
Synthesis of 2-((2-(Benzo[d]oxazol-2-yl)-2H-imidazol-4-yl)amino)-phenols from 2-((5H-1,2,3-Dithiazol-5-ylidene)amino)phenols through Unprecedented Formation of Imidazole Ring from Two Methanimino Groups

 ,
, 
Abstract
:
1. Introduction
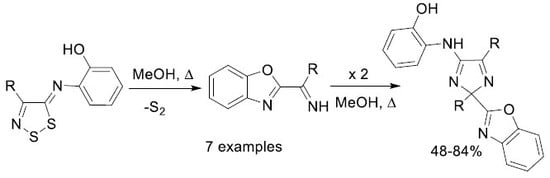
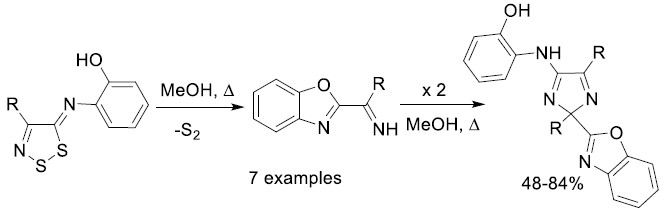
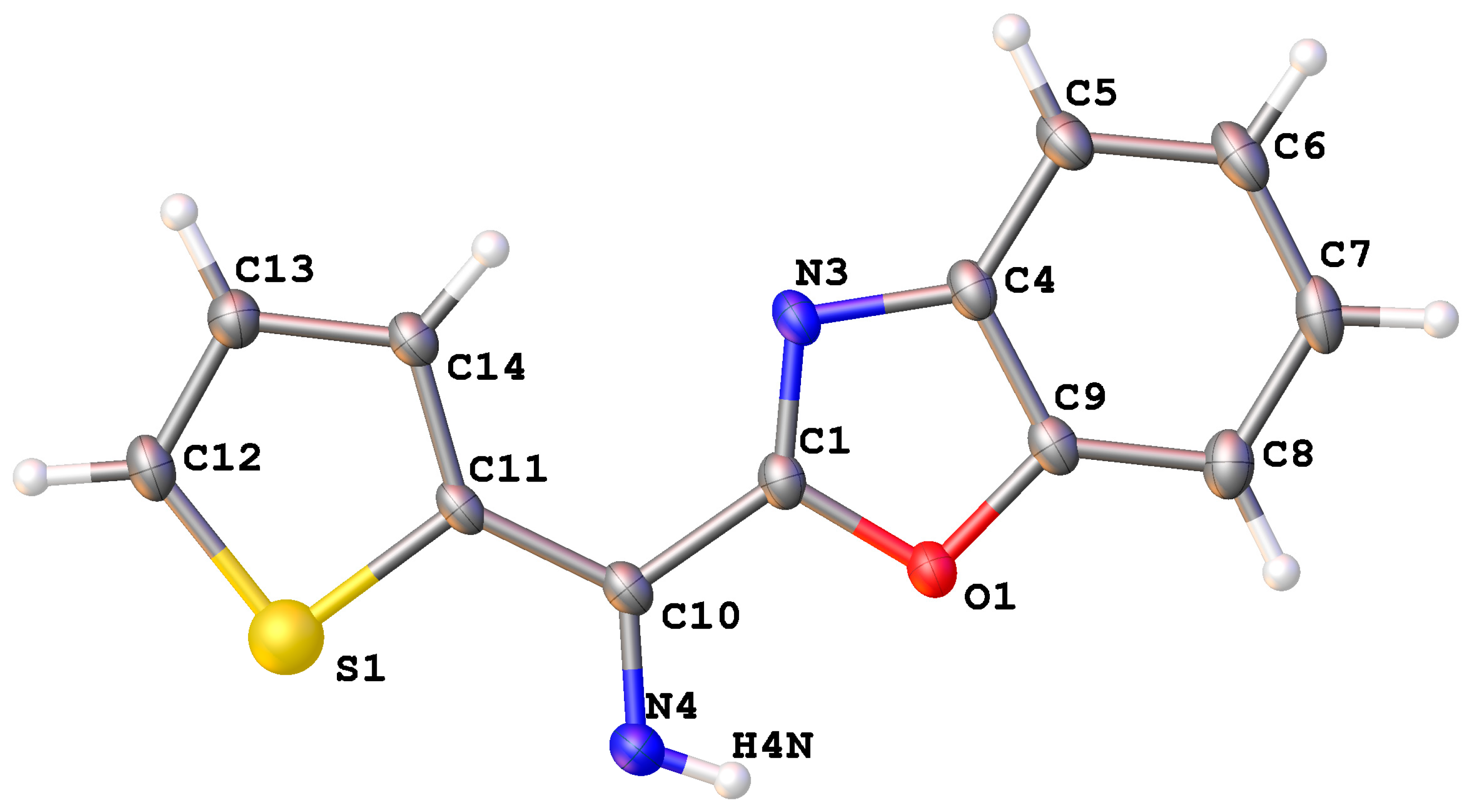
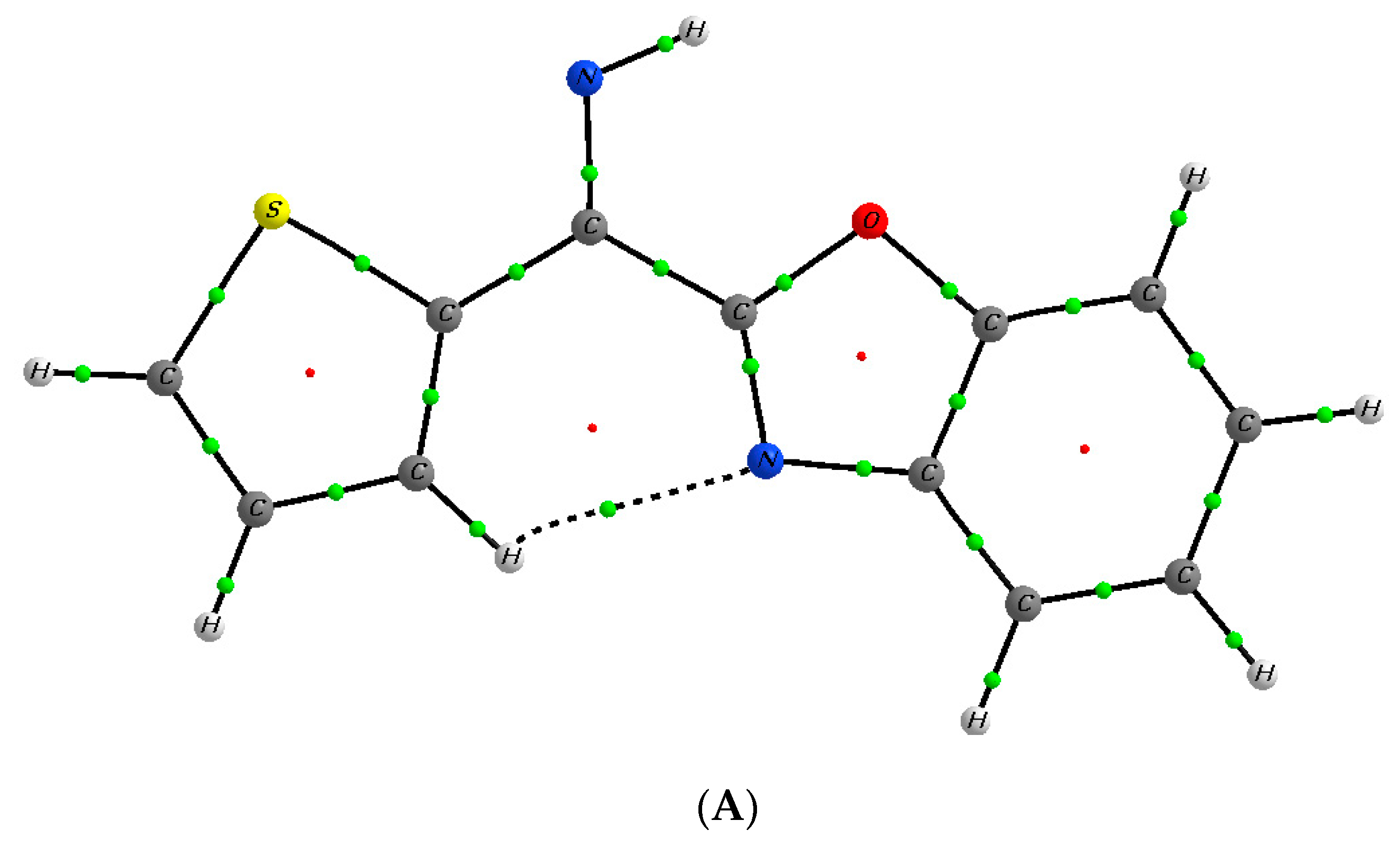
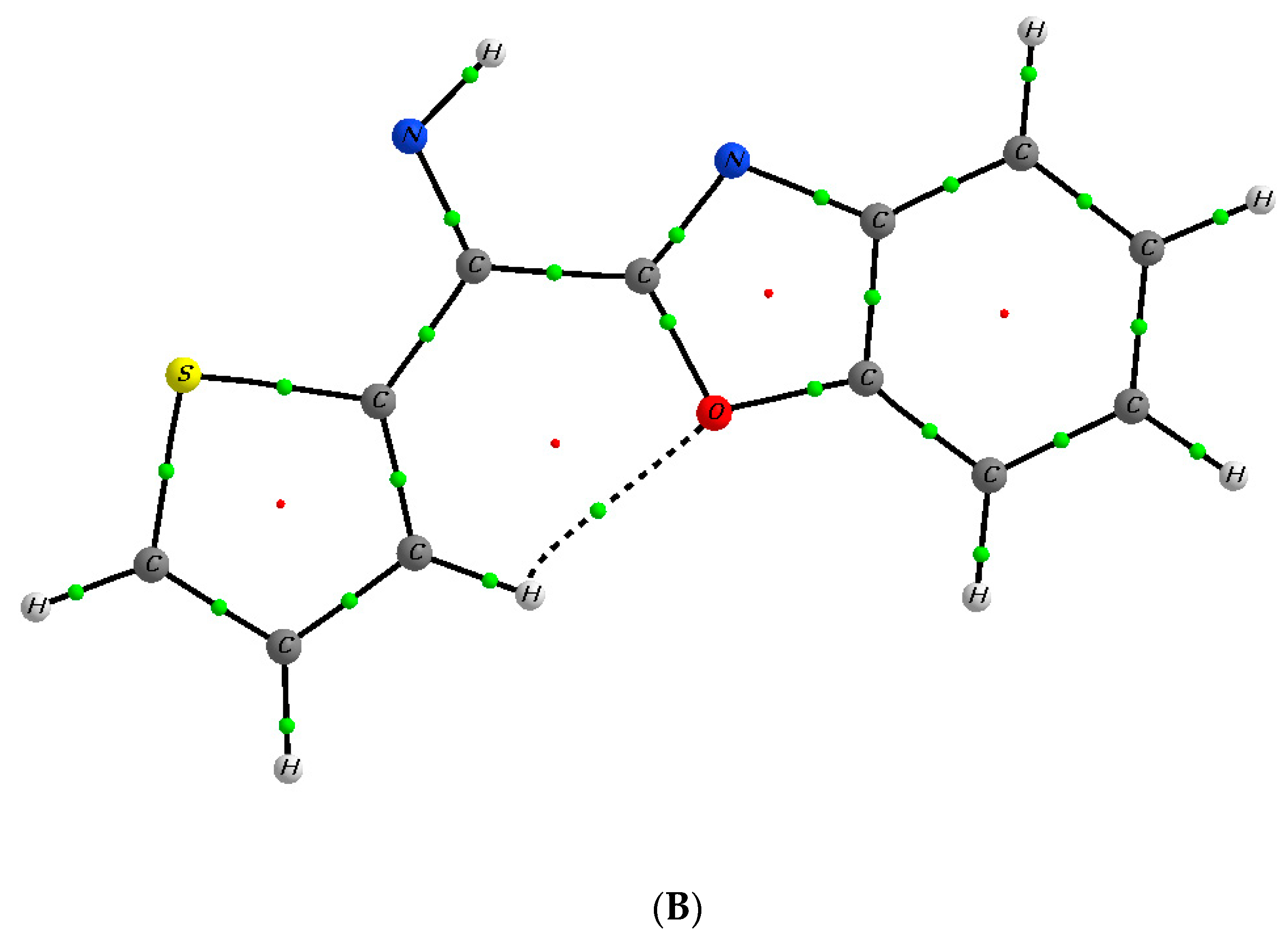
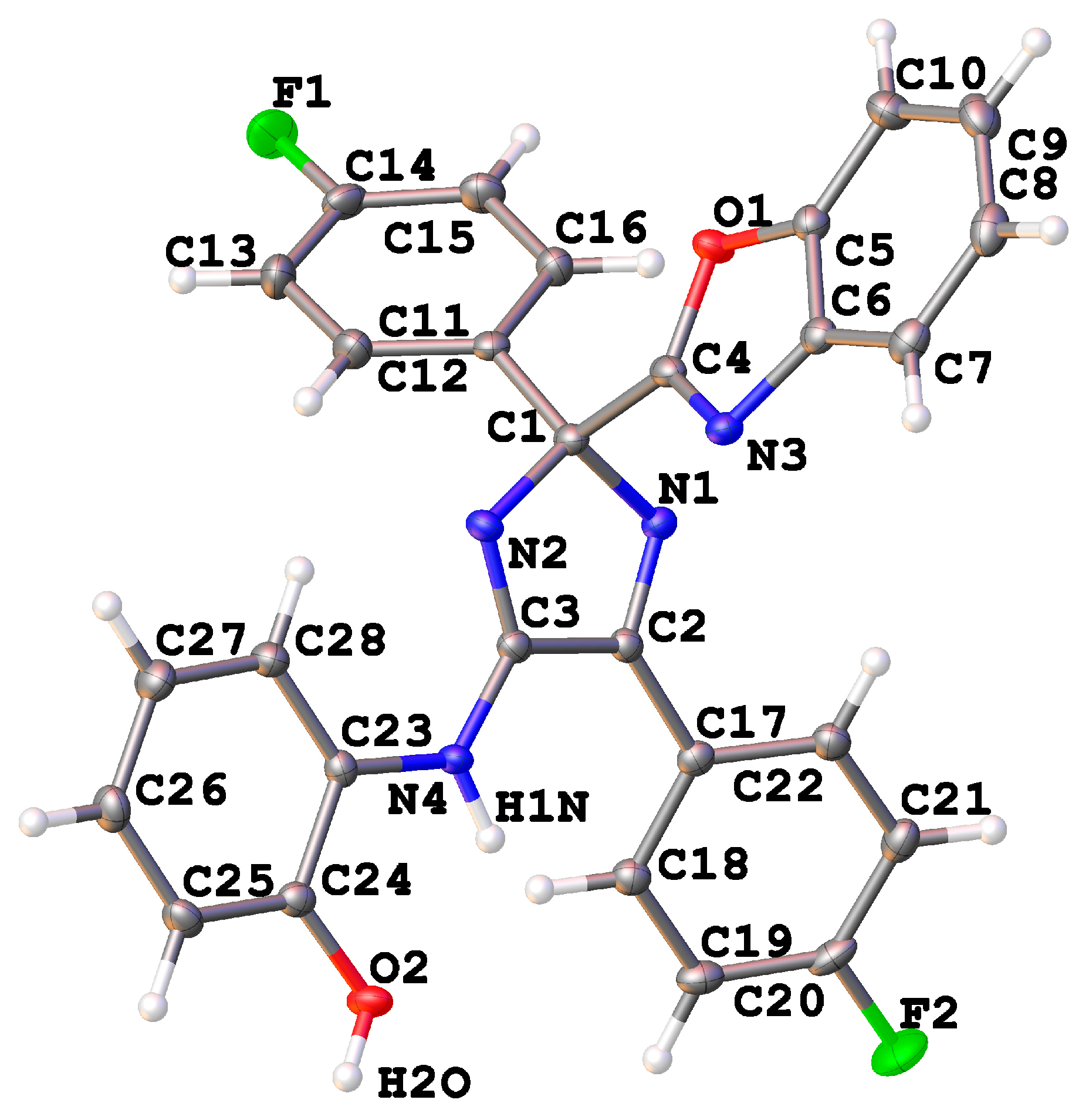
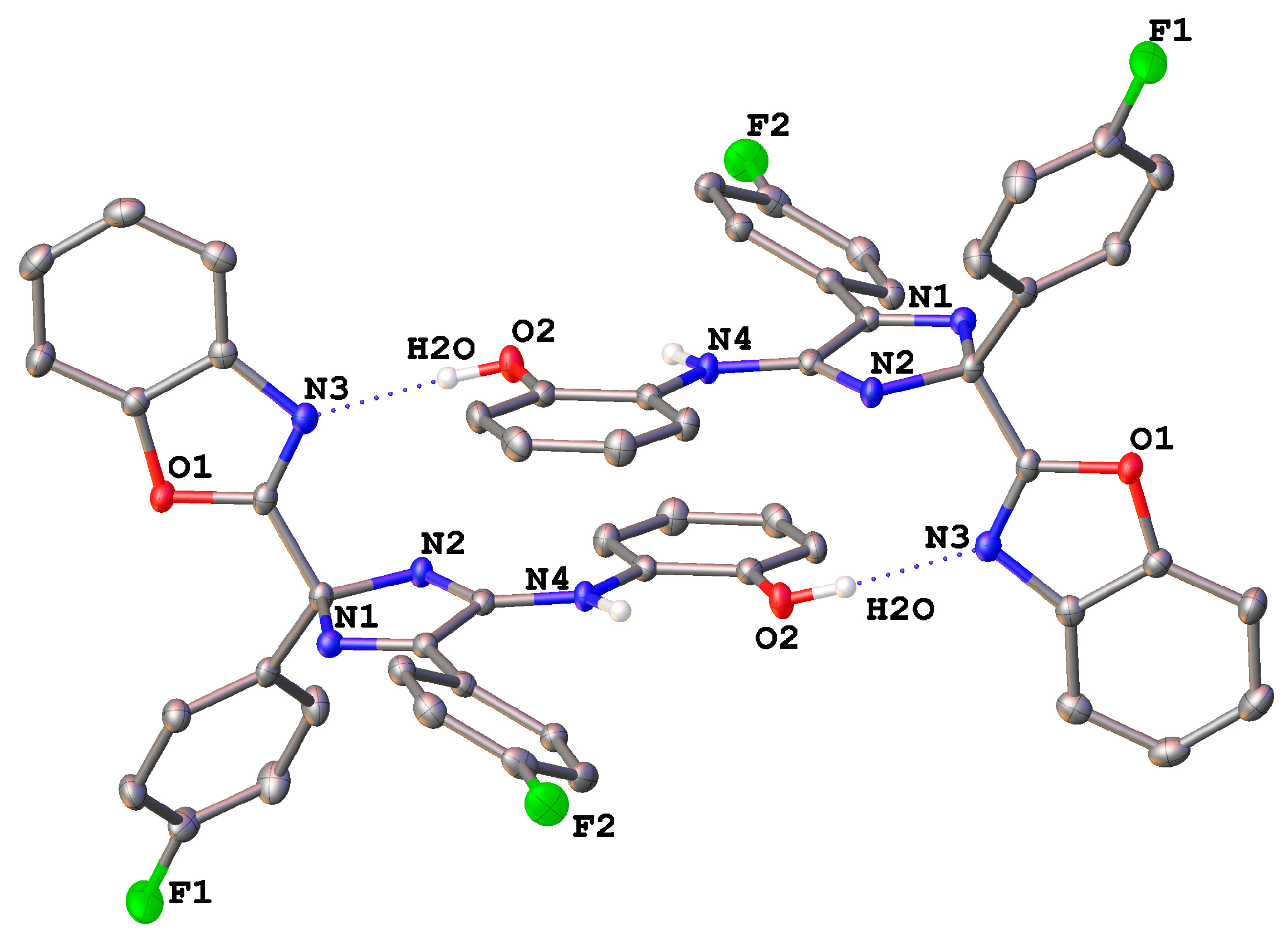
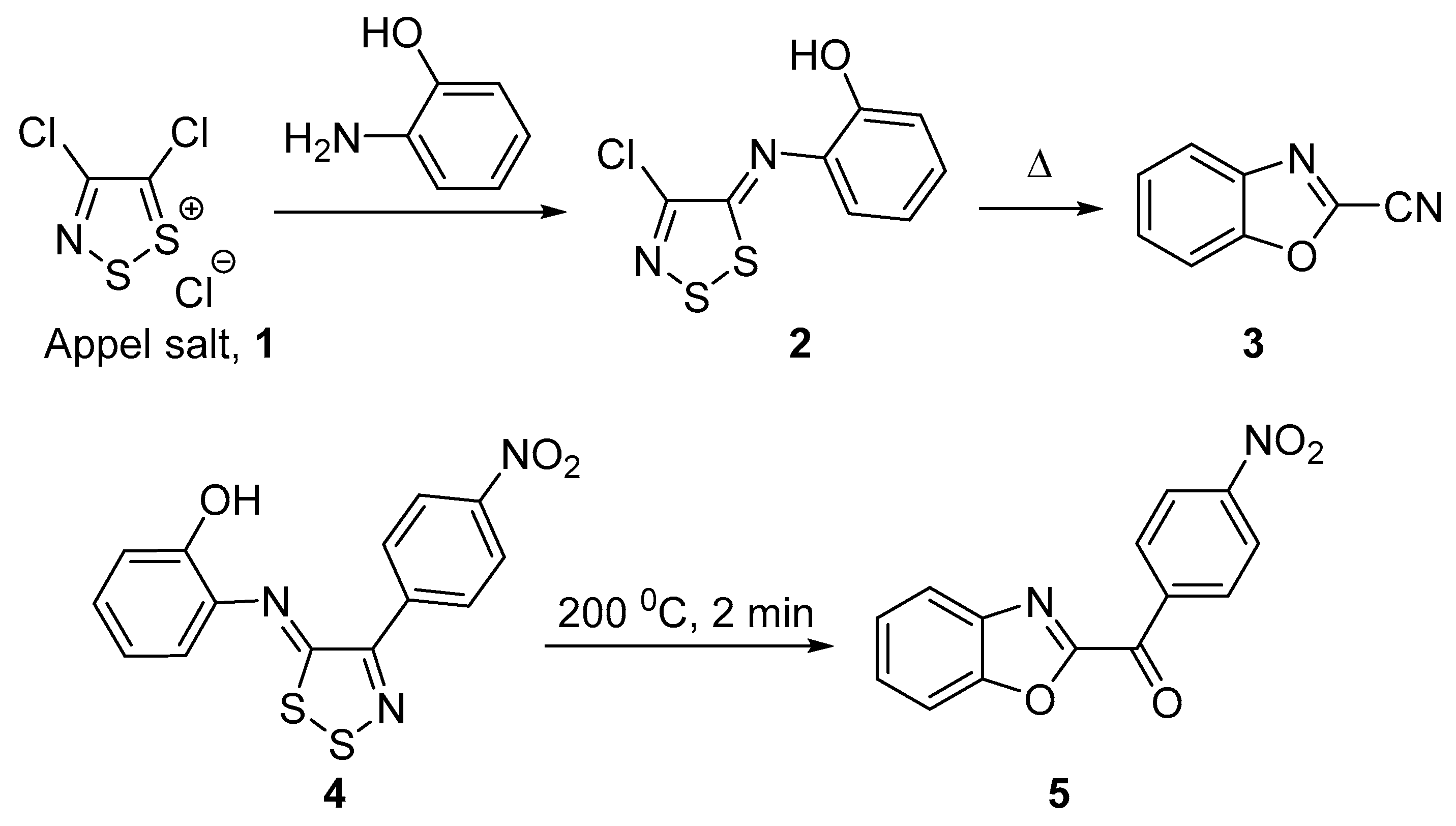
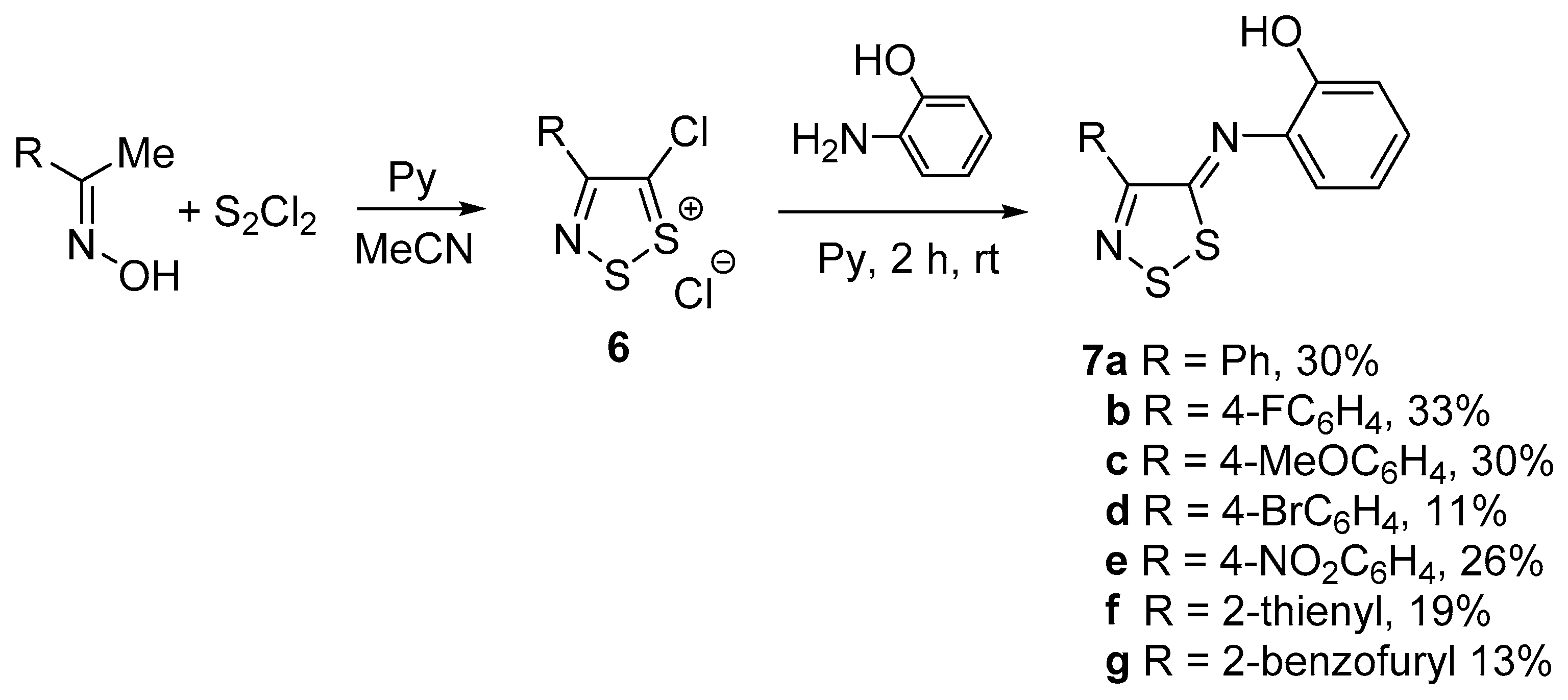
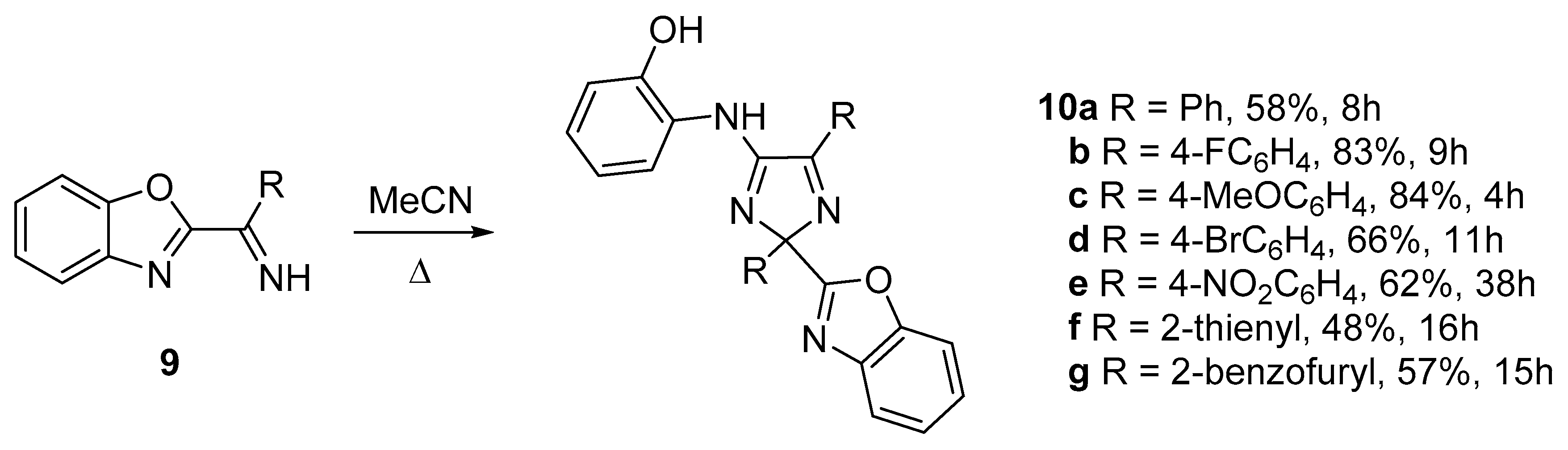
2. Results and Discussion
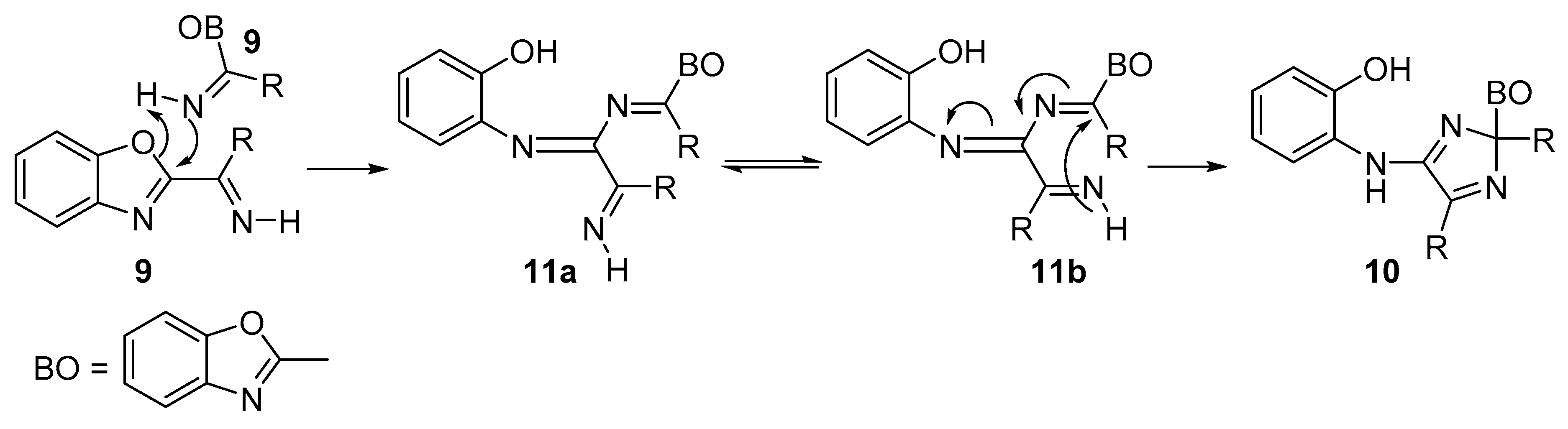
Mechanistic Rationale
3. Experimental Section
3.1. General Methods and Materials
3.2. General Procedure for the Synthesis of 2-((4-aryl-5H-1,2,3-dithiazol-5-ylidene)amino)phenols (7)
3.3. General Procedure for the Thermolysis of 2-((4-aryl(hetaryl)-5H-1,2,3-dithiazol-5-ylidene)amino)phenols 7 in Various Solvents
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Konstantinova, L.S.; Rakitin, O.A. Synthesis and properties of 1,2,3-dithiazoles. Russ. Chem. Rev. 2008, 77, 521–546. [Google Scholar] [CrossRef]
- Rakitin, O. 1,2-Oxa/thia-3-azoles. In Comprehensive Heterocyclic Chemistry III; Katritzky, A.R., Ramsden, C.A., Scriven, E.F.V., Taylor, R.J.K., Eds.; Elsevier BV: Amsterdam, The Netherlands, 2008; Volume 6, pp. 1–36. [Google Scholar]
- Rakitin, O.A.; Zibarev, A.V. Synthesis and Applications of 5-Membered Chalcogen-Nitrogen π-Heterocycles with Three Heteroatoms. Asian J. Org. Chem. 2018, 7, 2397–2416. [Google Scholar] [CrossRef]
