3.4. Chemistry
All reactions were carried out with magnetic stirring. Air or moisture sensitive reagents were handled under argon using either a glovebox or a Schlenk line. Organic extracts were dried over anhydrous MgSO4. Solutions were concentrated under reduced pressure at 40–50 °C using a rotary evaporator (Bruker, Rheinstetten, Germany).
Column chromatography was performed with silica gel (w/Ca, 0.1–0.3%), 60Å, 230–400 mesh particle size from Sigma-Aldrich GmbH (Steinheim, Germany). Solvent proportions are indicated in a volume/volume ratio.
Thin layer chromatography (TLC) was performed using aluminium sheets coated with silica gel 60 F254 (Merck KGaA, Darmstadt, Germany). Chromatograms were inspected under UV light (λ = 254 nm) and stained with molybdophosphoric acid (10% in ethanol), ninhydrin (0.2% in ethanol) or Dragendorff reagent.
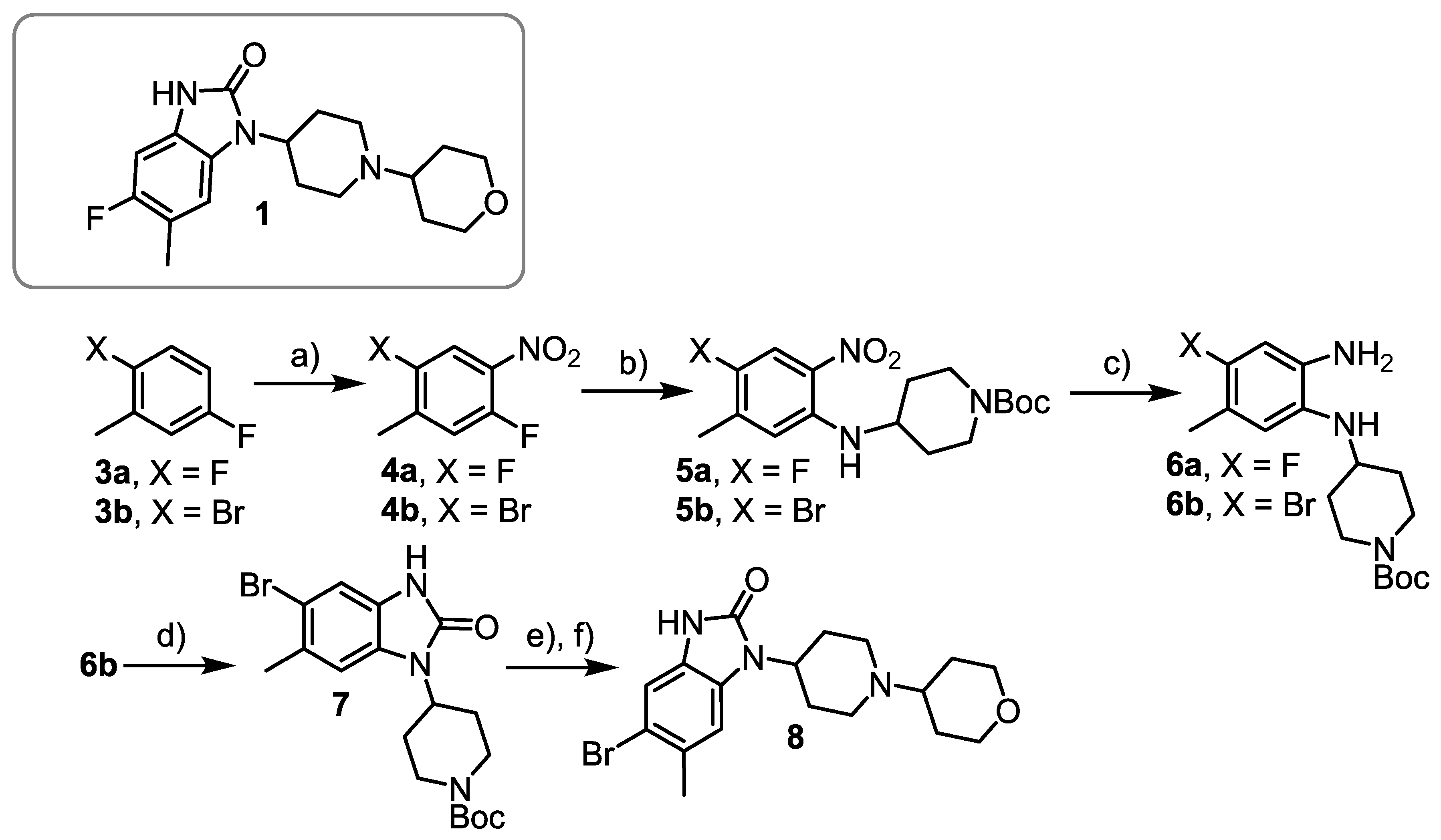
3.4.1. 2,5-Difluoro-4-nitrotoluene (4a)
KNO
3 (3.2 g, 31.2 mmol) was added to an ice-cold solution of 2,5-difluorotoluene (
3a) (4 g, 31.2 mmol) in concentrated H
2SO
4 (15 mL), the mixture was allowed to warm to ambient temperature and stirred at 28 °C overnight. The reaction mixture was then poured over ice and the resulting suspension was extracted with EtOAc (3 × 50 mL). The combined organic layers were concentrated under reduced pressure and the residue was purified by column chromatography (EtOAc/hexane = 1:15) to afford the title compound
4a [
9]. Yield: 3.45 g, 20 mmol (64%). Appearance: yellow crystalline solid [
9,
10].
