
Inside Perspective of the Synthetic and Computational Toolbox of JAK Inhibitors: Recent Updates

 ,
,  , ,
, ,  and
and
Abstract
:1. Introduction
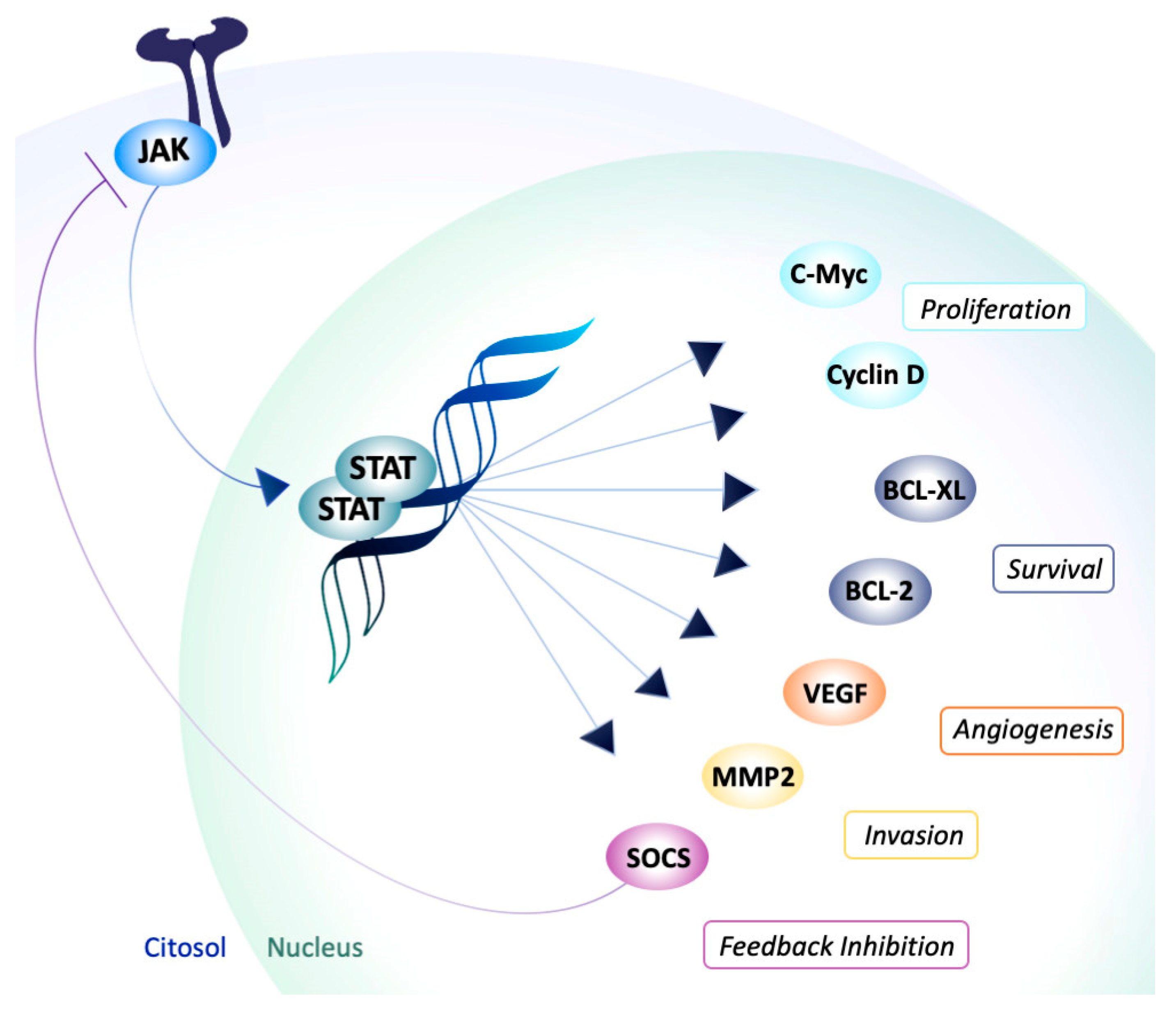
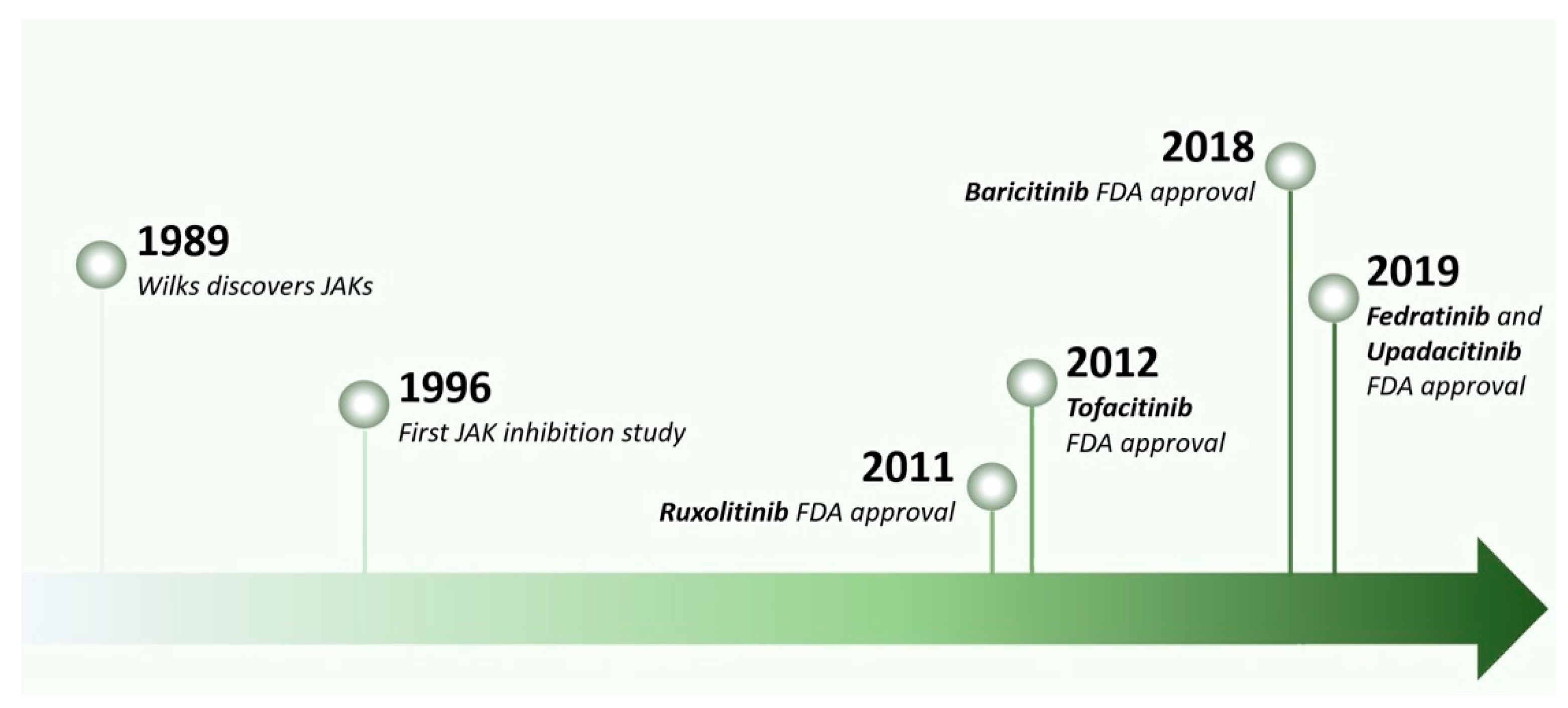
2. The Rise of JAK Inhibitors
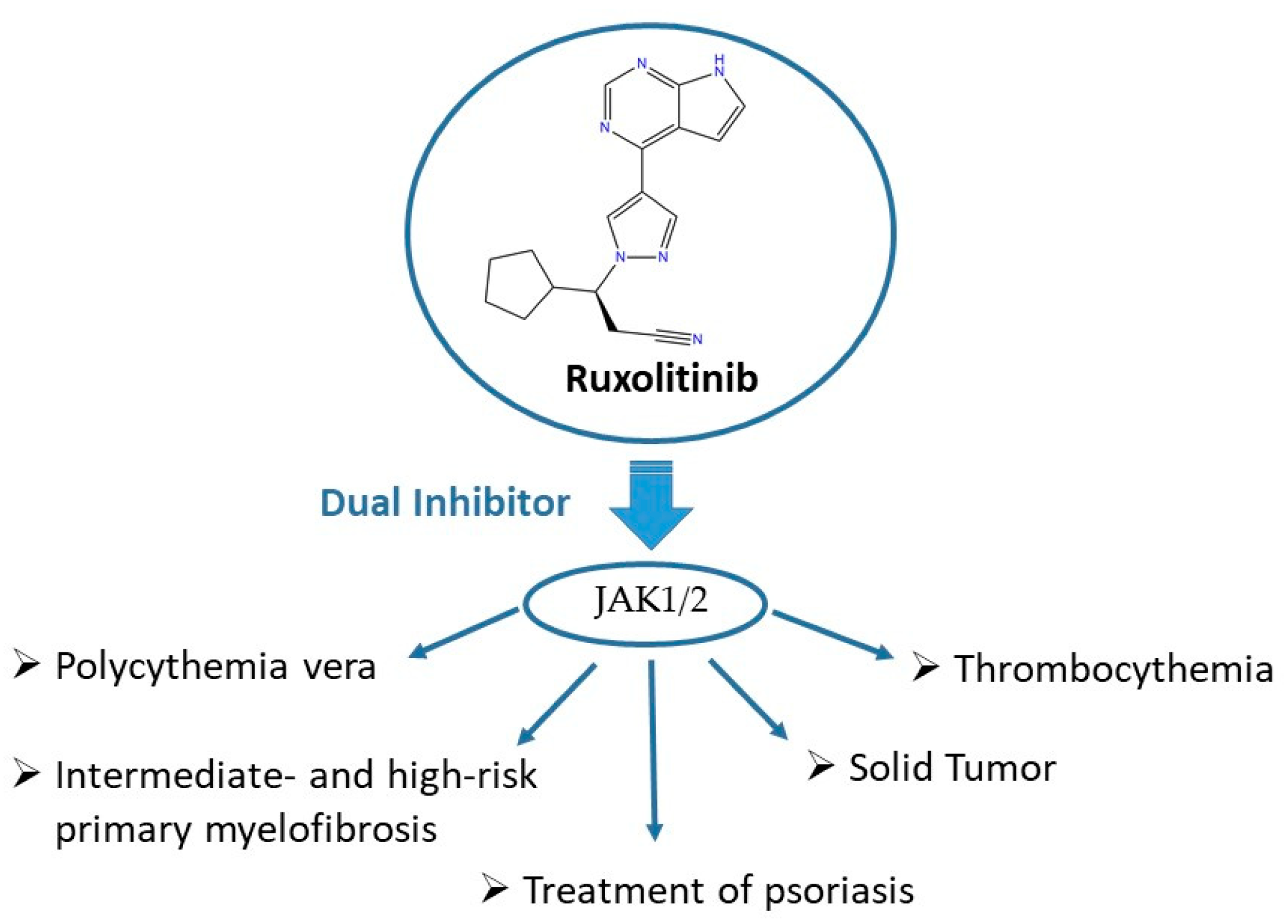
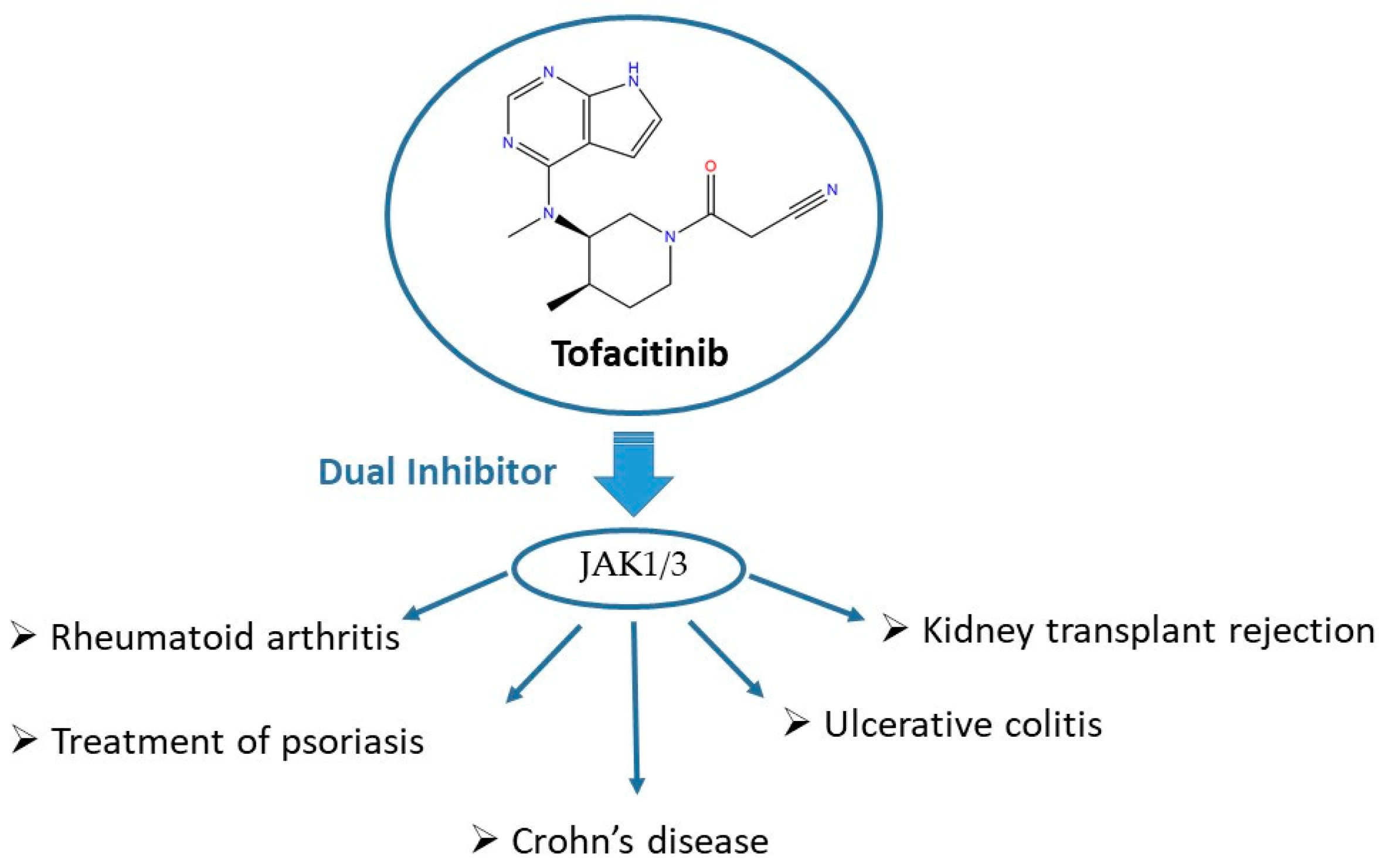
3. JAKis as Important Players in Multifactorial Diseases
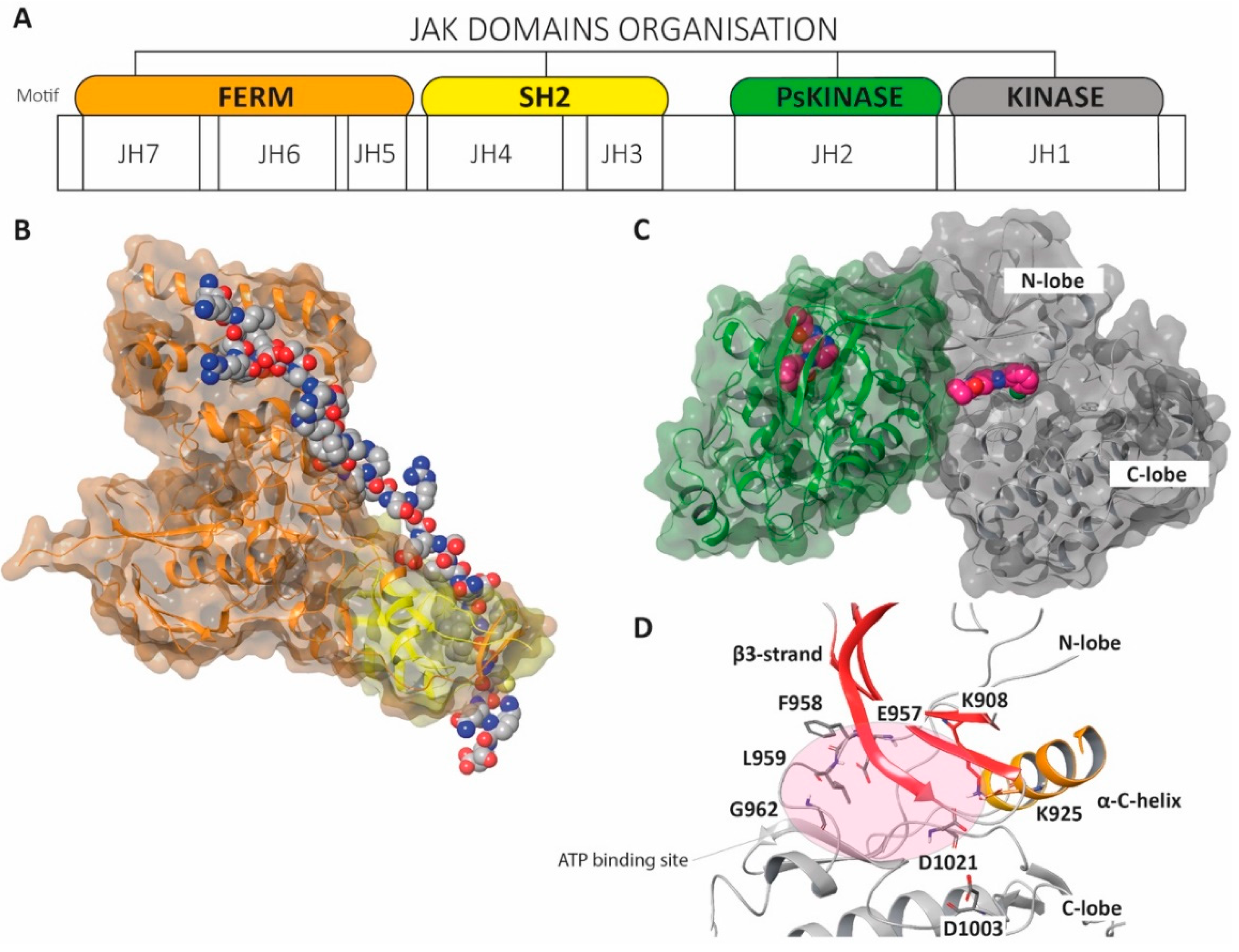
4. The Structure of Janus Kinases
- (a)
- JAK kinases are large, multidomain proteins (120–130 kDa) initially organized into seven different JAK homology (JH) domains;
- (b)
- Four domains are currently considered the most relevant: the N-terminal four-point-one, ezrin, radixin, moesin (FERM) domain (JH5/6/7), the Src homology 2 (SH2) or SH2-like (SH2L) domain (JH3/4), the pseudokinase domain (Ps-JH2) and the C-terminal tyrosine kinase domain (JH1);
- (c)
- The FERM and SH2 domains form a unique structural assemblage (Figure 8B);
- (d)
- JH1 represents the catalytic kinase domain (Figure 8C);
- (e)
- JH2 might negatively regulate JAK2 function by the phosphorylation of key residues within the JH2 domain;
- (f)
- JH3-JH4 stabilize the conformation of the JAKs;
- (g)
- JH5-JH7 directly interact with the intracellular domains of the cytokine receptor and with the JH1 domain.
5. Synthetic Strategies for the Development of Ruxolitinib and Tofacitinib
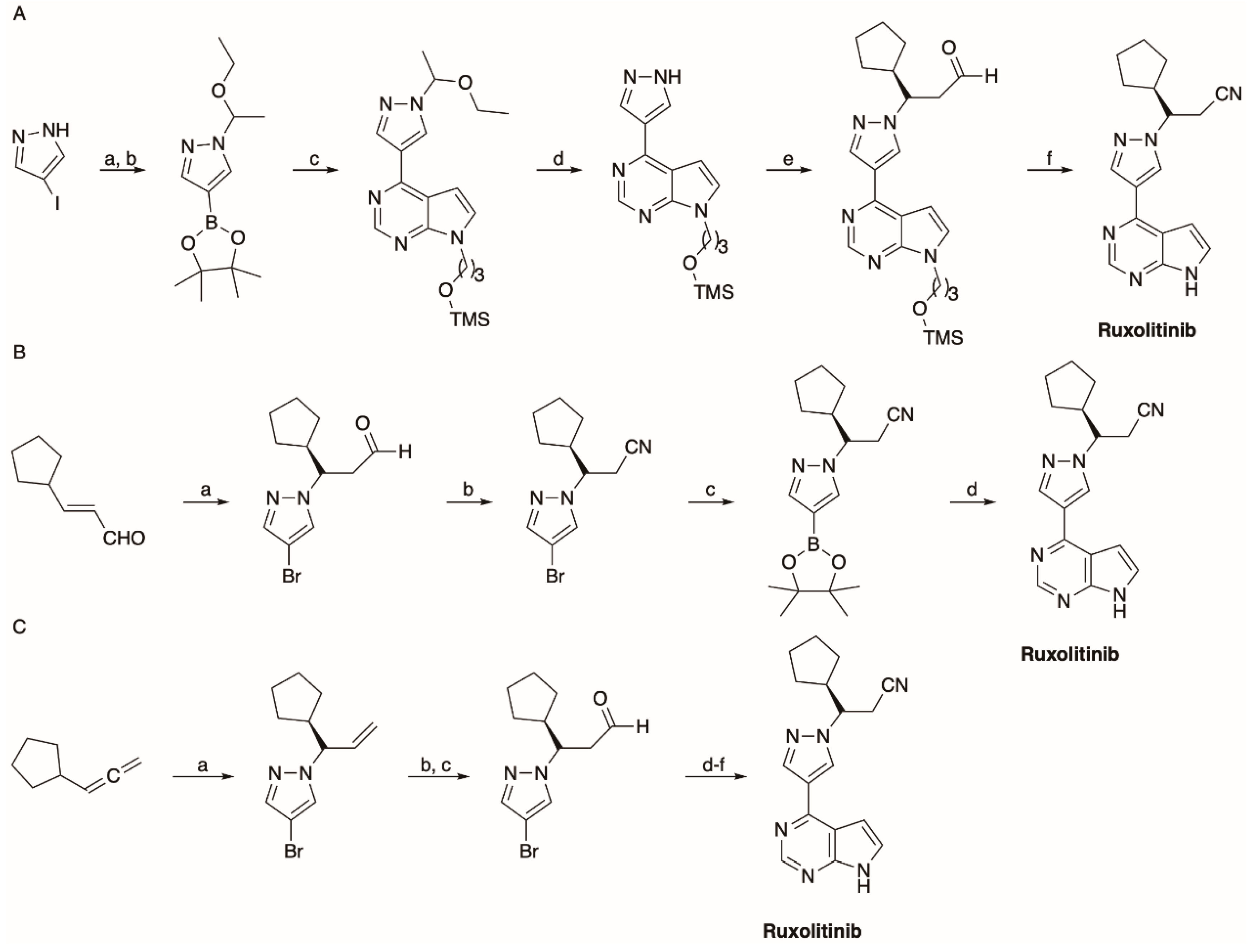
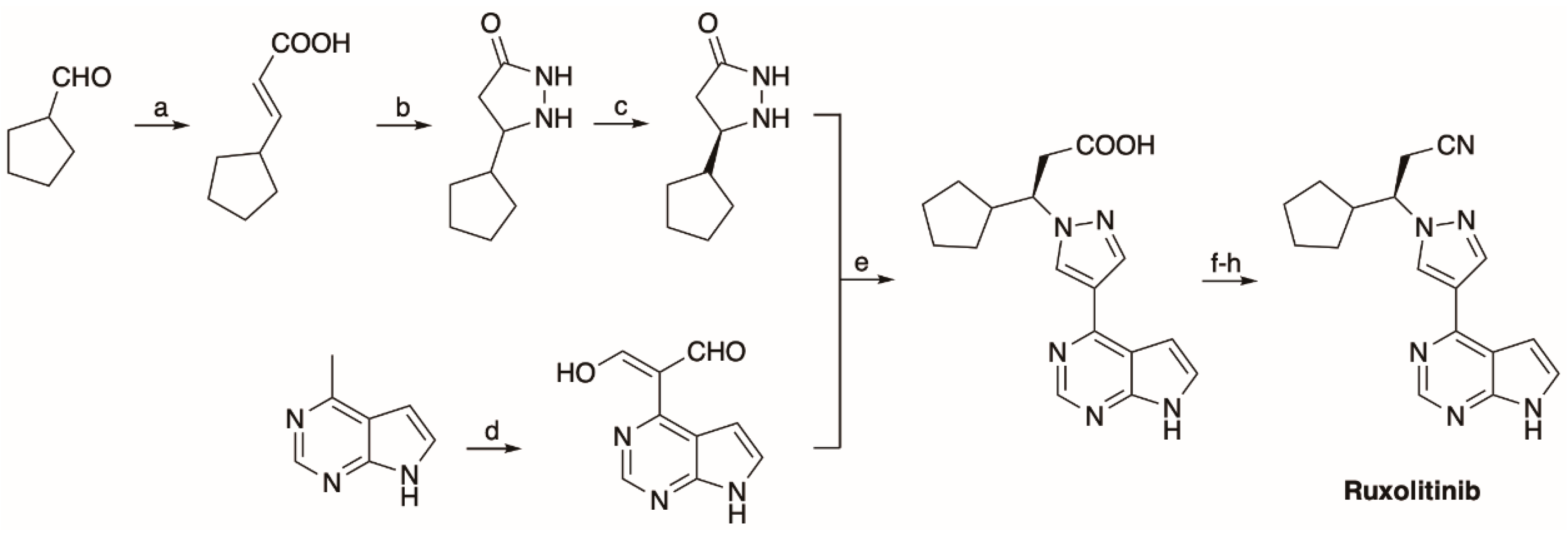
5.1. The Synthesis of Ruxolitinib
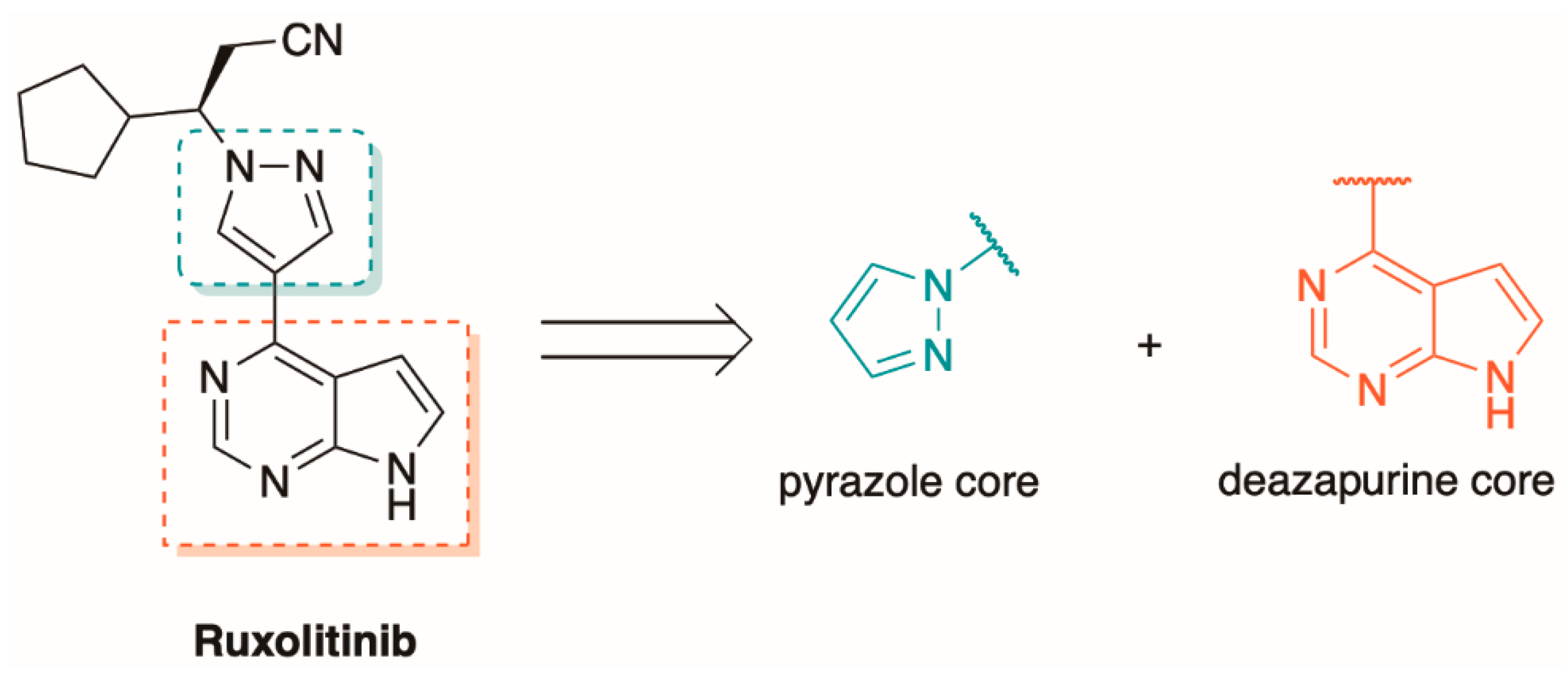
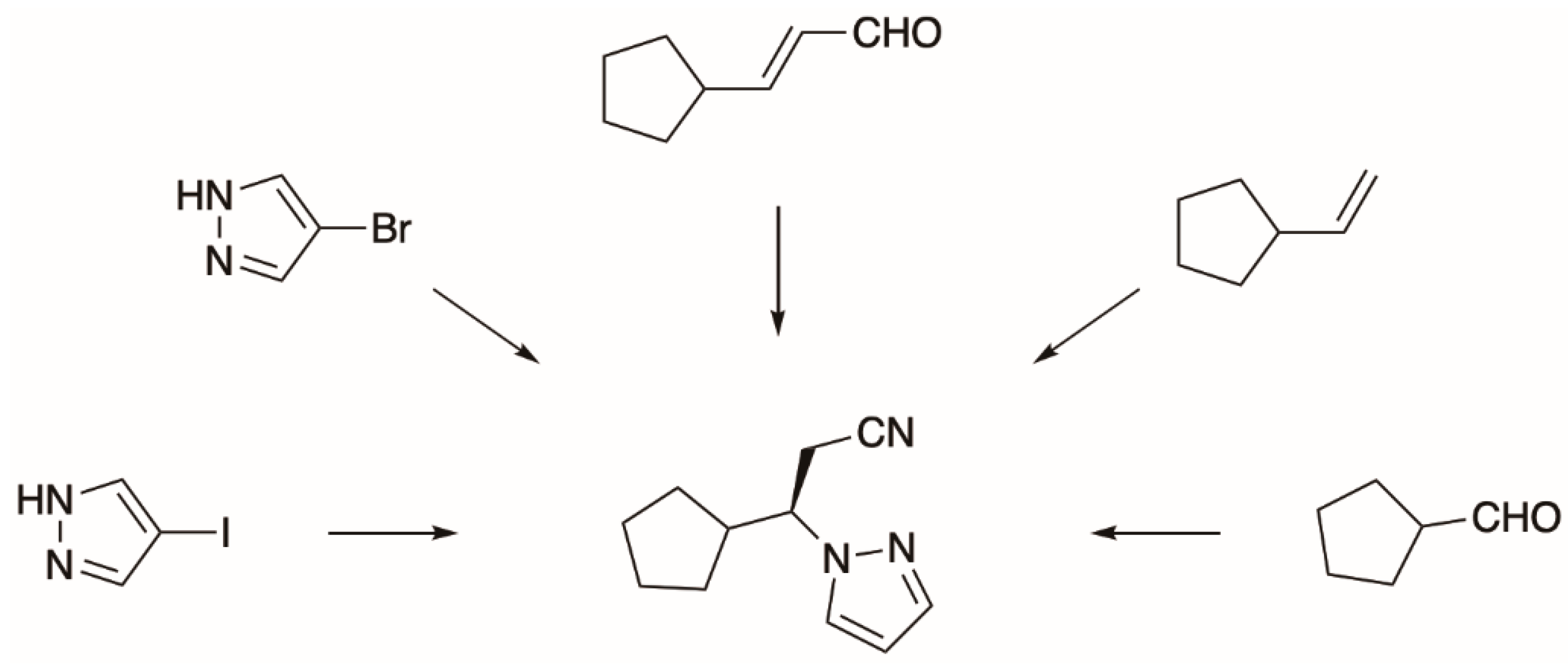
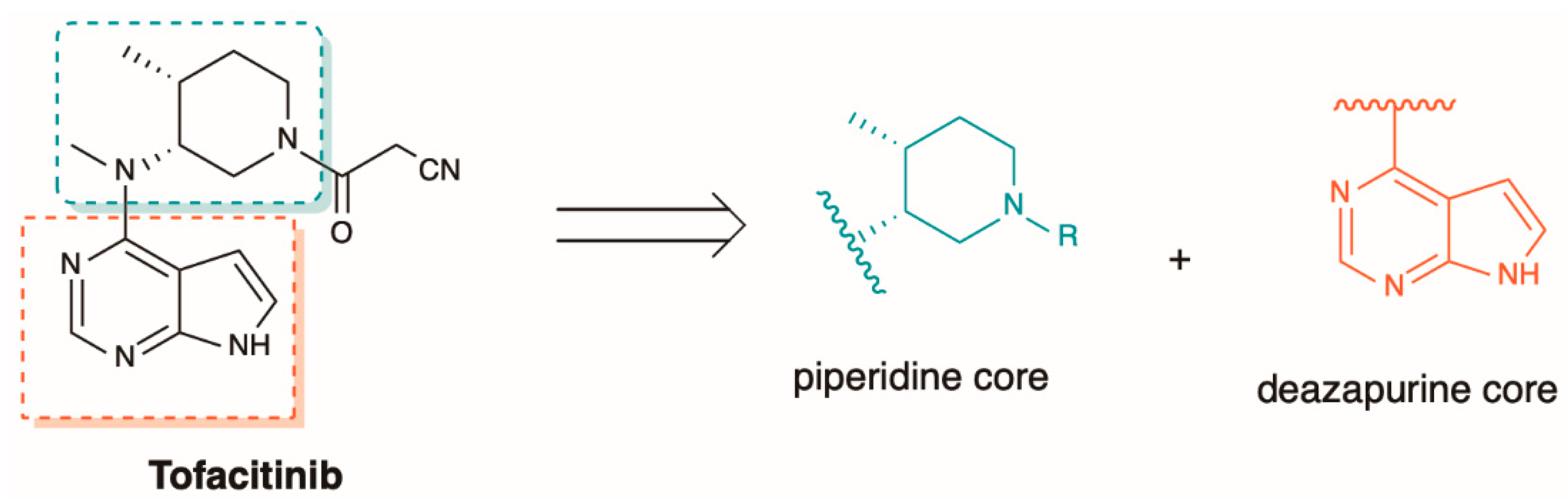
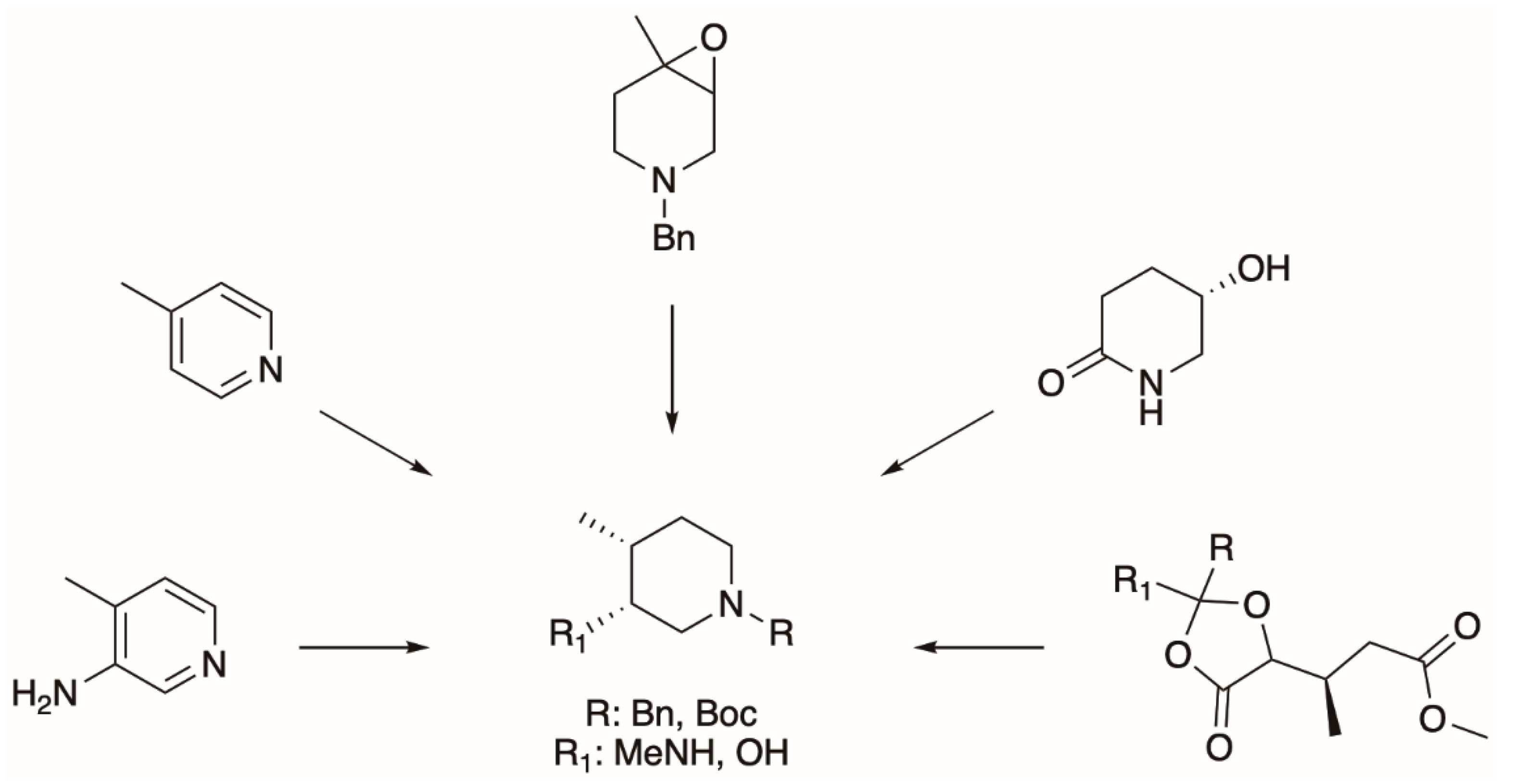
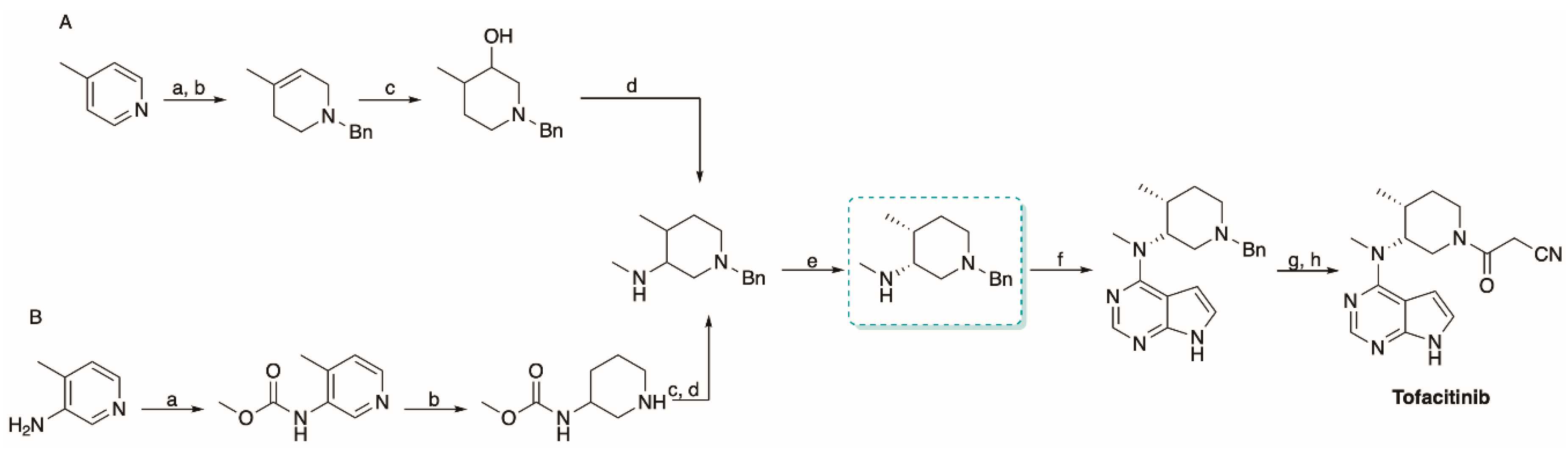
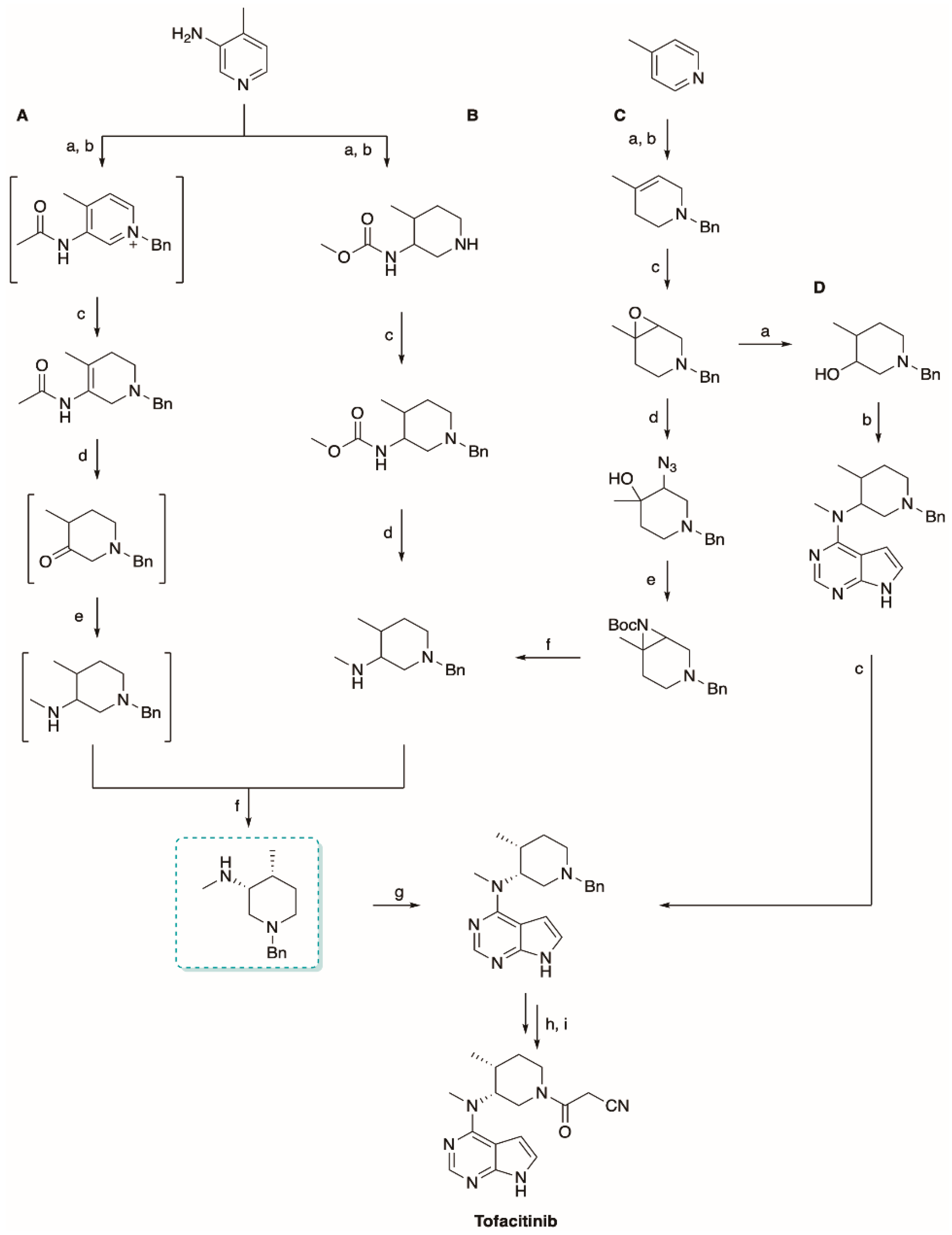
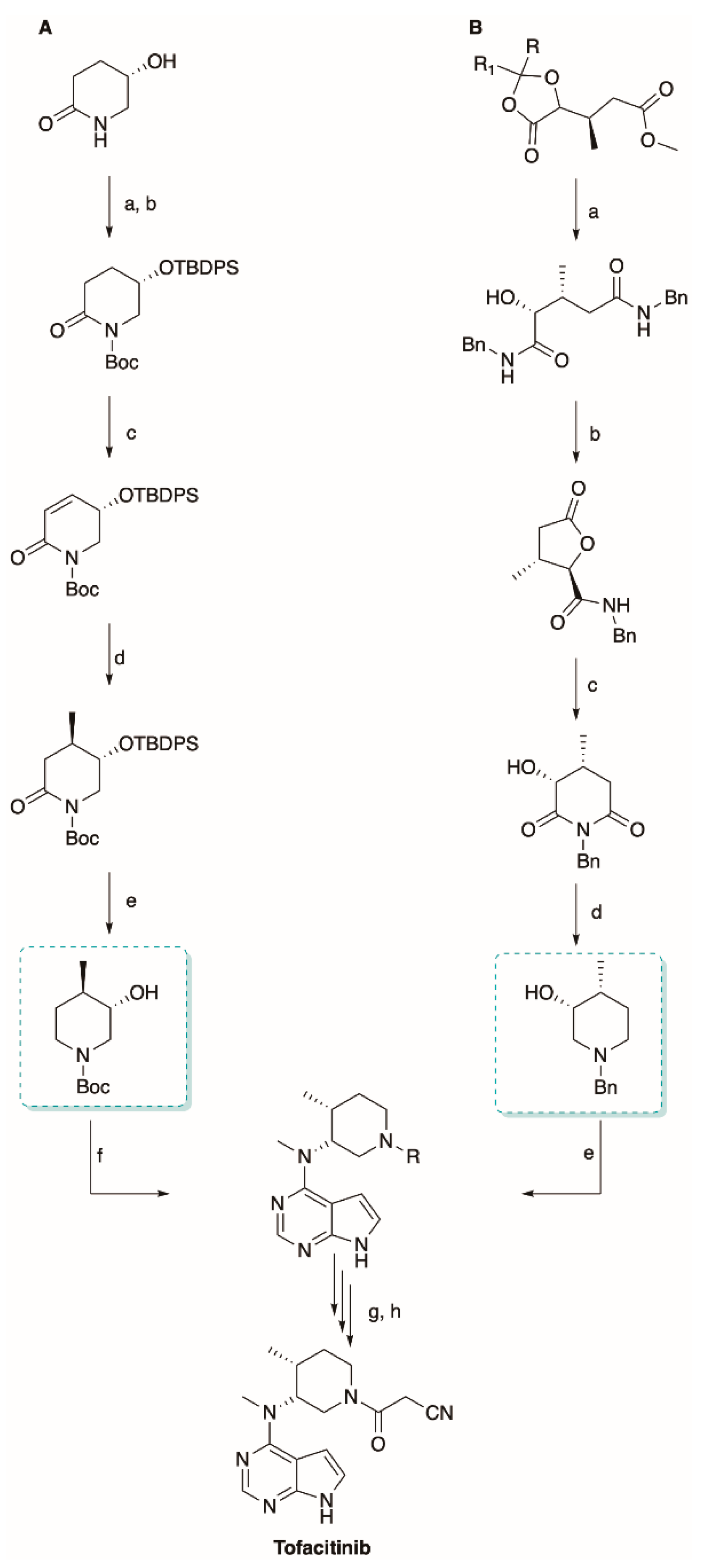
5.2. The Synthesis of Tofacitinib
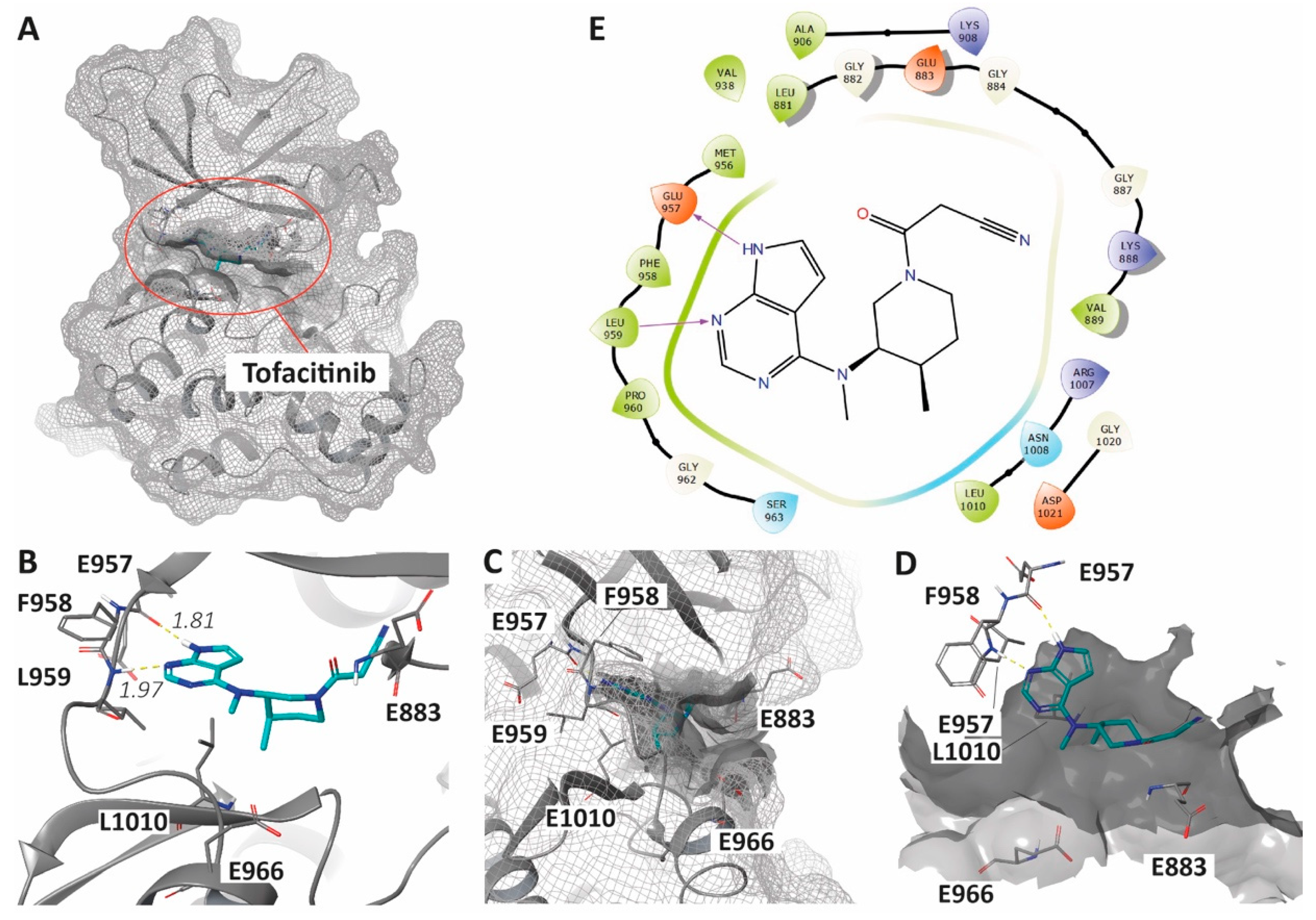
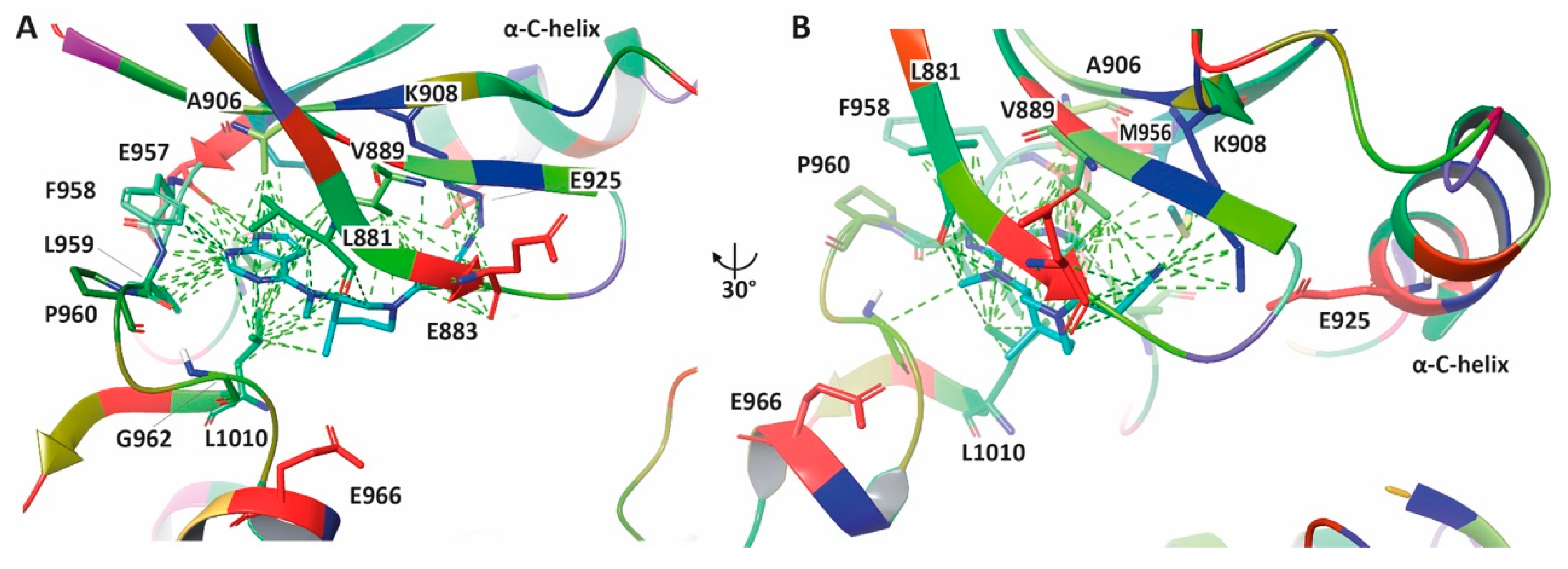
6. Enhancing JAKis Discovery through Structure-Based Drug Design Strategies
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Coricello, A.; El-Magboub, A.; Luna, M.; Ferrario, A.; Haworth, I.S.; Gomer, C.J.; Aiello, F.; Adams, J.D. Rational drug design and synthesis of new α-Santonin derivatives as potential COX-2 inhibitors. Bioorg. Med. Chem. Lett. 2018, 28, 993–996. [Google Scholar] [CrossRef]
- Perri, F.; Coricello, A.; Adams, J. Monoterpenoids: The Next Frontier in the Treatment of Chronic Pain? J 2020, 3, 195–214. [Google Scholar] [CrossRef]
- Coricello, A.; Adams, J.D.; Lien, E.J.; Nguyen, C.; Perri, F.; Williams, T.J.; Aiello, F. A Walk in Nature: Sesquiterpene Lactones as Multi-Target Agents Involved in Inflammatory Pathways. Curr. Med. Chem. 2020, 27, 1501–1514. [Google Scholar] [CrossRef]
- Mimmi, S.; Vecchio, E.; Iaccino, E.; Rossi, M.; Lupia, A.; Albano, F.; Chiurazzi, F.; Fiume, G.; Pisano, A.; Ceglia, S.; et al. Evidence of shared epitopic reactivity among independent B-cell clones in chronic lymphocytic leukemia patients. Leukemia 2016, 30, 2419–2422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iaccino, E.; Mimmi, S.; Dattilo, V.; Marino, F.; Candeloro, P.; Di Loria, A.; Marimpietri, D.; Pisano, A.; Albano, F.; Vecchio, E.; et al. Monitoring multiple myeloma by idiotype-specific peptide binders of tumor-derived exosomes. Mol. Cancer 2017, 16, 159. [Google Scholar] [CrossRef] [PubMed]
- Lupia, A.; Mimmi, S.; Iaccino, E.; Maisano, D.; Moraca, F.; Talarico, C.; Vecchio, E.; Fiume, G.; Ortuso, F.; Scala, G.; et al. Molecular modelling of epitopes recognized by neoplastic B lymphocytes in Chronic Lymphocytic Leukemia. Eur. J. Med. Chem. 2020, 185, 111838. [Google Scholar] [CrossRef]
- Zhao, Z.; Ye, C.; Dong, L. The off-label uses profile of tofacitinib in systemic rheumatic diseases. Int. Immunopharmacol. 2020, 83, 106480. [Google Scholar] [CrossRef]
- Alvarez, J.V.; Frank, D.A. Genome-wide analysis of STAT target genes: Elucidating the mechanism of STAT-mediated oncogenesis. Cancer Biol. Ther. 2004, 3, 1045–1050. [Google Scholar] [CrossRef] [Green Version]
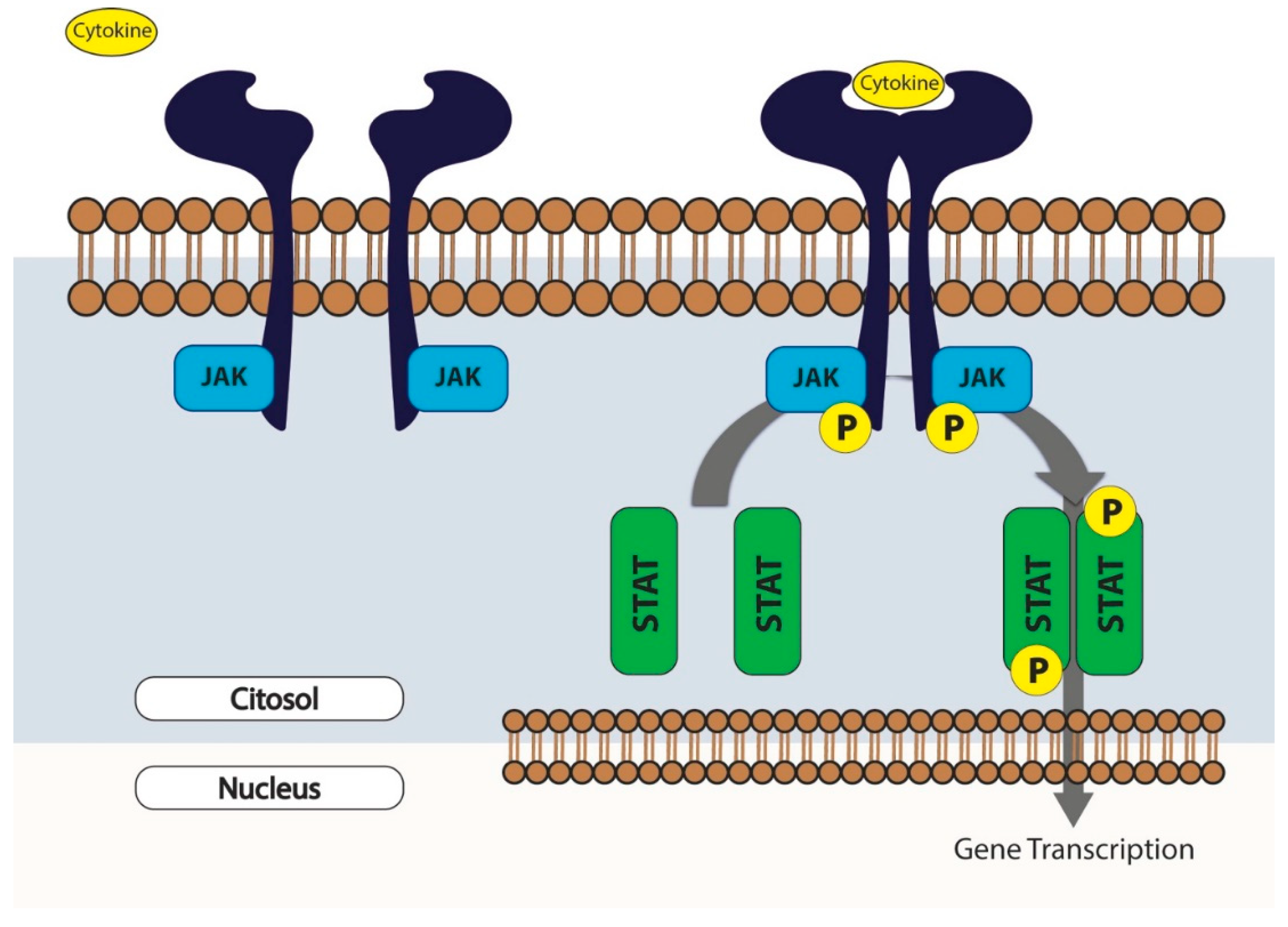
- Rawlings, J.S.; Rosler, K.M.; Harrison, D.A. The JAK/STAT signaling pathway. J. Cell Sci. 2004, 117, 1281–1283. [Google Scholar] [CrossRef] [Green Version]
- Tarantelli, C.; Lupia, A.; Stathis, A.; Bertoni, F. Is There a Role for Dual PI3K/mTOR Inhibitors for Patients Affected with Lymphoma? Int. J. Mol. Sci. 2020, 21, 1060. [Google Scholar] [CrossRef] [Green Version]
- O’Shea, J.J.; Kontzias, A.; Yamaoka, K.; Tanaka, Y.; Laurence, A. Janus kinase inhibitors in autoimmune diseases. Ann. Rheum. Dis. 2013, 72, ii111–ii115. [Google Scholar] [CrossRef] [PubMed]
