
Indole Derivative Interacts with Estrogen Receptor Beta and Inhibits Human Ovarian Cancer Cell Growth

 , ,
, ,  , and
, and
Abstract
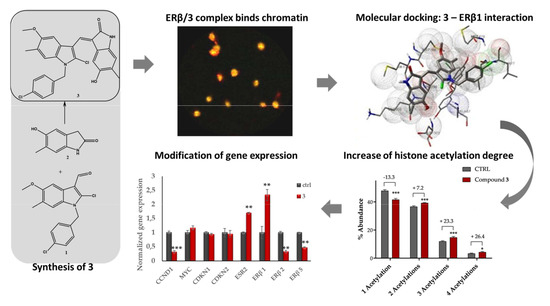
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Biological Activity
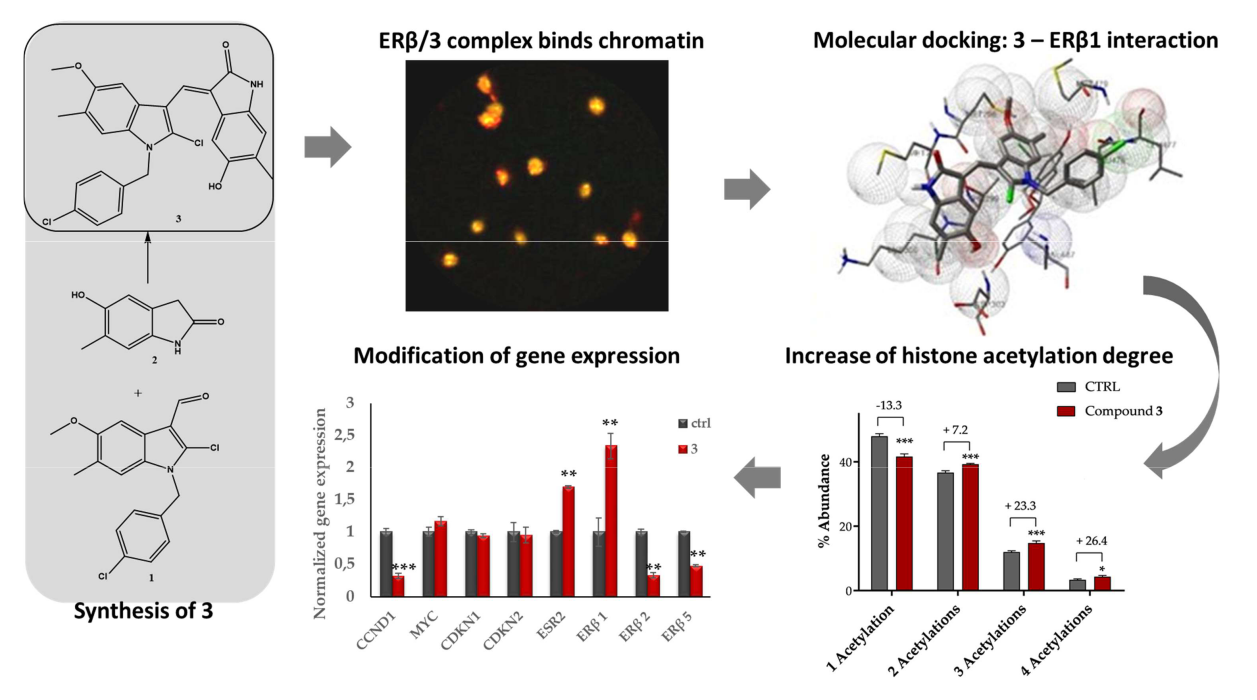
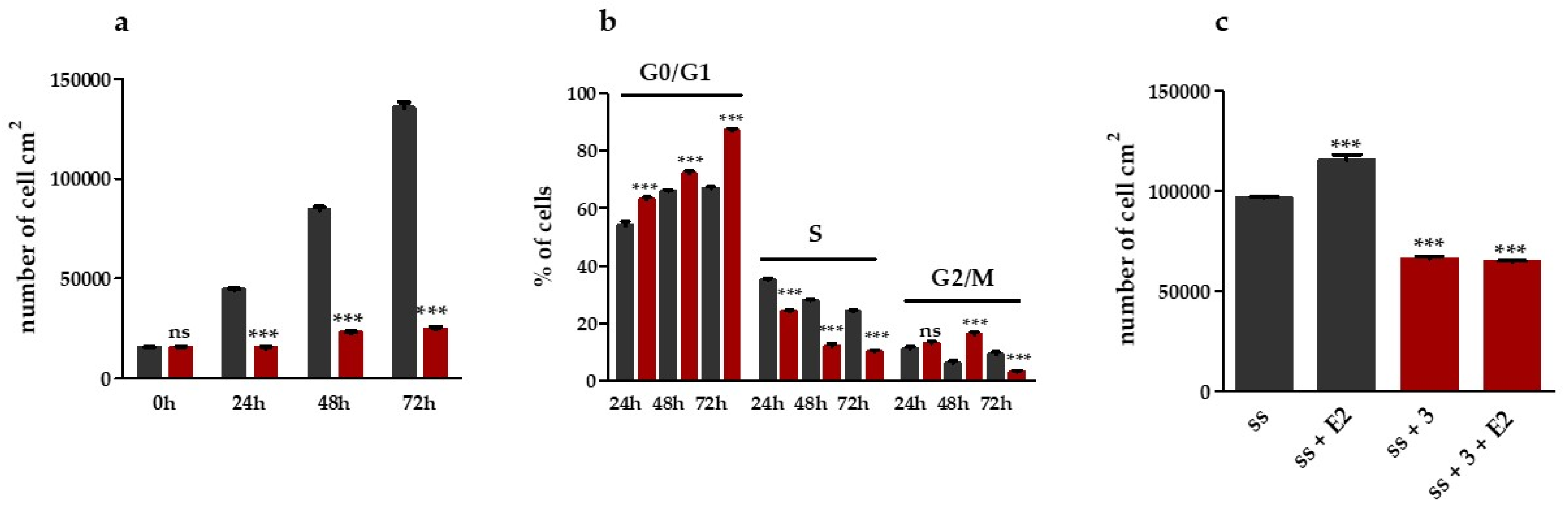
