
Synthesis and Characterization of New Organic Dyes Containing the Indigo Core

 , ,
, , 
 and
and
Abstract
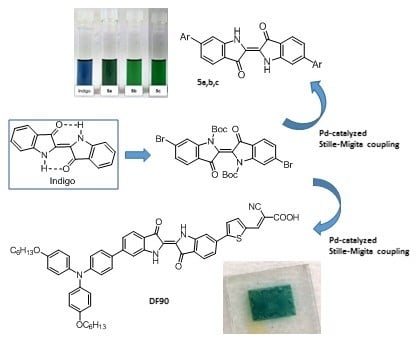
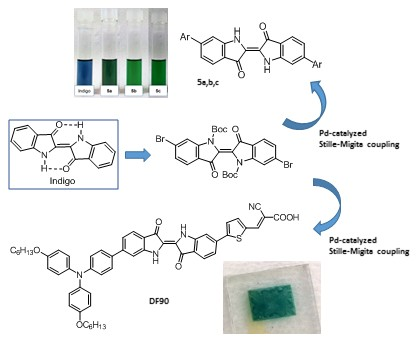
:
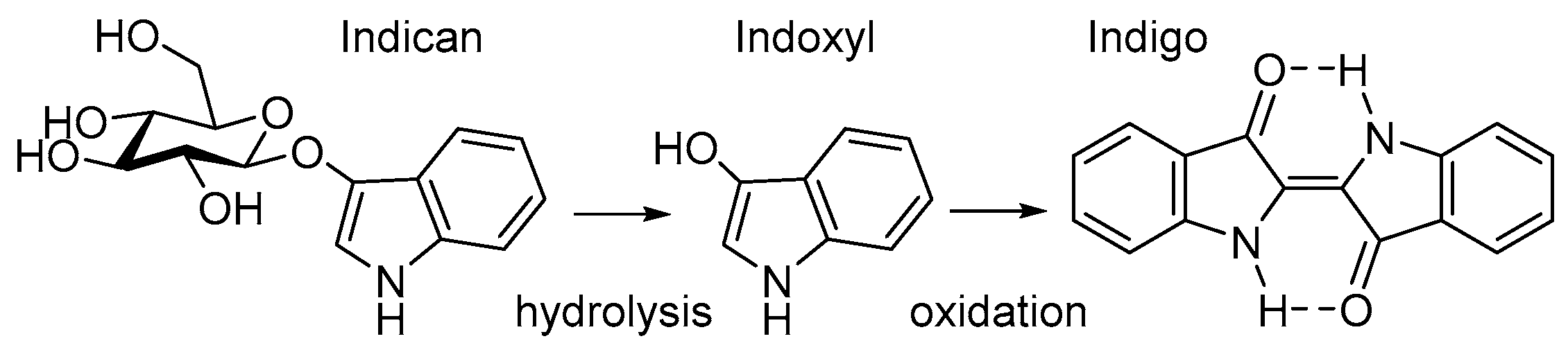
1. Introduction
2. Results and Discussion
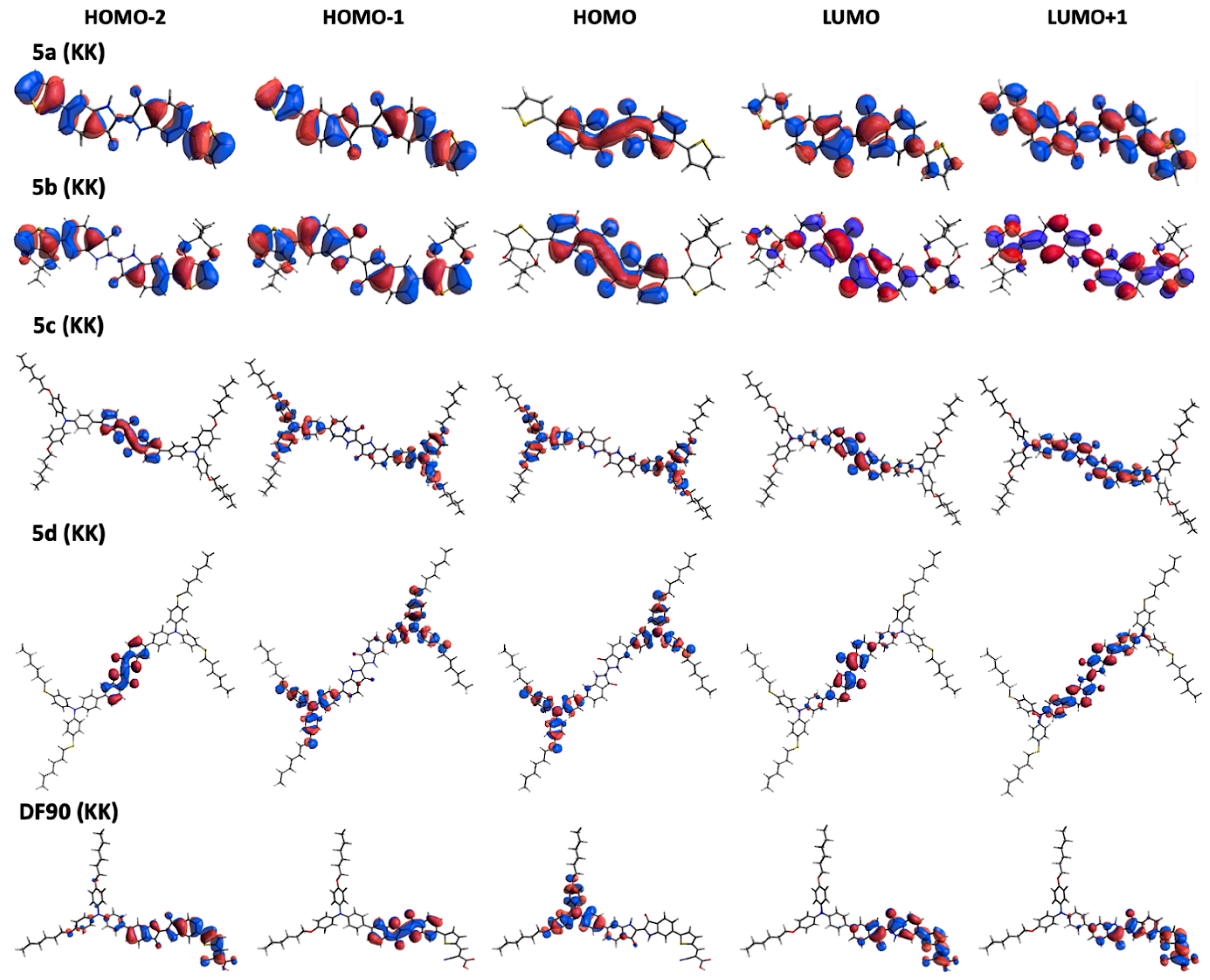
2.1. Computational Investigation
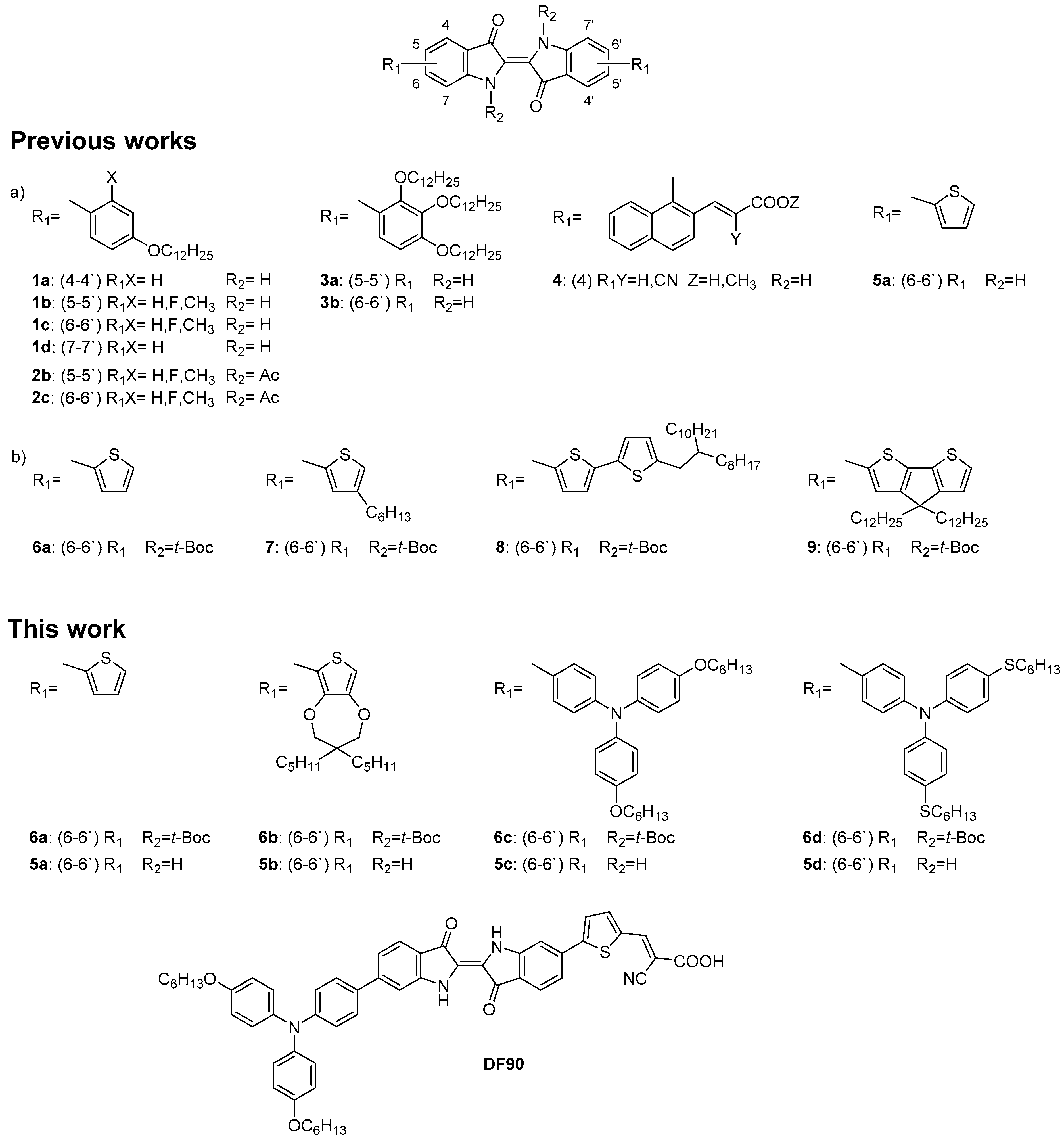
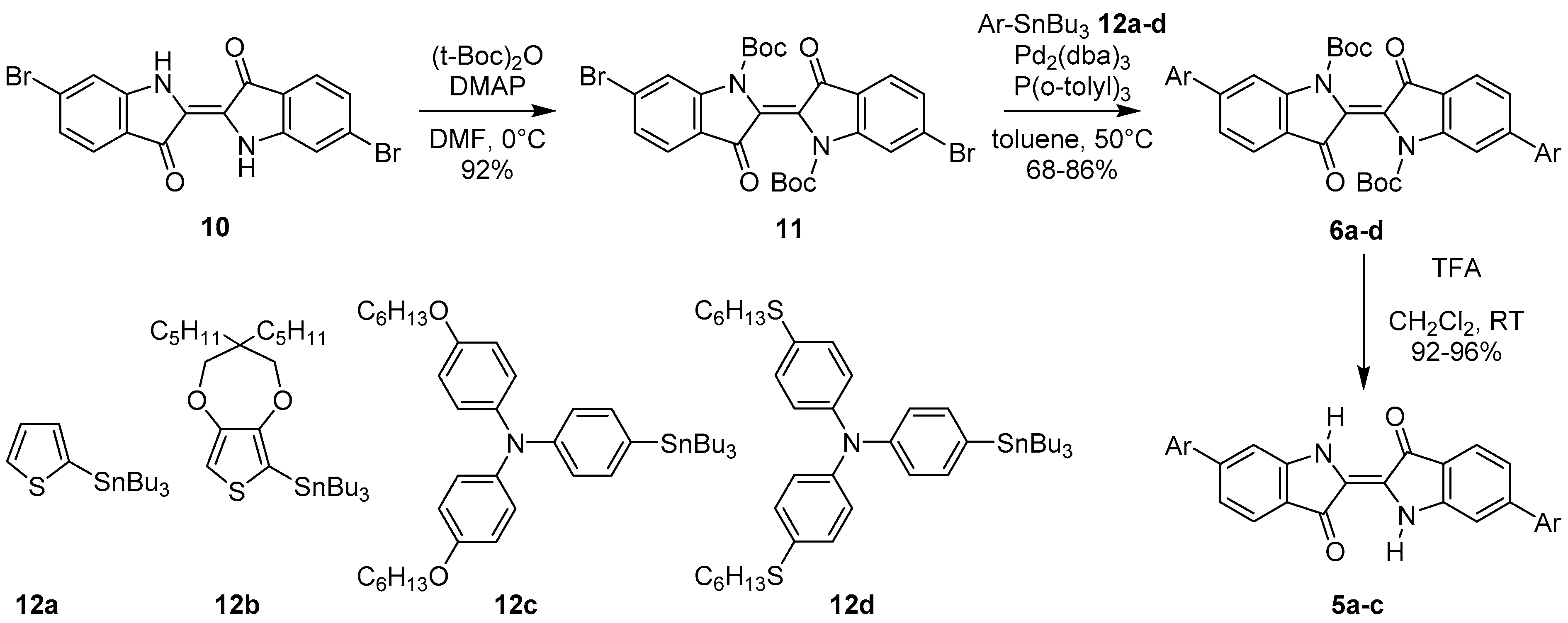
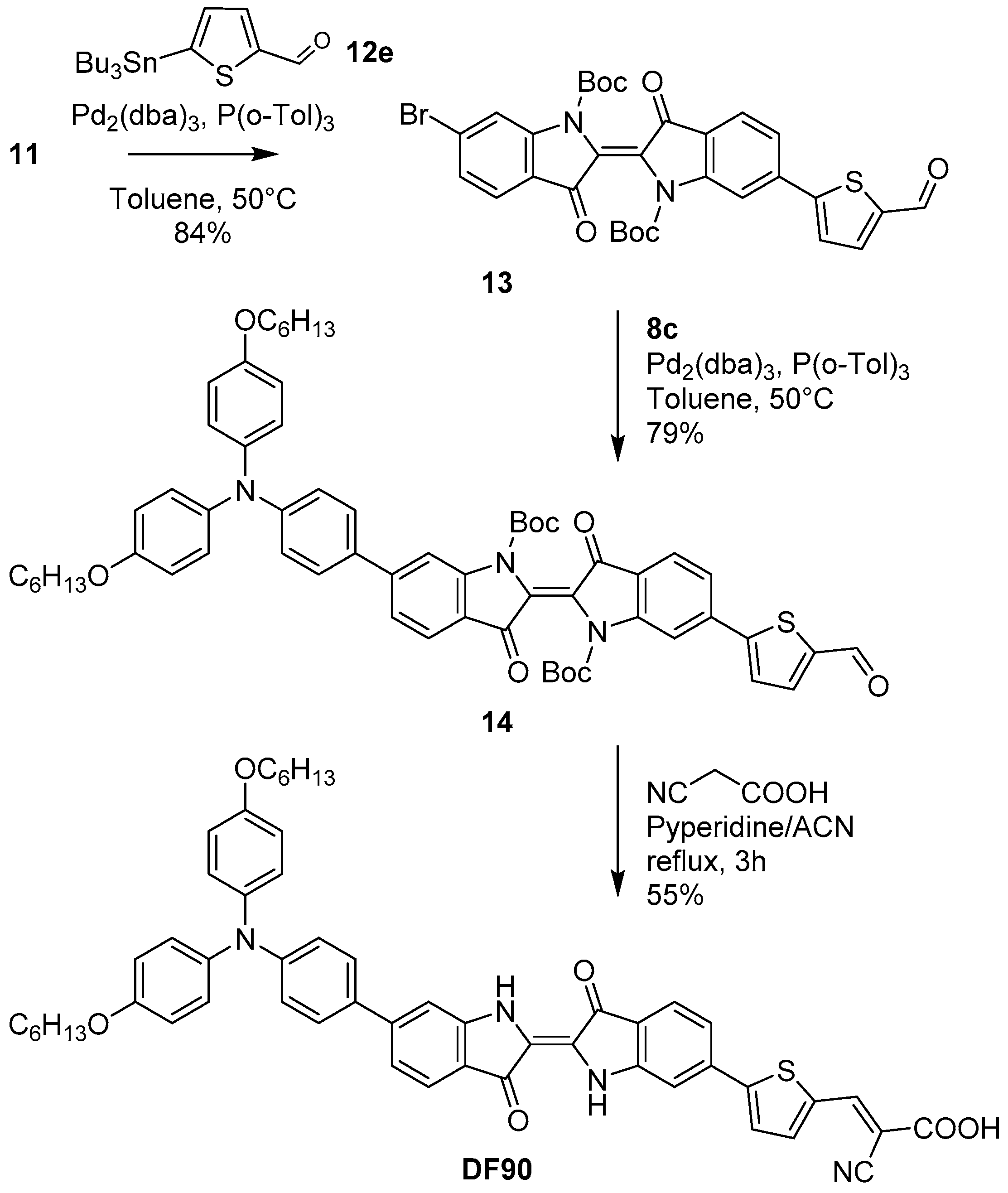
2.2. Synthesis of Dyes
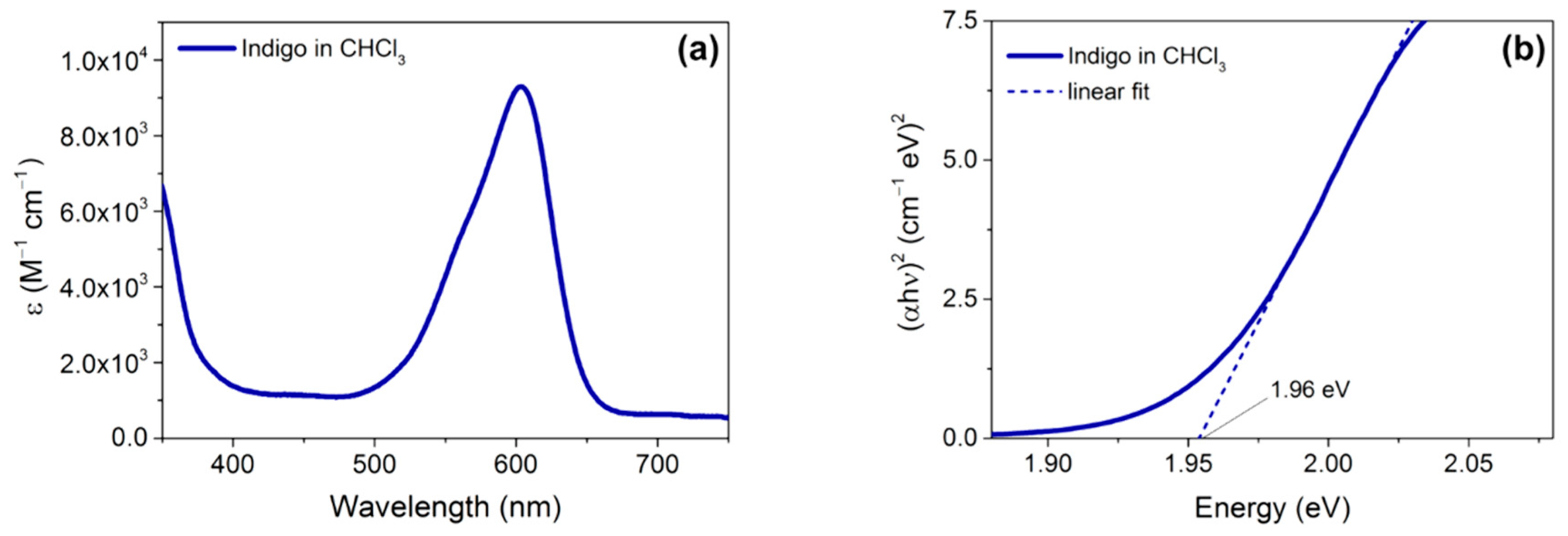
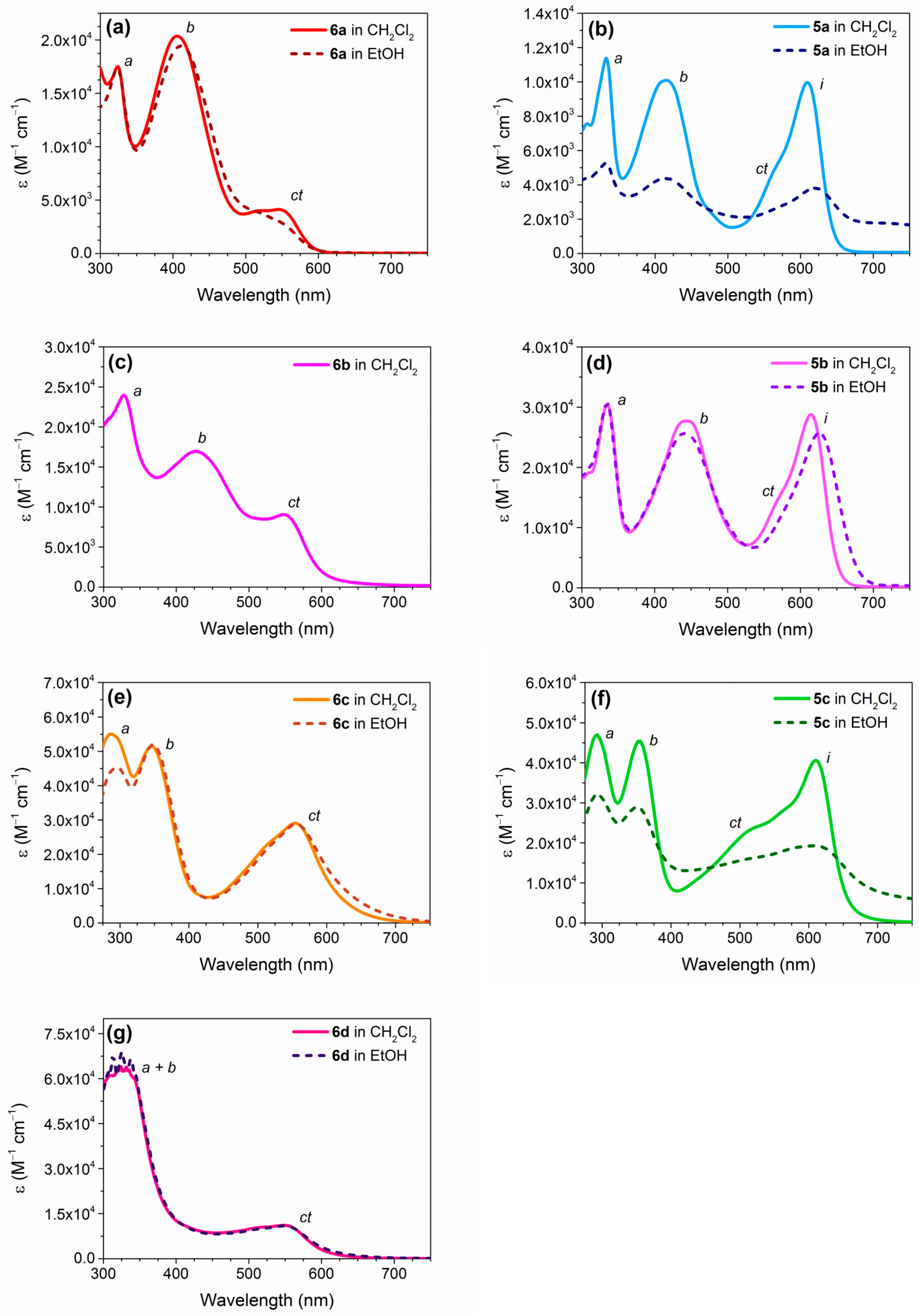
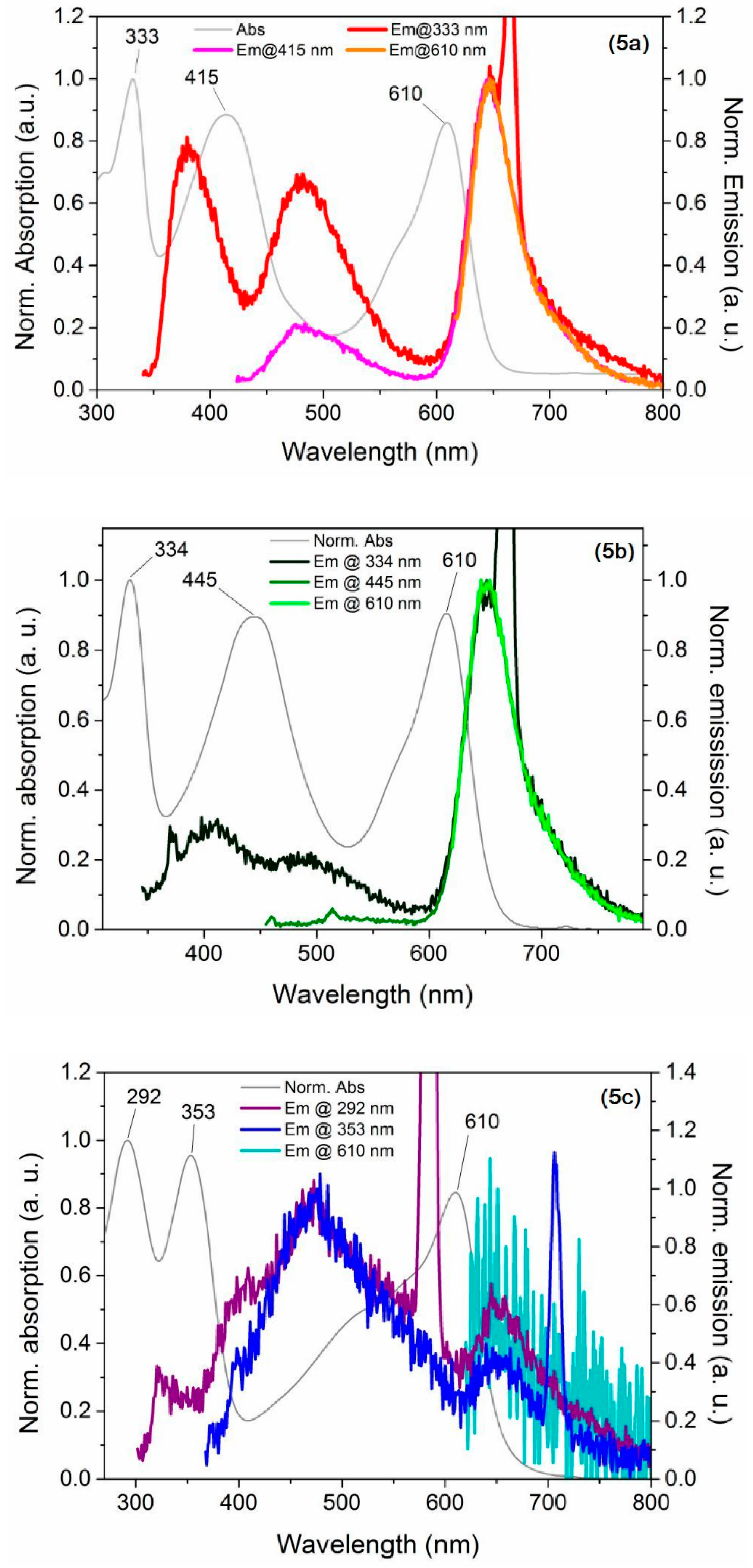
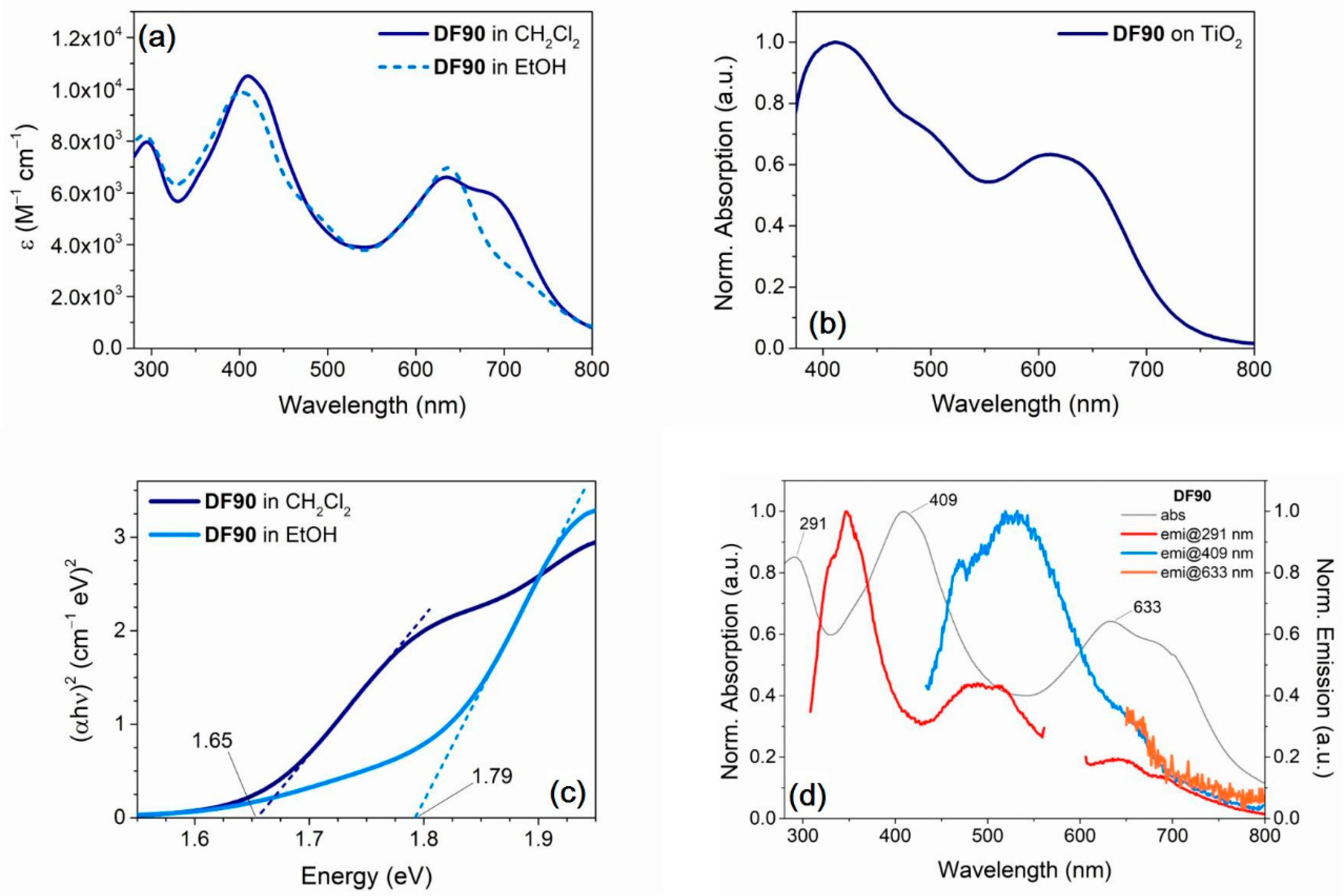
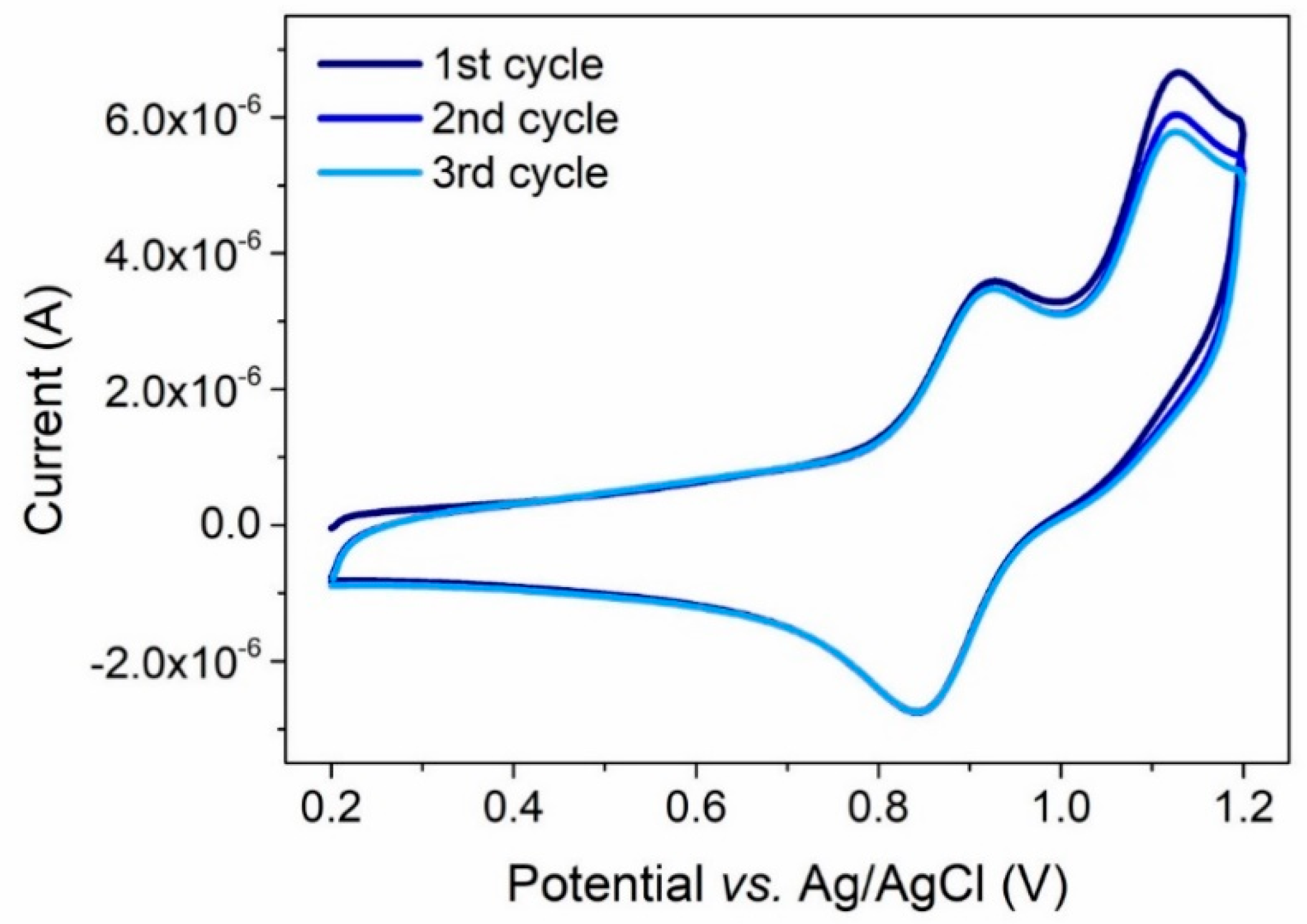
2.3. Optical and Electrochemical Properties
3. Experimental Section
3.1. General Information
3.2. Computational Details
3.3. Synthesis
3.3.1. Synthesis of Tributyl(3,3-dipentyl-3,4-dihydro-2H-thieno[3,4-b][1,4]dioxepin-6-yl)-stannane (12b)
3.3.2. General Procedure for Preparation of Compounds 6a–d
3.3.3. General Procedure for the Preparation of Compounds 5a–c
3.3.4. Synthesis of Dye DF90
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Editorial: Chemists go green to make better blue jeans. Nature 2018, 553, 128. [CrossRef] [PubMed] [Green Version]
- Baeyer, A.; Drewsen, V. Darstellung von Indigblau aus Orthonitrobenzaldehyd. Berichte der Dtsch. Chem. Gesellschaft 1882, 15, 2856–2864. [Google Scholar] [CrossRef] [Green Version]
- Baeyer, A. Ueber die Verbindungen der Indigogruppe. Berichte der Dtsch. Chem. Gesellschaft 1883, 16, 2188–2204. [Google Scholar] [CrossRef] [Green Version]
- Głowacki, E.D.; Voss, G.; Sariciftci, N.S. 25th Anniversary Article: Progress in Chemistry and Applications of Functional Indigos for Organic Electronics. Adv. Mater. 2013, 25, 6783–6800. [Google Scholar] [CrossRef] [PubMed]
