
A Novel Cytotoxic Conjugate Derived from the Natural Product Podophyllotoxin as a Direct-Target Protein Dual Inhibitor

 ,
,  , , , , and
, , , , and
Abstract
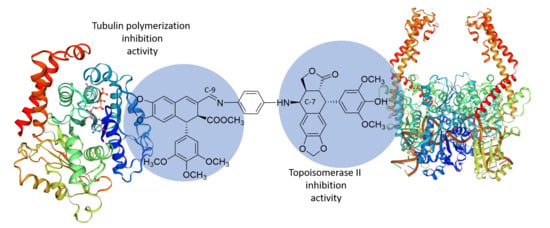
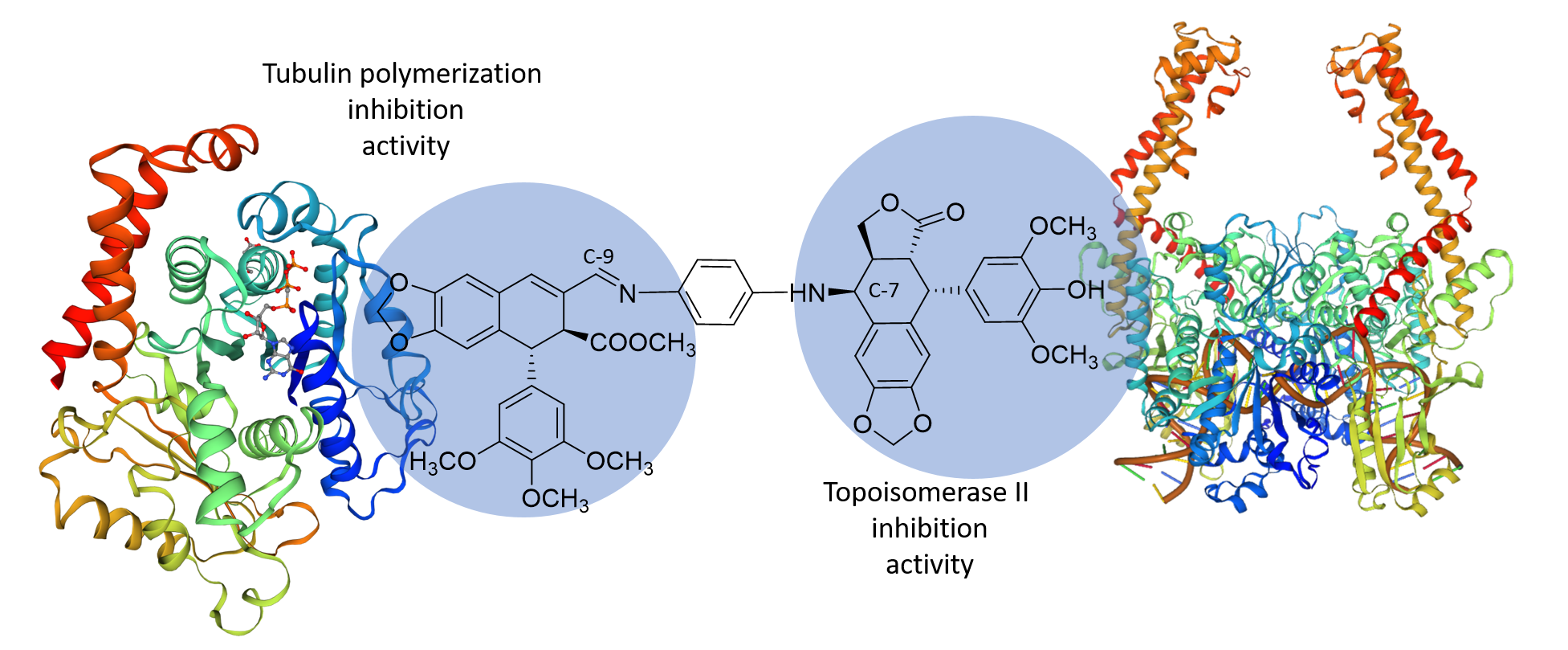
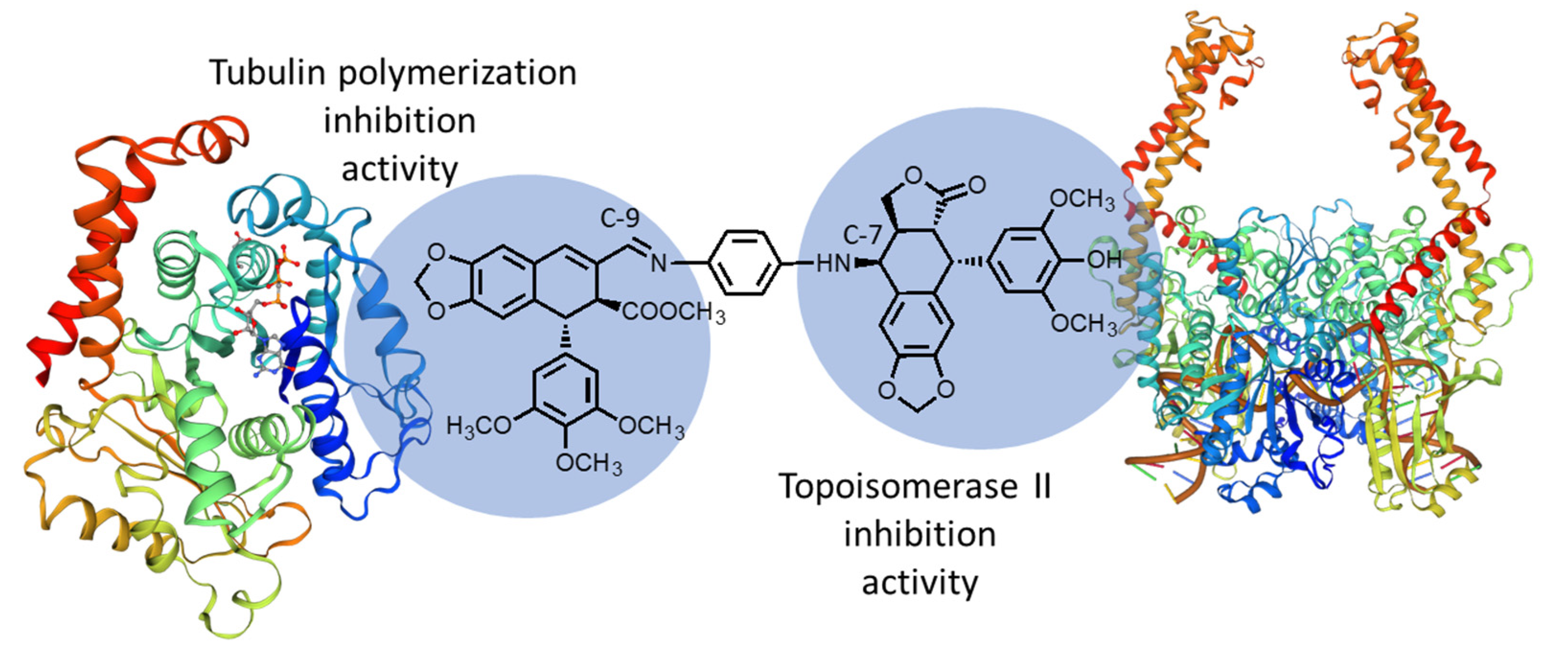
:
1. Introduction
2. Results
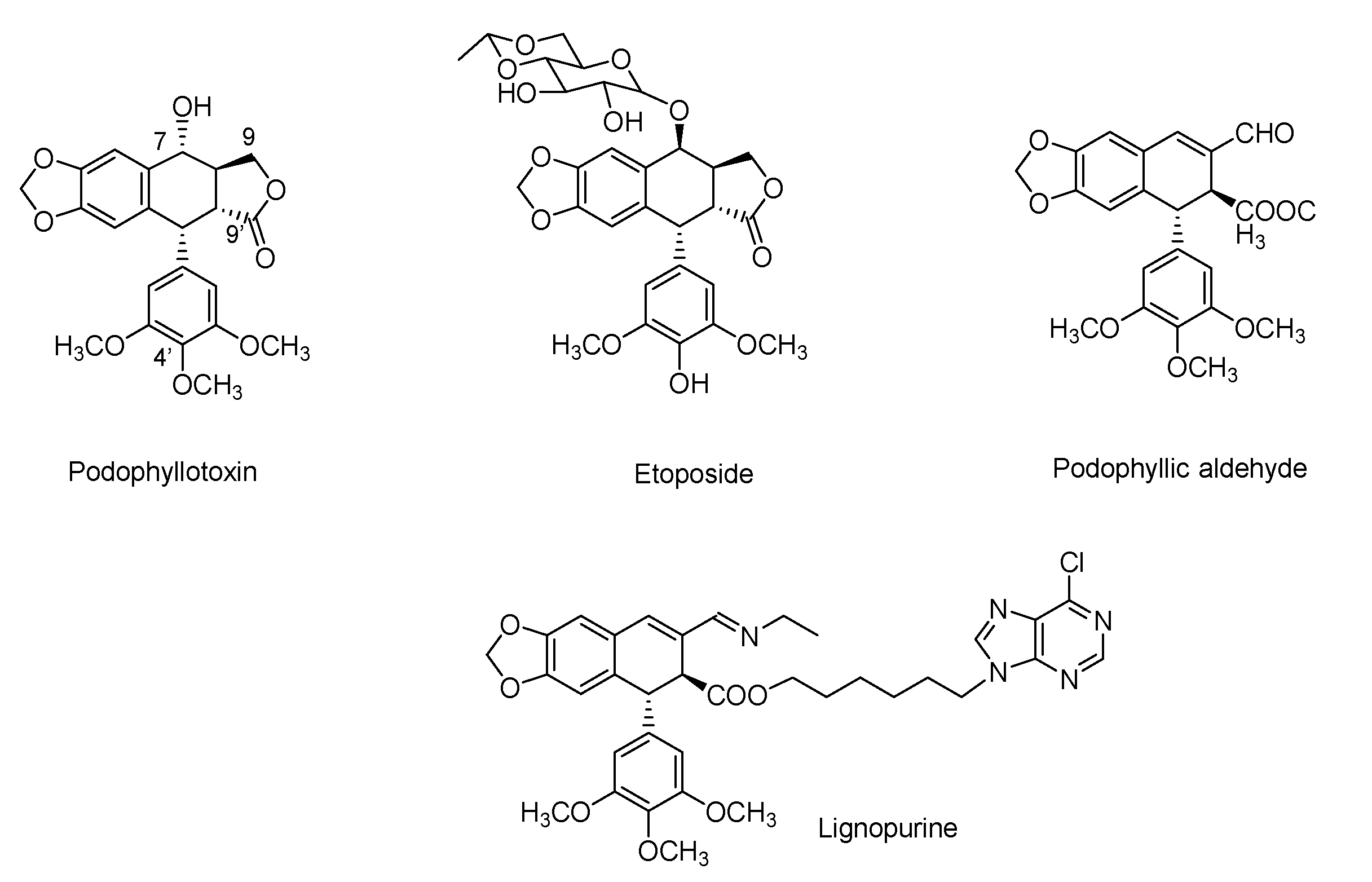
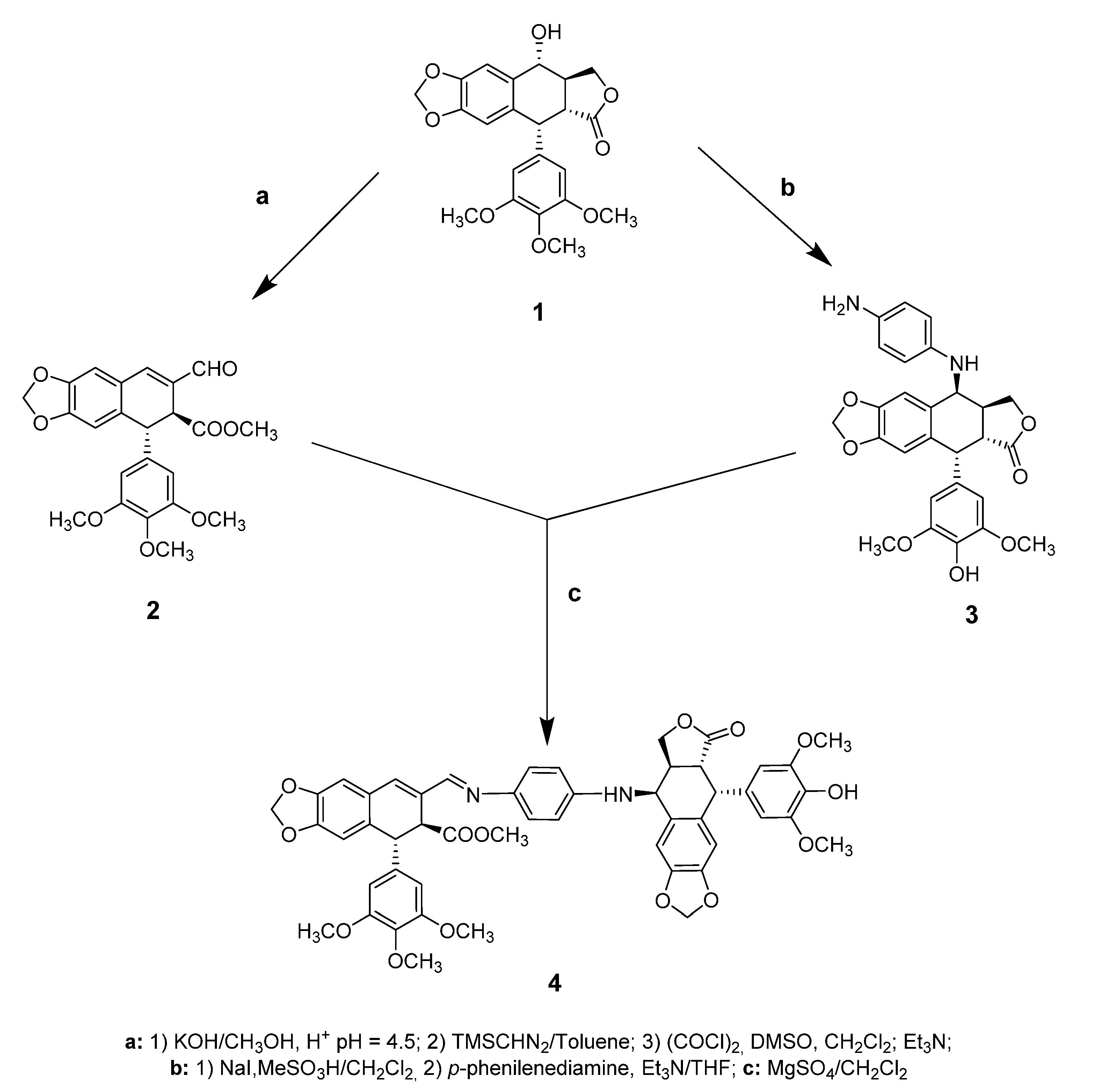
2.1. Chemistry
2.2. Biological Assays
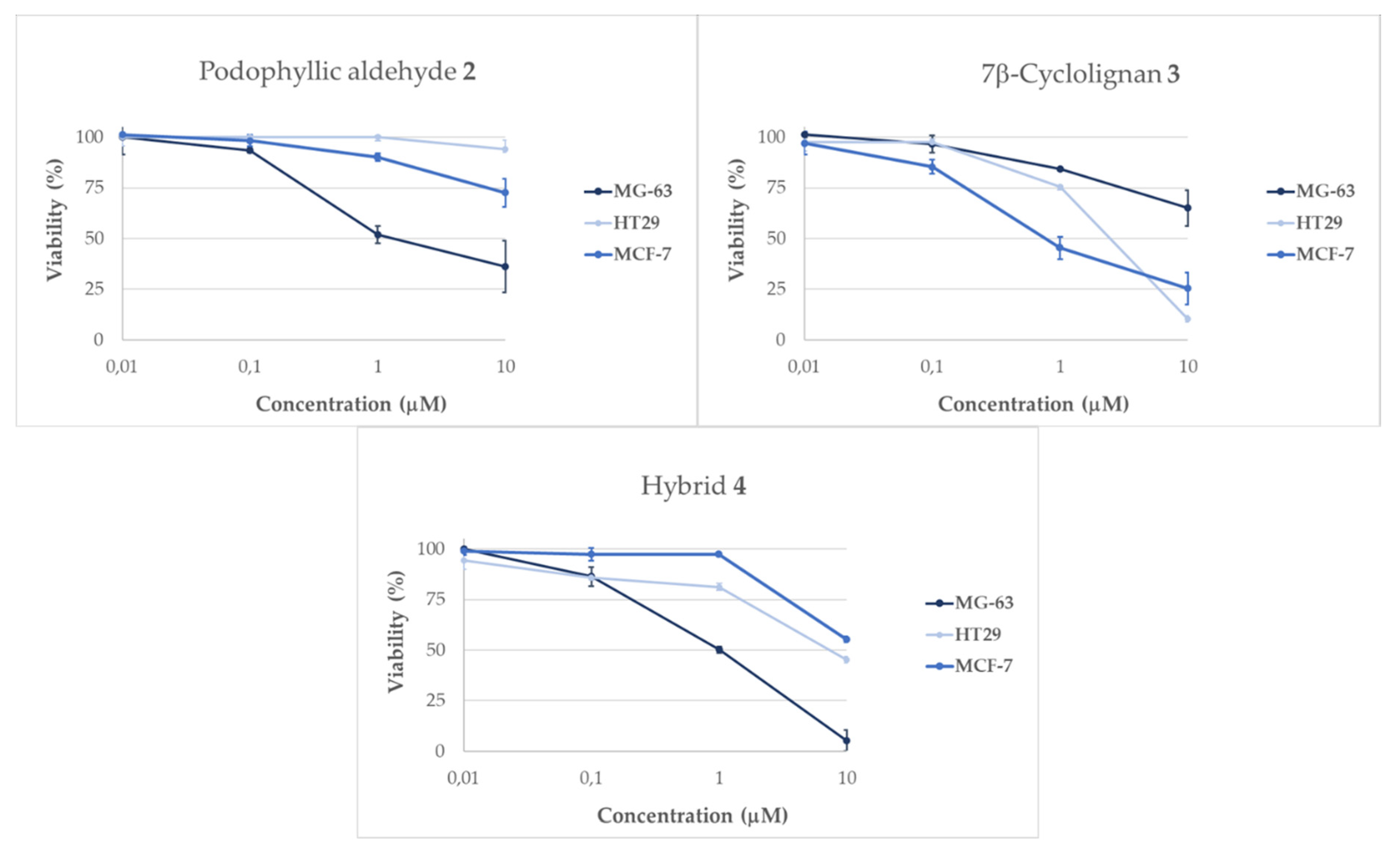
2.2.1. Evaluation of Cellular Viability
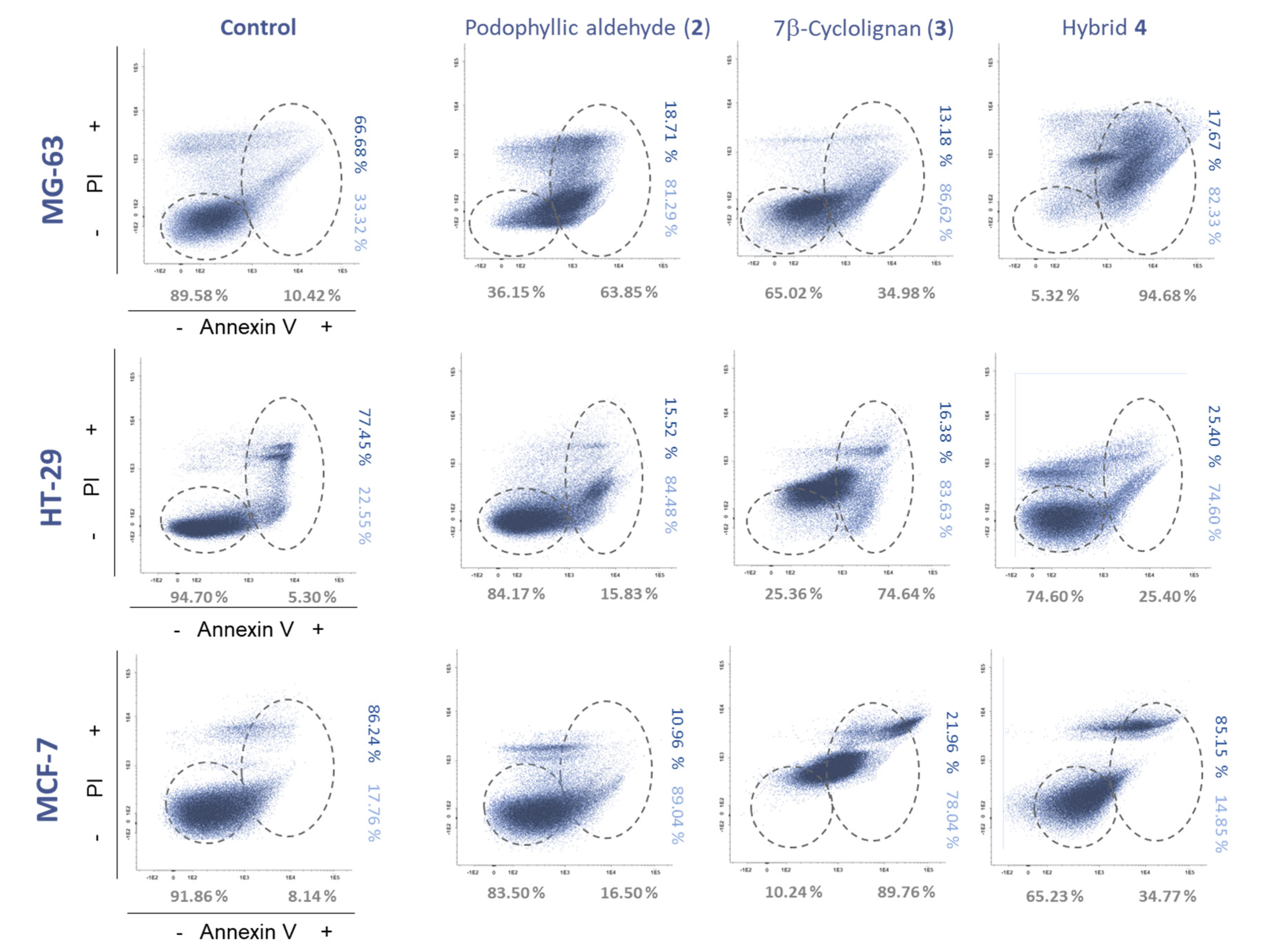
2.2.2. Apoptosis Evaluation
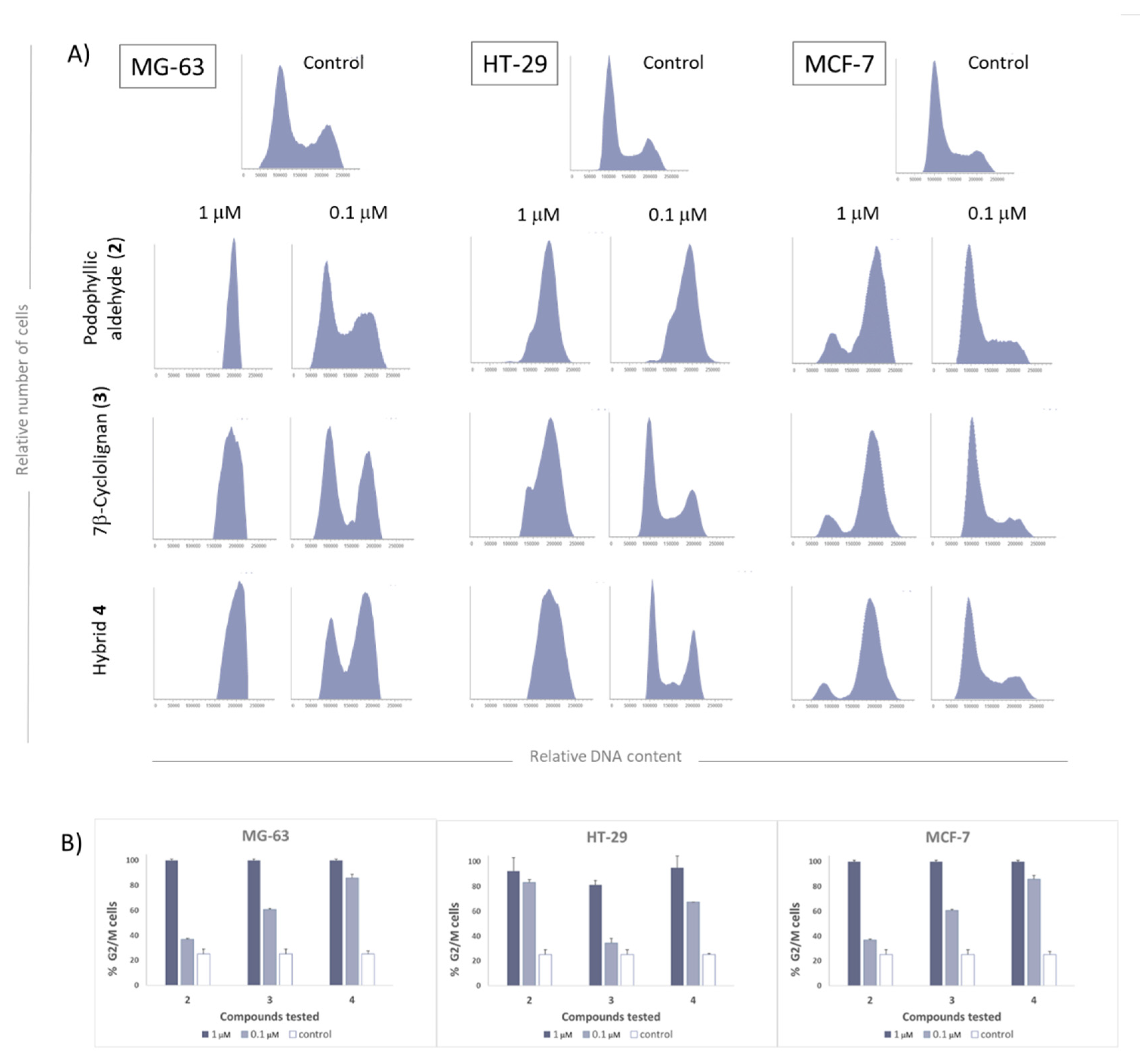
2.2.3. Cell Cycle Evaluation
2.3. Molecular Docking Studies
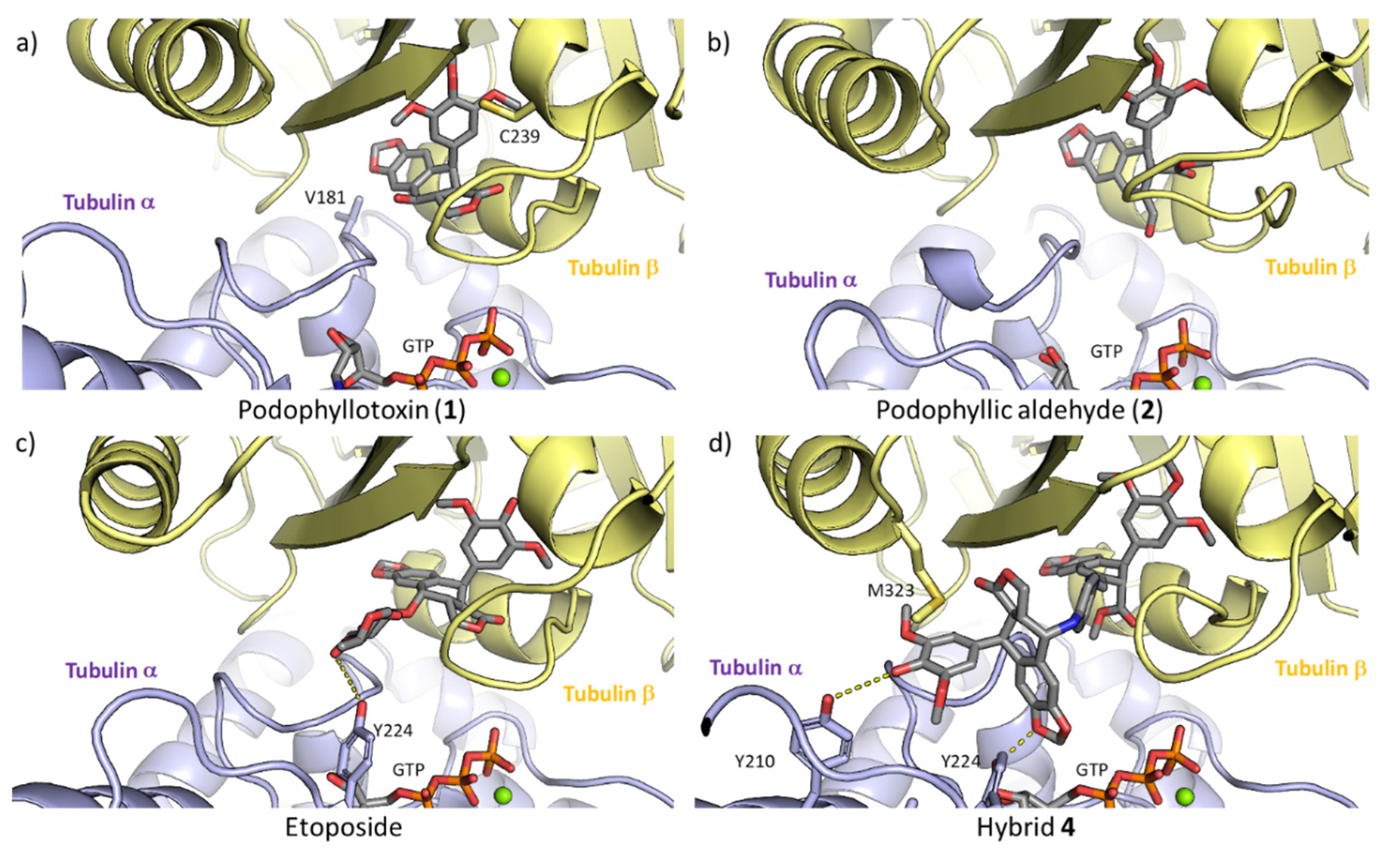
2.3.1 Tubulin Interaction
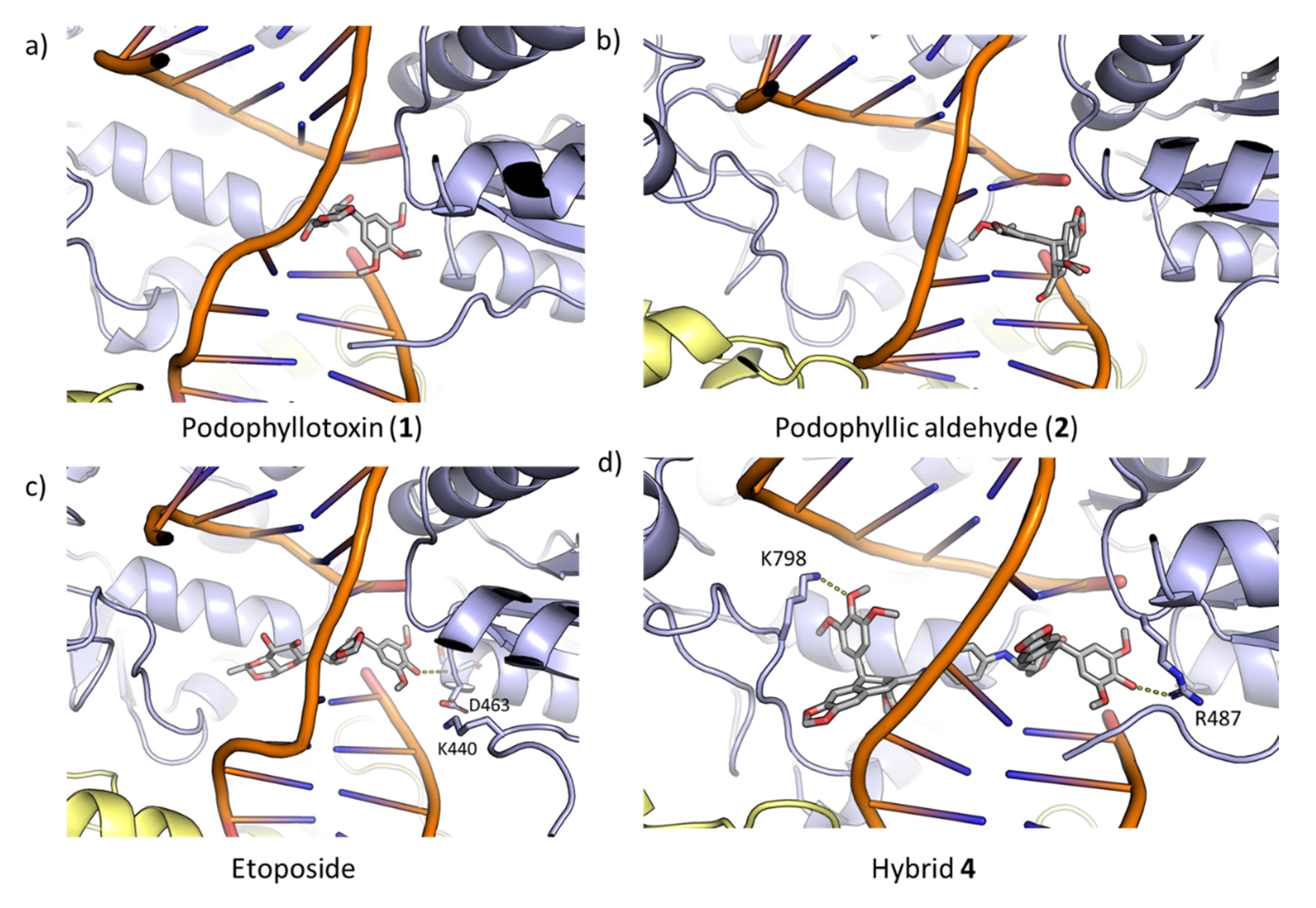
2.3.2 DNA-Topoisomerase II Interaction
3. Discussions
4. Materials and Methods
4.1. Chemistry
4.2. Biological Methods
4.2.1. Cell Lines and Culture Conditions
4.2.2. Flow Cytometry Assays
4.2.3. Cell Cycle Analysis
4.3. Computational Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Yuan, H.; Ma, Q.; Ye, L.; Piao, G. The traditional medicine and modern medicine from natural products. Molecules 2016, 21, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siu, D. Natural products and their role in cancer therapy. Med. Oncol. 2011, 28, 888–900. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, L.; Luo, Y.; Tian, M.; Zhang, S.Y.; Lu, R.; Wang, J.H.; Kasimu, R.; Li, X. Plant natural products: From traditional compounds to new emerging drugs in cancer therapy. Cell Prolif. 2014, 47, 506–515. [Google Scholar] [CrossRef]
- Gordaliza, M.; García, P.A.; Miguel del Corral, J.M.; Castro, M.A.; Gómez-Zurita, M.A. Podophyllotoxin: Distribution, sources, applications and new cytotoxic derivatives. Toxicon 2004, 44, 441–459. [Google Scholar] [CrossRef] [PubMed]
