
Poly-arginine-18 (R18) Confers Neuroprotection through Glutamate Receptor Modulation, Intracellular Calcium Reduction, and Preservation of Mitochondrial Function

 ,
,
Abstract
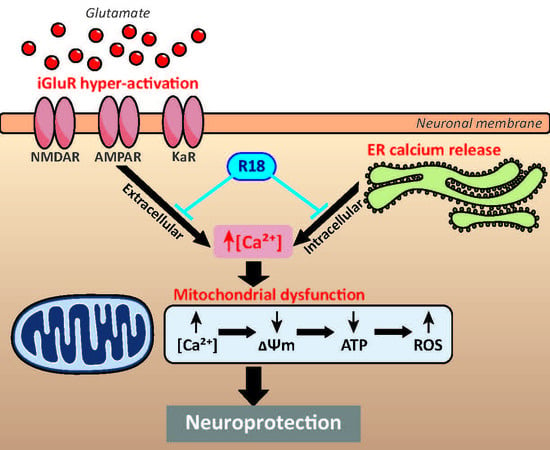
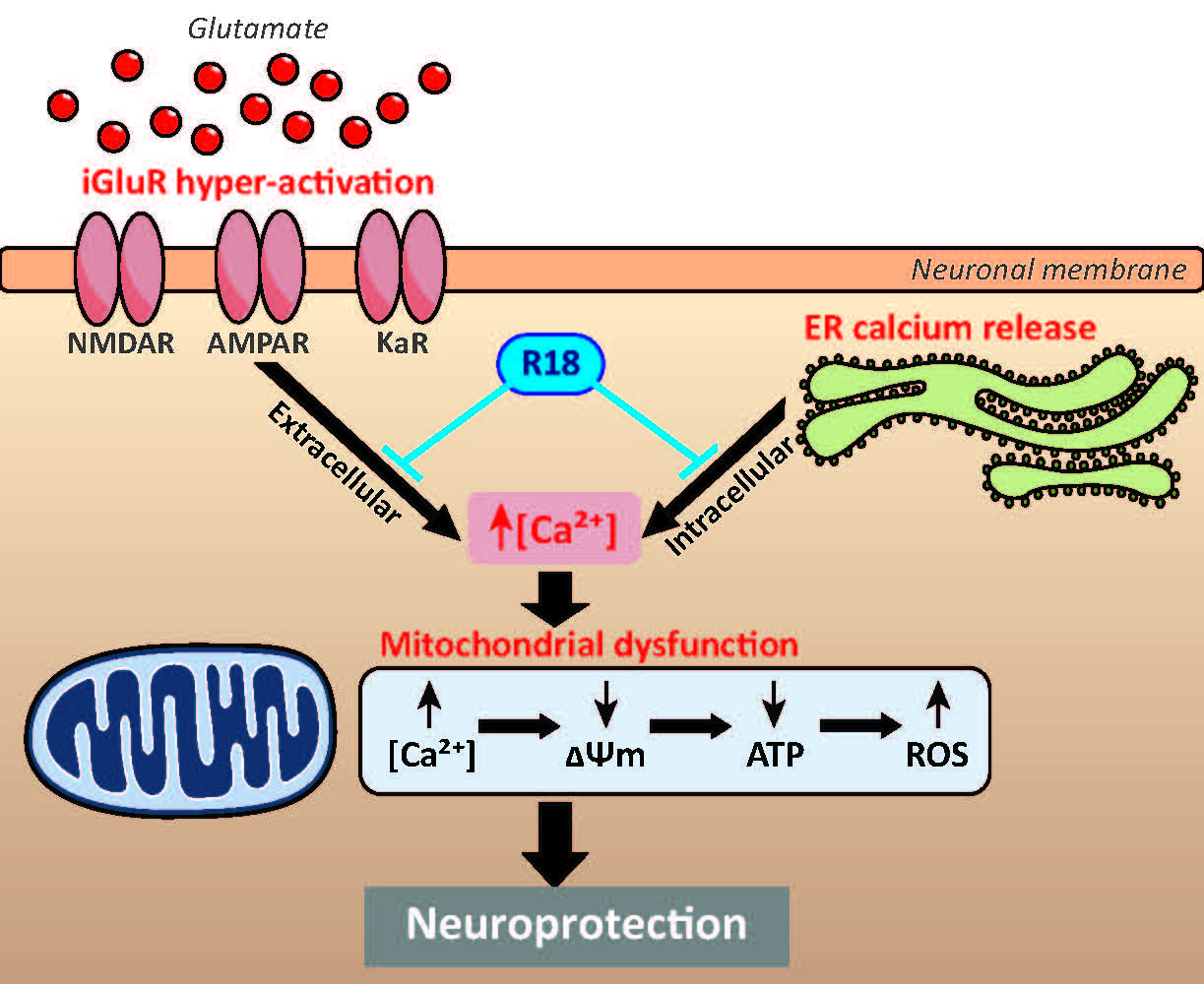
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
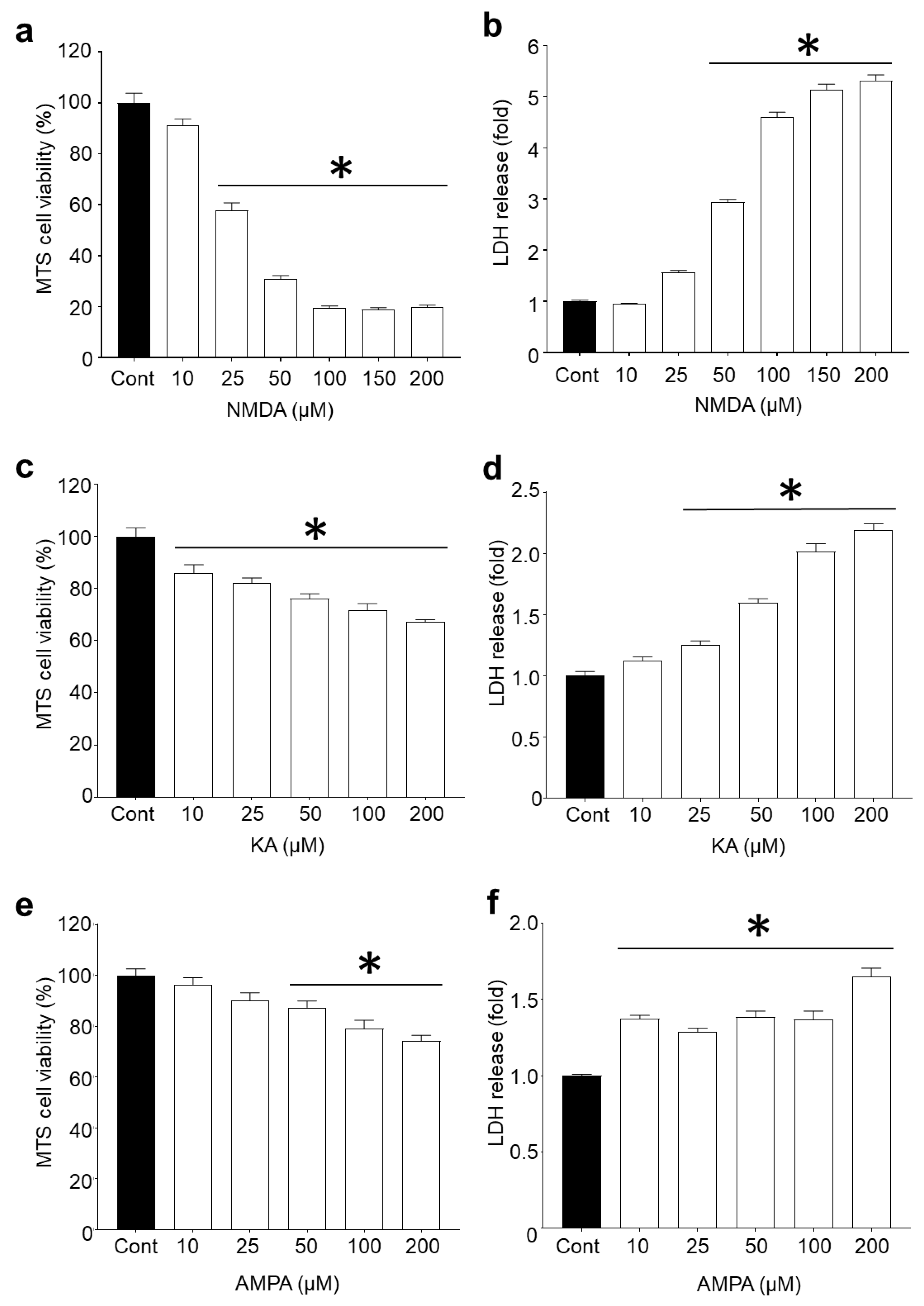
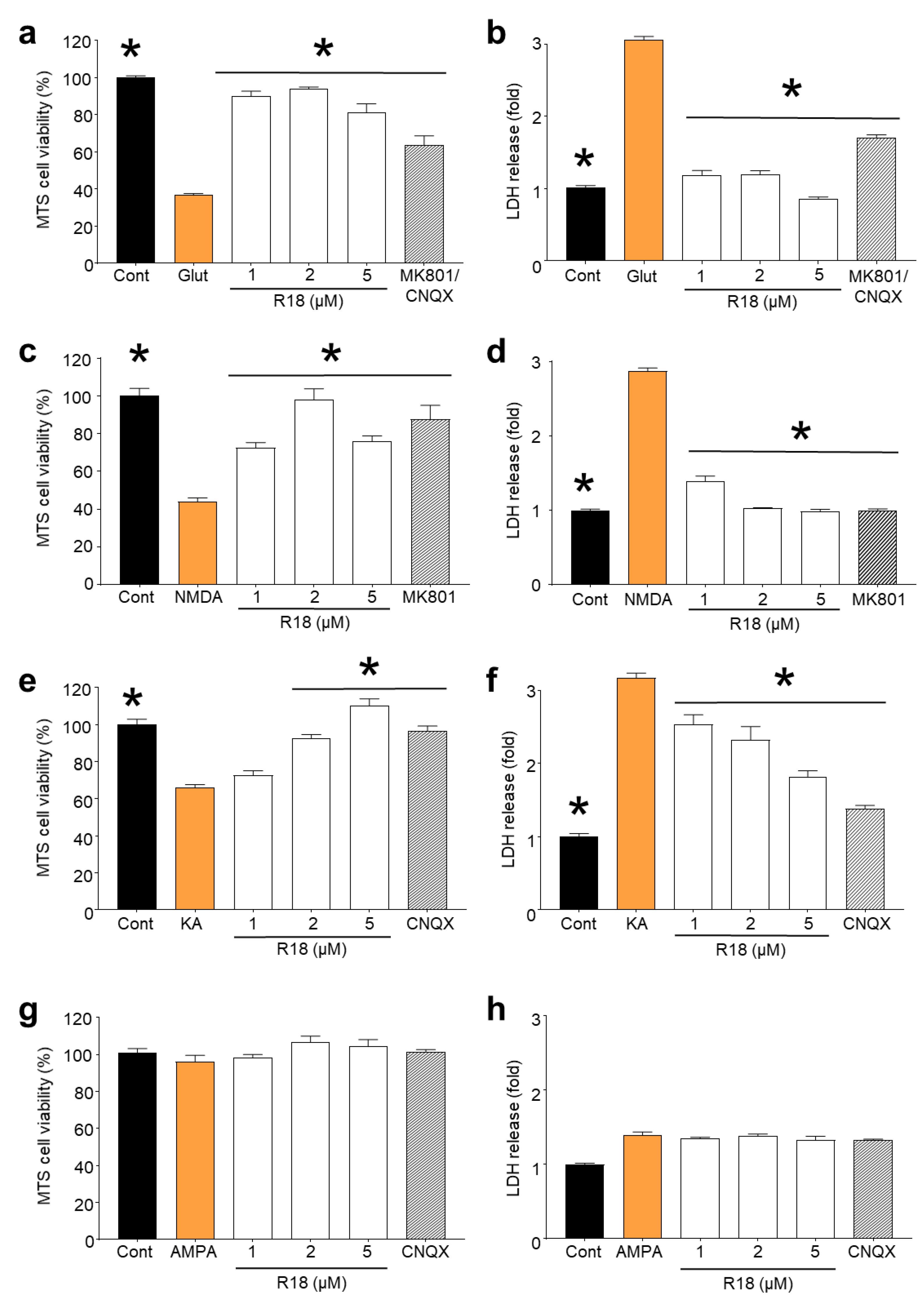
2.1. R18 is Neuroprotective against Different Ionotropic Glutamate Receptor Agonists
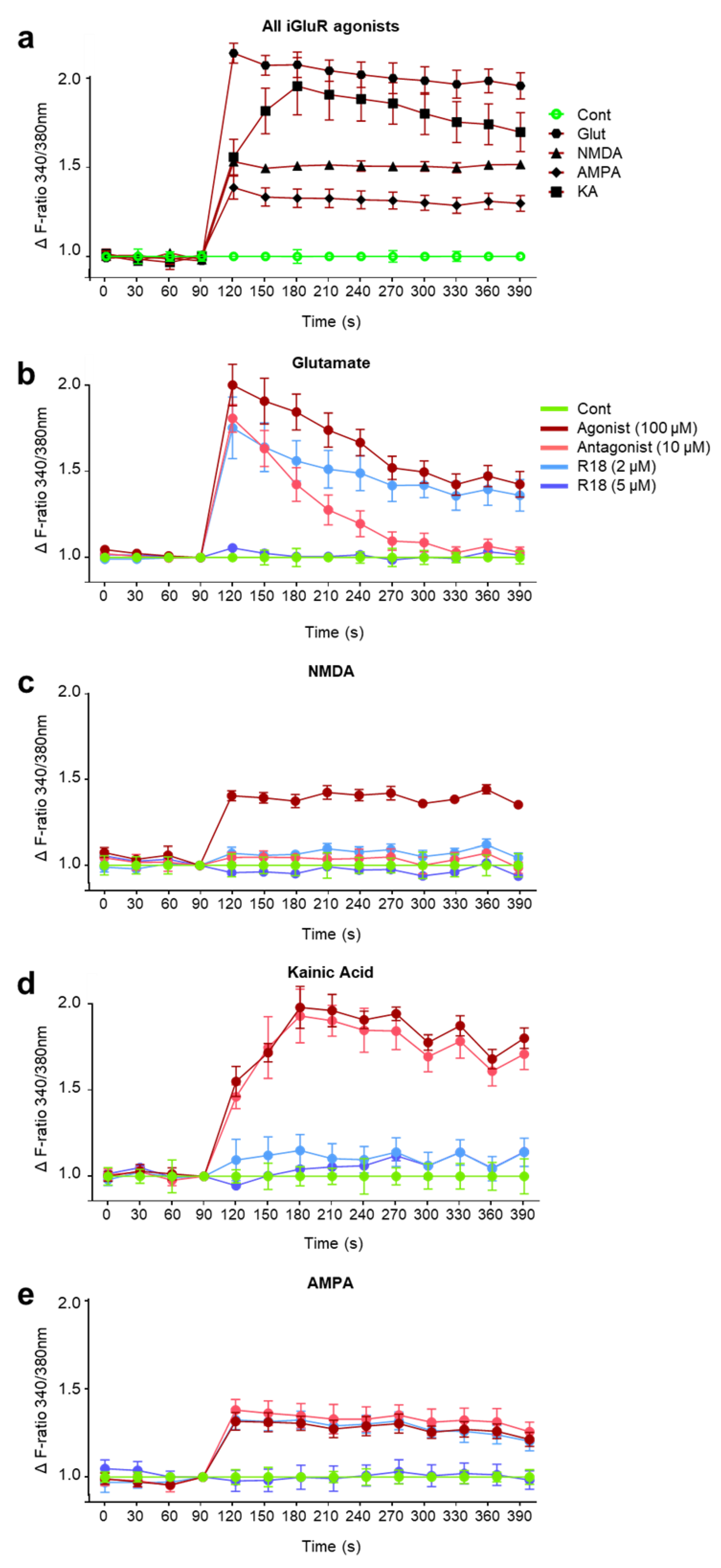
2.2. R18 Attenuates Excitotoxic Calcium Influx Stimulated by Ionotropic Glutamate Receptor Agonists
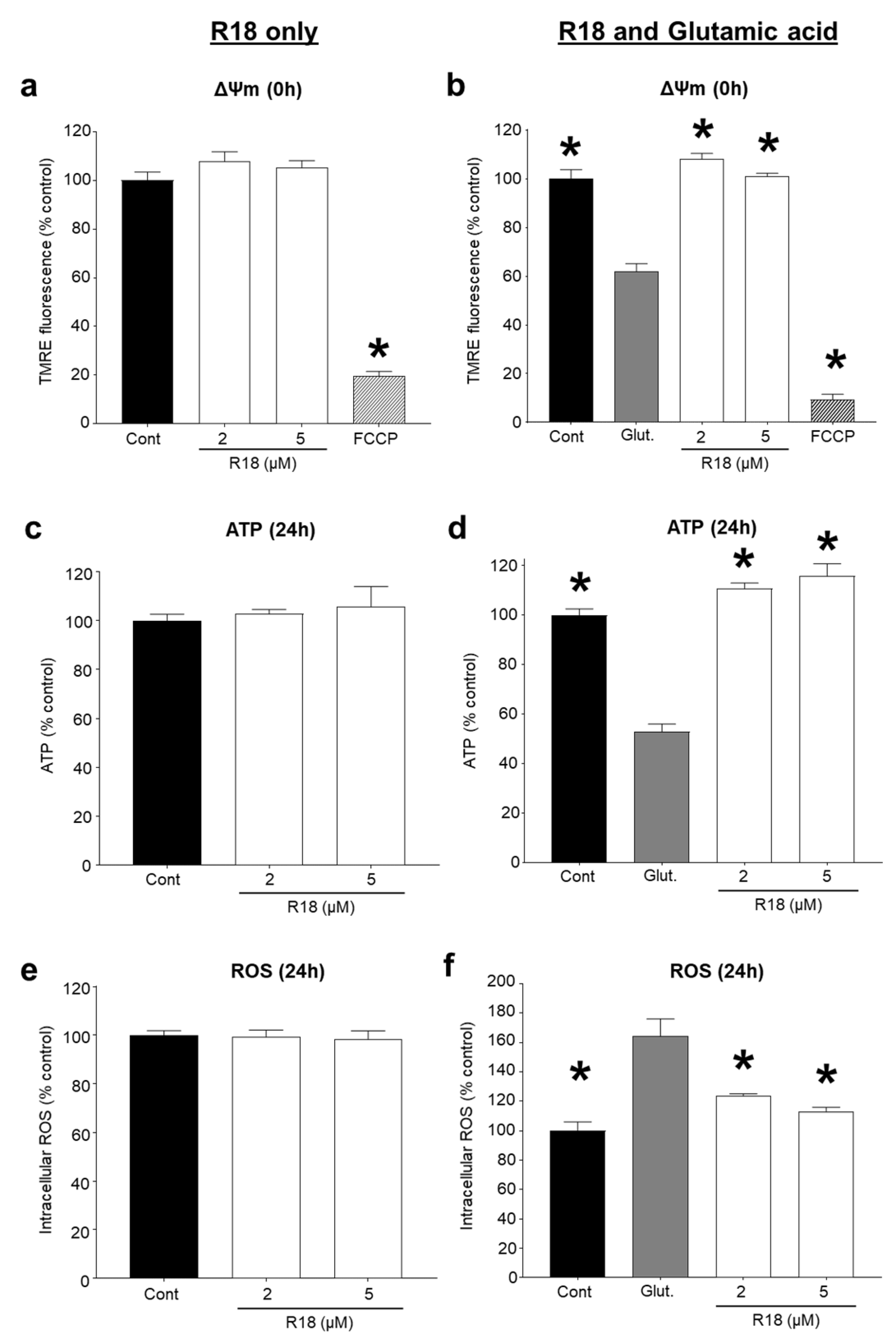
2.3. R18 Preserves Mitochondrial Bioenergetics after Glutamic Acid Exposure
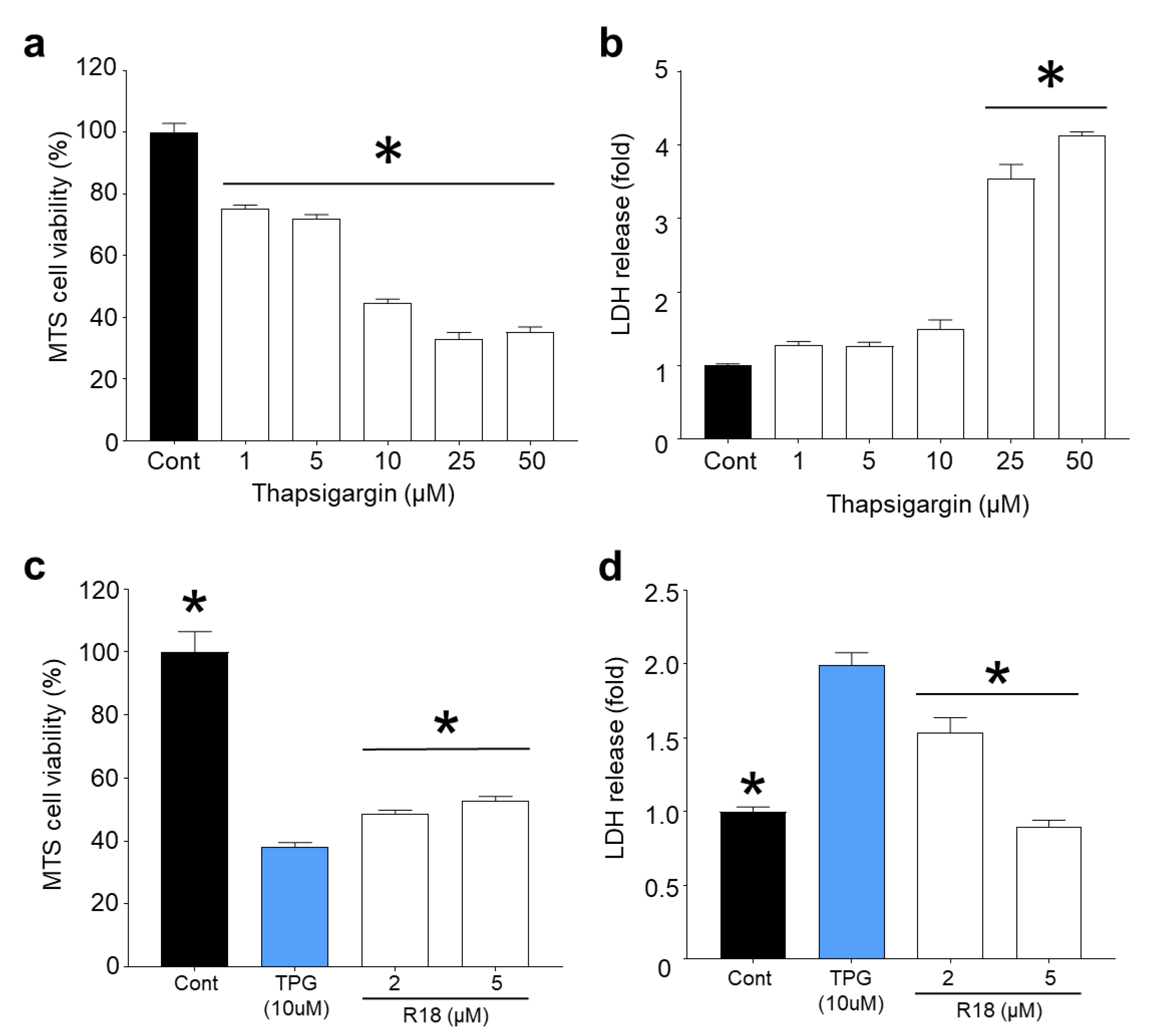
2.4. R18 Provides Neuroprotection against Thapsigargin-Induced Injury
3. Discussion
3.1. Protective Effects of R18 against iGluR agonist Induced Excitotoxicity
3.2. Protective Effects of R18 on Mitochondrial Function
3.3. Protective Effects of R18 against Thapsigargin Neurotoxicity
3.4. Potential Future Clinical Application of R18
4. Materials and Methods
4.1. Peptides
4.2. Primary Cortical Neuronal Cultures
4.3. iGluR agonist Excitotoxicity Model
4.4. Thapsigargin Intracellular Calcium Injury Model
4.5. MTS Cellular Viability and LDH Cytotoxicity Assays
4.6. Intracellular Calcium Kinetics
4.7. ATP Measurement in Cortical Neuronal Cultures
4.8. Reactive Oxidative Species (ROS) Detection
4.9. Mitochondrial Membrane Potential (ΔΨm)
4.10. Statistical Analysis
5. Conclusions
6. Patents
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Talantova, M.; Sanz-Blasco, S.; Zhang, X.; Xia, P.; Akhtar, M.W.; Okamoto, S.-I.; Dziewczapolski, G.; Nakamura, T.; Cao, G.; Pratt, A.E.; et al. Aβ induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc. Natl. Acad. Sci. USA 2013, 113, E2518–E2527. [Google Scholar] [CrossRef] [Green Version]
- De Felice, F.G.; Velasco, P.T.; Lambert, M.P.; Viola, K.; Fernandez, S.J.; Ferreira, S.T.; Klein, W.L. Aβ oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J. Biol. Chem. 2007, 282, 11590–11601. [Google Scholar] [CrossRef] [Green Version]