- Mavers, M.; Ruderman, E.M.; Perlman, H. Intracellular signal pathways: Potential for therapies. Curr. Rheumatol. Rep. 2009, 11, 378–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurence, A.; Pesu, M.; Silvennoinen, O.; O’Shea, J. JAK Kinases in Health and Disease: An Update. Open Rheumatol. J. 2012, 6, 232–244. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, S.R.; Ettinger, R.; Zhou, Y.J.; Gadina, M.; Lipsky, P.; Siegel, R.; Candotti, F.; O’Shea, J.J. Cytokines and their role in lymphoid development, differentiation and homeostasis. Curr. Opin. Allergy Clin. Immunol. 2002, 2, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Jasuja, H.; Chadha, N.; Singh, P.K.; Kaur, M.; Bahia, M.S.; Silakari, O. Putative dual inhibitors of Janus kinase 1 and 3 (JAK1/3): Pharmacophore based hierarchical virtual screening. Comput. Biol. Chem. 2018, 76, 109–117. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Plenge, R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity 2012, 36, 542–550. [Google Scholar] [CrossRef] [Green Version]
- Aringer, M.; Hofmann, S.R.; Frucht, D.M.; Chen, M.; Centola, M.; Morinobu, A.; Visconti, R.; Kastner, D.L.; Smolen, J.S.; O’Shea, J.J. Characterization and analysis of the proximal Janus kinase 3 promoter. J. Immunol. 2003, 170, 6057–6064. [Google Scholar] [CrossRef] [Green Version]
- Clark, J.D.; Flanagan, M.E.; Telliez, J.B. Discovery and development of Janus kinase (JAK) inhibitors for inflammatory diseases. J. Med. Chem. 2014, 57, 5023–5038. [Google Scholar] [CrossRef]
- Darnell, J.E., Jr.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef] [Green Version]
- Ihle, J.N.; Nosaka, T.; Thierfelder, W.; Quelle, F.W.; Shimoda, K. Jaks and Stats in cytokine signaling. Stem. Cells 1997, 15, 105–111; discussion 112. [Google Scholar] [CrossRef]
- Baker, S.J.; Rane, S.G.; Reddy, E.P. Hematopoietic cytokine receptor signaling. Oncogene 2007, 26, 6724–6737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meydan, N.; Grunberger, T.; Dadi, H.; Shahar, M.; Arpaia, E.; Lapidot, Z.; Leeder, J.S.; Freedman, M.; Cohen, A.; Gazit, A.; et al. Inhibition of acute lymphoblastic leukaemia by a Jak-2 inhibitor. Nature 1996, 379, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Quintás-Cardama, A.; Vaddi, K.; Liu, P.; Manshouri, T.; Li, J.; Scherle, P.A.; Caulder, E.; Wen, X.; Li, Y.; Waeltz, P.; et al. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: Therapeutic implications for the treatment of myeloproliferative neoplasms. Blood 2010, 115, 3109–3117. [Google Scholar] [CrossRef] [PubMed]
- Verstovsek, S.; Kantarjian, H.; Mesa, R.A.; Pardanani, A.D.; Cortes-Franco, J.; Thomas, D.A.; Estrov, Z.; Fridman, J.S.; Bradley, E.C.; Erickson-Viitanen, S.; et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N. Engl. J. Med. 2010, 363, 1117–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plosker, G.L. Ruxolitinib: A review of its use in patients with myelofibrosis. Drugs 2015, 75, 297–308. [Google Scholar] [CrossRef]
- Van Den Neste, E.; André, M.; Gastinne, T.; Stamatoullas, A.; Haioun, C.; Belhabri, A.; Reman, O.; Casasnovas, O.; Ghesquieres, H.; Verhoef, G.; et al. A phase II study of the oral JAK1/JAK2 inhibitor ruxolitinib in advanced relapsed/refractory Hodgkin lymphoma. Haematologica 2018, 103, 840–848. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Yoon, D.H.; Kang, H.J.; Hong, J.Y.; Lee, H.S.; Oh, S.Y.; Shin, H.J.; Kong, J.H.; Yi, J.H.; Sakamoto, K.; et al. Ruxolitinib shows activity against Hodgkin lymphoma but not primary mediastinal large B-cell lymphoma. BMC Cancer 2019, 19, 1080. [Google Scholar] [CrossRef] [Green Version]
- Spaner, D.E.; McCaw, L.; Wang, G.; Tsui, H.; Shi, Y. Persistent janus kinase-signaling in chronic lymphocytic leukemia patients on ibrutinib: Results of a phase I trial. Cancer Med. 2019, 8, 1540–1550. [Google Scholar] [CrossRef] [Green Version]
- Cingam, S.; Flatow-Trujillo, L.; Andritsos, L.A.; Arana Yi, C. Ruxolitinib In The Treatment of Polycythemia Vera: An Update On Health-Related Quality Of Life And Patient-Reported Outcomes. J. Blood Med. 2019, 10, 381–390. [Google Scholar] [CrossRef] [Green Version]
- Spoerl, S.; Mathew, N.R.; Bscheider, M.; Schmitt-Graeff, A.; Chen, S.; Mueller, T.; Verbeek, M.; Fischer, J.; Otten, V.; Schmickl, M.; et al. Activity of therapeutic JAK 1/2 blockade in graft-versus-host disease. Blood 2014, 123, 3832–3842. [Google Scholar] [CrossRef]
- Von Bubnoff, N.; Ihorst, G.; Grishina, O.; Röthling, N.; Bertz, H.; Duyster, J.; Finke, J.; Zeiser, R. Ruxolitinib in GvHD (RIG) study: A multicenter, randomized phase 2 trial to determine the response rate of Ruxolitinib and best available treatment (BAT) versus BAT in steroid-refractory acute graft-versus-host disease (aGvHD) (NCT02396628). BMC Cancer 2018, 18, 1132. [Google Scholar] [CrossRef] [PubMed]
- Traynor, K. FDA approves tofacitinib for rheumatoid arthritis. Am. J. Health Syst. Pharm. 2012, 69, 2120. [Google Scholar] [CrossRef]
- Scott, L.J. Tofacitinib: A review of its use in adult patients with rheumatoid arthritis. Drugs 2013, 73, 857–874. [Google Scholar] [CrossRef] [PubMed]
