-elicited changes in NO vs. superoxide production by eNOS. Am. J. Physiol. Circ. Physiol. 2008, 294, H1530–H1540. [Google Scholar] [CrossRef] [Green Version]
- Kar, S.; Kavdia, M. Endothelial NO and O2-production rates differentially regulate oxidative, nitroxidative, and nitrosative stress in the microcirculation. Free Radic. Biol. Med. 2013, 63, 161–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Lin, S.C.; Nadershahi, A.; Watts, S.W.; Sarkar, R. Role of redox signaling and poly (adenosine diphosphate-ribose) polymerase activation in vascular smooth muscle cell growth inhibition by nitric oxide and peroxynitrite. J. Vasc. Surg. 2008, 47, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Stasch, J.-P.; Schmidt, P.M.; Nedvetsky, P.; Nedvetskaya, T.Y.; Arun Kumar, H.S.; Meurer, S.; Deile, M.; Taye, A.; Knorr, A.; Lapp, H.; et al. Targeting the heme-oxidized nitric oxide receptor for selective vasodilatation of diseased blood vessels. J. Clin. Investig. 2006, 116, 2552–2561. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.; Allahdadi, K.J.; Tostes, R.C.; Webb, R.C. Augmented S-nitrosylation contributes to impaired relaxation in angiotensin II hypertensive mouse aorta. J. Hypertens. 2011, 29, 2359–2368. [Google Scholar] [CrossRef] [Green Version]
- Crassous, P.-A.; Couloubaly, S.; Huang, C.; Zhou, Z.; Baskaran, P.; Kim, D.D.; Papapetropoulos, A.; Fioramonti, X.; Durán, W.N.; Beuve, A. Soluble guanylyl cyclase is a target of angiotensin II-induced nitrosative stress in a hypertensive rat model. Am. J. Physiol. Circ. Physiol. 2012, 303, H597–H604. [Google Scholar] [CrossRef] [Green Version]
- Cai, H.; Griendling, K.; Harrison, D.G. The vascular NAD(P)H oxidases as therapeutic targets in cardiovascular diseases. Trends Pharmacol. Sci. 2003, 24, 471–478. [Google Scholar] [CrossRef]
- Mihm, M.J.; Wattanapitayakul, S.K.; Piao, S.-F.; Hoyt, D.G.; Bauer, J.A. Effects of angiotensin II on vascular endothelial cells: Formation of receptor-mediated reactive nitrogen species. Biochem. Pharmacol. 2003, 65, 1189–1197. [Google Scholar] [CrossRef]
- Satoh, M.; Fujimoto, S.; Arakawa, S.; Yada, T.; Namikoshi, T.; Haruna, Y.; Horike, H.; Sasaki, T.; Kashihara, N. Angiotensin II type 1 receptor blocker ameliorates uncoupled endothelial nitric oxide synthase in rats with experimental diabetic nephropathy. Nephrol. Dial. Transplant. 2008, 23, 3806–3813. [Google Scholar] [CrossRef] [Green Version]
- Kase, H.; Hashikabe, Y.; Uchida, K.; Nakanishi, N.; Hattori, Y. Supplementation with tetrahydrobiopterin prevents the cardiovascular effects of angiotensin II-induced oxidative and nitrosative stress. J. Hypertens. 2005, 23, 1375–1382. [Google Scholar] [CrossRef] [PubMed]
- Nie, H.; Wu, J.-L.; Zhang, M.; Xu, J.; Zou, M.-H. Endothelial Nitric Oxide Synthase-Dependent Tyrosine Nitration of Prostacyclin Synthase in Diabetes In Vivo. Diabetes 2006, 55, 3133–3141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kannel, W.B. Prevalence and implications of uncontrolled systolic hypertension. Drugs Aging 2003, 20, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Berk, B.C. Vascular smooth muscle growth: Autocrine growth mechanisms. Physiol. Rev. 2001, 81, 999–1030. [Google Scholar] [CrossRef]
- Zalba, G.; Beaumont, F.J.; José, G.S.; Fortuño, A.; Díez, J. Is the balance between nitric oxide and superoxide altered in spontaneously hypertensive rats with endothelial dysfunction? Nephrol. Dial. Transplant. 2001, 16, 2–5. [Google Scholar] [CrossRef]
- Somoza, B.; González, C.; Cachofeiro, V.; Lahera, V.; Fernandez-Alfonso, M.S. Chronic l-arginine treatment reduces vascular smooth muscle cell hypertrophy through cell cycle modifications in spontaneously hypertensive rats. J. Hypertens. 2004, 22, 751–758. [Google Scholar] [CrossRef]