- Jacquemin, D.; Perpète, E.A.; Scuseria, G.E.; Ciofini, I.; Adamo, C. TD-DFT Performance for the Visible Absorption Spectra of Organic Dyes: Conventional versus Long-Range Hybrids. J. Chem. Theory Comput. 2008, 4, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Tatsch, E.; Schrader, B. Near-infrared fourier transform Raman spectroscopy of indigoids. J. Raman Spectrosc. 1995, 26, 467–473. [Google Scholar] [CrossRef]
- Konarev, D.V.; Zorina, L.V.; Batov, M.S.; Khasanov, S.S.; Otsuka, A.; Yamochi, H.; Kitagawa, H.; Lyubovskaya, R.N. Optical and magnetic properties of trans -indigo˙−radical anions. Magnetic coupling between trans-indigo˙-(S = 1/2) mediated by intermolecular hydrogen N–H⋯O=C bonds. New J. Chem. 2019, 43, 7350–7354. [Google Scholar] [CrossRef]
- Serrano-Andrés, L.; Roos, B.O. A Theoretical Study of the Indigoid Dyes and Their Chromophore. Chem.-A Eur. J. 1997, 3, 717–725. [Google Scholar] [CrossRef]
- Amat, A.; Rosi, F.; Miliani, C.; Sgamellotti, A.; Fantacci, S. Theoretical and experimental investigation on the spectroscopic properties of indigo dye. J. Mol. Struct. 2011, 993, 43–51. [Google Scholar] [CrossRef]
- Irimia-Vladu, M.; Głowacki, E.D.; Troshin, P.A.; Schwabegger, G.; Leonat, L.; Susarova, D.K.; Krystal, O.; Ullah, M.; Kanbur, Y.; Bodea, M.A.; et al. Indigo - A Natural Pigment for High Performance Ambipolar Organic Field Effect Transistors and Circuits. Adv. Mater. 2012, 24, 375–380. [Google Scholar] [CrossRef]
- Klimovich, I.V.; Leshanskaya, L.I.; Troyanov, S.I.; Anokhin, D.V.; Novikov, D.V.; Piryazev, A.A.; Ivanov, D.A.; Dremova, N.N.; Troshin, P.A. Design of indigo derivatives as environment-friendly organic semiconductors for sustainable organic electronics. J. Mater. Chem. C 2014, 2, 7621–7631. [Google Scholar] [CrossRef]
- Alexy, M.; Voss, G.; Heinze, J. Optochemical sensor for determining ozone based on novel soluble indigo dyes immobilised in a highly permeable polymeric film. Anal. Bioanal. Chem. 2005, 382, 1628–1641. [Google Scholar] [CrossRef] [PubMed]
- Brunet, J.; Spinelle, L.; Ndiaye, A.; Dubois, M.; Monier, G.; Varenne, C.; Pauly, A.; Lauron, B.; Guerin, K.; Hamwi, A. Physical and chemical characterizations of nanometric indigo layers as efficient ozone filter for gas sensor devices. Thin Solid Films 2011, 520, 971–977. [Google Scholar] [CrossRef]