- Besson, T.; Rees, C.W.; Cottenceau, G.; Pons, A.-M. Antimicrobial evaluation of 3,1-benzoxazin-4-ones, 3,1-benzothiazin-4-ones, 4-alkoxyquinazolin-2-carbonitriles and N-arylimino-1,2,3-dithiazoles. Bioorg. Med. Chem. Lett. 1996, 6, 2343–2348. [Google Scholar] [CrossRef]
- Oppedisano, F.; Catto, M.; Koutentis, P.A.; Nicolotti, O.; Pochini, L.; Koyioni, M.; Introcaso, A.; Michaelidou, S.S.; Carotti, A.; Indiveri, C. Inactivation of the glutamine/amino acid transporter ASCT2 by 1,2,3-dithiazoles: Proteoliposomes as a tool to gain insights in the molecular mechanism of action and of antitumor activity. Toxicol. Appl. Pharmacol. 2012, 265, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Charalambous, A.; Koyioni, M.; Antoniades, I.; Pegeioti, D.; Eleftheriou, I.; Michaelidou, S.S.; Amelichev, S.A.; Konstantinova, L.S.; Rakitin, O.A.; Koutentis, P.A.; et al. 1,2,3-Dithiazoles–new reversible melanin synthesis inhibitors: A chemical genomics study. MedChemComm 2015, 6, 935–946. [Google Scholar] [CrossRef]
- Asquith, C.R.M.; Konstantinova, L.S.; Laitinen, T.; Meli, M.L.; Poso, A.; Rakitin, O.A.; Hofmann-Lehmann, R.; Hilton, S.T. Evaluation of Substituted 1,2,3-Dithiazoles as Inhibitors of the Feline Immunodeficiency Virus (FIV) Nucleocapsid Protein via a Proposed Zinc Ejection Mechanism. ChemMedChem 2016, 11, 2119–2126. [Google Scholar] [CrossRef]
- Asquith, C.R.M.; Meili, T.; Rakitin, O.A.; Baranovsky, I.V.; Konstantinova, L.S.; Poso, A.; Rakitin, O.A.; Hofmann-Lehmann, R. Synthesis and comparison of substituted 1,2,3-dithiazole and 1,2,3-thiaselenazole as inhibitors of the feline immunodeficiency virus (FIV) nucleocapsid protein as a model for HIV infection. Bioorganic Med. Chem. Lett. 2019, 29, 1765–1768. [Google Scholar] [CrossRef]