- Zeron, M.M.; Hansson, O.; Chen, N.; Wellington, C.L.; Leavitt, B.R.; Brundin, P.; Hayden, M.R.; Raymond, L.A. Increased sensitivity to N-methyl-D-aspartate receptor-mediated excitotoxicity in a mouse model of Huntington’s disease. Neuron 2002, 14, 849–860. [Google Scholar] [CrossRef] [Green Version]
- Shehadeh, J.; Fernandes, H.B.; Zeron Mullins, M.M.; Graham, R.K.; Leavitt, B.R.; Hayden, M.R.; Raymond, L.A. Striatal neuronal apoptosis is preferentially enhanced by NMDA receptor activation in YAC transgenic mouse model of Huntington disease. Neurobiol. Dis. 2006, 21, 392–403. [Google Scholar] [CrossRef]
- Helton, T.D.; Otsuka, T.; Lee, M.-C.; Mu, Y.; Ehlers, M.D. Pruning and loss of excitatory synapses by the parkin ubiquitin ligase. Proc. Natl. Acad. Sci. USA 2008, 105, 19492–19497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zádori, D.; Klivényi, P.; Plangár, I.; Toldi, J.; Vécsei, L. Endogenous neuroprotection in chronic neurodegenerative disorders: With particular regard to the kynurenines. J. Cell. Mol. Med. 2011, 15, 701–717. [Google Scholar] [CrossRef] [PubMed]
- Carriedo, S.G.; Yin, H.Z.; Weiss, J.H. Motor neurons are selectively vulnerable to AMPA/kainate receptor-mediated injury in vitro. J. Neurosci. 1996, 16, 4069–4079. [Google Scholar] [CrossRef] [Green Version]
- Van Damme, P.; Van den Bosch, L.; Van Houtte, E.; Callewaert, G.; Robberecht, W. GluR2-dependent properties of AMPA receptors determine the selective vulnerability of motor neurons to excitotoxicity. J. Neurophysiol. 2002, 88, 1279–1287. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.; Tymianski, M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch. Eur. J. Physiol. 2010, 460, 525–542. [Google Scholar] [CrossRef]
- Duncan, R.S.; Goad, D.L.; Grillo, M.A.; Kaja, S.; Payne, A.J.; Koulen, P. Control of intracellular calcium signaling as a neuroprotective strategy. Molecules 2010, 15, 1168–1195. [Google Scholar] [CrossRef] [Green Version]
- MacDougall, G.; Anderton, R.S.; Mastaglia, F.L.; Knuckey, N.W.; Meloni, B.P. Mitochondria and neuroprotection in stroke: Cationic arginine-rich peptides (CARPs) as a novel class of mitochondria-targeted neuroprotective therapeutics. Neurobiol. Dis. 2018, 121, 17–33. [Google Scholar] [CrossRef] [Green Version]