1H-NMR: 7.78 (dd,
J = 8.4, 6.3 Hz, 1H), 7.16 (dd,
J = 10.9, 6.1 Hz, 1H), 2.39 (d,
J = 1.8 Hz, 3H).
3.4.2. tert-Butyl 4-[(4-fluoro-5-methyl-2-nitrophenyl)amino] piperidine-1-carboxylate (5a)
Diisopropylethylamine (0.87 mL, 0.65g, 5 mmol, 1 eq) was added to a solution of
4a (0.87 g, 5 mmol, 1 eq) and 4-amino-1-
N-Boc-piperidine (1 g, 5 mmol, 1 eq) in DMF (8 mL) at 40 °C and the resulting solution was stirred at 80 °C for 38 h. After removal of the solvent under reduced pressure and addition of water, the resulting mixture was extracted with CH
2Cl
2 (3 × 40 mL). The combined organic fractions were dried and concentrated under reduced pressure. The residue was purified by column chromatography (EtOAc/hexane = 1:15) to afford the title compound
5a [
9]. Yield: 1.12 g, 3.16 mmol (63%). Appearance: orange-red solid.
1H-NMR: 8.02 (br, 1H), 7.84 (d,
J = 10.0 Hz, 1H), 6.67 (d,
J = 6.3 Hz, 1H), 4.21–3.85 (m, 2H), 3.79–3.55 (m, 1H), 3.08 (t,
J = 11.1 Hz, 2H), 2.32 (s, 3H), 2.15–1.97 (m, 2H), 1.63–1.52 (m, 2H), 1.49 (s, 9H).
13C-NMR: 169.52, 152.27 (d,
J = 353.3 Hz), 153.07, 141.35, 115.32 (d,
J = 3.0 Hz), 111.70 (d,
J = 27.0 Hz), 79.91, 49.40, 42.09, 36.47, 31.78, 28.41.
3.4.3. tert-Butyl 4-([2-amino-4-fluoro-5-methylphenyl]amino)piperidine-1-carboxylate
Hydrazine hydrate (700 µL, 680 mg, 14 mmol, 10 eq) was slowly added to a suspension of Raney nickel (0.7 mL, 50% aq. suspension) in a solution of
5a (0.5 g, 1.4 mmol, 1 eq) in EtOH (30 mL). The mixture was stirred at 45 °C for 2 h and filtered over Celite. After removal of volatiles under reduced pressure and addition of saturated NaHCO
3 (30 mL), the resulting emulsion was extracted with CH
2Cl
2 (3 × 30 mL). The combined organic layers were dried and concentrated under reduced pressure. The residue was purified by column chromatography (EtOAc/hexane = 1:1) followed by crystallization from hexane to afford the title compound
tert-butyl-4-([2-amine-4-fluoro-5-methylphenyl]amine)piperidine-1-carboxylate [
9]. Yield: 0.32 g, 0.98 mmol (70%). Appearance: brown solid.
1H-NMR: 6.47 (d,
J = 7.5 Hz, 1H), 6.42 (d,
J = 10.5 Hz, 1H, H-6′), 4.10 (d,
J = 7.2 Hz, 1H), 4.03 (d,
J = 11.8 Hz, 2H), 3.45 (t,
J = 13.4 Hz, 2H), 3.25 (ddd,
J = 13.8, 9.9, 3.7 Hz, 1H), 2.91 (t,
J = 11.4 Hz, 2H), 2.15 (s, 3H), 1.97 (dd,
J = 9.1, 3.7 Hz, 2H), 1.47 (s, 9H), 1.41–1.29 (m, 2H).
13C-NMR: 157.29, 154.49 (d,
J = 48.0 Hz), 135.75 (d,
J = 9.8 Hz), 130.64, 118.46 (d,
J = 5.7 Hz), 103.66 (d,
J = 26.1 Hz), 79.54, 51.24, 32.58, 28.44, 22.65, 14.06.
3.4.4. 2-Bromo-5-fluoro-4-nitrotoluene (4b)
KNO
3 (2 g, 20 mmol, 1 eq) was added to an ice-cold solution of
3b (3.78 g, 20 mmol, 1 eq) in concentrated H
2SO
4 (15 mL). The mixture was allowed to reach ambient temperature and stirred overnight. Afterwards, the reaction mixture was poured over ice and extracted with EtOAc (3 × 50 mL). The combined organic fractions were dried, concentrated under reduced pressure and the remaining brown-red oil purified by column chromatography (CH
2Cl
2/MeOH = 9:1) to afford the title compound
4b [
10]. Yield: 4.25 g, 14.4 mmol (72%). Appearance: yellow oil.
1H-NMR: 8.14 (d,
J = 7.1 Hz, 1H), 7.18 (d,
J = 11.4 Hz, 1H), 2.44 (s, 3H).
