
Synthesis of “All-Cis” Trihydroxypiperidines from a Carbohydrate-Derived Ketone: Hints for the Design of New β-Gal and GCase Inhibitors

 , ,
, ,
Abstract
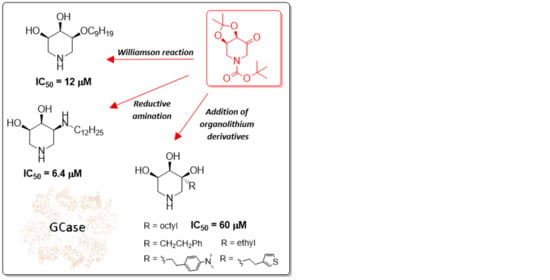
:
1. Introduction
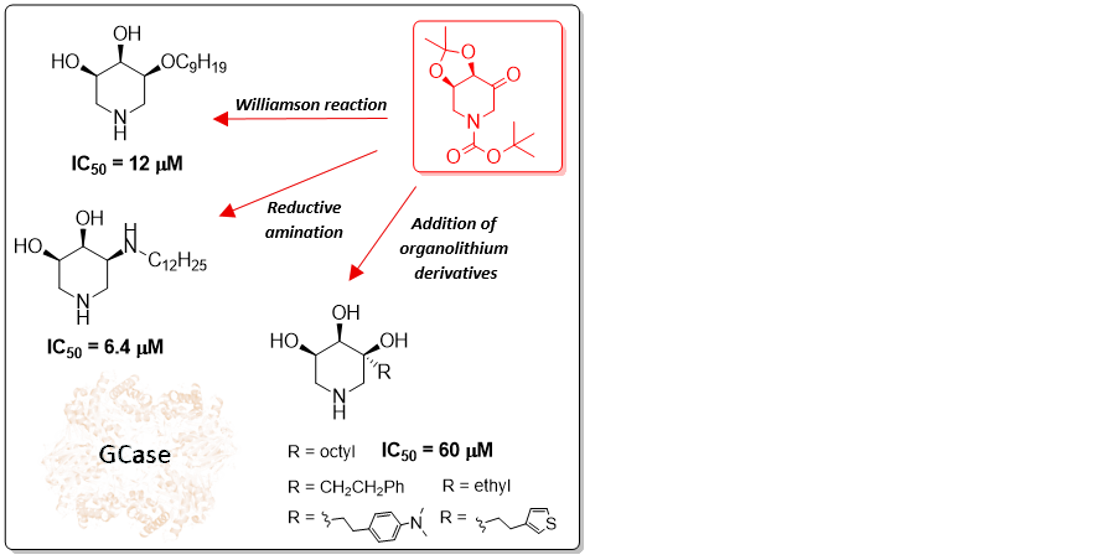
2. Results and Discussion
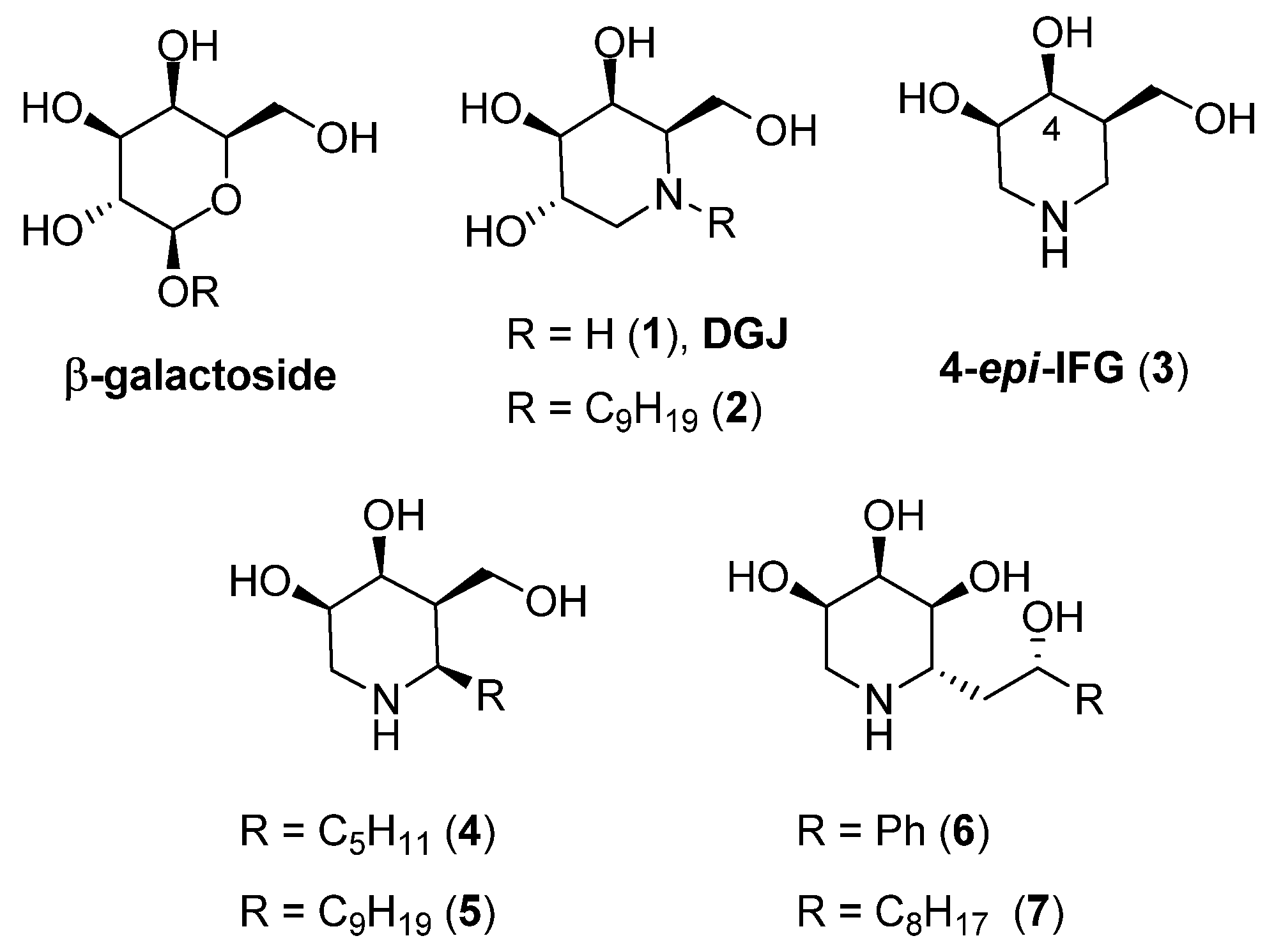