- Beer, L.; Cordes, A.W.; Haddon, R.C.; Itkis, M.E.; Oakley, R.T.; Reed, R.W.; Robertson, C.M. A π-stacked 1,2,3-dithiazolyl radical. Preparation and solid state characterization of (Cl2C3NS)(ClC2NS2). Chem. Commun. 2002, 1872–1873. [Google Scholar] [CrossRef]
- Rakitin, O.A. Stable heterocyclic radicals. Russ. Chem. Rev. 2011, 80, 647–659. [Google Scholar] [CrossRef]
- Lekin, K.; Phan, H.; Winter, S.M.; Wong, J.W.L.; Leitch, A.A.; Laniel, D.; Yong, W.; Secco, R.A.; Tse, J.S.; Desgreniers, S.; et al. Heat, Pressure and Light-Induced Interconversion of Bisdithiazolyl Radicals and Dimers. J. Am. Chem. Soc. 2014, 136, 8050–8062. [Google Scholar] [CrossRef]
- Yu, X.; Mailman, A.; Lekin, K.; Assoud, A.; Robertson, C.M.; Noll, B.; Campana, C.F.; Howard, J.A.K.; Dube, P.A.; Oakley, R.T. Semiquinone-Bridged Bisdithiazolyl Radicals as Neutral Radical Conductors. J. Am. Chem. Soc. 2012, 134, 2264–2275. [Google Scholar] [CrossRef] [PubMed]
- Konstantinova, L.S.; Baranovsky, I.V.; Irtegova, I.G.; Bagryanskaya, I.Y.; Shundrin, L.A.; Zibarev, A.V.; Rakitin, O.A. Fused 1,2,3-Dithiazoles: Convenient Synthesis, Structural Characterization, and Electrochemical Properties. Molecules 2016, 21, 596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barclay, T.M.; Beer, L.; Cordes, A.W.; Haddon, R.C.; Itkis, M.I.; Oakley, R.T.; Preuss, K.E.; Reed, R.W. Trans-4,4′-Dichloro-1,1′,2,2′,3,3′-tetrathiadiazafulvalene (DC-TAF) and Its 1:1 Radical Cation Salts [DC-TAF][X]: Preparation and Solid-State Properties of BF4-, ClO4-, and FSO3-Derivatives. J. Am. Chem. Soc. 1999, 121, 6657–6663. [Google Scholar] [CrossRef]
- Appel, R.; Janssen, H.; Siray, M.; Knoch, F. Synthese und Reaktionen des 4,5-Dichlor-1,2,3-dithiazolium-chlorids. Eur. J. Inorg. Chem. 1985, 118, 1632–1643. [Google Scholar] [CrossRef]