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Montecucco, A.; Zanetta, F.; Biamonti, G. Molecular mechanisms of etoposide. EXCLI J. 2015, 14, 95–108. [Google Scholar] [CrossRef]
- Castro, M.A.; García, P.A.; Hernández, A.P.; Díez, D. An overview on heterocyclic podophyllotoxin derivatives. Targets Heterocycl. Syst. 2015, 19, 28–61. [Google Scholar]
- Yu, X.; Che, Z.; Xu, H. Recent Advances in the chemistry and biology of podophyllotoxins. Chem. Eur. J. 2017, 23, 4467–4526. [Google Scholar] [CrossRef]
- Castro, M.A.; Miguel del Corral, J.M.; Gordaliza, M.; García, P.A.; Gómez-Zurita, M.A.; García-Grávalos, M.D.; de la Iglesia-Vicente, J.; Gajate, C.; An, F.; Mollinedo, F.; et al. Synthesis and biological evaluation of new selective cytotoxic cyclolignans derived from podophyllotoxin. J. Med. Chem. 2004, 47, 1214–1222. [Google Scholar] [CrossRef]
- Castro, M.A.; Miguel del Corral, J.M.; García, P.A.; Rojo, M.V.; de la Iglesia-Vicente, J.; Mollinedo, F.; Cuevas, C.; San Feliciano, A. Synthesis and biological evaluation of new podophyllic aldehyde derivatives with cytotoxic and apoptosis-inducing activities. J. Med. Chem. 2010, 53, 983–993. [Google Scholar] [CrossRef] [PubMed]
- Castro, M.A.; Miguel del Corral, J.M.; García, P.A.; Rojo, M.V.; Bento, A.C.; Mollinedo, F.; Francesch, A.M.; San Feliciano, A. Lignopurines: A new family of hybrids between cyclolignans and purines. Synthesis and biological evaluation. Eur. J. Med. Chem. 2012, 58, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Hernández, A.P.; Díez, P.; García, P.A.; Miguel del Corral, J.M.; Pérez-Andrés, M.; Díez, D.; San Feliciano, A.; Fuentes, M.; Castro, M.A. New hybrids