2.1.1. Compound 3 Inhibits IGROV1 Ovarian Cancer Cells Proliferation and Progression in the Cell Cycle
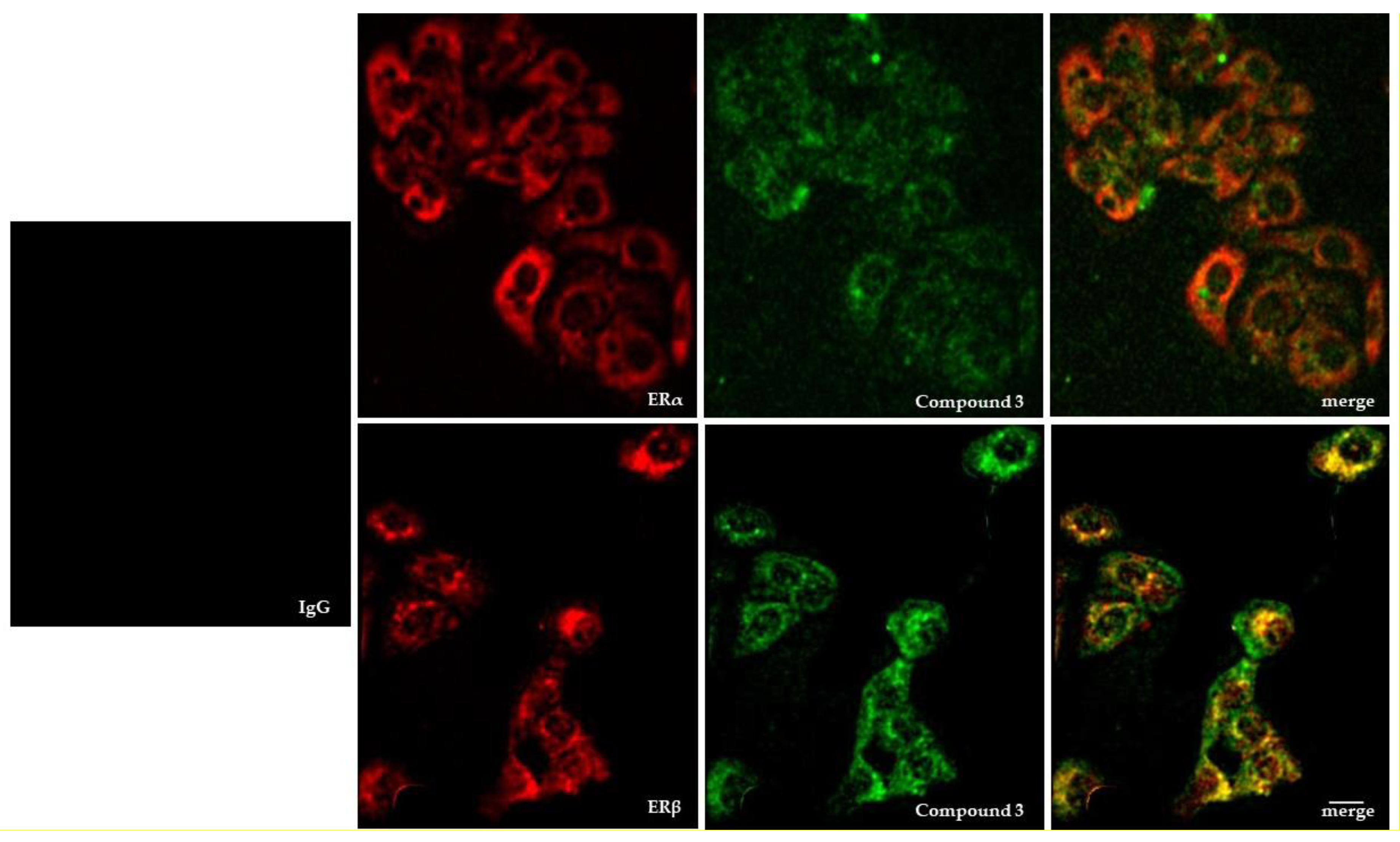
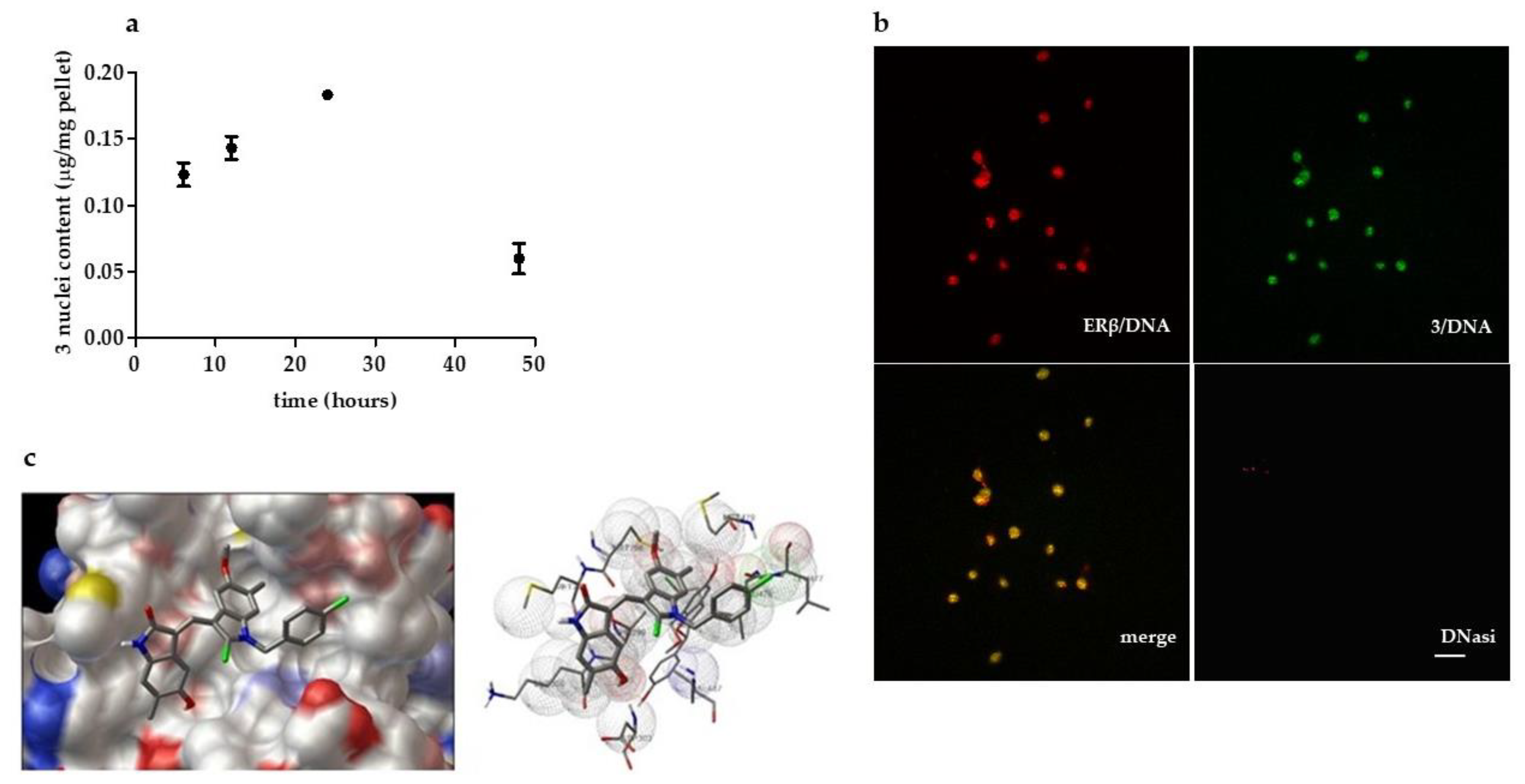
2.1.2. Compound 3: Nuclear Localization, ERβ on Chromatin Colocalization, and Molecular Docking
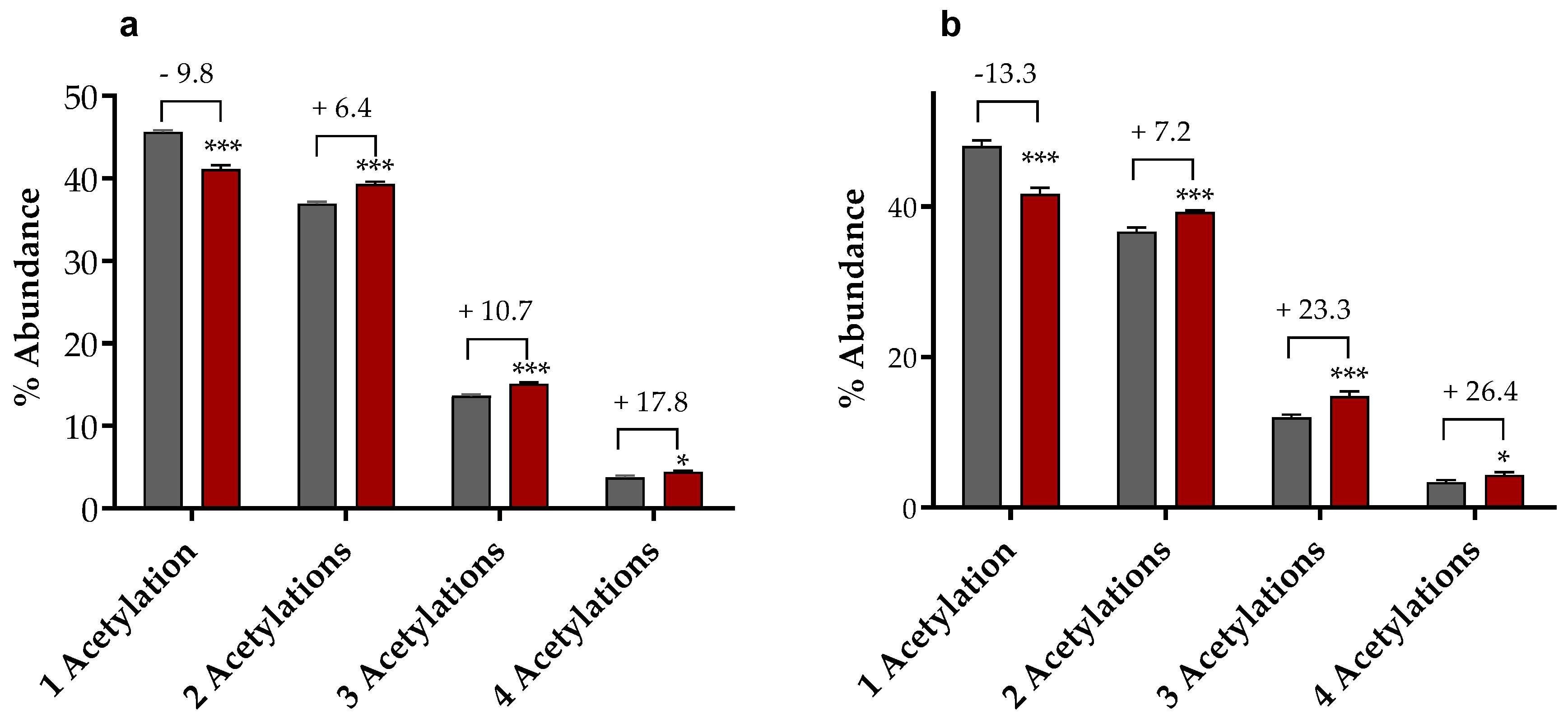