- Vuitton, L.; Koch, S.; Peyrin-Biroulet, L. Janus kinase inhibition with tofacitinib: Changing the face of inflammatory bowel disease treatment. Curr. Drug Targets 2013, 14, 1385–1391. [Google Scholar] [CrossRef] [PubMed]
- Panés, J.; Vermeire, S.; Lindsay, J.O.; Sands, B.E.; Su, C.; Friedman, G.; Zhang, H.; Yarlas, A.; Bayliss, M.; Maher, S.; et al. Tofacitinib in Patients with Ulcerative Colitis: Health-Related Quality of Life in Phase 3 Randomised Controlled Induction and Maintenance Studies. J. Crohn Colitis 2018, 12, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Jabbari, A.; Sansaricq, F.; Cerise, J.; Chen, J.C.; Bitterman, A.; Ulerio, G.; Borbon, J.; Clynes, R.; Christiano, A.M.; Mackay-Wiggan, J. An Open-Label Pilot Study to Evaluate the Efficacy of Tofacitinib in Moderate to Severe Patch-Type Alopecia Areata, Totalis, and Universalis. J. Invest. Dermatol. 2018, 138, 1539–1545. [Google Scholar] [CrossRef] [Green Version]
- Cheng, M.W.; Kehl, A.; Worswick, S.; Goh, C. Successful Treatment of Severe Alopecia Areata With Oral or Topical Tofacitinib. J. Drugs Dermatol. 2018, 17, 800–803. [Google Scholar]
- Gupta, A.K.; Cernea, M.; Lynde, C.W. Tofacitinib in the Treatment of Rheumatoid Arthritis and Chronic Plaque Psoriasis. Skin Therapy Lett. 2017, 22, 1–7. [Google Scholar]
- Valenzuela, F.; Korman, N.J.; Bissonnette, R.; Bakos, N.; Tsai, T.F.; Harper, M.K.; Ports, W.C.; Tan, H.; Tallman, A.; Valdez, H.; et al. Tofacitinib in patients with moderate-to-severe chronic plaque psoriasis: Long-term safety and efficacy in an open-label extension study. Br. J. Dermatol. 2018, 179, 853–862. [Google Scholar] [CrossRef] [Green Version]
- Paik, J.; Deeks, E.D. Tofacitinib: A Review in Psoriatic Arthritis. Drugs 2019, 79, 655–663. [Google Scholar] [CrossRef]
- Mogul, A.; Corsi, K.; McAuliffe, L. Baricitinib: The Second FDA-Approved JAK Inhibitor for the Treatment of Rheumatoid Arthritis. Ann. Pharmacother. 2019, 53, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Guttman-Yassky, E.; Silverberg, J.I.; Nemoto, O.; Forman, S.B.; Wilke, A.; Prescilla, R.; de la Peña, A.; Nunes, F.P.; Janes, J.; Gamalo, M.; et al. Baricitinib in adult patients with moderate-to-severe atopic dermatitis: A phase 2 parallel, double-blinded, randomized placebo-controlled multiple-dose study. J. Am. Acad. Dermatol. 2019, 80, 913–921.e919. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.J.; Furie, R.A.; Tanaka, Y.; Kalunian, K.C.; Mosca, M.; Petri, M.A.; Dörner, T.; Cardiel, M.H.; Bruce, I.N.; Gomez, E.; et al. Baricitinib for systemic lupus erythematosus: A double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 2018, 392, 222–231. [Google Scholar] [CrossRef]
- Tuttle, K.R.; Brosius, F.C., 3rd; Adler, S.G.; Kretzler, M.; Mehta, R.L.; Tumlin, J.A.; Tanaka, Y.; Haneda, M.; Liu, J.; Silk, M.E.; et al. JAK1/JAK2 inhibition by baricitinib in diabetic kidney disease: Results from a Phase 2 randomized controlled clinical trial. Nephrol. Dial. Transpl. 2018, 33, 1950–1959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panchapakesan, U.; Pollock, C. Drug repurposing in kidney disease. Kidney Int. 2018, 94, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Blair, H.A. Fedratinib: First Approval. Drugs 2019, 79, 1719–1725. [Google Scholar] [CrossRef]
- Burmester, G.R.; Kremer, J.M.; Van den Bosch, F.; Kivitz, A.; Bessette, L.; Li, Y.; Zhou, Y.; Othman, A.A.; Pangan, A.L.; Camp, H.S. Safety and efficacy of upadacitinib in patients with rheumatoid arthritis and inadequate response to conventional synthetic disease-modifying anti-rheumatic drugs (SELECT-NEXT): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2018, 391, 2503–2512. [Google Scholar] [CrossRef]
- Duggan, S.; Keam, S.J. Upadacitinib: First Approval. Drugs 2019, 79, 1819–1828. [Google Scholar] [CrossRef]
- Van der Heijde, D.; Song, I.H.; Pangan, A.L.; Deodhar, A.; van den Bosch, F.; Maksymowych, W.P.; Kim, T.H.; Kishimoto, M.; Everding, A.; Sui, Y.; et al. Efficacy and safety of upadacitinib in patients with active ankylosing spondylitis (SELECT-AXIS 1): A multicentre, randomised, double-blind, placebo-controlled, phase 2/3 trial. Lancet 2019, 394, 2108–2117. [Google Scholar] [CrossRef]
- Takeuchi, T.; Tanaka, Y.; Tanaka, S.; Kawakami, A.; Iwasaki, M.; Katayama, K.; Rokuda, M.; Izutsu, H.; Ushijima, S.; Kaneko, Y.; et al. Efficacy and safety of peficitinib (ASP015K) in patients with rheumatoid arthritis and an inadequate response to methotrexate: Results of a phase III randomised, double-blind, placebo-controlled trial (RAJ4) in Japan. Ann. Rheum. Dis. 2019, 78, 1305–1319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genovese, M.C.; Kalunian, K.; Gottenberg, J.E.; Mozaffarian, N.; Bartok, B.; Matzkies, F.; Gao, J.; Guo, Y.; Tasset, C.; Sundy, J.S.; et al. Effect of Filgotinib vs Placebo on Clinical Response in Patients With Moderate to Severe Rheumatoid Arthritis Refractory to Disease-Modifying Antirheumatic Drug Therapy: The FINCH 2 Randomized Clinical Trial. JAMA 2019, 322, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Van der Heijde, D.; Baraliakos, X.; Gensler, L.S.; Maksymowych, W.P.; Tseluyko, V.; Nadashkevich, O.; Abi-Saab, W.; Tasset, C.; Meuleners, L.; Besuyen, R.; et al. Efficacy and safety of filgotinib, a selective Janus kinase 1 inhibitor, in patients with active ankylosing spondylitis (TORTUGA): Results from a randomised, placebo-controlled, phase 2 trial. Lancet 2018, 392, 2378–2387. [Google Scholar] [CrossRef] [Green Version]