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 566–567. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.; Barrow, S.; Voronina, S.; Chvanov, M.; Petersen, O.H.; Tepikin, A. Modulation of calcium signalling by mitochondria. Biochim. Biophys. Acta-Bioenerg. 2009, 1787, 1374–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bano, D.; Nicotera, P. Ca2+ signals and neuronal death in brain ischemia. Stroke 2007, 38, 674–676. [Google Scholar] [CrossRef] [Green Version]
- Nikonenko, I.; Bancila, M.; Bloc, A.; Muller, D.; Bijlenga, P. Inhibition of T-type calcium channels protects neurons from delayed ischemia-induced damage. Mol. Pharmacol. 2005, 68, 84–89. [Google Scholar] [CrossRef] [Green Version]
- Oliver, M.W.; Shacklock, J.A.; Kessler, M.; Lynch, G.; Baimbridge, K.G. The glycine site modulates NMDA-mediated changes of intracellular free calcium in cultures of hippocampal neurons. Neurosci. Lett. 1990, 114, 197–202. [Google Scholar] [CrossRef]
- Hossmann, K.A. Pathophysiology and therapy of experimental stroke. Cell. Mol. Neurobiol. 2006, 26, 1057–1083. [Google Scholar] [CrossRef] [PubMed]
- Meloni, B.; Mastaglia, F.; Knuckey, N. Cationic arginine-rich peptides (CARPs): A novel class of neuroprotective agents with a multimodal mechanism of action. Front. Neurol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Meloni, B.P.; Milani, D.; Cross, J.L.; Clark, V.W.; Edwards, A.B.; Anderton, R.S.; Blacker, D.J.; Knuckey, N.W. Assessment of the neuroprotective effects of arginine-rich protamine peptides, poly-arginine peptides (R12-Cyclic, R22) and arginine–tryptophan-containing peptides following in vitro excitotoxicity and/or permanent middle cerebral artery occlusion in rats. NeuroMolecular Med. 2017, 19, 271–285. [Google Scholar] [CrossRef]
- Milani, D.; Bakeberg, M.C.; Cross, J.L.; Clark, V.W.; Anderton, R.S.; Blacker, D.J.; Knuckey, N.W.; Meloni, B.P. Comparison of neuroprotective efficacy of poly-arginine R18 and R18D (D-enantiomer) peptides following permanent middle cerebral artery occlusion in the Wistar rat and in vitro toxicity studies. PLoS ONE 2018, 13, 1–20. [Google Scholar] [CrossRef]