3.4.5. tert-Butyl 4-[(4-bromo-5-methyl-2-nitrophenyl)amino]piperidine-1-carboxylate (5b)
DIEA (0.87 mL, 0.65 g, 5 mmol, 1 eq) was added to a solution of 4b (1.5 g, 5 mmol, 1 eq) and 4-amino-1-N-Boc-piperidine (1 g, 5 mmol, 1 eq) in DMF (8 mL) at 40 °C. The resulting mixture was stirred at 80 °C for 38 h and concentrated under reduced pressure. H2O (40 mL) was added to the oily residue and the resulting suspension was extracted with CH2Cl2 (3 × 30 mL). The combined organic fractions were dried and concentrated under reduced pressure. The residue was purified by column chromatography (EtOAc/hexane = 1:15) to afford the title compound. Yield: 1.6 g, 3.6 mmol (72%). Appearance: orange solid. 1H-NMR: (300 MHz, CDCl3) δ (ppm) = 8.35 (s, 1H), 8.06 (br, 1H), 6.74 (s, 1H), 4.02 (d, J = 13.6 Hz, 2H), 3.75–3.57 (m, 1H), 3.21–3.02 (m, 2H), 2.41 (s, 3H), 2.15–1.97 (m, 2H), 1.61–1.45 (m, 2H), 1.47 (s, 9H). 13C-NMR: 154.65, 147.22, 143.26, 130.65, 129.94, 115.06, 109.92, 79.98, 49.33, 42.11, 31.71, 28.45, 23.87.
3.4.6. tert-Butyl 4-[(2-amino-4-bromo-5-methylphenyl)amino]piperidine-1-carboxylate (6b)
Hydrazine hydrate (0.94 mL, 0.97 g, 18.83 mmol, 3 eq) was slowly added to a suspension of Raney nickel (6 mL; 50% suspension in H2O) in a solution of 5b (2.6 g, 6.28 mmol, 1 eq) in ethanol (30 mL). The mixture was stirred at 45 °C for 2 h and filtered over Celite. After removal of volatiles under reduced pressure and addition of saturated NaHCO3 (30 mL), the resulting emulsion was extracted with CH2Cl2 (3 × 30 mL). The combined organic layers were dried and concentrated under reduced pressure. The residue was purified by column chromatography (EtOAc/hexane = 1:1) followed by crystallization from hexane to afford the title compound. Yield: 0.71 g, 1.85 mmol (29%). Appearance: orange solid. 1H-NMR: 6.91 (s, 1H), 6.52 (s, 1H), 4.05 (d, J = 12.1 Hz, 2H), 3.36 (td, J = 9.9, 4.9 Hz, 1H), 3.25 (s, 2H), 2.97 (t, J = 11.4 Hz, 2H), 2.30 (s, 3H), 2.03 (d, J = 10.5 Hz, 2H), 1.49 (s, 9H), 1.41 (dd, J = 17.3, 7.3 Hz, 2H), 1.30 (dd, J = 12.3, 3.7 Hz, 1H); NH2-Group is unobservable. 13C-NMR: 135.57, 133.82, 129.18, 120.47, 115.20, 79.61, 50.15, 42.41, 31.71, 28.44, 22.30. MS: m/z: [M + H]+ calcd: 384.1; found: 384.1.
3.4.7. tert-Butyl 4-(5-bromo-6-methyl-2-oxo-2,3-dihydro-1H-benz[d]imidazol-1-yl)-piperidine-1-carboxylate (7)
Et
3N (1.15 mL, 0.83 g, 8.25 mmol, 2.01 eq) was added dropwise to a solution of
6b (1.58 g, 4.1 mmol, 1 eq) and triphosgene (0.41 g, 1.37 mmol, 0.33 eq) in THF (10 mL). The reaction mixture was stirred at 45 °C for 2 h. After removal of volatiles under reduced pressure and addition of saturated NaHCO
3 (30 mL), the resulting emulsion was extracted with EtOAc (3 × 30 mL). The combined organic fraction was dried and concentrated under reduced pressure. The residue was purified by column chromatography (EtOAc/hexane = 1:1) to afford the known title compound
7 [
20], which was directly used for the next step. Yield: 1.59 g, 3.87 mmol (94%). Appearance: white solid. MS:
m/z: [2M + H]
+2 calcd: 409.6; found: 410.1.
3.4.8. Deprotection of 7
Procedure A
An excess of 4 m HCl in EtOAc was added to a solution of 7 (0.27 g, 0.66 mmol) in CH2Cl2 (2 mL) and the resulting mixture was stirred at ambient temperature for 1 h. After removal of volatiles under reduced pressure and addition of 0.1 m NaOH (50 mL), the resulting mixture was extracted with CH2Cl2 (3 × 30 mL). The combined organic fraction was dried and concentrated under reduced pressure to afford the crude amine hydrochloride, which was used for the next step without any purification and characterization. Yield: 0.18 g, 0.6 mmol (90% crude). Appearance: white solid.
Procedure B
TFA (30 mL) was slowly added to a solution of 7 (1.55 g, 3.78 mmol) in CH2Cl2 (30 mL) and the reaction mixture was stirred at ambient temperature for 1 h. After concentration under reduced pressure and addition of saturated NaHCO3 (30 mL) the resulting mixture was extracted with CH2Cl2 (3 × 30 mL). The combined organic fraction was dried and concentrated under reduced pressure to afford the crude amine trifluoroacetate, which was used for the next step without any purification and characterization. Yield: 1.44 g, ≤3.78 mmol (100% crude). Appearance: white solid.