- Yao, M.; Kuratani, K.; Kojima, T.; Takeichi, N.; Senoh, H.; Kiyobayashi, T. Indigo carmine: An organic crystal as a positive-electrode material for rechargeable sodium batteries. Sci. Rep. 2015, 4, 3650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, M.; Araki, M.; Senoh, H.; Yamazaki, S.; Sakai, T.; Yasuda, K. Indigo Dye as a Positive-electrode Material for Rechargeable Lithium Batteries. Chem. Lett. 2010, 39, 950–952. [Google Scholar] [CrossRef]
- Porada, J.H.; Neudörfl, J.-M.; Blunk, D. Planar and distorted indigo as the core motif in novel chromophoric liquid crystals. New J. Chem. 2015, 39, 8291–8301. [Google Scholar] [CrossRef] [Green Version]
- Porada, J.H.; Blunk, D. Phasmidic indigoid liquid crystals. J. Mater. Chem. 2010, 20, 2956–2958. [Google Scholar] [CrossRef]
- Rajan, A.K.; Cindrella, L. Studies on new natural dye sensitizers from Indigofera tinctoria in dye-sensitized solar cells. Opt. Mater. (Amst). 2019, 88, 39–47. [Google Scholar] [CrossRef]
- Kalyanasundaram, K. Dye-sensitized Solar Cells; EFPL Press: Lausanne, Switzerland, 2010; ISBN 9781439808665. [Google Scholar]
- O’Regan, B.; Grätzel, M. A low-cost, high-efficiency solar cell based on dye-sensitized colloidal TiO2 films. Nature 1991, 353, 737–740. [Google Scholar] [CrossRef]
- Calogero, G.; Bartolotta, A.; Di Marco, G.; Di Carlo, A.; Bonaccorso, F. Vegetable-based dye-sensitized solar cells. Chem. Soc. Rev. 2015, 44, 3244–3294. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, X.; Wang, W.; Gurzadyan, G.G.; Li, J.; Li, X.; An, J.; Yu, Z.; Wang, H.; Cai, B.; et al. 13.6% Efficient Organic Dye-Sensitized Solar Cells by Minimizing Energy Losses of the Excited State. ACS Energy Lett. 2019, 4, 943–951. [Google Scholar] [CrossRef]
- Mishra, A.; Fischer, M.K.R.; Bäuerle, P. Metal-Free Organic Dyes for Dye-Sensitized Solar Cells: From Structure: Property Relationships to Design Rules. Angew. Chemie Int. Ed. 2009, 48, 2474–2499. [Google Scholar] [CrossRef]