- Kim, K. Synthesis and Reactions of 1,2,3-Dithiazoles. Sulfur Rep. 1998, 21, 147–207. [Google Scholar] [CrossRef]
- Koyioni, M.; Manoli, M.; Koutentis, P.A. The Reaction of DABCO with 4-Chloro-5H-1,2,3-dithiazoles: Synthesis and Chemistry of 4-[N-(2-Chloroethyl)piperazin-1-yl]-5H-1,2,3-dithiazoles. J. Org. Chem. 2015, 81, 615–631. [Google Scholar] [CrossRef]
- Kalogirou, A.S.; Michaelidou, S.S.; Koyioni, M.; Koutentis, P.A. Ring transformations of 2-hydroxy-(4-chloro-5H-1,2,3-dithiazol-5-ylideneamino)arenes. Tetrahedron 2015, 71, 7181–7190. [Google Scholar] [CrossRef]
- Kalogirou, A.S.; Michaelidou, S.S.; White, A.J.; Koutentis, P.A. Transformation of 2-(4-chloro-5H-1,2,3-dithiazol-5-ylideneamino)-6-ethoxy-4-phenylpyridine-3,5-dicarbonitrile into 4-aminopyrido[2,3-d]pyrimidines and 2-(pyrid-2-yl)guanidines. Tetrahedron 2015, 71, 1799–1807. [Google Scholar] [CrossRef]
- Koyioni, M.; Manoli, M.; Manolis, M.J.; Koutentis, P.A. Reinvestigating the Reaction of 1H-Pyrazol-5-amines with 4,5-Dichloro-1,2,3-dithiazolium Chloride: A Route to Pyrazolo[3,4-c]isothiazoles and Pyrazolo[3,4-d]thiazoles. J. Org. Chem. 2014, 79, 4025–4037. [Google Scholar] [CrossRef]
- Konstantinova, L.S.; Rakitin, O.A.; Rees, C.W.; Torroba, T.; White, A.J.P.; Williams, D.J. 1,2,4-Thiadiazole 4-oxides. J. Chem. Soc. Perkin Trans. 1999, 1, 2243–2248. [Google Scholar] [CrossRef]
- Clarke, D.; Emayan, K.; Rees, C.W. New synthesis of isothiazoles from primary enamines. J. Chem. Soc. Perkin Trans. 1 1998, 77–82. [Google Scholar] [CrossRef]
- Lee, H.; Kim, K. A facile synthesis of 2-cyano-4H-3,1-benzothiazines and 2-cyano-4H-3,1-benzoxazines. Heteroat. Chem. 1993, 4, 263–270. [Google Scholar] [CrossRef]