3.4.9. 5-Bromo-6-methyl-1-[1-(tetrahydro-2H-pyran-4-yl)piperidin-4-yl]-1,3-dihydro-2H-benz[d]imidazol-2-one (8). Protocol A
NaBH3CN (1.54 g, 24.5 mmol, 7 eq) was added to a solution of 7 (1.08 g, 3.5 mmol, 1 eq), tetrahydro-4H-pyran-4-one (2.45 g, 24.5 mmol, 7 eq), Bu4NBr (0.79 g, 2.45 mmol, 0.7 eq) and Et3N (1.46 mL, 1.06 g, 10.5 mmol, 3 eq) in CH2Cl2 (80 mL) and the reaction mixture was stirred for 24 h. After concentration under reduced pressure and addition of 0.1 n NaOH (50 mL), the resulting emulsion was extracted with CH2Cl2 (3 × 30 mL). The combined organic fraction was dried and concentrated under reduced pressure. The residue was purified by column chromatography (CH2Cl2/MeOH = 9:1) to afford the title compound. Yield: 83 mg, 0.21 mmol (6%). Appearance: white solid. 1H-NMR [(CD3)2SO + TFA; mixture of 8 and 8·TFA]: 7.22 (s, 1H), 7.16 (s, 1H), 4.57–4.51 (m, 1H), 4.00–3.78 (m, 2H), 3.63–3.61 (m, 2H), 3.61–3.51 (m, 1H), 3.49–3.38 (m, 2H), 3.25–3.19 (m, 2H), 2.72–2.63 (m, 2H), 2.36 (s, 3H), 1.98 (m, 4H), 1.76–1.171 (m, 2H). 13C-NMR [(CD3)2SO + TFA; mixture of 8 and 8·TFA]: 154.05, 153.93; 129.28, 129.24; 128.99, 128.41; 128.26, 115.39, 112.50, 110.86, 65.99, 62.08, 48.34, 47.54, 27.41, 26.26, 22.88; TFA: 158.86 (q, J = 37.4 Hz, CO2H), 115.76 (q, J = 289.9 Hz, CF3) 120.07, 117.20, 114.32, 112.50, MS: m/z: [2 M + MeOH + H]+2 calcd: 396.6; found: 396.1.
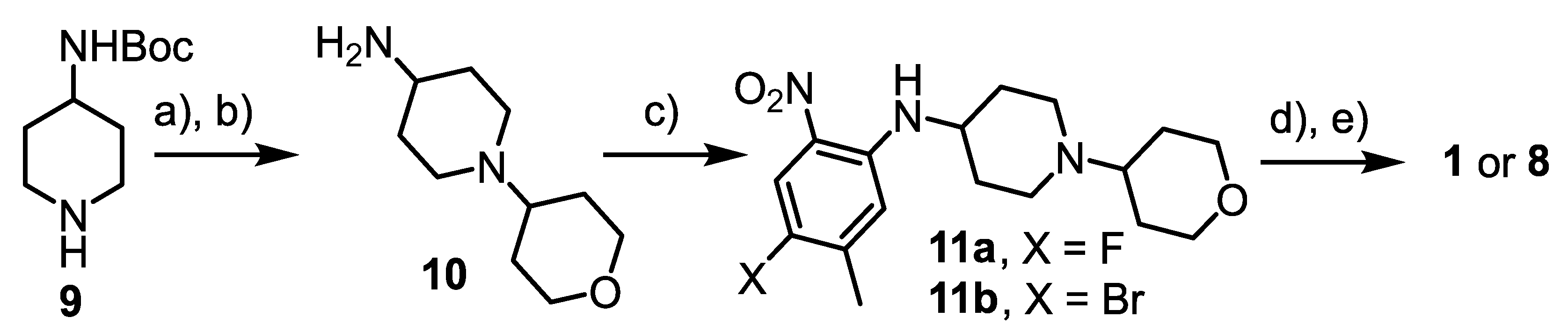
3.4.10. tert-Butyl [1-(tetrahydro-2H-pyran-4-yl)piperidin-4-yl]carbamate
NaBH3CN (0.16 g, 2.5 mmol, 1 eq) was added to a solution of 4-N-Boc-aminopiperidine (0.5 g, 2.5 mmol, 1 eq), tetrahydropyran-4-one (0.3 g, 3 mmol, 1.2 eq), acetic acid (0.23 mL, 0.24 g, 4 mmol, 1.6 eq), and Bu4NBr (97 mg, 0.3 mmol, 0.12 eq) in CH2Cl2 (20 mL) and the reaction mixture was stirred for 2 days. Thereafter, the mixture was washed with saturated K2CO3 (10 mL), the organic layer was dried and concentrated under reduced pressure. The residue was purified by column chromatography (CH2Cl2/MeOH = 9:1) to afford the title compound. Yield: 0.34 g, 1.44 mmol (57%). Appearance: white solid. 1H-NMR: 4.47 (s, 1H), 4.02 (d, J = 10.7 Hz, 2H), 3.44 (s, 1H), 3.37 (dd, J = 11.7, 9.7 Hz, 2H), 2.95 (d, J = 12.4 Hz, 2H), 2.50 (d, J = 11.1 Hz, 1H), 2.29 (t, J = 10.6 Hz, 2H), 1.98 (d, J = 10.9 Hz, 2H), 1.79 (d, J = 9.9 Hz, 2H), 1.71–1.57 (m, 2H), 1.55 (d, J = 7.1 Hz, 2H), 1.45 (s, 9H). 13C-NMR: 144.39, 67.49, 61.23, 48.03, 32.57, 29.30, 28.44. MS: m/z: [M + H]+ calcd: 285.2 found: 285.2.