derived from podophyllic aldehyde and diterpenylhydroquinones with selectivity toward osteosarcoma cells. ACS Med. Chem. Lett. 2018, 9, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Ojima, I. Modern molecular approaches to drug design and discovery. Acc. Chem. Res. 2008, 41, 2–3. [Google Scholar] [CrossRef] [Green Version]
- Fortin, S.; Berube, G. Advances in the development of hybrid anticancer drugs. Expert Opin. Drug Discover. 2013, 8, 1029–1047. [Google Scholar] [CrossRef]
- Tietze, L.F.; Bell, H.P.; Chandrasekhar, S. Natural product hybrids as new leads for drug discovery. Angew. Chem. 2003, 42, 3996–4028. [Google Scholar] [CrossRef]
- Bansal, Y.; Silakari, O. Multifunctional compounds: Smart molecules for multifactorial diseases. Eur. J. Med. Chem. 2014, 76, 31–42. [Google Scholar] [CrossRef]
- Yang, R.; Ding, H.; Wu, Y.; Xiao, Q. Synthesis podophyllotoxin derivatives via “click chemistry”. Adv. Mat. Res. 2014, 881–883, 400–404. [Google Scholar] [CrossRef]
- Banday, A.H.; Kulkarni, V.V.; Hruby, V.J. Design, synthesis, and biological and docking studies of novel epipodophyllotoxin-chalcone hybrids as potential anticancer agents. MedChemComm 2015, 6, 94–104. [Google Scholar] [CrossRef]
- Ye, D.; Shi, Q.; Leung, C.-H.; Kim, S.-W.; Park, S.-Y.; Gullen, E.A.; Jiang, Z.L.; Zhu, H.; Morris-Natschke, S.L.; Cheng, Y.-C.; et al. Antitumor agents 294. Novel E-ring-modified camptothecin–4β-anilino-4′-O-demethyl-epipodophyllotoxin conjugates as DNA topoisomerase I inhibitors and cytotoxic agents. Bioorg. Med. Chem. 2012, 20, 4489–4494. [Google Scholar] [CrossRef] [Green Version]