2.1.3. Mass Spectrometry-Based Assessment of Histone Acetylation
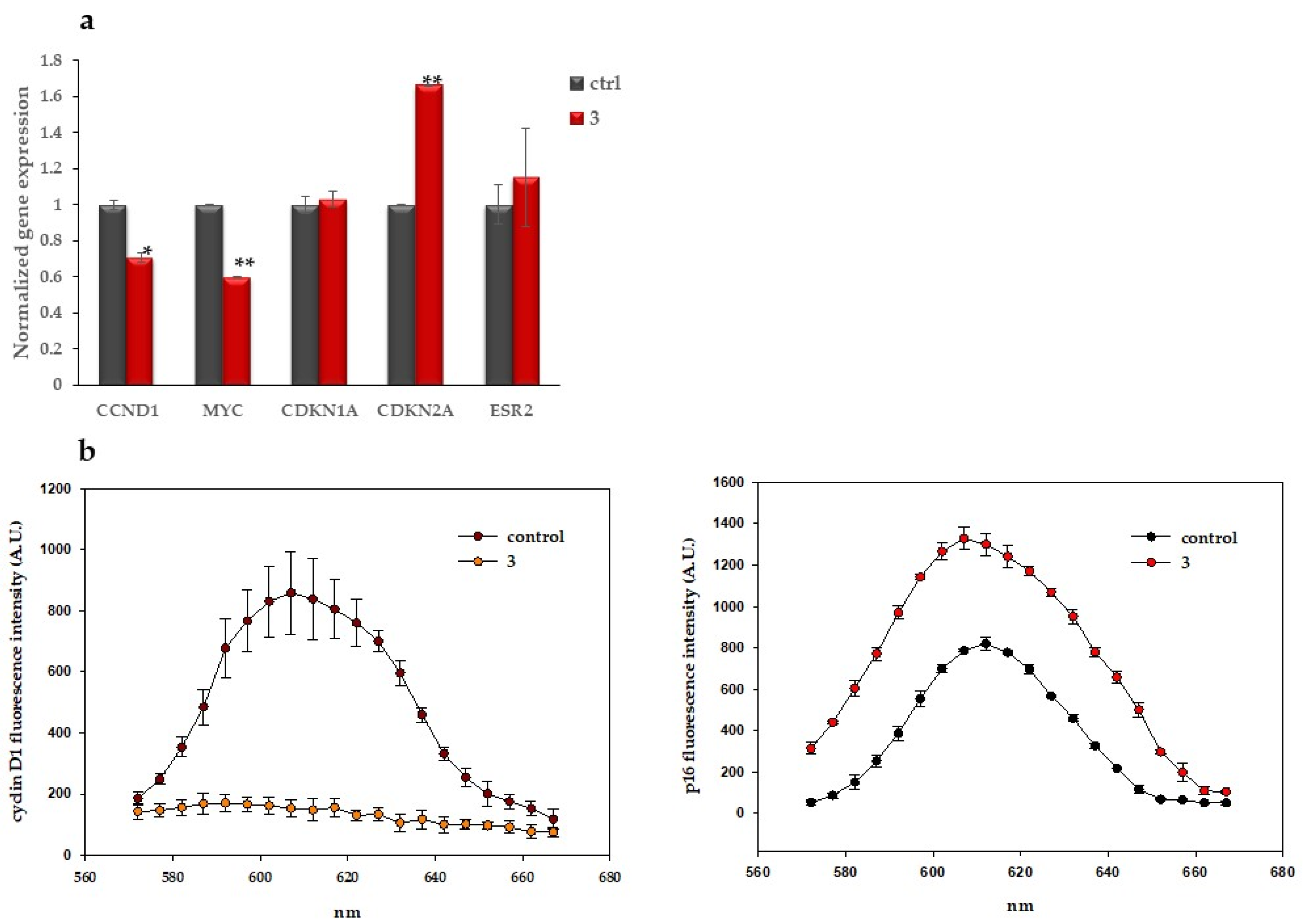
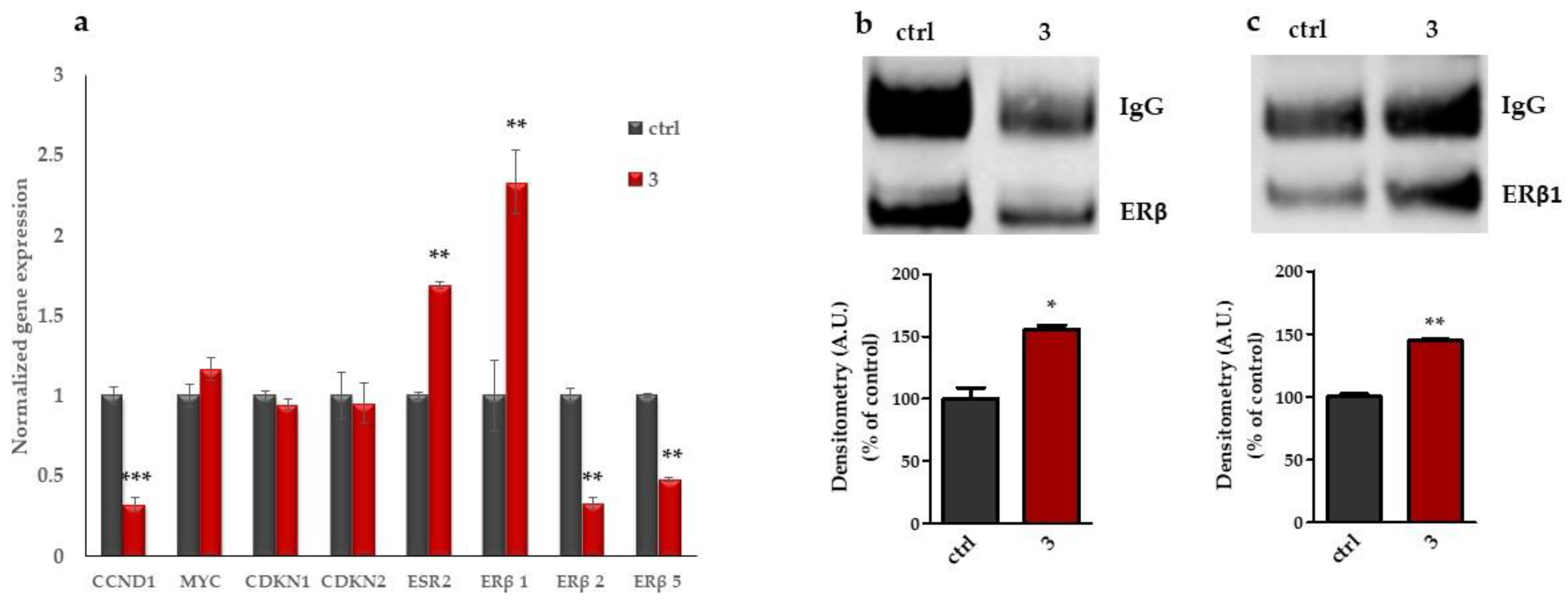
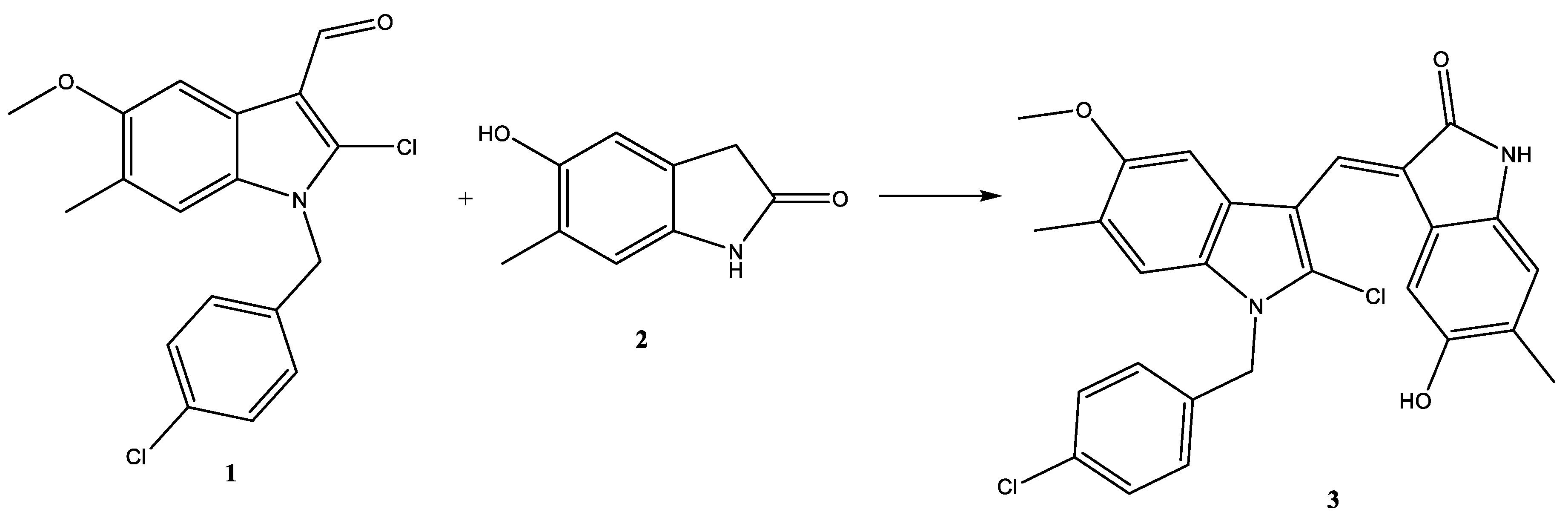
2.1.4. Compound 3 Induces Gene and Protein Expression Modulations
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Chemicals
4.1.1. Proliferation Assays and Cell Cycle Analysis
4.1.2. Confocal Microscopy
4.1.3. Immunoprecipitation and Western Blot Analysis
4.1.4. LC-MS Based Assessment of 3 in IGROV1 Nuclei
4.1.5. MS-Based Determination of Histones Acetylation Degree
4.1.6. Molecular Docking
4.1.7. Quantitative RT-PCR
4.1.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vargas, A.N. Natural history of ovarian cancer. Ecancermedicalscience 2014, 8, 465. [Google Scholar] [PubMed]
- National Cancer Institute. Surveillance, Epidemiology, and End Results Program. Cancer stat facts: Ovarian cancer. Available online: https://seer.cancer.gov/statfacts/html/ovary.html (accessed on 30 December 2018).