- Ooyama, Y.; Harima, Y. Molecular Designs and Syntheses of Organic Dyes for Dye-Sensitized Solar Cells. European J. Org. Chem. 2009, 2009, 2903–2934. [Google Scholar] [CrossRef]
- Obotowo, I.N.; Obot, I.B.; Ekpe, U.J. Organic sensitizers for dye-sensitized solar cell (DSSC): Properties from computation, progress and future perspectives. J. Mol. Struct. 2016, 1122, 80–87. [Google Scholar] [CrossRef]
- Brogdon, P.; Cheema, H.; Delcamp, J.H. Near-Infrared-Absorbing Metal-Free Organic, Porphyrin, and Phthalocyanine Sensitizers for Panchromatic Dye-Sensitized Solar Cells. ChemSusChem 2018, 11, 86–103. [Google Scholar] [CrossRef] [PubMed]
- Burke, A.; Schmidt-Mende, L.; Ito, S.; Grätzel, M. A novel blue dye for near-IR ‘dye-sensitised’ solar cell applications. Chem. Commun. 2007, 234–236. [Google Scholar] [CrossRef] [Green Version]
- Paek, S.; Choi, H.; Kim, C.; Cho, N.; So, S.; Song, K.; Nazeeruddin, M.K.; Ko, J. Efficient and stable panchromatic squaraine dyes for dye-sensitized solar cells. Chem. Commun. 2011, 47, 2874. [Google Scholar] [CrossRef]
- Shi, Y.; Hill, R.B.M.; Yum, J.-H.; Dualeh, A.; Barlow, S.; Grätzel, M.; Marder, S.R.; Nazeeruddin, M.K. A High-Efficiency Panchromatic Squaraine Sensitizer for Dye-Sensitized Solar Cells. Angew. Chem. Int. Ed. 2011, 50, 6619–6621. [Google Scholar] [CrossRef]
- Jradi, F.M.; Kang, X.; O’Neil, D.; Pajares, G.; Getmanenko, Y.A.; Szymanski, P.; Parker, T.C.; El-Sayed, M.A.; Marder, S.R. Near-Infrared Asymmetrical Squaraine Sensitizers for Highly Efficient Dye Sensitized Solar Cells: The Effect of π-Bridges and Anchoring Groups on Solar Cell Performance. Chem. Mater. 2015, 27, 2480–2487. [Google Scholar] [CrossRef]
- Yum, J.-H.; Holcombe, T.W.; Kim, Y.; Rakstys, K.; Moehl, T.; Teuscher, J.; Delcamp, J.H.; Nazeeruddin, M.K.; Grätzel, M. Blue-Coloured Highly Efficient Dye-Sensitized Solar Cells by Implementing the Diketopyrrolopyrrole Chromophore. Sci. Rep. 2013, 3, 2446. [Google Scholar] [CrossRef] [Green Version]