- Mease, P.; Coates, L.C.; Helliwell, P.S.; Stanislavchuk, M.; Rychlewska-Hanczewska, A.; Dudek, A.; Abi-Saab, W.; Tasset, C.; Meuleners, L.; Harrison, P.; et al. Efficacy and safety of filgotinib, a selective Janus kinase 1 inhibitor, in patients with active psoriatic arthritis (EQUATOR): Results from a randomised, placebo-controlled, phase 2 trial. Lancet 2018, 392, 2367–2377. [Google Scholar] [CrossRef]
- Vermeire, S.; Schreiber, S.; Petryka, R.; Kuehbacher, T.; Hebuterne, X.; Roblin, X.; Klopocka, M.; Goldis, A.; Wisniewska-Jarosinska, M.; Baranovsky, A.; et al. Clinical remission in patients with moderate-to-severe Crohn’s disease treated with filgotinib (the FITZROY study): Results from a phase 2, double-blind, randomised, placebo-controlled trial. Lancet 2017, 389, 266–275. [Google Scholar] [CrossRef]
- Papp, K.; Gordon, K.; Thaçi, D.; Morita, A.; Gooderham, M.; Foley, P.; Girgis, I.G.; Kundu, S.; Banerjee, S. Phase 2 Trial of Selective Tyrosine Kinase 2 Inhibition in Psoriasis. N. Engl. J. Med. 2018, 379, 1313–1321. [Google Scholar] [CrossRef]
- Mascarenhas, J.; Hoffman, R.; Talpaz, M.; Gerds, A.T.; Stein, B.; Gupta, V.; Szoke, A.; Drummond, M.; Pristupa, A.; Granston, T.; et al. Pacritinib vs. Best Available Therapy, Including Ruxolitinib, in Patients With Myelofibrosis: A Randomized Clinical Trial. JAMA Oncol. 2018, 4, 652–659. [Google Scholar] [CrossRef]
- Schmieder, G.J.; Draelos, Z.D.; Pariser, D.M.; Banfield, C.; Cox, L.; Hodge, M.; Kieras, E.; Parsons-Rich, D.; Menon, S.; Salganik, M.; et al. Efficacy and safety of the Janus kinase 1 inhibitor PF-04965842 in patients with moderate-to-severe psoriasis: Phase II, randomized, double-blind, placebo-controlled study. Br. J. Dermatol. 2018, 179, 54–62. [Google Scholar] [CrossRef]
- Kheirkhah, A.; Di Zazzo, A.; Satitpitakul, V.; Fernandez, M.; Magilavy, D.; Dana, R. A Pilot Randomized Trial on Safety and Efficacy of a Novel Topical Combined Inhibitor of Janus Kinase 1/3 and Spleen Tyrosine Kinase for GVHD-Associated Ocular Surface Disease. Cornea 2017, 36, 799–804. [Google Scholar] [CrossRef]
- Mascarenhas, J.O.; Talpaz, M.; Gupta, V.; Foltz, L.M.; Savona, M.R.; Paquette, R.; Turner, A.R.; Coughlin, P.; Winton, E.; Burn, T.C.; et al. Primary analysis of a phase II open-label trial of INCB039110, a selective JAK1 inhibitor, in patients with myelofibrosis. Haematologica 2017, 102, 327–335. [Google Scholar] [CrossRef]
- Markham, A.; Keam, S.J. Peficitinib: First Global Approval. Drugs 2019, 79, 887–891. [Google Scholar] [CrossRef]
- El Jammal, T.; Gerfaud-Valentin, M.; Sève, P.; Jamilloux, Y. Inhibition of JAK/STAT signaling in rheumatologic disorders: The expanding spectrum. Jt. Bone Spine 2020, 87, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Fragoulis, G.E.; McInnes, I.B.; Siebert, S. JAK-inhibitors. New players in the field of immune-mediated diseases, beyond rheumatoid arthritis. Rheumatology (Oxford) 2019, 58, i43–i54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venugopal, S.; Bar-Natan, M.; Mascarenhas, J.O. JAKs to STATs: A tantalizing therapeutic target in acute myeloid leukemia. Blood Rev. 2020, 40, 100634. [Google Scholar] [CrossRef]
- Hosseini, A.; Gharibi, T.; Marofi, F.; Javadian, M.; Babaloo, Z.; Baradaran, B. Janus kinase inhibitors: A therapeutic strategy for cancer and autoimmune diseases. J. Cell Physiol. 2020. [Google Scholar] [CrossRef]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005, 365, 1054–1061. [Google Scholar] [CrossRef]
- Perner, F.; Perner, C.; Ernst, T.; Heidel, F.H. Roles of JAK2 in Aging, Inflammation, Hematopoiesis and Malignant Transformation. Cells 2019, 8, 854. [Google Scholar] [CrossRef] [Green Version]
- Vicente, C.; Schwab, C.; Broux, M.; Geerdens, E.; Degryse, S.; Demeyer, S.; Lahortiga, I.; Elliott, A.; Chilton, L.; La Starza, R.; et al. Targeted sequencing identifies associations between IL7R-JAK mutations and epigenetic modulators in T-cell acute lymphoblastic leukemia. Haematologica 2015, 100, 1301–1310. [Google Scholar] [CrossRef] [Green Version]
- Fattizzo, B.; Rosa, J.; Giannotta, J.A.; Baldini, L.; Fracchiolla, N.S. The Physiopathology of T-Cell Acute Lymphoblastic Leukemia: Focus on Molecular Aspects. Front. Oncol. 2020, 10, 273. [Google Scholar] [CrossRef] [Green Version]