3.4.11. 1-(Tetrahydro-2H-pyran-4-yl)piperidine-4-amine (10)
A solution of anhydrous HCl in EtOAc was prepared by addition of AcCl (2.25 mL, 2.48 g, 31.6 mmol, 4.9 eq) to an ice-cold solution of MeOH (1.34 mL, 1.06 g, 33 mmol, 5.2 eq) in EtOAc (8.25 mL). The resulting solution was added to a solution of tert-butyl [1-(tetrahydro-2H-pyran-4-yl)piperidin-4-yl]carbamate (1.83 g, 6.4 mmol, 1 eq) in CH2Cl2 (5 mL) and the mixture was stirred for 2 h. Volatiles were removed under reduced pressure, the residue was taken up in 0.1 m NaOH (50 mL) and the resulting mixture was extracted with CH2Cl2 (3 × 30 mL). The combined organic fraction was dried and concentrated under reduced pressure to afford the title compound, which was directly used for the next step. Yield: 1.1 g, 5.96 mmol (93%). Appearance: yellow solid. 1H-NMR (CD3OD + TFA; mixture of 10·TFA and 10·2 TFA): 4.10–4.04 (m, 2H), 3.75–3.72 (m, 2H), 3.59–3.41 (m, 4H), 3.25–3.19 (m, 2H), 2.36–2.34 (m, 2H), 2.19–2.05 (m, 4H), 1.91–1.81 (m, 2H). MS: m/z: [M + H]+ calcd: 185.2; found: 185.4.
3.4.12. N-(4-Fluoro-5-methyl-2-nitrophenyl)-1-(tetrahydro-2H-pyran-4-yl)piperidine-4-amine (11a)
A solution of 10 (0.184 g, 1 mmol, 1 eq), 4a (0.173 g, 1 mmol, 1 eq) and DIEA (0.175 mL, 0.13 g, 5.8 mmol, 5.8 eq) in DMF (5 mL) was stirred at 70 °C for 16 h. After removal of volatiles under reduced pressure and addition of H2O (30 mL), the resulting mixture was extracted with CH2Cl2 (3 × 10 mL). The combined organic fraction was dried and concentrated under reduced pressure. The residue was purified by column chromatography (CHCl3/acetone = 1:5) to afford the title compound which was directly used for the next step without any characterization. Yield: 0.19 g, 0.58 mmol (58%). Appearance: red-orange solid.
3.4.15. N-(4-Bromo-5-methyl-2-nitrophenyl)-1-(tetrahydro-2H-pyran-4-yl)piperidine-4-amine (11b)
A solution of 10 (1.07 g, 5.8 mmol, 1 eq), 4b (1.36 g, 5.8 mmol, 1 eq) and DIEA (1 mL, 0.74 g, 5.8 mmol, 1 eq) in DMF (15 mL) was stirred at 70 °C for 16 h. After removal of volatiles under reduced pressure, the residue was taken up with H2O (50 mL), and the mixture was extracted with CH2Cl2 (3 × 10 mL). The combined organic fraction was dried and concentrated under reduced pressure affording the title compound (1.5 g, 66%). 1H-NMR (CDCl3): 8.35 (s, 1H),8.10 (t, J = 10.6 Hz, 1H), 6.74 (s, 1H), 4.07–4.04 (m, 2H), 3.57–3.55 (m, 1H), 3.43–3.38 (m, 2H), 2.93–2.91 (m, 2H), 2.59–2.45 (m, 3H), 2.41 (s, 3H), 2.19–2.05 (m, 2H), 1.79–1.68 (m, 6H). 13C-NMR (CDCl3): 147.06, 143.5, 130.60, 129.85, 115.13, 109.62, 67.54, 61.08, 49.12, 47.17, 31.93, 29.38, 23.82. MS: m/z: [M + H]+ calcd: 398.1; found: 398.1.
3.4.16. 4-Bromo-5-methyl-N-(1-(tetrahydro-2H-pyran-4-yl)piperidin-4-yl)benzene-1,2-diamine
Zn powder (2.66 g, 40.7 mmol, 10.7 eq) was added to a stirred solution of crude 11b (1.5 g, max. 3.8 mmol, 1 eq) and NH4Cl (2.18 g, 3.8 mmol, 1 eq) in a mixture of EtOAc/EtOH 1:1 (50 mL). The reaction mixture was stirred overnight, then diluted with EtOAc (100 mL) and filtered over Celite. The filtrate was concentrated under reduced pressure. The residue was purified by column chromatography (MeOH/CH2Cl2, 1:6) followed by recrystallization from hexane to afford the title compound. Yield: 1.26 g, 3.42 mmol (58% over two steps). Appearance: brown solid. 1H-NMR: 6.90 (s, 1H), 6.51 (s, 1H), 4.05 (d, J = 8.7 Hz, 2H), 3.40 (t, J = 10.9 Hz, 2H), 3.25 (s, 2H), 2.96 (d, J = 11.6 Hz, 2H), 2.52 (s, 1H), 2.45–2.32 (m, 2H), 2.29 (s, 3H), 2.09 (d, J = 12.0 Hz, 2H), 1.81 (s, 1H), 1.76 (s, 2H), 1.69–1.34 (m, 4H). 13C-NMR: 135.96, 133.65, 129.18, 120.37, 119.87, 115.00, 67.65, 61.12, 50.22, 47.94, 32.83, 29.50, 22.39. MS: m/z: [M + H]+ calcd: 368.1; found: 368.1.