- Liyanage, N.P.; Yella, A.; Nazeeruddin, M.; Grätzel, M.; Delcamp, J.H. Thieno[3,4- b]pyrazine as an Electron Deficient π-Bridge in D–A−π– A DSCs. ACS Appl. Mater. Interfaces 2016, 8, 5376–5384. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhu, W.-H.; Zakeeruddin, S.M.; Grätzel, M. Insight into D–A−π–A Structured Sensitizers: A Promising Route to Highly Efficient and Stable Dye-Sensitized Solar Cells. ACS Appl. Mater. Interfaces 2015, 7, 9307–9318. [Google Scholar] [CrossRef] [PubMed]
- Dessì, A.; Sinicropi, A.; Mohammadpourasl, S.; Basosi, R.; Taddei, M.; Fabrizi de Biani, F.; Calamante, M.; Zani, L.; Mordini, A.; Bracq, P.; et al. New Blue Donor–Acceptor Pechmann Dyes: Synthesis, Spectroscopic, Electrochemical, and Computational Studies. ACS Omega 2019, 4, 7614–7627. [Google Scholar] [CrossRef]
- Abdullah, M.I.; Janjua, M.R.S.A.; Mahmood, A.; Ali, S.; Ali, M. Quantum Chemical Designing of Efficient Sensitizers for Dye Sensitized Solar Cells. Bull. Korean Chem. Soc. 2013, 34, 2093–2098. [Google Scholar] [CrossRef] [Green Version]
- Cervantes-Navarro, F.; Glossman-Mitnik, D. Density functional theory study of indigo and its derivatives as photosensitizers for dye-sensitized solar cells. J. Photochem. Photobiol. A Chem. 2013, 255, 24–26. [Google Scholar] [CrossRef]
- Hosseinnezhad, M.; Moradian, S.; Gharanjig, K. Synthesis and Characterization of Two New Organic Dyes for Dye-Sensitized Solar Cells. Synth. Commun. 2014, 44, 779–787. [Google Scholar] [CrossRef]
- Głowacki, E.D.; Apaydin, D.H.; Bozkurt, Z.; Monkowius, U.; Demirak, K.; Tordin, E.; Himmelsbach, M.; Schwarzinger, C.; Burian, M.; Lechner, R.T.; et al. Air-stable organic semiconductors based on 6,6′-dithienylindigo and polymers thereof. J. Mater. Chem. C 2014, 2, 8089–8097. [Google Scholar] [CrossRef]
- Pina, J.; Alnady, M.; Eckert, A.; Scherf, U.; Seixas de Melo, J.S. Alternating donor–acceptor indigo-cyclopentadithiophene copolymers: Competition between excited state conformational relaxation, energy transfer and excited state proton transfer. Mater. Chem. Front. 2018, 2, 281–290. [Google Scholar] [CrossRef]