- Kan, Z.; Zheng, H.; Liu, X.; Li, S.; Barber, T.D.; Gong, Z.; Gao, H.; Hao, K.; Willard, M.D.; Xu, J.; et al. Whole-genome sequencing identifies recurrent mutations in hepatocellular carcinoma. Genome Res. 2013, 23, 1422–1433. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Luo, C.; Gu, Q.; Xu, Q.; Wang, G.; Sun, H.; Qian, Z.; Tan, Y.; Qin, Y.; Shen, Y.; et al. Activating JAK1 mutation may predict the sensitivity of JAK-STAT inhibition in hepatocellular carcinoma. Oncotarget 2016, 7, 5461–5469. [Google Scholar] [CrossRef] [Green Version]
- Hin Tang, J.J.; Hao Thng, D.K.; Lim, J.J.; Toh, T.B. JAK/STAT signaling in hepatocellular carcinoma. Hepat. Oncol. 2020, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gavegnano, C.; Brehm, J.H.; Dupuy, F.P.; Talla, A.; Ribeiro, S.P.; Kulpa, D.A.; Cameron, C.; Santos, S.; Hurwitz, S.J.; Marconi, V.C.; et al. Novel mechanisms to inhibit HIV reservoir seeding using Jak inhibitors. PLoS Pathog. 2017, 13, e1006740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gavegnano, C.; Savarino, A.; Owanikoko, T.; Marconi, V.C. Crossroads of Cancer and HIV-1: Pathways to a Cure for HIV. Front. Immunol. 2019, 10, 2267. [Google Scholar] [CrossRef] [PubMed]
- Stebbing, J.; Krishnan, V.; de Bono, S.; Ottaviani, S.; Casalini, G.; Richardson, P.J.; Monteil, V.; Lauschke, V.M.; Mirazimi, A.; Youhanna, S.; et al. Mechanism of baricitinib supports artificial intelligence-predicted testing in COVID-19 patients. EMBO Mol. Med. 2020. [Google Scholar] [CrossRef]
- Richardson, P.; Griffin, I.; Tucker, C.; Smith, D.; Oechsle, O.; Phelan, A.; Stebbing, J. Baricitinib as potential treatment for 2019-nCoV acute respiratory disease. Lancet 2020, 395, e30–e31. [Google Scholar] [CrossRef] [Green Version]
- Spinelli, F.R.; Conti, F.; Gadina, M. HiJAKing SARS-CoV-2? The potential role of JAK inhibitors in the management of COVID-19. Sci. Immunol. 2020, 5. [Google Scholar] [CrossRef]
- Yanuck, S.F.; Pizzorno, J.; Messier, H.; Fitzgerald, K.N. Evidence Supporting a Phased Immuno-physiological Approach to COVID-19 From Prevention Through Recovery. Integr. Med. (Encinitas) 2020, 19, 8–35. [Google Scholar]
- Wilks, A.F. Two putative protein-tyrosine kinases identified by application of the polymerase chain reaction. Proc. Natl. Acad. Sci. USA 1989, 86, 1603–1607. [Google Scholar] [CrossRef] [Green Version]
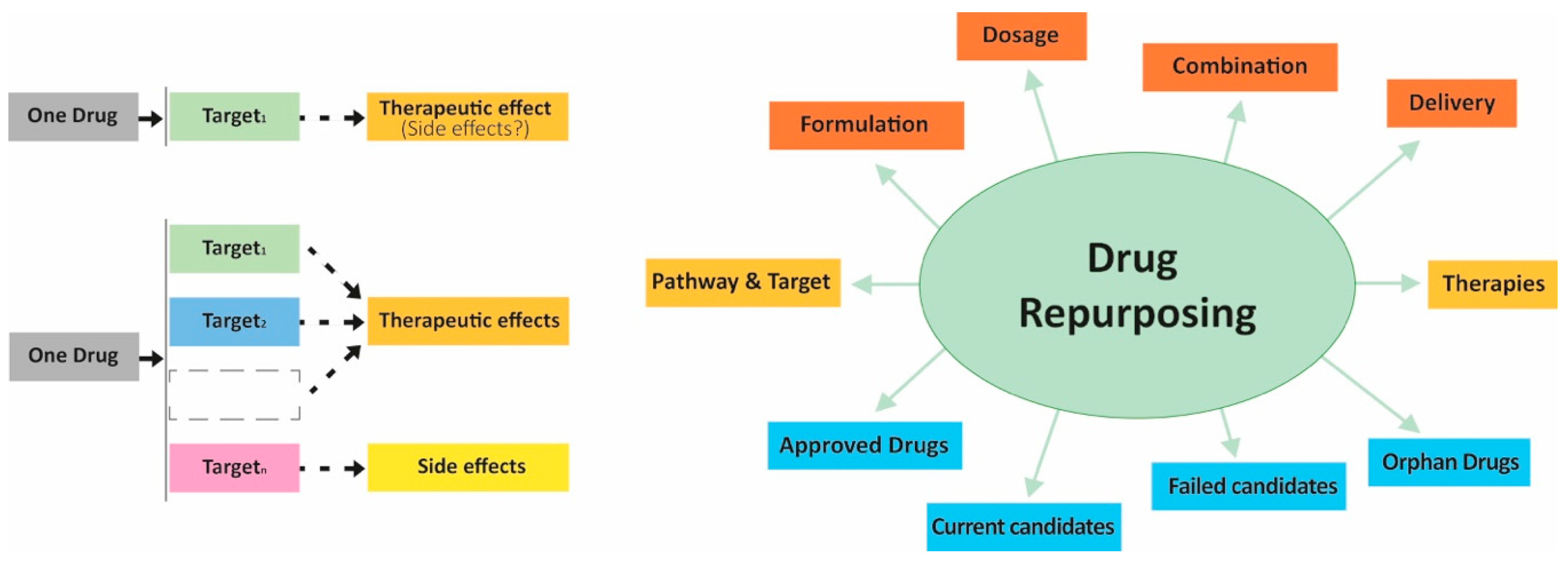
- Ramsay, R.R.; Popovic-Nikolic, M.R.; Nikolic, K.; Uliassi, E.; Bolognesi, M.L. A perspective on multi-target drug discovery and design for complex diseases. Clin. Transl. Med. 2018, 7, 3. [Google Scholar] [CrossRef] [Green Version]
- Maruca, A.; Lanzillotta, D.; Rocca, R.; Lupia, A.; Costa, G.; Catalano, R.; Moraca, F.; Gaudio, E.; Ortuso, F.; Artese, A.; et al. Multi-Targeting Bioactive Compounds Extracted from Essential Oils as Kinase Inhibitors. Molecules 2020, 25, 2174. [Google Scholar] [CrossRef]
- Bagetta, D.; Maruca, A.; Lupia, A.; Mesiti, F.; Catalano, R.; Romeo, I.; Moraca, F.; Ambrosio, F.A.; Costa, G.; Artese, A.; et al. Mediterranean products as promising source of multi-target agents in the treatment of metabolic syndrome. Eur. J. Med. Chem. 2020, 186, 111903. [Google Scholar] [CrossRef]
- Maruca, A.; Catalano, R.; Bagetta, D.; Mesiti, F.; Ambrosio, F.A.; Romeo, I.; Moraca, F.; Rocca, R.; Ortuso, F.; Artese, A.; et al. The Mediterranean Diet as source of bioactive compounds with multi-targeting anti-cancer profile. Eur. J. Med. Chem. 2019, 181, 111579. [Google Scholar] [CrossRef]
- Maruca, A.; Moraca, F.; Rocca, R.; Molisani, F.; Alcaro, F.; Gidaro, M.; Alcaro, S.; Costa, G.; Ortuso, F. Chemoinformatic Database Building and in Silico Hit-Identification of Potential Multi-Targeting Bioactive Compounds Extracted from Mushroom Species. Molecules 2017, 22, 1571. [Google Scholar] [CrossRef] [Green Version]
- Alcaro, S.; Bolognesi, M.L.; García-Sosa, A.T.; Rapposelli, S. Editorial: Multi-Target-Directed Ligands (MTDL) as Challenging Research Tools in Drug Discovery: From Design to Pharmacological Evaluation. Front. Chem. 2019, 7, 71. [Google Scholar] [CrossRef] [PubMed]
- Catalano, R.; Rocca, R.; Juli, G.; Costa, G.; Maruca, A.; Artese, A.; Caracciolo, D.; Tagliaferri, P.; Alcaro, S.; Tassone, P.; et al. A drug repurposing screening reveals a novel epigenetic activity of hydroxychloroquine. Eur. J. Med. Chem. 2019, 183, 111715. [Google Scholar] [CrossRef]
- Alcaro, S.; Ortuso, F. Multi-target drug discovery: An opportunity for novel and repurposed bioactive compounds. Eur. J. Med. Chem. 2020, 192, 112188. [Google Scholar] [CrossRef] [PubMed]
- Catalogna, G.; Moraca, F.; D’Antona, L.; Dattilo, V.; Perrotti, G.; Lupia, A.; Costa, G.; Ortuso, F.; Iuliano, R.; Trapasso, F.; et al. Review about the multi-target profile of resveratrol and its implication in the SGK1 inhibition. Eur. J. Med. Chem. 2019, 183, 111675. [Google Scholar] [CrossRef]