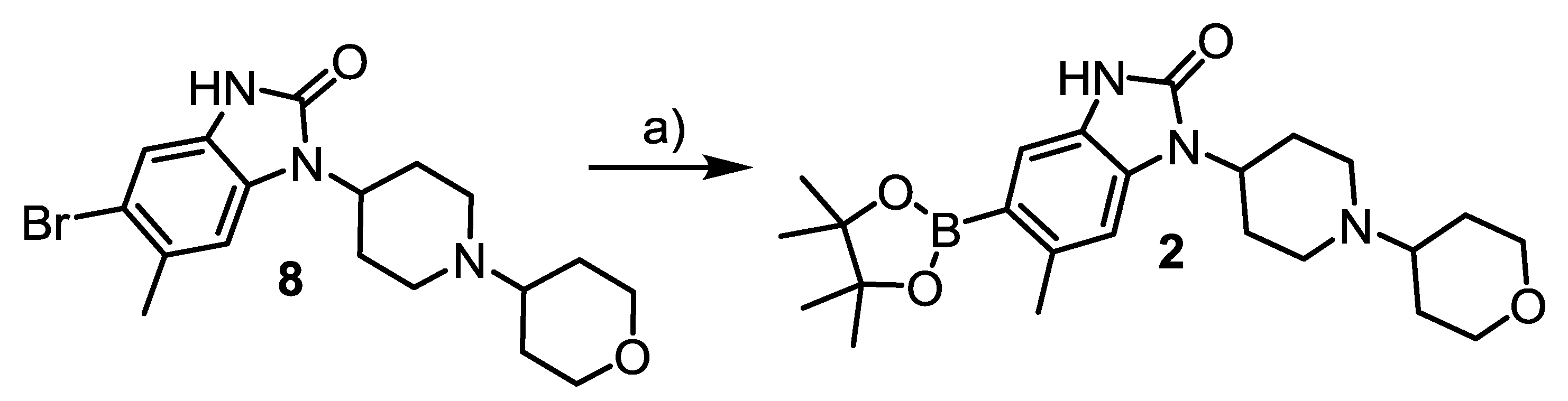
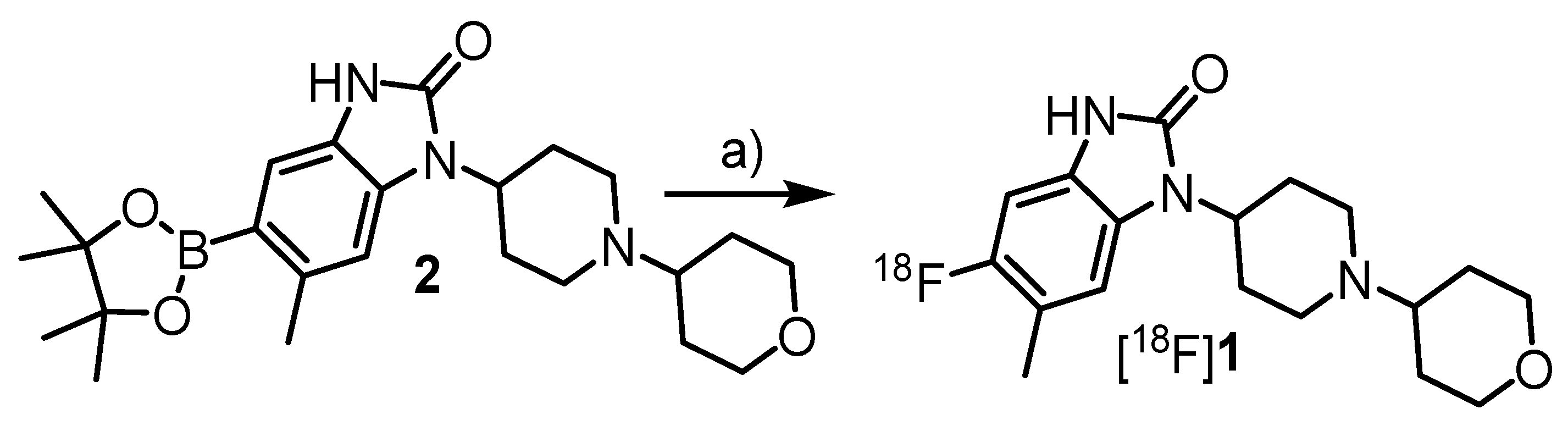
3.4.18. 6-Methyl-1-[1-(tetrahydro-2H-pyran-4-yl)piperidin-4-yl]-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3-dihydro-2H-benz[d]imidazol-2-one (2)
A suspension KOAc (0.39 g, 3.9 mmol, 3 eq) in a solution of 8 (0.51 g, 1.3 mmol, 1 eq), Pd(dppf)Cl2 (47.5 mg, 65 µmol, 0.05 eq) and bis(pinacolato)diboron (0.41 g, 1.63 mmol, 1.25 eq) in anhydrous dioxane (6 mL) was stirred at 110 °C for 3 h. After removal of the volatiles under reduced pressure and addition of 0.1 n NaOH (5 mL), the mixture was extracted with CH2Cl2 (3 × 20 mL). The combined organic fraction was dried and concentrated under reduced pressure. The residue was purified by column chromatography (MeOH/CH2Cl2 = 1:10) and recrystallized from CH2Cl2/hexane to afford the title compound. Yield: 0.28 g, 0.62 mmol (48%). Appearance: white solid. 1H-NMR: 10.04 (s, 1H), 7.53 (d, J = 19.9 Hz, 1H), 7.12 (s, 1H), 4.37 (s, 1H), 4.09 (d, J = 9.5 Hz, 2H), 3.43 (t, J = 11.4 Hz, 2H), 3.16 (s, 2H), 2.60 (s, 3H), 2.69–2.25 (m, 2H), 2.05–1.55 (m, 6H), 1.33 (s, 12H). 13C-NMR: 155.44, 125.42, 116.74, 113.08, 111.02, 109.18, 83.35, 67.63, 48.83, 29.70, 24.89, 22.35; C-Bpin was not observed. MS: m/z: [M + H]+ calcd: 442.3; found: 442.3.
3.5. Radiochemistry
3.5.1. General
[18F]Fluoride was produced by the 18O (p,n) 18F reaction by bombardment of enriched [18O] water with 17 MeV protons at the BC1710 cyclotron (The Japan Steel Works, Tokyo, Japan) of the INM-5 (Forschungszentrum Jülich).
Radioactivity was measured using a CRC-55tR Dose Calibrator from Capintec, Inc. (Florham Park, Netherlands).
All radiosyntheses were carried out in 5 mL V-Vials (Wheaton) equipped with PTFE wing coated stir bars using anhydrous DMA (Aldrich), anhydrous
nBuOH and anhydrous MeOH dried over molecular sieves (both Acros Organics, Geel, Belgium). Cu(OTf)