- Passarella, D.; Giardini, A.; Peretto, B.; Fontana, G.; Sacchetti, A.; Silvani, A.; Ronchi, C.; Cappelletti, G.; Cartelli, D.; Borlak, J.; et al. Inhibitors of tubulin polymerization: Synthesis and biological evaluation of hybrids of vindoline, anhydrovinblastine and vinorelbine with thiocolchicine, podophyllotoxin and baccatin III. Bioorg. Med. Chem. 2008, 16, 6269–6285. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, J.; Liu, L.; Zheng, C.; Wang, Y.; Chen, Y.; Wei, G. Podophyllotoxin-pterostilbene fused conjugates as potential multifunctional antineoplastic agents against human uveal melanoma cells. RSC Adv. 2017, 7, 10601–10608. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Wang, J.; Liu, L.; Zheng, C.; Wang, Y. Synthesis and antiproliferative activity of novel all-trans-retinoic acid-podophyllotoxin conjugate towards human gastric cancer cells. Molecules 2017, 22, 628. [Google Scholar] [CrossRef] [Green Version]
- Kamal, A.; Nayak, V.L.; Bagul, C.; Vishnuvardhan, M.V.; Mallareddy, A. Investigation of the mechanism and apoptotic pathway induced by 4β-cinnamido linked podophyllotoxins against human lung cancer cells A549. Apoptosis 2015, 20, 1518–1529. [Google Scholar] [CrossRef]
- Kamal, A.; Tamboli, J.R.; Vishnuvardhan, M.V.; Adil, S.F.; Nayak, V.L.; Ramakrishna, S. Synthesis and anticancer activity of heteroaromatic linked 4beta-amido podophyllotoxins as apoptotic inducing agents. Bioorg. Med. Chem. Lett. 2013, 23, 273–280. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, L.; Zheng, C.; Wang, Y.; Nie, X.; Shi, D.; Chen, Y.; Wei, G.; Wang, J. Synthesis and biological evaluation of novel podophyllotoxin-NSAIDs conjugates as multifunctional anti-MDR agents against resistant human hepatocellular carcinoma Bel-7402/5-FU cells. Eur. J. Med. Chem. 2017, 131, 81–91. [Google Scholar] [CrossRef]