- Cabassi, A.; Dumont, E.C.; Girouard, H.; Bouchard, J.F.; Jossec, M.L.; Lamontagne, D.; Besner, J.G.; De Champlain, J. Effects of chronic N-acetylcysteine treatment on the actions of peroxinitrite on aortic vascular reactivity in hypertensive rats. J. Hypertens. 2001, 19, 1233–1244. [Google Scholar] [CrossRef] [Green Version]
- Saternos, H.C.; Almarghalani, D.A.; Gibson, H.M.; Meqdad, M.A.; Antypas, R.B.; Lingireddy, A.; AbouAlaiwi, W.A. Distribution and function of the muscarinic receptor subtypes in the cardiovascular system. Physiol. Genom. 2018, 50, 1–9. [Google Scholar] [CrossRef]
- Hang, P.; Zhao, J.; Qi, J.; Wang, Y.; Wu, J.; Du, Z. Novel insights into the pervasive role of M3 muscarinic receptor in cardiac diseases. Curr. Drug Targets 2013, 14, 372–377. [Google Scholar]
- Schmidt, P.; Youhnovski, N.; Daiber, A.; Balan, A.; Arsic, M.; Bachschmid, M.; Przybylski, M.; Ullrich, V. Specific nitration at tyrosine 430 revealed by high resolution mass spectrometry as basis for redox regulation of bovine prostacyclin synthase. J. Biol. Chem. 2003, 278, 12813–12819. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, S.; Gross, C.M.; Kumar, S.; Datar, S.; Oishi, P.; Kalkan, G.; Schreiber, C.; Fratz, S.; Fineman, J.R.; Black, S.M.; et al. Attenuated vasodilatation in lambs with endogenous and exogenous activation of cGMP signaling: Role of protein kinase G nitration. J. Cell. Physiol. 2011, 226, 3104–3113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmerman, M.C.; Lazartigues, E.; Sharma, R.V.; Davisson, R.L. Hypertension Caused by Angiotensin II Infusion Involves Increased Superoxide Production in the Central Nervous System. Circ. Res. 2004, 95, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Francis, J.; Davisson, R. Emerging Concepts in Hypertension. Antioxid. Redox Signal. 2014, 20, 69–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, C.X.; Nabeebaccus, A.A.; Shah, A.; Camargo, L.L.; Filho, S.V.; Lopes, L. Endoplasmic Reticulum Stress and Nox-Mediated Reactive Oxygen Species Signaling in the Peripheral Vasculature: Potential Role in Hypertension. Antioxid. Redox Signal. 2014, 20, 121–134. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolic Disorders | Alterations |
|---|---|
| Diabetes mellitus | ↑ONOO−, 3-nitrotyrosine, iNOS, NAD(P)H oxidase, O2− (uncoupled eNOS), HbA1c, ICAM, VCAM [81,88,89,90] |
| Diabetic retinopathy | ↑Aldose reductase activity [97] ↓GSH, Ascorbate, Taurine, Na/KATP-ase activity [114] ○PARP-1 [97] ●HO-1 [101] |
| Obesity | ↑iNOS, NO [101] |
| Fatty liver | ↓ApoB100, Acyl-coenzyme-A expressions [105] |
| Cardiovascular disorders | ↑3-nitrotyrosine, ONOO-, iNOS [107] ↓Intracellular Ca2+, Ca2p, ATP, PARP-1 [108] |
| Endothelial dysfunction | ↑O2−, ONOO−, iNOS, Interleukins, VCAM, ICAM, P-seletins, PKC, NFKB [14,129] ●SOD [61] |
| Hypertension | ↑ONOO−, COX-2 activity, NAD(P)H expression [20,128,137] ↓L-arg, NO, PGI2 [128,129,141]. |
| ○Activation ●Inactivation ↑Increase ↓Decrease | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pérez-Torres, I.; Manzano-Pech, L.; Rubio-Ruíz, M.E.; Soto, M.E.; Guarner-Lans, V. Nitrosative Stress and Its Association with Cardiometabolic Disorders. Molecules 2020, 25, 2555. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25112555
Pérez-Torres I, Manzano-Pech L, Rubio-Ruíz ME, Soto ME, Guarner-Lans V. Nitrosative Stress and Its Association with Cardiometabolic Disorders. Molecules. 2020; 25(11):2555. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25112555
Chicago/Turabian StylePérez-Torres, Israel, Linaloe Manzano-Pech, María Esther Rubio-Ruíz, María Elena Soto, and Verónica Guarner-Lans. 2020. "Nitrosative Stress and Its Association with Cardiometabolic Disorders" Molecules 25, no. 11: 2555. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25112555