- Milani, D.; Cross, J.L.; Anderton, R.S.; Blacker, D.J.; Knuckey, N.W.; Meloni, B.P. Neuroprotective efficacy of poly-arginine R18 and NA-1 (TAT-NR2B9c) peptides following transient middle cerebral artery occlusion in the rat. Neurosci. Res. 2017, 114, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Milani, D.; Cross, J.L.; Anderton, R.S.; Blacker, D.J.; Knuckey, N.W.; Meloni, B.P. Delayed 2-h post-stroke administration of R18 and NA-1 (TAT-NR2B9c) peptides after permanent and/or transient middle cerebral artery occlusion in the rat. Brain Res. Bull. 2017, 135, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Milani, D.; Knuckey, N.W.; Anderton, R.S.; Cross, J.L.; Meloni, B.P. The R18 Polyarginine Peptide Is More Effective Than the TAT-NR2B9c (NA-1) Peptide when administered 60 min after permanent middle cerebral artery occlusion in the rat. Stroke Res. Treat. 2016. [Google Scholar] [CrossRef] [PubMed]
- Meloni, B.P.; Chen, Y.; Harrison, K.A.; Nashed, J.Y.; Blacker, D.J.; South, S.M.; Anderton, R.S.; Mastaglia, F.L.; Winterborn, A.; Knuckey, N.W.; et al. Poly-arginine peptide-18 (R18) reduces brain injury and improves functional outcomes in a non-human primate stroke model. Neurotherapeutics 2009, in press. [Google Scholar]
- Edwards, A.; Feindel, K.; Cross, J.; Anderton, R.; Clark, V.; Knuckey, N.; Meloni, B. Neuroprotective efficacy of poly-arginine-18 (R18) peptides using an in vivo model of perinatal hypoxic ischaemic encephalopathy (HIE). J. Cereb. Blood Flow Metab. 2017, 37, 18–19. [Google Scholar]
- Edwards, A.B.; Cross, J.L.; Anderton, R.S.; Knuckey, N.W.; Meloni, B.P. Poly-arginine R18 and R18D (D-enantiomer) peptides reduce infarct volume and improves behavioural outcomes following perinatal hypoxic-ischaemic encephalopathy in the P7 rat. Mol. Brain 2018. [Google Scholar] [CrossRef]
- Chiu, L.S.; Anderton, R.S.; Cross, J.L.; Clark, V.W.; Edwards, A.B.; Knuckey, N.W.; Meloni, B.P. Assessment of R18, COG1410, and APP96-110 in excitotoxicity and traumatic brain injury. Transl. Neurosci. 2017, 8, 147–157. [Google Scholar] [CrossRef] [Green Version]
- Chiu, L.S.; Anderton, R.S.; Cross, J.L.; Clark, V.W.; Knuckey, N.W.; Meloni, B.P. Poly-arginine peptide R18D reduces neuroinflammation and functional deficits following traumatic brain injury in the long-evans rat. Int. J. Pept. Res. Ther. 2019, 1–10. [Google Scholar] [CrossRef] [Green Version]
- MacDougall, G.; Anderton, R.S.; Edwards, A.B.; Knuckey, N.W.; Meloni, B.P. The neuroprotective peptide poly-arginine-12 (R12) reduces cell surface levels of NMDA NR2B receptor subunit in cortical neurons; Investigation into the involvement of endocytic mechanisms. J. Mol. Neurosci. 2017, 61, 1–12. [Google Scholar] [CrossRef]