- Konstantinova, L.S.; Rakitin, O.A.; Rees, C.W.; Sivadasan, S.; Torroba, T. New route to 2-cyanobenzimidazoles. Tetrahedron 1998, 54, 9639–9650. [Google Scholar] [CrossRef]
- Lee, H.-S.; Chang, Y.-G.; Kim, K. A facile synthesis of 3-substituted 2-cyanoquinazolin-4(3H)-ones and 3-alkyl-2-cyanothieno[3,2-d]pyrimidin-4(3H)-onesvia1,2,3-dithiazoles. J. Heterocycl. Chem. 1998, 35, 659–668. [Google Scholar] [CrossRef]
- English, R.F.; Rakitin, O.A.; Rees, C.W.; Vlasova, O.G. Conversion of imino-1,2,3-dithiazoles into 2-cyanobenzothiazoles, cyanoimidoyl chlorides and diatomic sulfur. J. Chem. Soc. Perkin Trans. 1997, 1, 201–206. [Google Scholar] [CrossRef]
- Koutentis, P.A.; Rees, C.W. Reactions of tetracyanoethylene oxide with 1,2,3-dithiazoles. J. Chem. Soc. Perkin Trans. 1998, 1, 2505–2510. [Google Scholar] [CrossRef]
- Emayan, K.; Rees, C.W. The reaction of acetophenone oximes with disulfur dichloride; 4-aryl-5-arylimino-1,2,3-dithiazoles and pentathiepino[6,7-c]pyrrole. Bull. Soc. Chim. Belg. 1997, 106, 605–611. [Google Scholar] [CrossRef]
- Koutentis, P.A.; Koyioni, M.; Michaelidou, S.S. The conversion of [(4-chloro-5H-1,2,3-dithiazol-5-ylidene)amino]azines into azine fused thiazole-2-carbonitriles. Org. Biomol. Chem. 2013, 11, 621–629. [Google Scholar] [CrossRef]
- Koyioni, M.; Manoli, M.; Koutentis, P.A. Synthesis of Fused 1,2,4-Dithiazines and 1,2,3,5-Trithiazepines. J. Org. Chem. 2014, 79, 9717–9727. [Google Scholar] [CrossRef]
- Konstantinova, L.S.; Bol’shakov, O.I.; Obruchnikova, N.V.; Laborie, H.; Tanga, A.; Sopena, V.; Lanneluc, I.; Picot, L.; Sable, S.; Thiéry, V.; et al. One-pot synthesis of 5-phenylimino, 5-thieno or 5-oxo-1,2,3-dithiazoles and evaluation of their antimicrobial and antitumor activity. Bioorg. Med. Chem. Lett. 2009, 19, 136–141. [Google Scholar] [CrossRef]