2(py)
4 was prepared according to the literature [
21] and stored under ambient conditions without any precautions.
Radiolabeling experiments were performed using AREX-9 Digital Pro hot plate equipped with VTF digital thermometer (VELP SCIENTIFICA, Usmate Velate MB, Italy), vacuum pump Laboport (KNF, Freiburg im Breisgau, Germany) in the customized hot cell installed by Von Gahlen Nederland B.V (Zevenaar, Netherlands).
Radiosyntheses were carried out under synthetic air (80% N2 + 20% O2) or Ar.
Sep-Pak Plus C18 cartridges and Sep-Pak Accell Plus QMA carbonate plus light cartridges, 46 mg sorbent per cartridge, (both Waters GmbH, Eschborn, Germany) were applied.
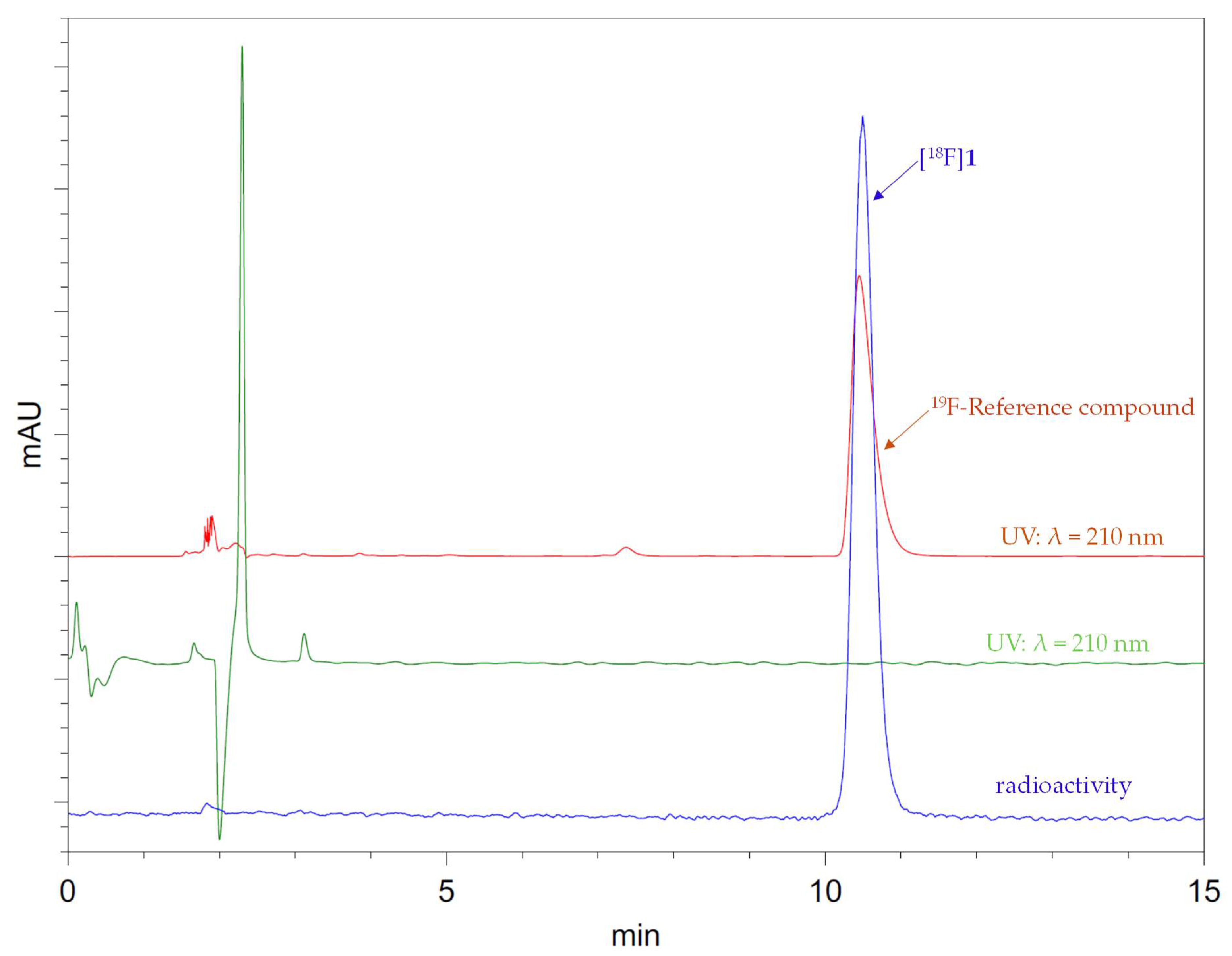
HPLC analyses and preparative separations were carried out using an Ultimate® 3000 HPLC system from Thermo Fisher Scientific (Sunnyvale, CA, USA) with variable wavelength detector coupled in series with a HERM LB 500 radio-flow monitor (Berthold Technologies, Bad Wildbad, Germany). The unselective adsorption of 18F− onto HPLC columns was determined to be <10% in each case. The UV and radioactivity detectors were connected in sequence, giving a time delay of 0.1–0.9 min between the corresponding responses, depending on the flow rate. The identity of [18F]1 was confirmed by the co-injection of the non-radioactive reference compound.
Analytical HPLC
Column: Luna C18 (2), 250 × 4.6 mm (Phenomenex, Aschaffenburg, Germany); eluent: 20% MeCN (0.1% TFA); flow rate: 1.5 mL/min.
Preparative HPLC
Column: Luna C18 (2), 250 × 10 mm (Phenomenex, Aschaffenburg, Germany); eluent: 25% MeCN (0.1% TFA); flow rate: 4 mL/min.
3.5.2. Processing [18F]fluoride