- Rigobello, M.P.; Barzon, E.; Marin, O.; Bindoli, A. Effect of polycation peptides on mitochondrial permeability transition. Biochem. Biophys. Res. Commun. 1995, 280, 15579–15586. [Google Scholar] [CrossRef]
- Nicholls, D. Mitochondrial dysfunction and glutamate excitotoxicity studied in primary neuronal cultures. Curr. Mol. Med. 2005, 4, 149–177. [Google Scholar] [CrossRef] [PubMed]
- Starkov, A.A.; Chinopoulos, C.; Fiskum, G. Mitochondrial calcium and oxidative stress as mediators of ischemic brain injury. Cell Calcium 2004, 36, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Lytton, J.; Westlin, M.; Hanley, M.R. Thapsigargin inhibits the sarcoplasmic or endoplasmic reticulum Ca-ATPase family of calcium pumps. J. Biol. Chem. 1991, 266, 17067–17071. [Google Scholar] [PubMed]
- Sehgal, P.; Szalai, P.; Olesen, C.; Praetorius, H.A.; Nissen, P.; Christensen, S.B.; Engedal, N.; Møller, J.V. Inhibition of the sarco/endoplasmic reticulum (ER) Ca2+-ATPase by thapsigargin analogs induces cell death via ER Ca2+ depletion and the unfolded protein response. J. Biol. Chem. 2017, 292, 19656–19673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meloni, B.P.; Brookes, L.M.; Clark, V.W.; Cross, J.L.; Edwards, A.B.; Anderton, R.S.; Hopkins, R.M.; Hoffmann, K.; Knuckey, N.W. Poly-arginine and arginine-rich peptides are neuroprotective in stroke models. J. Cereb. Blood Flow Metab. 2015, 35, 993–1004. [Google Scholar] [CrossRef]
- Ferrer-Montiel, A.V.; Merino, J.M.; Blondelle, S.E.; Perez-Payà, E.; Houghten, R.A.; Montal, M. Selected peptides targeted to the NMDA receptor channel protect neurons from excitotoxic death. Nat. Biotechnol. 1998, 16, 286–291. [Google Scholar] [CrossRef]
- Planells-Cases, R.; Aracil, A.; Merino, J.M.; Gallar, J.; Pérez-Payá, E.; Belmonte, C.; González-Ros, J.M.; Ferrer-Montiel, A.V. Arginine-rich peptides are blockers of VR-1 channels with analgesic activity. FEBS Lett. 2000, 481, 131–136. [Google Scholar] [CrossRef] [Green Version]
- Davoli, E.; Sclip, A.; Cecchi, M.; Cimini, S.; Carrà, A.; Salmona, M.; Borsello, T. Determination of tissue levels of a neuroprotectant drug: The cell permeable JNK inhibitor peptide. J. Pharmacol. Toxicol. Methods 2014, 70, 55–61. [Google Scholar] [CrossRef]