- Guan, X.W.; Xu, X.H.; Feng, S.L.; Tang, Z.B.; Chen, S.W.; Hui, L. Synthesis of hybrid 4-deoxypodophyllotoxin-5-fluorouracil compounds that inhibit cellular migration and induce cell cycle arrest. Bioorg. Med. Chem. Lett. 2016, 26, 1561–1566. [Google Scholar] [CrossRef]
- León, I.E.; Díez, P.; Baran, E.J.; Etcheverry, S.B.; Fuentes, M. Decoding the anticancer activity of VO-clioquinol compound: The mechanism of action and cell death pathways in human osteosarcoma cells. Metallomics 2017, 9, 891–901. [Google Scholar] [CrossRef]
- León, I.E.; Díez, P.; Etcheverry, S.B.; Fuentes, M. Deciphering the effect of an oxovanadium(IV) complex with the flavonoid chrysin (VOChrys) on intracellular cell signalling pathways in an osteosarcoma cell line. Metallomics 2016, 8, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Barba-Vicente, V.; Parra, M.J.A.; Boyero-Benito, J.F.; Auria-Soro, C.; Juanes-Velasco, P.; Landeira-Vinuela, A.; Furones-Cuadrado, A.; Hernández, A.P.; Manzano-Roman, R.; Fuentes, M. Detection of human p53 in-vitro expressed in a transcription-translation cell-free system by a novel conjugate based on cadmium sulphide nanoparticles. Nanomaterials 2020, 10, 984. [Google Scholar] [CrossRef]
- Sánchez-Paradinas, S.; Pérez-Andrés, M.; Almendral-Parra, M.J.; Rodríguez-Fernández, E.; Milian, A.; Palacio, F.; Orfao, A.; Criado, J.J.; Fuentes, M. Enhanced cytotoxic activity of bile acid cisplatin derivatives by conjugation with gold nanoparticles. J. Inorg. Biochem. 2014, 131, 8–11. [Google Scholar] [CrossRef] [PubMed]
- Díez, P.; González-Muñoz, M.; González-González, M.; Degano, R.M.; Jara-Acevedo, R.; Sánchez-Paradinas, S.; Pinol, R.; Murillo, J.L.; Lou, G.; Palacio, F.; et al. Functional insights into the cellular response triggered by a bile-acid platinum compound conjugated to biocompatible ferric nanoparticles