- Rokosz, L.L.; Beasley, J.R.; Carroll, C.D.; Lin, T.; Zhao, J.; Appell, K.C.; Webb, M.L. Kinase inhibitors as drugs for chronic inflammatory and immunological diseases: Progress and challenges. Expert Opin. Ther. Targets 2008, 12, 883–903. [Google Scholar] [CrossRef]
- Banerjee, S.; Biehl, A.; Gadina, M.; Hasni, S.; Schwartz, D.M. JAK-STAT Signaling as a Target for Inflammatory and Autoimmune Diseases: Current and Future Prospects. Drugs 2017, 77, 521–546. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Schwartz, D.M.; Villarino, A.V.; Gadina, M.; McInnes, I.B.; Laurence, A. The JAK-STAT pathway: Impact on human disease and therapeutic intervention. Annu. Rev. Med. 2015, 66, 311–328. [Google Scholar] [CrossRef] [Green Version]
- Vaddi, K.; Sarlis, N.J.; Gupta, V. Ruxolitinib, an oral JAK1 and JAK2 inhibitor, in myelofibrosis. Expert Opin. Pharmacother. 2012, 13, 2397–2407. [Google Scholar] [CrossRef] [PubMed]
- Elli, E.M.; Baratè, C.; Mendicino, F.; Palandri, F.; Palumbo, G.A. Mechanisms Underlying the Anti-inflammatory and Immunosuppressive Activity of Ruxolitinib. Front. Oncol. 2019, 9, 1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abedin, S.M.; Hamadani, M. Ruxolitinib: A potential treatment for corticosteroid refractory acute graft-versus-host disease. Expert Opin. Investig. Drugs 2020, 29, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Gunawan, A.; Harrington, P.; Garcia-Curto, N.; McLornan, D.; Radia, D.; Harrison, C. Ruxolitinib for the Treatment of Essential Thrombocythemia. Hemasphere 2018, 2, e56. [Google Scholar] [CrossRef]
- Mesa, R.A. Ruxolitinib, a selective JAK1 and JAK2 inhibitor for the treatment of myeloproliferative neoplasms and psoriasis. IDrugs 2010, 13, 394–403. [Google Scholar]
- Tavallai, M.; Booth, L.; Roberts, J.L.; Poklepovic, A.; Dent, P. Rationally Repurposing Ruxolitinib (Jakafi (®)) as a Solid Tumor Therapeutic. Front. Oncol. 2016, 6, 142. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Wei, J.; Zou, L.; Jiang, T.; Wang, G.; Chen, L.; Huang, L.; Meng, F.; Wang, N.; Zhou, X.; et al. Ruxolitinib in treatment of severe coronavirus disease 2019 (COVID-19): A multicenter, single-blind, randomized controlled trial. J. Allergy Clin. Immunol. 2020. [Google Scholar] [CrossRef]
- Galimberti, S.; Baldini, C.; Baratè, C.; Ricci, F.; Balducci, S.; Grassi, S.; Ferro, F.; Buda, G.; Benedetti, E.; Fazzi, R.; et al. The CoV-2 outbreak: How hematologists could help to fight Covid-19. Pharmacol. Res. 2020, 157, 104866. [Google Scholar] [CrossRef]
- Dhillon, S. Tofacitinib: A Review in Rheumatoid Arthritis. Drugs 2017, 77, 1987–2001. [Google Scholar] [CrossRef]
- Hodge, J.A.; Kawabata, T.T.; Krishnaswami, S.; Clark, J.D.; Telliez, J.B.; Dowty, M.E.; Menon, S.; Lamba, M.; Zwillich, S. The mechanism of action of tofacitinib—An oral Janus kinase inhibitor for the treatment of rheumatoid arthritis. Clin. Exp. Rheumatol. 2016, 34, 318–328. [Google Scholar]
- Ju, W.; Zhang, M.; Jiang, J.K.; Thomas, C.J.; Oh, U.; Bryant, B.R.; Chen, J.; Sato, N.; Tagaya, Y.; Morris, J.C.; et al. CP-690,550, a therapeutic agent, inhibits cytokine-mediated Jak3 activation and proliferation of T cells from patients with ATL and HAM/TSP. Blood 2011, 117, 1938–1946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wrobleski, S.; Pitts, W. Advances in the Discovery of Small Molecule JAK3 Inhibitors. Annu. Rep. Med. Chem. 2009, 44, 247–264. [Google Scholar] [CrossRef]
- Lam, C.; Ferguson, I.D.; Mariano, M.C.; Lin, Y.T.; Murnane, M.; Liu, H.; Smith, G.A.; Wong, S.W.; Taunton, J.; Liu, J.O.; et al. Repurposing tofacitinib as an anti-myeloma therapeutic to reverse growth-promoting effects of the bone marrow microenvironment. Haematologica 2018, 103, 1218–1228. [Google Scholar] [CrossRef] [Green Version]
- Changelian, P.S.; Flanagan, M.E.; Ball, D.J.; Kent, C.R.; Magnuson, K.S.; Martin, W.H.; Rizzuti, B.J.; Sawyer, P.S.; Perry, B.D.; Brissette, W.H.; et al. Prevention of organ allograft rejection by a specific Janus kinase 3 inhibitor. Science 2003, 302, 875–878. [Google Scholar] [CrossRef]
- Williams, N.K.; Bamert, R.S.; Patel, O.; Wang, C.; Walden, P.M.; Wilks, A.F.; Fantino, E.; Rossjohn, J.; Lucet, I.S. Dissecting specificity in the Janus kinases: The structures of JAK-specific inhibitors complexed to the JAK1 and JAK2 protein tyrosine kinase domains. J. Mol. Biol. 2009, 387, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Alicea-Velázquez, N.L.; Boggon, T.J. The use of structural biology in Janus kinase targeted drug discovery. Curr. Drug Targets 2011, 12, 546–555. [Google Scholar] [CrossRef]
- Kisseleva, T.; Bhattacharya, S.; Braunstein, J.; Schindler, C.W. Signaling through the JAK/STAT pathway, recent advances and future challenges. Gene 2002, 285, 1–24. [Google Scholar] [CrossRef]
- Lupardus, P.J.; Ultsch, M.; Wallweber, H.; Bir Kohli, P.; Johnson, A.R.; Eigenbrot, C. Structure of the pseudokinase-kinase domains from protein kinase TYK2 reveals a mechanism for Janus kinase (JAK) autoinhibition. Proc. Natl. Acad. Sci. USA 2014, 111, 8025–8030. [Google Scholar] [CrossRef] [Green Version]