- Matta, C.F.; Boyd, R.J. The Quantum Theory of Atoms in Molecules: From Solid State to DNA and Drug Design; Wiley-VCH Verlag GmbH & Co. KgaA: Weinheim, Germany, 2007. [Google Scholar] [CrossRef]
- Espinosa, E.; Molins, E.; LeComte, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Wang, X.; Xu, D.; Miao, C.; Zhang, Q.; Sun, W. N-Bromosuccinimide as an oxidant for the transition-metal-free synthesis of 2-aminobenzoxazoles from benzoxazoles and secondary amines. Org. Biomol. Chem. 2014, 12, 3108–3113. [Google Scholar] [CrossRef] [PubMed]
- Wertz, S.; Kodama, S.; Studer, A. Amination of Benzoxazoles and 1,3,4-Oxadiazoles Using 2,2,6,6-Tetramethylpiperidine-N-oxoammonium Tetrafluoroborate as an Organic Oxidant. Angew. Chem. Int. Ed. 2011, 50, 11511–11515. [Google Scholar] [CrossRef] [PubMed]
- Wagh, Y.S.; Tiwari, N.J.; Bhanage, B.M. Metal-free synthesis of 2-aminobenzoxazoles using hypervalent iodine reagent. Tetrahedron Lett. 2013, 54, 1290–1293. [Google Scholar] [CrossRef]
- Cook, S.; Jefferies, L.; Weber, S. Iron-Catalyzed C–N Bond Formation via the Beckmann Rearrangement. Synlett 2014, 26, 331–334. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2007, 64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Perdew, J.P.; Ernzerhof, M.; Burke, K. Rationale for mixing exact exchange with density functional approximations. J. Chem. Phys. 1996, 105, 9982–9985. [Google Scholar] [CrossRef]