using quantitative proteomic approaches. Nanoscale 2017, 9, 9960–9972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordaliza, M.; Castro, M.A.; Miguel del Corral, J.M.; López-Vazquez, M.L.; García, P.A.; García-Grávalos, M.D.; San Feliciano, A. Synthesis and antineoplastic activity of cyclolignan aldehydes. Eur. J. Med. Chem. 2000, 35, 691–698. [Google Scholar] [CrossRef]
- Castro, M.A.; Miguel del Corral, J.M.; Gordaliza, M.; García, P.A.; Gómez-Zurita, M.A.; San Feliciano, A. Synthesis and cytotoxic evaluation of C-9 oxidized podophyllotoxin derivatives. Bioorg. Med. Chem. 2007, 15, 1670–1678. [Google Scholar] [CrossRef]
- Wang, L.; Yang, F.; Yang, X.; Guan, X.; Hu, C.; Liu, T.; He, Q.; Yang, B.; Hu, Y. Synthesis and biological evaluation of new 4β-anilino-4′-O-demethyl-4-desoxypodophyllotoxin derivatives as potential antitumor agents. Eur. J. Med. Chem. 2011, 46, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravelli, R.B.G.; Gigant, B.; Curmi, P.A.; Jourdain, I.; Lachkar, S.; Sobel, A.; Knossow, M. Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain. Nature 2004, 428, 198–202. [Google Scholar] [CrossRef]
- Wang, Y.-R.; Chen, S.-F.; Wu, C.-C.; Liao, Y.-W.; Lin, T.-S.; Liu, K.-T.; Chen, Y.-S.; Li, T.-K.; Chien, T.-C.; Chan, N.-L. Producing irreversible topoisomerase ii-mediated DNA breaks by site-specific Pt(II)-methionine coordination chemistry. Nucleic Acids Res. 2017, 45, 10861–10871. [Google Scholar] [CrossRef] [Green Version]
- Yadav, A.A.; Chee, G.-L.; Wu, X.; Patel, D.; Yalowich, J.C.; Hasinoff, B.B. Structure-based design, synthesis and biological testing of piperazine-linked bis-epipodophyllotoxin etoposide analogs. Bioorg. Med. Chem. 2015, 23, 3542–3551. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.-C.; Li, T.-K.; Farh, L.; Lin, L.-Y.; Lin, T.-S.; Yu, Y.-J.; Yen, T.-J.; Chiang, C.-W.; Chan, N.-L. Structural basis of type II topoisomerase inhibition by the anticancer drug etoposide. Science 2011, 333, 459. [Google Scholar] [CrossRef] [Green Version]