- Babon, J.J.; Lucet, I.S.; Murphy, J.M.; Nicola, N.A.; Varghese, L.N. The molecular regulation of Janus kinase (JAK) activation. Biochem. J. 2014, 462, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Roskoski, R., Jr. Janus kinase (JAK) inhibitors in the treatment of inflammatory and neoplastic diseases. Pharmacol. Res. 2016, 111, 784–803. [Google Scholar] [CrossRef]
- Taylor, S.S.; Keshwani, M.M.; Steichen, J.M.; Kornev, A.P. Evolution of the eukaryotic protein kinases as dynamic molecular switches. Philos. Trans. R Soc. Lond. B Biol. Sci. 2012, 367, 2517–2528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, V.; Shammas, R.M.; Sauk, J.S.; Padua, D. Evaluating tofacitinib citrate in the treatment of moderate-to-severe active ulcerative colitis: Design, development and positioning of therapy. Clin. Exp. Gastroenterol. 2019, 12, 179–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, D.M.; Kanno, Y.; Villarino, A.; Ward, M.; Gadina, M.; O’Shea, J.J. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat. Rev. Drug Discov. 2017, 17, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Q.; Meloni, D.; Pan, Y.; Xia, M.; Rodgers, J.; Shepard, S.; Li, M.; Galya, L.; Metcalf, B.; Yue, T.Y.; et al. Enantioselective synthesis of Janus kinase inhibitor INCB018424 via an organocatalytic aza-Michael reaction. Org. Lett. 2009, 11, 1999–2002. [Google Scholar] [CrossRef] [PubMed]
- Haydl, A.M.; Xu, K.; Breit, B. Regio- and enantioselective synthesis of N-substituted pyrazoles by rhodium-catalyzed asymmetric addition to allenes. Angew. Chem. Int. Ed. Engl. 2015, 54, 7149–7153. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, A.; Zhou, Z.; Yang, L.; Yao, H.; Zhu, X.; Wang, H. Synthesis Process of Ruxolitinib. US Patent 10562904B2, 29 December 2016. [Google Scholar]
- Ripin, D.H.B.; Abele, S.; Cai, W.; Blumenkopf, T.; Casavant, J.M.; Doty, J.L.; Flanagan, M.; Koecher, C.; Laue, K.W.; McCarthy, K.; et al. Development of a Scaleable Route for the Production of cis-N-Benzyl-3-methylamino-4-methylpiperidine. Org. Process Res. Dev. 2003, 7, 115–120. [Google Scholar] [CrossRef]
- Cai, W.; Colony, J.L.; Frost, H.; Hudspeth, J.P.; Kendall, P.M.; Krishnan, A.M.; Makowski, T.; Mazur, D.J.; Phillips, J.; Ripin, D.H.B.; et al. Investigation of Practical Routes for the Kilogram-Scale Production of cis-3-Methylamino-4-methylpiperidines. Org. Process Res. Dev. 2005, 9, 51–56. [Google Scholar] [CrossRef]
- Patil, Y.S.; Bonde, N.L.; Kekan, A.S.; Sathe, D.G.; Das, A. An Improved and Efficient Process for the Preparation of Tofacitinib Citrate. Org. Process Res. Dev. 2014, 18, 1714–1720. [Google Scholar] [CrossRef]
- Zhi, S.; Liu, D.; Ying, L.; Liu, B.; Wang, D.; Chen, L. An Efficient Method for Synthesis of Tofacitinib Citrate. J. Heterocycl. Chem. 2015, 53. [Google Scholar] [CrossRef]
- Srishylam, V.; Mulakayala, D.; Mulakayala, N. An Efficient and Alternative Method for Synthesis of Tofacitinib. Der Pharma Chem. 2018, 10, 49–71. [Google Scholar]
- Maricán, A.; Simirgiotis, M.J.; Santos, L.S. Asymmetric total synthesis of Tofacitinib. Tetrahedron. Lett. 2013, 54, 5096–5098. [Google Scholar] [CrossRef]
- Liao, H.-C.; Uang, B.-J. Formal asymmetric synthesis of (+)-tofacitinib. Tetrahedron Asymmetry 2017, 28, 105–109. [Google Scholar] [CrossRef]
- Li, C.; Wan, F.; Chen, Y.; Peng, H.; Tang, W.; Yu, S.; McWilliams, J.C.; Mustakis, J.; Samp, L.; Maguire, R.J. Stereoelectronic Effects in Ligand Design: Enantioselective Rhodium-Catalyzed Hydrogenation of Aliphatic Cyclic Tetrasubstituted Enamides and Concise Synthesis of (R)-Tofacitinib. Angew. Chem. Int. Ed. Engl. 2019, 58, 13573–13583. [Google Scholar] [CrossRef]
- Verzijl, G.K.M.; Schuster, C.; Dax, T.; de Vries, A.H.M.; Lefort, L. Asymmetric Synthesis of a Key Intermediate for Tofacitinib via a Dynamic Kinetic Resolution-Reductive Amination Protocol. Org. Process Res. Dev. 2018, 22, 1817–1822. [Google Scholar] [CrossRef]
- Price, K.E.; Larrivée-Aboussafy, C.; Lillie, B.M.; McLaughlin, R.W.; Mustakis, J.; Hettenbach, K.W.; Hawkins, J.M.; Vaidyanathan, R. Mild and Efficient DBU-Catalyzed Amidation of Cyanoacetates. Org. Lett. 2009, 11, 2003–2006. [Google Scholar] [CrossRef]
- DiMasi, J.A.; Grabowski, H.G.; Hansen, R.W. Innovation in the pharmaceutical industry: New estimates of R&D costs. J. Health Econ. 2016, 47, 20–33. [Google Scholar] [CrossRef] [Green Version]
- Lavecchia, A.; Di Giovanni, C. Virtual screening strategies in drug discovery: A critical review. Curr. Med. Chem. 2013, 20, 2839–2860. [Google Scholar] [CrossRef]
- Jorgensen, W.L. The many roles of computation in drug discovery. Science 2004, 303, 1813–1818. [Google Scholar] [CrossRef]