- Liu, C.; Xu, W.; Xue, Q.; Cai, P.; Ying, L.; Huang, F.; Cao, Y. Nanowires of indigo and isoindigo-based molecules with thermally removable groups. Dye. Pigment. 2016, 125, 54–63. [Google Scholar] [CrossRef]
- Liu, C.; Dong, S.; Cai, P.; Liu, P.; Liu, S.; Chen, J.; Liu, F.; Ying, L.; Russell, T.P.; Huang, F.; et al. Donor–Acceptor Copolymers Based on Thermally Cleavable Indigo, Isoindigo, and DPP Units: Synthesis, Field Effect Transistors, and Polymer Solar Cells. ACS Appl. Mater. Interfaces 2015, 7, 9038–9051. [Google Scholar] [CrossRef]
- Ma, C.; Li, H.; Yang, Y.; Li, D.; Liu, Y. TD-DFT study on electron transfer mobility and intramolecular hydrogen bond of substituted indigo derivatives. Chem. Phys. Lett. 2015, 638, 72–77. [Google Scholar] [CrossRef]
- Pina, J.; Sarmento, D.; Accoto, M.; Gentili, P.L.; Vaccaro, L.; Adelino, G.; Seixas De Melo, J.S. Excited-State Proton Transfer in Indigo. J. Phys. Chem. B 2017, 121, 2308–2318. [Google Scholar] [CrossRef]
- Dessì, A.; Bartolini, M.; Calamante, M.; Zani, L.; Mordini, A.; Reginato, G. Extending the Conjugation of Pechmann Lactone Thienyl Derivatives: A New Class of Small Molecules for Organic Electronics Application. Synthesis (Sttutgart) 2018, 50, 1284–1292. [Google Scholar] [CrossRef] [Green Version]
- Baran, P.S.; Shenvi, R.A. Total Synthesis of (±)-Chartelline C. J. Am. Chem. Soc. 2006, 128, 14028–14029. [Google Scholar] [CrossRef]
- Gao, P.; Tsao, H.N.; Grätzel, M.; Nazeeruddin, M.K. Fine-tuning the Electronic Structure of Organic Dyes for Dye-Sensitized Solar Cells. Org. Lett. 2012, 14, 4330–4333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacquemin, D.; Preat, J.; Wathelet, V.; Perpète, E.A. Substitution and chemical environment effects on the absorption spectrum of indigo. J. Chem. Phys. 2006, 124, 074104. [Google Scholar] [CrossRef] [PubMed]