- Bielack, S.S.; Hecker-Nolting, S.; Blattmann, C.; Kager, L. Advances in the management of osteosarcoma. F1000Research 2016, 5, 2767. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bienert, S.; Waterhouse, A.; de Beer, T.A.; Tauriello, G.; Studer, G.; Bordoli, L.; Schwede, T. The SWISS-MODEL Repository—New features and functionality. Nucleic Acids Res. 2016, 45, D313–D319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, B.R.; Brooks, C.L., 3rd; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [Green Version]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.M.; Mittal, J.; Feig, M.; MacKerell, A.D. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone ϕ, ψ and side-chain χ1 and χ2 dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [Green Version]
- Hart, K.; Foloppe, N.; Baker, C.M.; Denning, E.J.; Nilsson, L.; MacKerell, A.D. Optimization of the CHARMM additive force field for DNA: Improved treatment of the BI/BII conformational equilibrium. J. Chem. Theory Comput. 2012, 8, 348–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [Green Version]
- CGenFF Interface at Paramchem.org. Available online: https://cgenff.umaryland.edu (accessed on 1 January 2020).
- Pérez-Pertejo, Y.; Escudero-Martínez, J.M.; Reguera, R.M.; Balaña-Fouce, R.; García, P.A.; Jambrina, P.G.; San Feliciano, A.; Castro, M.-Á. Antileishmanial activity of terpenylquinones on Leishmania infantum and their effects on Leishmania topoisomerase IB. Int. J. Parasitol. Drugs Drug Resist. 2019, 11, 70–79. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 2 and 4 are available from the authors P.A.G and M.A.C. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | ΔG | Protomers Interaction |