- Medvedev, M.G.; Bushmarinov, I.S.; Sun, J.; Perdew, J.P.; Lyssenko, K.A. Density functional theory is straying from the path toward the exact functional. Science 2017, 355, 49–52. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Boominathan, S.S.K.; Hu, W.-P.; Senadi, G.C.; Vandavasi, J.K.; Wang, J.-J. A one-pot hypoiodite catalysed oxidative cycloetherification approach to benzoxazoles. Chem. Commun. 2014, 50, 6726–6728. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; He, Y.; Zhang, X.; Guo, S.; Wang, Y. Synthesis of heteroaryl ketones via tandem reaction of 1,1-dibromoethenes. Tetrahedron 2011, 67, 6369–6374. [Google Scholar] [CrossRef]
- Gravenfors, Y.; Viklund, J.; Blid, J.; Ginman, T.; Karlström, S.; Kihlström, J.; Kolmodin, K.; Lindström, J.; Von Berg, S.; Von Kieseritzky, F.; et al. New Aminoimidazoles as β-Secretase (BACE-1) Inhibitors Showing Amyloid-β (Aβ) Lowering in Brain. J. Med. Chem. 2012, 55, 9297–9311. [Google Scholar] [CrossRef]
- Blid, J.; Ginman, T.; Gravenfors, Y.; Karlström, S.; Kolmodin, K.; Lindstroem, J.; Plobeck, N.; Rahm, F.; Swahn, B.-M.; Viklund, J.; et al. Preparation of phenylimidazolamine derivatives for use in treatment of neurodegenerative diseases. Patent: 2011, WO2011/2407. Chem. Abstr. 2011, 154, 109611. [Google Scholar]
Sample Availability: Samples of the compounds are available from the authors. |












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 9f | 10b | |
|---|---|---|
| CCDC number | 1993040 | 1993041 |
| Empirical formula | C28H18F2N4O2 | C12H8N2OS |
| Formula weight | 480.46 | 228.26 |
| T, K | 120 | 120 |
| Crystal system | Monoclinic | Monoclinic |
| Space group | P21/n | P21/n |
| Z (Z’) | 4 (1) | 4(1) |
| a, Å | 12.3650(12) | 6.1355(5) |
| b, Å | 12.9901(13) | 7.5454(6) |
| c, Å | 14.3314(14) | 22.0813(19) |
| α, ° | 90 | 90 |
| β, ° | 107.871(2) | 94.779(2) |
| γ, ° | 90 | 90 |
| V, Å3 | 2190.9(4) | 1018.70(15) |