- Chen, G.G.; Zeng, Q.; Tze, G.M. Estrogen and its receptors in cancer. Med. Res. Rev. 2008, 28, 954–974. [Google Scholar] [CrossRef] [PubMed]
- Li, A.J.; Baldwin, R.L.; Karlan, B.Y. Estrogen and progesterone receptor subtype expression in normal and malignant ovarian epithelial cell cultures. Am. J. Obstet. Gynecol. 2003, 189, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Matthews, J.; Gustafsson, J.A. Estrogen signaling: a subtle balance between ER alpha and ER beta. Mol. Interv. 2003, 3, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Pujol, P.; Rey, J.-M.; Nirde, P.; Roger, P.; Gastaldi, M.; Laffargue, F.; Rochefort, H.; Maudelonde, T. Differential expression of estrogen receptor-alpha and -beta messenger RNAs as a potential marker of ovarian carcinogenesis. Cancer Res. 1998, 58, 5367–5373. [Google Scholar] [PubMed]
- Jia, M.; Dahlman, K.; Gustafsson, J.Å. Estrogen receptor alpha and beta in health and disease. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 557–568. [Google Scholar] [CrossRef]
- Liu, M.M.; Albanese, C.; Anderson, C.M.; Hilty, K.; Webb, P.; Uht, R.M.; Price, R.H., Jr.; Pestell, R.G.; Kushner, P.J. Opposing action of estrogen receptors alpha and beta on cyclin D1 gene expression. J. Biol. Chem. 2002, 277, 24353–24360. [Google Scholar] [CrossRef] [Green Version]
- Planas-Silva, M.D.; Weinberg, R.A. Estrogen-dependent cyclin E-cdk2 activation through p21 redistribution. Mol. Cell Biol. 1997, 17, 4059–4069. [Google Scholar] [CrossRef] [Green Version]
- Worsley, S.D.; Ponder, B.A.; Davies, B.R. Overexpression of cyclin D1 in epithelial ovarian cancers. Gynecol. Oncol. 1997, 64, 189–195. [Google Scholar] [CrossRef]
- Moore, J.T.; McKee, D.D.; Slentz-Kesler, K.; Moore, L.B.; Jones, S.A.; Horne, E.L.; Su, J.L.; Kliewer, S.A.; Lehmann, J.M.; Willson, T.M. Cloning and characterization of human estrogen receptor beta isoforms. Biochem. Biophys. Res. Commun. 1998, 247, 75–78. [Google Scholar] [CrossRef]
- Suzuki, F.; Akahira, J.; Miura, I.; Suzuki, T.; Ito, K.; Hayashi, S.; Sasano, H.; Yaegashi, N. Loss of estrogen receptor beta isoform expression and its correlation with aberrant DNA methylation of the 5′-untranslated region in human epithelial ovarian carcinoma. Cancer Sci. 2008, 99, 2365–2372. [Google Scholar] [CrossRef] [PubMed]
- Ciucci, A.; Zannoni, G.F.; Travaglia, D.; Petrillo, M.; Scambia, G.; Gallo, D. Prognostic significance of the estrogen receptor beta (ERbeta) isoforms ERbeta1, ERbeta2, and ERbeta5 in advanced serous ovarian cancer. Gynecol. Oncol. 2014, 132, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.K.L.; Siu, M.K.Y.; Jiang, Y.; Wang, J.; Wang, Y.; Leung, T.H.Y.; Liu, S.S.; Cheung, A.N.Y.; Ngan, H.Y.S. Differential expression of estrogen receptor subtypes and variants in ovarian cancer: Effects on cell invasion, proliferation and prognosis. BMC Cancer 2017, 17, 606. [Google Scholar] [CrossRef] [PubMed]
- De Stefano, I.; Zannoni, G.F.; Prisco, M.G.; Fagotti, A.; Tortorella, L.; Vizzielli, G.; Mencaglia, L.; Scambia, G.; Gallo, D. Cytoplasmic expression of estrogen receptor beta (ERb) predicts poor clinical outcome in advanced serous ovarian cancer. Gynecol. Oncol. 2011, 122, 573–579. [Google Scholar] [CrossRef]
- Bardin, A.; Boulle, N.; Lazennec, G.; Vignon, F.; Pujol, P. Loss of ERbeta expression as a common step in estrogen-dependent tumor progression. Endocr. Relat. Cancer 2004, 11, 537–551. [Google Scholar] [CrossRef] [Green Version]
- Rutherford, T.; Brown, W.D.; Sapi, E.; Aschkenazi, S.; Munoz, A.; Mor, G. Absence of estrogen receptor-beta expression in metastatic ovarian cancer. Obstet. Gynecol. 2000, 96, 417–421. [Google Scholar]
- Bossard, C.; Busson, M.; Vindrieux, D.; Gaudin, F.; Machelon, V.; Brigitte, M.; Jacquard, C.; Pillon, A.; Balaguer, P.; Balabanian, K.; et al. Potential role of estrogen receptor beta as a tumor suppressor of epithelial ovarian cancer. PLoS ONE 2012, 7, e44787. [Google Scholar] [CrossRef] [Green Version]
- Fekete, T.; Raso, E.; Pete, I.; Tegze, B.; Liko, I.; Munkacsy, G.; Sipos, N.; Rigo, J., Jr.; Gyorffy, B. Meta-analysis of gene expression profiles associated with histological classification and survival in 829 ovarian cancer samples. Int. J. Cancer 2012, 131, 95–105. [Google Scholar] [CrossRef] [Green Version]