|---|---|---|
| Podophyllotoxin (1) | −9.5 | −496 ± 70 |
| Podophyllic aldehyde (2) | −8.5 | −449 ± 80 |
| Etoposide | −10.3 | −440 ± 80 |
| Hybrid 4 | −12.5 | −353 ± 77 |
| Compound | ΔG | Interaction Energy Ligand-Protein | Interaction Energy Ligand-DNA |
|---|---|---|---|
| Podophyllotoxin (1) | −9.7 | −25 ± 11 | −41 ± 9 |
| Podophyllic aldehyde (2) | −9.4 | −27 ± 13 | −50 ± 9 |
| Etoposide | −12.4 | −37 ± 10 | −54 ± 10 |
| Hybrid 4 | −12.8 | −62 ± 18 | −67 ± 12 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernández, Á.-P.; Díez, P.; García, P.A.; Pérez-Andrés, M.; Ortega, P.; Jambrina, P.G.; Díez, D.; Castro, M.Á.; Fuentes, M. A Novel Cytotoxic Conjugate Derived from the Natural Product Podophyllotoxin as a Direct-Target Protein Dual Inhibitor. Molecules 2020, 25, 4258. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25184258
Hernández Á-P, Díez P, García PA, Pérez-Andrés M, Ortega P, Jambrina PG, Díez D, Castro MÁ, Fuentes M. A Novel Cytotoxic Conjugate Derived from the Natural Product Podophyllotoxin as a Direct-Target Protein Dual Inhibitor. Molecules. 2020; 25(18):4258. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25184258
Chicago/Turabian StyleHernández, Ángela-Patricia, Paula Díez, Pablo A. García, Martín Pérez-Andrés, Pablo Ortega, Pablo G. Jambrina, David Díez, María Ángeles Castro, and Manuel Fuentes. 2020. "A Novel Cytotoxic Conjugate Derived from the Natural Product Podophyllotoxin as a Direct-Target Protein Dual Inhibitor" Molecules 25, no. 18: 4258. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25184258