- Costa, G.; Rocca, R.; Corona, A.; Grandi, N.; Moraca, F.; Romeo, I.; Talarico, C.; Gagliardi, M.G.; Ambrosio, F.A.; Ortuso, F.; et al. Novel natural non-nucleoside inhibitors of HIV-1 reverse transcriptase identified by shape- and structure-based virtual screening techniques. Eur. J. Med. Chem. 2019, 161, 1–10. [Google Scholar] [CrossRef]
- Rocca, R.; Moraca, F.; Costa, G.; Talarico, C.; Ortuso, F.; Da Ros, S.; Nicoletto, G.; Sissi, C.; Alcaro, S.; Artese, A. In Silico Identification of Piperidinyl-amine Derivatives as Novel Dual Binders of Oncogene c-myc/c-Kit G-quadruplexes. ACS Med. Chem. Lett. 2018, 9, 848–853. [Google Scholar] [CrossRef] [PubMed]
- Maruca, A.; Ambrosio, F.A.; Lupia, A.; Romeo, I.; Rocca, R.; Moraca, F.; Talarico, C.; Bagetta, D.; Catalano, R.; Artese, A.; et al. Computer-based techniques for lead identification and optimization I: Basics. Phys. Sci. Rev. 2019, 4. [Google Scholar] [CrossRef]
- Lupia, A.; Moraca, F.; Bagetta, D.; Maruca, A.; Ambrosio, F.; Rocca, R.; Catalano, R.; Romeo, I.; Talarico, C.; Ortuso, F.; et al. Computer-based techniques for lead identification and optimization II: Advanced search methods. Phys. Sci. Rev. 2020, 1, 333–360. [Google Scholar] [CrossRef]
- Al-Asri, J.; Wolber, G. Discovery of novel α-amylase inhibitors using structure-based drug design. J. Cheminform. 2014, 6 (Suppl. 1), 50. [Google Scholar] [CrossRef] [Green Version]
- Simov, V.; Deshmukh, S.V.; Dinsmore, C.J.; Elwood, F.; Fernandez, R.B.; Garcia, Y.; Gibeau, C.; Gunaydin, H.; Jung, J.; Katz, J.D.; et al. Structure-based design and development of (benz)imidazole pyridones as JAK1-selective kinase inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 1803–1808. [Google Scholar] [CrossRef] [PubMed]
- Hansen, B.B.; Jepsen, T.H.; Larsen, M.; Sindet, R.; Vifian, T.; Burhardt, M.N.; Larsen, J.; Seitzberg, J.G.; Carnerup, M.A.; Jerre, A.; et al. Fragment-Based Discovery of Pyrazolopyridones as JAK1 Inhibitors with Excellent Subtype Selectivity. J. Med. Chem. 2020, 63, 7008–7032. [Google Scholar] [CrossRef]
- Ritzén, A.; Sørensen, M.D.; Dack, K.N.; Greve, D.R.; Jerre, A.; Carnerup, M.A.; Rytved, K.A.; Bagger-Bahnsen, J. Fragment-Based Discovery of 6-Arylindazole JAK Inhibitors. ACS Med. Chem. Lett. 2016, 7, 641–646. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Chen, C.J.; Yu, R.N.; Wang, Z.J.; Zhang, T.T.; Zhang, D.Y. Structure-based design and synthesis of 1H-pyrazolo[3,4-d]pyrimidin-4-amino derivatives as Janus kinase 3 inhibitors. Bioorg. Med. Chem. 2018, 26, 4774–4786. [Google Scholar] [CrossRef]
- Yu, R.N.; Chen, C.J.; Shu, L.; Yin, Y.; Wang, Z.J.; Zhang, T.T.; Zhang, D.Y. Structure-based design and synthesis of pyrimidine-4,6-diamine derivatives as Janus kinase 3 inhibitors. Bioorg. Med. Chem. 2019, 27, 1646–1657. [Google Scholar] [CrossRef]
- Shu, L.; Chen, C.; Huan, X.; Huang, H.; Wang, M.; Zhang, J.; Yan, Y.; Liu, J.; Zhang, T.; Zhang, D. Design, synthesis, and pharmacological evaluation of 4- or 6-phenyl-pyrimidine derivatives as novel and selective Janus kinase 3 inhibitors. Eur. J. Med. Chem. 2020, 191, 112148. [Google Scholar] [CrossRef]
- Bajusz, D.; Ferenczy, G.G.; Keserű, G.M. Discovery of Subtype Selective Janus Kinase (JAK) Inhibitors by Structure-Based Virtual Screening. J. Chem. Inf. Model. 2016, 56, 234–247. [Google Scholar] [CrossRef] [PubMed]
- Egyed, A.; Bajusz, D.; Keserű, G.M. The impact of binding site waters on the activity/selectivity trade-off of Janus kinase 2 (JAK2) inhibitors. Bioorg. Med. Chem. 2019, 27, 1497–1508. [Google Scholar] [CrossRef] [PubMed]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
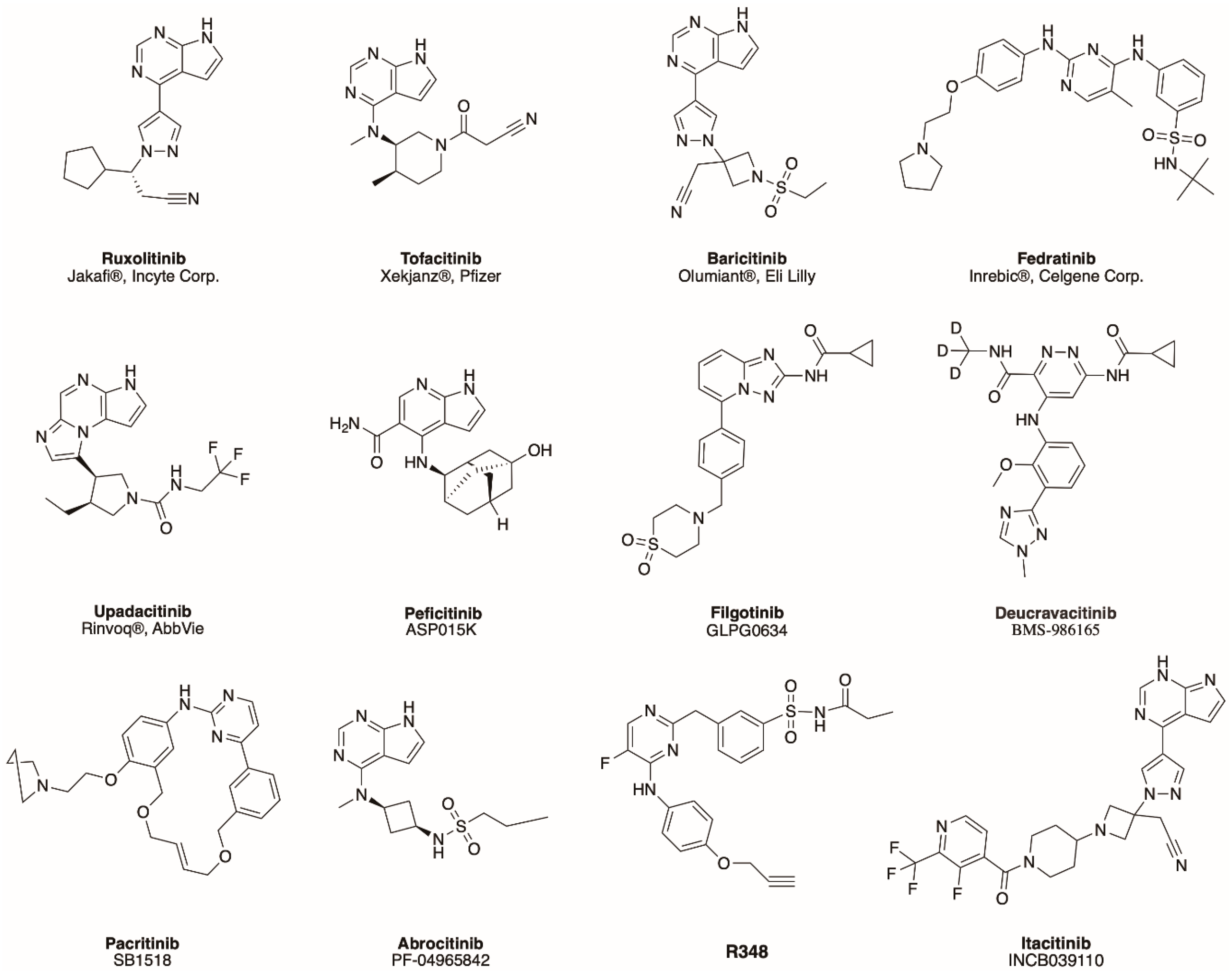
| Name/Alternative Name | Target | Target Disease | References |
|---|---|---|---|
| Peficitinib */ASP015K | pan-JAK | Rheumatoid Arthritis | [50] |
| Filgotinib/GLPG0634 | JAK1 | Rheumatoid Arthritis, Active Ankylosing Spondylitis, Psoriatic Arthritis, Crohn’s disease | [51,52,53,54] |
| Deucravacitinib/ BMS-986165 | TYK2 | Psoriasis | [55] |
| Pacritinib/SB1518 | JAK2 | Myelofibrosis | [56] |
| Abrocitinib/PF-04965842 | JAK1 | Psoriasis | [57] |
| R348 | JAK3 | GvHD-associated ocular surface disease | [58] |
| Itacitinib/INCB039110 | JAK1 | Myelofibrosis | [59] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coricello, A.; Mesiti, F.; Lupia, A.; Maruca, A.; Alcaro, S. Inside Perspective of the Synthetic and Computational Toolbox of JAK Inhibitors: Recent Updates. Molecules 2020, 25, 3321. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25153321
Coricello A, Mesiti F, Lupia A, Maruca A, Alcaro S. Inside Perspective of the Synthetic and Computational Toolbox of JAK Inhibitors: Recent Updates. Molecules. 2020; 25(15):3321. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25153321
Chicago/Turabian StyleCoricello, Adriana, Francesco Mesiti, Antonio Lupia, Annalisa Maruca, and Stefano Alcaro. 2020. "Inside Perspective of the Synthetic and Computational Toolbox of JAK Inhibitors: Recent Updates" Molecules 25, no. 15: 3321. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25153321