- Veenema, G.H.; Ottow, E.; Weinmann, H. Subtype-Selective estrogens in Nuclear Receptors as Drug Targets. In Methods and Principles in Medicinal Chemistry Series; Mannhold, R., Kubinyi, H., Folkers, G., Eds.; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008; Chapter 4; Volume 39. [Google Scholar]
- Mersereau, J.E.; Levy, N.; Staub, R.E.; Baggett, S.; Zogric, T.; Chow, S.; Ricke, W.A.; Tagliaferri, M.; Cohen, I.; Bjeldanes, L.F.; et al. Liquiritigenin is a plant-derived highly selective estrogen receptor β agonist. Mol. Cell. Endocrin. 2008, 283, 49–57. [Google Scholar] [CrossRef] [Green Version]
- Lattrich, C.; Schüler, S.; Häring, J.; Skrzypczak, M.; Ortmann, O.; Treeck, O. Effects of a combined treatment with tamoxifen and estrogen receptor β agonists on human breast cancer cell lines. Arch. Gynecol. Obstet. 2014, 289, 163–171. [Google Scholar] [CrossRef]
- Manas, E.S.; Unwalla, R.-J.; Xu, Z.B.; Malamas, M.S.; Miller, C.P.; Harris, H.A.; Hsiao, C.; Akopian, T.; Hum, W.-T.; Malakian, K.; et al. Structure-Based Design of Estrogen Receptor-β Selective Ligands. J. Am. Chem. Soc. 2004, 126, 15106–15119. [Google Scholar] [CrossRef] [PubMed]
- Gribble, G.W. Indole Ring Synthesis: From Natural Products to Drug Discovery; John Wiley & Sons: Chichester, UK, 2016. [Google Scholar]
- Andreani, A.; Burnelli, S.; Granaiola, M.; Leoni, A.; Locatelli, A.; Morigi, R.; Rambaldi, M.; Varoli, L.; Calonghi, N.; Cappadone, C.; et al. Substituted E-3-(2-chloro-3-indolylmethylene)1,3-dihydroindol-2-ones with antitumor activity. Effect on the cell cycle and apoptosis. J. Med. Chem. 2007, 50, 3167–3172. [Google Scholar] [CrossRef]
- Naldi, M.; Andrisano, V.; Fiori, J.; Calonghi, N.; Pagnotta, E.; Parolin, C.; Pieraccini, G.; Masotti, L. Histone proteins determined in a human colon cancer by high-performance liquid chromatography and mass spectrometry. J. Chromatogr. A 2006, 1129, 73–81. [Google Scholar] [CrossRef]
- Naldi, M.; Calonghi, N.; Masotti, L.; Parolin, C.; Valente, S.; Mai, A.; Andrisano, V. Histone post-translational modifications by HPLC-ESI-MS after HT29 cell treatment with histone deacetylase inhibitors. Proteomics 2009, 9, 5437–5445. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.K.; Leung, T.H.; Chan, D.W.; Wei, N.; Lau, G.T.; Liu, S.S.; Siu, M.K.; Ngan, H.Y. Targeting estrogen receptor subtypes (ERα and ERβ) with selective ERmodulators in ovarian cancer. J. Endocrinol. 2014, 221, 325–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazennec, G. Estrogen receptor beta, a possible tumor suppressor involved in ovarian carcinogenesis. Cancer Lett. 2006, 231, 151–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paruthiyil, S.; Parmar, H.; Kerekatte, V.; Cunha, G.R.; Firestone, G.L.; Leitman, D.C. Estrogen receptor β inhibits human breast cancer cell proliferation and tumor formation by causing a G2 cell cycle arrest. Cancer Res. 2004, 64, 423–428. [Google Scholar] [CrossRef] [Green Version]
- Schüler-Toprak, S.; Moehle, C.; Skrzypczak, M.; Ortmann, O.; Treeck, O. Effect of estrogen receptor β agonists on proliferation and gene expression of ovarian cancer cells. BMC Cancer 2017, 17, 319. [Google Scholar] [CrossRef]
- Serrano, M.; Hannon, G.J.; Beach, D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature 1993, 366, 704–707. [Google Scholar] [CrossRef]
- Qin, Y.; Liu, J.Y.; Li, B.; Sun, Z.L.; Sun, Z.F. Association of low p16INK4a and p15INK4b mRNAs expression with their CpG islands methylation with human hepatocellular carcinogenesis. World J. Gastroenterol. 2004, 10, 1276–1280. [Google Scholar] [CrossRef]
- Kim, B.N.; Yamamoto, H.; Ikeda, K.; Damdinsuren, B.; Sugita, Y.; Ngan, C.Y.; Fujie, Y.; Ogawa, M.; Hata, T.; Ikeda, M.; et al. Methylation and expression of p16INK4 tumor suppressor gene in primary colorectal cancer tissues. Int. J. Oncol. 2005, 26, 1217–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, S.J.; Zhang, G.F.; Zhang, R.P. High CpG island methylation of p16 gene and loss of p16 protein expression associate with the development and progression of tetralogy of Fallot. J. Genet. 2016, 95, 831–837. [Google Scholar] [CrossRef] [PubMed]
- Sharpless, N.E. INK4a/ARF: A multifunctional tumor suppressor locus. Mutat. Res. 2005, 576, 22–38. [Google Scholar] [CrossRef] [PubMed]
- Moselhy, S.S.; Kumosani, T.A.; Kamal, I.H.; Jalal, J.A.; Jabaar, H.S.; Dalol, A. Hypermethylation of P15, P16, and E-cadherin genes in ovarian cancer. Toxicol. Ind. Health. 2015, 31, 924–930. [Google Scholar] [CrossRef] [PubMed]