- Brittain, J.M.; Chen, L.; Wilson, S.M.; Brustovetsky, T.; Gao, X.; Ashpole, N.M.; Molosh, A.I.; You, H.; Hudmon, A.; Shekhar, A.; et al. Neuroprotection against traumatic brain Injury by a peptide derived from the Collapsin Response Mediator Protein 2 (CRMP2). J. Biol. Chem. 2011, 286, 37778–37792. [Google Scholar] [CrossRef] [Green Version]
- Cook, D.R.; Gleichman, A.J.; Cross, S.A.; Doshi, S.; Ho, W.; Jordan-Sciutto, K.L.; Lynch, D.R.; Kolson, D.L. NMDA receptor modulation by the neuropeptide apelin: Implications for excitotoxic injury. J. Neurochem. 2011, 118, 1113–1123. [Google Scholar] [CrossRef] [Green Version]
- François-Moutal, L.; Wang, Y.; Moutal, A.; Cottier, K.E.; Melemedjian, O.K.; Yang, X.; Wang, Y.; Ju, W.; Largent-Milnes, T.M.; Khanna, M.; et al. A membrane-delimited N-myristoylated CRMP2 peptide aptamer inhibits CaV2.2 trafficking and reverses inflammatory and postoperative pain behaviors. Pain 2015, 156, 1247–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herce, H.D.; Garcia, A.E.; Cardoso, M.C. Fundamental molecular mechanism for the cellular uptake of guanidinium-rich molecules. J. Am. Chem. Soc. 2014, 136. [Google Scholar] [CrossRef] [PubMed]
- Fotin-Mleczek, M. Cationic cell-penetrating peptides interfere with TNF signalling by induction of TNF receptor internalization. J. Cell Sci. 2005, 118, 3339–3351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weng, X.C.; Gai, X.D.; Zheng, J.Q.; Li, J. Agmatine blocked voltage-gated calcium channel in cultured rat hippocampal neurons. Acta Pharmacol. Sin. 2003, 25, 281–285. [Google Scholar]
- Keana, J.F.W.; McBurney, R.N.; Scherz, M.W.; Fischer, J.B.; Hamilton, P.N.; Smith, S.M.; Server, A.C.; Finkbeiner, S.; Stevens, C.F.; Jahr, C.; et al. Synthesis and characterization of a series of diarylguanidines that are noncompetitive N-methyl-D-aspartate receptor antagonists with neuroprotective properties. Proc. Natl. Acad. Sci. USA 1989, 86, 5631–5635. [Google Scholar] [CrossRef] [Green Version]
- Cerrato, C.P.; Pirisinu, M.; Vlachos, E.N.; Langel, Ü. Novel cell-penetrating peptide targeting mitochondria. FASEB J. 2015, 29, 4589–4599. [Google Scholar] [CrossRef]