Aqueous [18F]fluoride (0.02–7 GBq) was loaded onto an anion-exchange resin (QMA cartridge) from the male side. The resin was flushed with MeOH (2 mL) and dried with 5–10 mL of air from the male side. 18F− was slowly eluted into the reaction vial from the female side using a solution of Et4NHCO3 (2 mg, 10 μmol) in MeOH (0.5 mL). It should be noted that flushing with MeOH was carried out from the male side whereas drying and 18F− elution were carried out from the female side. If the QMA cartridge had been loaded, flushed, and eluted from the female side only, sometimes a significant amount of [18F]fluoride remained on the resin, probably because QMA light cartridges have a single frit on the male side but four frits on the female side. MeOH was evaporated using a flow of air at 80 °C within 10 min. Using this procedure, recovery of 18F− amounted to ≥85%.
3.5.3. 5-[18F]Fluoro-6-methyl-1-(1-(tetrahydro-2H-pyran-4-yl)piperidin-4-yl)-1,3-dihydro-2H-benz-[d]-imidazol-2-one ([18F]1)
A solution of the pinacol boronate precursor
2 (7.2 mg, 16 μmol, 1 eq) and Cu(py)
4(OTf)
2 (13.6 mg, 20 μmol, 1.25 eq) in DMA/
nBuOH 2:1 (750 μL) was added to [
18F]Et
4NF, and the resulting solution was heated at 110 °C for 10 min under air or argon. For the determination of RCC, the reaction mixture was cooled to <40 °C, 0.1% TFA (1 mL) was added and the mixture was stirred for 30 s. HPLC analysis demonstrated that [
18F]
1 formed in a RCC of 96 ± 3% (
n = 3). For isolation of [
18F]
1, the reaction mixture was concentrated at 110 °C for 10 min, the residue was taken up in 25% MeCN (0.1% TFA) and the mixture was purified by preparative HPLC. The fraction containing [
18F]
1 was diluted with a 10-fold volume of H
2O and loaded onto a C18 cartridge. The cartridge was washed with 5% MeCN (10 mL), H
2O (5 mL) and dried with air (10 mL). The tracer was eluted with EtOH (1 mL). EtOH was evaporated at 90 °C and the residue was taken up in saline for injection affording [
18F]
1 as a ready-to-use solution in 13–19% n.d.c RCY (
Table 1).

 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}