- Di Vinci, A.; Perdelli, L.; Banelli, B.; Salvi, S.; Casciano, I.; Gelvi, I.; Allemanni, G.; Margallo, E.; Gatteschi, B.; Romani, M. p16(INK4a) promoter methylation and protein expression in breast fibroadenoma and carcinoma. Int. J. Cancer 2005, 114, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Shima, K.; Nosho, K.; Baba, Y.; Cantor, M.; Meyerhardt, J.A.; Giovannucci, E.L.; Fuchs, C.S.; Ogino, S. Prognostic significance of CDKN2A (p16) promoter methylation and loss of expression in 902 colorectal cancers: Cohort study and literature review. Int. J. Cancer 2011, 128, 1080–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruan, J.; Xu, P.; Fan, W.; Deng, Q.; Yu, M. Quantitative assessment of aberrant P16INK4a methylation in ovarian cancer: a meta-analysis based on literature and TCGA datasets. Cancer Manag. Res. 2018, 10, 3033–3046. [Google Scholar] [CrossRef] [Green Version]
- Treeck, O.; Pfeiler, G.; Mitter, D.; Lattrich, C.; Piendl, G.; Ortmann, O. Estrogen receptor {beta}1 exerts antitumoral effects on SK-OV-3 ovarian cancer cells. J. Endocrinol. 2007, 193, 421–433. [Google Scholar] [CrossRef]
- Zhu, J.; Hua, K.; Sun, H.; Yu, Y.; Jin, H.; Feng, Y. Re-expression of estrogen receptor beta inhibits the proliferation and migration of ovarian clear cell adenocarcinoma cells. Oncol. Rep. 2011, 26, 1497–1503. [Google Scholar]
- Micheletti, G.; Calonghi, N.; Farruggia, G.; Strocchi, E.; Palmacci, V.; Telese, D.; Bordoni, S.; Frisco, G.; Boga, C. Synthesis of Novel Structural Hybrids between Aza-Heterocycles and Azelaic Acid Moiety with a Specific Activity on Osteosarcoma Cells. Molecules 2020, 25, 404. [Google Scholar] [CrossRef] [Green Version]
- Gladden, A.B.; Diehl, J.A. The cyclin D1-dependent kinase associates with the pre- replication complex and modulates RB-MCM7 binding. J. Biol. Chem. 2003, 278, 9754–9760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parolin, C.; Calonghi, N.; Presta, E.; Boga, C.; Caruana, P.; Naldi, M.; Andrisano, V.; Masotti, L.; Sartor, G. Mechanism and stereoselectivity of HDAC I inhibition by (R)-9-hydroxystearic acid in colon cancer. Biochim. Biophys. Acta 2012, 1821, 1334–1340. [Google Scholar] [CrossRef] [PubMed]
- Calonghi, N.; Pagnotta, E.; Parolin, C.; Molinari, C.; Boga, C.; Dal Piaz, F.; Brusa, G.L.; Santucci, M.A.; Masotti, L. Modulation of apoptotic signalling by 9-hydroxystearic acid in osteosarcoma cells. Biochim. Biophys. Acta 2007, 1771, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Olmo, S.; Gotti, R.; Naldi, M.; Andrisano, V.; Calonghi, N.; Parolin, C.; Masotti, L.; Cavrini, V. Analysis of human histone H4 by capillary electrophoresis in a pullulan-coated capillary, LC-ESI-MS and MALDI-TOF-MS. Anal. Bioanal. Chem. 2008, 390, 1881–1888. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Jirawatnotai, S.; Hu, Y.; Michowski, W.; Elias, J.E.; Becks, L.; Bienvenu, F.; Zagozdzon, A.; Goswami, T.; Wang, Y.E.; Clark, A.B.; et al. A function for cyclin D1 in DNA repair uncovered by interactome analyses in human cancers. Nature 2011, 474, 230–234. [Google Scholar] [CrossRef] [Green Version]
- Di Sante, G.; Di Rocco, A.; Pupo, C.; Casimiro, M.C.; Pestell, R.G. Hormone-induced DNA damage response and repair mediated by cyclin D1 in breast and prostate cancer. Oncotarget 2017, 8, 81803–81812. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verardi, L.; Fiori, J.; Andrisano, V.; Locatelli, A.; Morigi, R.; Naldi, M.; Bertucci, C.; Strocchi, E.; Boga, C.; Micheletti, G.; et al. Indole Derivative Interacts with Estrogen Receptor Beta and Inhibits Human Ovarian Cancer Cell Growth. Molecules 2020, 25, 4438. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25194438
Verardi L, Fiori J, Andrisano V, Locatelli A, Morigi R, Naldi M, Bertucci C, Strocchi E, Boga C, Micheletti G, et al. Indole Derivative Interacts with Estrogen Receptor Beta and Inhibits Human Ovarian Cancer Cell Growth. Molecules. 2020; 25(19):4438. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25194438
Chicago/Turabian StyleVerardi, Laura, Jessica Fiori, Vincenza Andrisano, Alessandra Locatelli, Rita Morigi, Marina Naldi, Carlo Bertucci, Elena Strocchi, Carla Boga, Gabriele Micheletti, and et al. 2020. "Indole Derivative Interacts with Estrogen Receptor Beta and Inhibits Human Ovarian Cancer Cell Growth" Molecules 25, no. 19: 4438. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25194438