- Birk, A.V.; Liu, S.; Soong, Y.; Mills, W.; Singh, P.; Warren, J.D.; Seshan, S.V.; Pardee, J.D.; Szeto, H.H. The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. J. Am. Soc. Nephrol. 2013, 24, 1250–1261. [Google Scholar] [CrossRef]
- Birk, A.V.; Chao, W.M.; Liu, S.; Soong, Y.; Szeto, H.H. Disruption of cytochrome c heme coordination is responsible for mitochondrial injury during ischemia. Biochim. Biophys. Acta 2015, 1847, 1075–1084. [Google Scholar] [CrossRef] [Green Version]
- Szeto, H.H. Mitochondria-targeted cytoprotective peptides for ischemia-reperfusion injury. Antioxidants Redox Signal. 2008, 10, 601–619. [Google Scholar] [CrossRef]
- Szeto, H.H.; Liu, S.; Soong, Y.; Wu, D.; Darrah, S.F.; Cheng, F.-Y.; Zhao, Z.; Ganger, M.; Tow, C.Y.; Seshan, S.V. Mitochondria-targeted peptide accelerates ATP recovery and reduces ischemic kidney injury. J. Am. Soc. Nephrol. 2011, 22, 1041–1052. [Google Scholar] [CrossRef] [Green Version]
- Aluganti Narasimhulu, C.; Selvarajan, K.; Brown, M.; Parthasarathy, S. Cationic peptides neutralize Ox-LDL, prevent its uptake by macrophages, and attenuate inflammatory response. Atherosclerosis 2014, 236, 133–141. [Google Scholar] [CrossRef]
- Yildiz, G.; Demiryürek, A.T.; Sahin-Erdemli, I.; Kanzik, I. Comparison of antioxidant activities of aminoguanidine, methylguanidine and guanidine by luminol-enhanced chemiluminescence. Br. J. Pharmacol. 1998, 124, 905–910. [Google Scholar] [CrossRef] [Green Version]
- Wascher, T.C.; Posch, K.; Wallner, S.; Hermetter, A.; Kostner, G.M.; Graier, W.F. Vascular effects of L-arginine: Anything beyond a substrate for the NO-synthase? Biochem. Biophys. Res. Commun. 1997, 234, 35–38. [Google Scholar] [CrossRef]
- Lass, A.; Suessenbacher, A.; Wölkart, G.; Mayer, B.; Brunner, F. Functional and analytical evidence for scavenging of oxygen radicals by L-arginine. Mol. Pharmacol. 2002, 61, 1081–1088. [Google Scholar] [CrossRef]
- Giardino, I.; Fard, A.K.; Hatchell, D.L.; Brownlee, M. Aminoguanidine inhibits reactive oxygen species formation, lipid peroxidation, and oxidant-induced apoptosis. Diabetes 1998, 47, 1114–1120. [Google Scholar] [CrossRef]
- Lawler, J.M.; Barnes, W.S.; Wu, G.; Song, W.; Demaree, S. Direct antioxidant properties of creatine. Biochem. Biophys. Res. Commun. 2002, 290, 47–52. [Google Scholar] [CrossRef]