- Dähne, S.; Leupold, D. Coupling Principles in Organic Dyes. Angew. Chem. Int. Ed. Eng. 1966, 5, 984–993. [Google Scholar] [CrossRef]
- Shimizu, M.; Hiyama, T. Organic Fluorophores Exhibiting Highly Efficient Photoluminescence in the Solid State. Chem. Asian J. 2010, 5, 1516–1531. [Google Scholar] [CrossRef]
- Zhang, L.; Cole, J.M. Dye aggregation in dye-sensitized solar cells. J. Mater. Chem. A 2017, 5, 19541–19559. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.J.; Chow, T.J. Highly efficient triarylene conjugated dyes for sensitized solar cells. J. Mater. Chem. 2011, 21, 9523. [Google Scholar] [CrossRef]
- Boschloo, G.; Hagfeldt, A. Characteristics of the Iodide/Triiodide Redox Mediator in Dye-Sensitized Solar Cells. Acc. Chem. Res. 2009, 42, 1819–1826. [Google Scholar] [CrossRef] [PubMed]
- Hagfeldt, A.; Boschloo, G.; Sun, L.; Kloo, L.; Pettersson, H. Dye-Sensitized Solar Cells. Chem. Rev. 2010, 110, 6595–6663. [Google Scholar] [CrossRef] [PubMed]
- Honenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. B 1964, 136, 864–871. [Google Scholar] [CrossRef] [Green Version]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, 1134–1138. [Google Scholar] [CrossRef] [Green Version]
- Parr, R.G.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Adamo, C.; Jacquemin, D. The calculations of excited-state properties with Time-Dependent Density Functional Theory. Chem. Soc. Rev. 2013, 42, 845–856. [Google Scholar] [CrossRef] [PubMed]
- Laurent, A.D.; Adamo, C.; Jacquemin, D. Dye chemistry with time-dependent density functional theory. Phys. Chem. Chem. Phys. 2014, 16, 14334–14356. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Samples of the compounds 5a–c and 6a–c available from the authors. |












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | Excited States | λamax (nm) | Eexc (eV) | f | Contribution (%) |
|---|---|---|---|---|---|
| 5a | 1 | 604 | 2.05 | 0.54 | 100% H→L |
| 5b | 1 | 610 | 2.03 | 0.58 | 100% H→L |
| 5c | 1 | 760 | 1.63 | 0.86 | 99% H→L |
| 3 | 594 | 2.08 | 0.32 | 99% H-2→L | |