| Dcalc,gcm−3 | 1.457 | 1.488 |
| μ, cm−1 | 1.06 | 2.93 |
| F(000) | 992 | 472 |
| 2θmax, ° | 58 | 58 |
| Reflections collected | 25153 | 11900 |
| Reflections unique (Rint) | 5821 (0.0410) | 2710 (0.0357) |
| Reflections with I > 2σ(I) | 4558 | 2443 |
| Variables/restraints | 333 | 149 |
| R1 | 0.0425 | 0.0357 |
| wR2 | 0.1092 | 0.0962 |
| GOF | 1.024 | 1.036 |
| Largest difference in peak/hole (e/Å3) | 0.328/−0.242 | 0.415/−0.342 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baranovsky, I.V.; Konstantinova, L.S.; Tolmachev, M.A.; Popov, V.V.; A. Lyssenko, K.; Rakitin, O.A. Synthesis of 2-((2-(Benzo[d]oxazol-2-yl)-2H-imidazol-4-yl)amino)-phenols from 2-((5H-1,2,3-Dithiazol-5-ylidene)amino)phenols through Unprecedented Formation of Imidazole Ring from Two Methanimino Groups. Molecules 2020, 25, 3768. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25173768
Baranovsky IV, Konstantinova LS, Tolmachev MA, Popov VV, A. Lyssenko K, Rakitin OA. Synthesis of 2-((2-(Benzo[d]oxazol-2-yl)-2H-imidazol-4-yl)amino)-phenols from 2-((5H-1,2,3-Dithiazol-5-ylidene)amino)phenols through Unprecedented Formation of Imidazole Ring from Two Methanimino Groups. Molecules. 2020; 25(17):3768. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25173768
Chicago/Turabian StyleBaranovsky, Ilia V., Lidia S. Konstantinova, Mikhail A. Tolmachev, Vadim V. Popov, Konstantin A. Lyssenko, and Oleg A. Rakitin. 2020. "Synthesis of 2-((2-(Benzo[d]oxazol-2-yl)-2H-imidazol-4-yl)amino)-phenols from 2-((5H-1,2,3-Dithiazol-5-ylidene)amino)phenols through Unprecedented Formation of Imidazole Ring from Two Methanimino Groups" Molecules 25, no. 17: 3768. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25173768