- Courderot-Masuyer, C.; Dalloz, F.; Maupoil, V.; Rochette, L. Antioxidant properties of aminoguanidine. Fundam. Clin. Pharmacol. 1999, 13, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Gering, I.; Kutzsche, J.; Nagel-Steger, L.; Willbold, D. Toward the mode of action of the clinical stage all-D-enantiomeric peptide RD2 on Aβ42 aggregation. ACS Chem. Neurosci. 2019, 10, 4800–4809. [Google Scholar] [CrossRef]
- Meloni, B.P.; Majda, B.T.; Knuckey, N.W. Establishment of neuronal in vitro models of ischemia in 96-well microtiter strip-plates that result in acute, progressive and delayed neuronal death. Neuroscience 2001, 108, 17–26. [Google Scholar] [CrossRef]
- Lewerenz, J.; Maher, P. Chronic glutamate toxicity in neurodegenerative diseases - What is the evidence? Front. Neurosci. 2015, 16, 469. [Google Scholar] [CrossRef]
- Carvalho, C.; Moreira, P.I. Oxidative stress: A major player in cerebrovascular alterations associated to neurodegenerative events. Front. Physiol. 2018, 3, 806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Sample Availability: Samples of the compound R18 are available from the authors. |





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MacDougall, G.; Anderton, R.S.; Trimble, A.; Mastaglia, F.L.; Knuckey, N.W.; Meloni, B.P. Poly-arginine-18 (R18) Confers Neuroprotection through Glutamate Receptor Modulation, Intracellular Calcium Reduction, and Preservation of Mitochondrial Function. Molecules 2020, 25, 2977. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25132977
MacDougall G, Anderton RS, Trimble A, Mastaglia FL, Knuckey NW, Meloni BP. Poly-arginine-18 (R18) Confers Neuroprotection through Glutamate Receptor Modulation, Intracellular Calcium Reduction, and Preservation of Mitochondrial Function. Molecules. 2020; 25(13):2977. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25132977
Chicago/Turabian StyleMacDougall, Gabriella, Ryan S. Anderton, Amy Trimble, Frank L. Mastaglia, Neville W. Knuckey, and Bruno P. Meloni. 2020. "Poly-arginine-18 (R18) Confers Neuroprotection through Glutamate Receptor Modulation, Intracellular Calcium Reduction, and Preservation of Mitochondrial Function" Molecules 25, no. 13: 2977. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25132977