| 5d | 1 | 746 | 1.62 | 0.76 | 99% H→L |
| 3 | 596 | 2.08 | 0.36 | 99% H-2→L | |
| DF90 | 1 | 857 | 1.44 | 0.32 | 99% H→L |
| 2 | 645 | 1.92 | 0.41 | 97% H-1→L | |
| DF90 # | 1 | 543 | 2.28 | 0.66 | 80% H-1→L |
| Dye | λmax CH2Cl2 [nm] | ε CH2Cl2 [× 104 M−1 cm−1] | E0-0 CH2Cl2 [eV] a | λmax EtOH [nm] | ε EtOH [× 104 M−1 cm−1] | E0-0 EtOH [eV] a |
|---|---|---|---|---|---|---|
| Indigo | 604 b | 0.93 b | 1.96 b | - | - | - |
| 6a | 324 | 1.75 | 2.14 | 323 | 1.74 | 2.14 |
| 406 | 2.04 | 411 | 1.94 | |||
| 546 | 0.41 | 545 | 0.3 | |||
| 5a | 333 | 1.14 | 1.95 | 333 | 0.52 | 1.85 |
| 415 | 1.01 | 414 | 0.44 | |||
| 610 | 1 | 620 | 0.38 | |||
| 6b | 328 | 2.4 | 2.11 | - c | - c | - c |
| 426 | 1.7 | |||||
| 548 | 0.9 | |||||
| 5b | 335 | 3.05 | 1.94 | 335 | 3.06 | 1.87 |
| 445 | 2.77 | 441 | 2.56 | |||
| 614 | 2.88 | 626 | 2.57 | |||
| 6c | 290 | 5.5 | 2.06 | 296 | 4.52 | 2.03 |
| 346 | 5.15 | 347 | 5.2 | |||
| 554 | 2.9 | 556 | 2.88 | |||
| 5c | 293 | 4.7 | 1.93 | 293 | 3.2 | 1.82 |
| 355 | 4.55 | 351 | 2.89 | |||
| 610 | 4.06 | 608 | 1.93 | |||
| 6d | 324 | 6.4 | 2.1 | 325 | 6.89 | 2.09 |
| 548 | 1.11 | 549 | 1.09 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franchi, D.; Calamante, M.; Coppola, C.; Mordini, A.; Reginato, G.; Sinicropi, A.; Zani, L. Synthesis and Characterization of New Organic Dyes Containing the Indigo Core. Molecules 2020, 25, 3377. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25153377
Franchi D, Calamante M, Coppola C, Mordini A, Reginato G, Sinicropi A, Zani L. Synthesis and Characterization of New Organic Dyes Containing the Indigo Core. Molecules. 2020; 25(15):3377. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25153377
Chicago/Turabian StyleFranchi, Daniele, Massimo Calamante, Carmen Coppola, Alessandro Mordini, Gianna Reginato, Adalgisa Sinicropi, and Lorenzo Zani. 2020. "Synthesis and Characterization of New Organic Dyes Containing the Indigo Core" Molecules 25, no. 15: 3377. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25153377