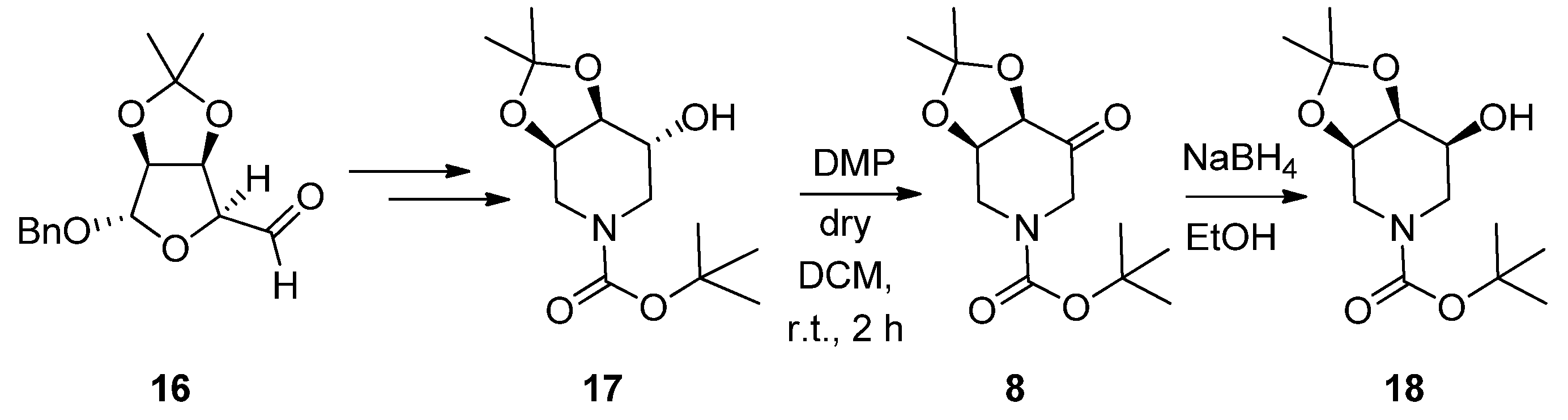
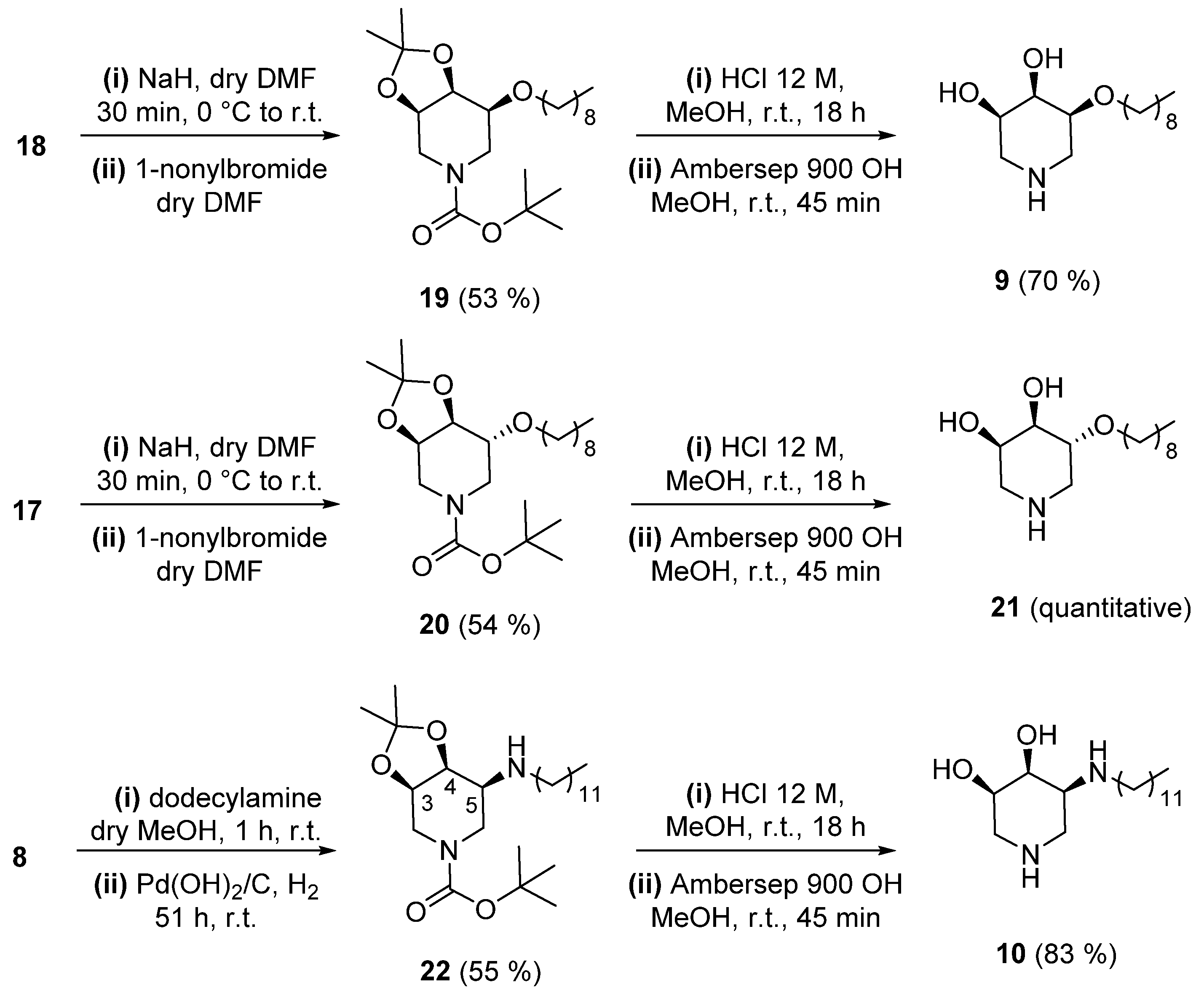
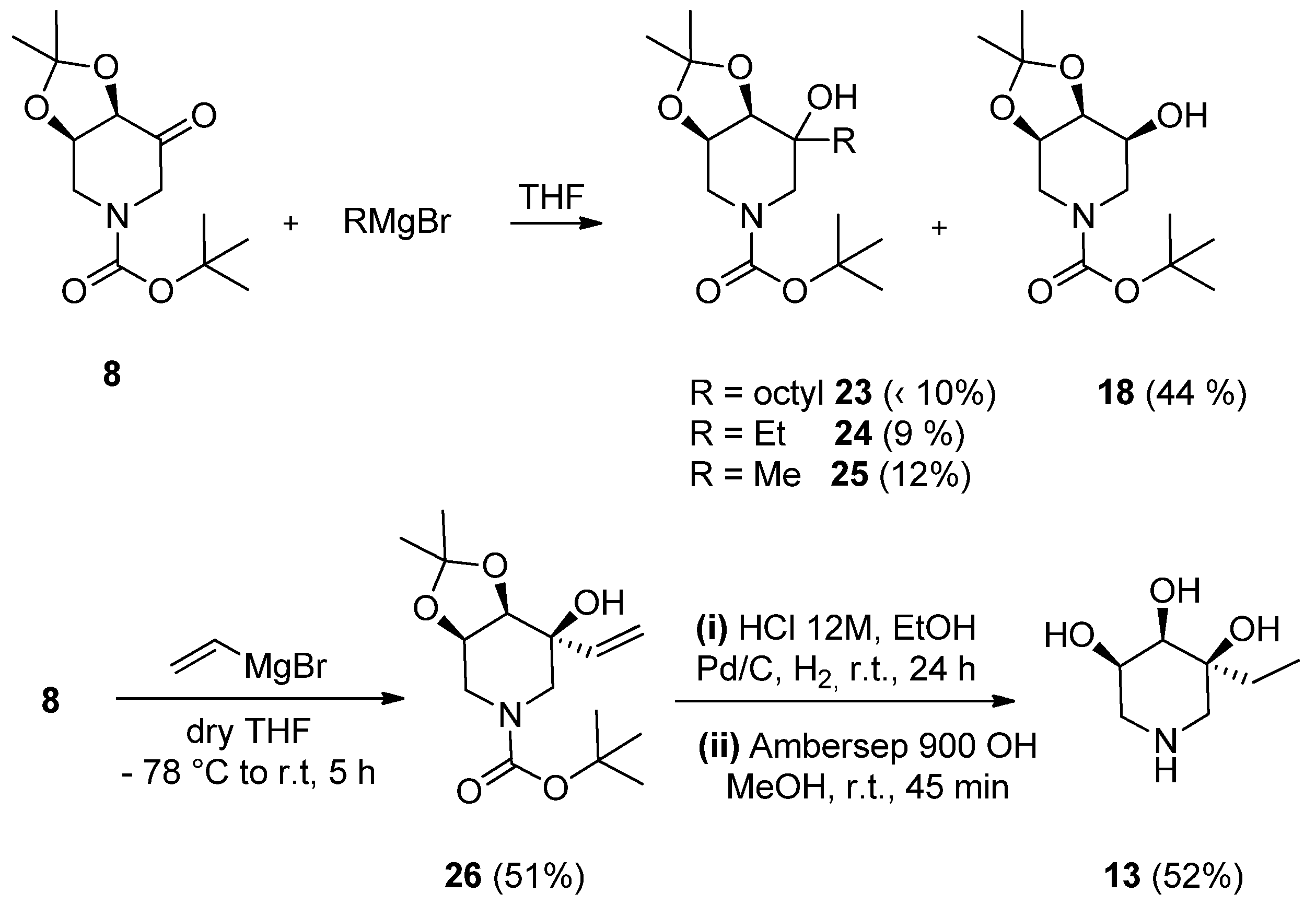
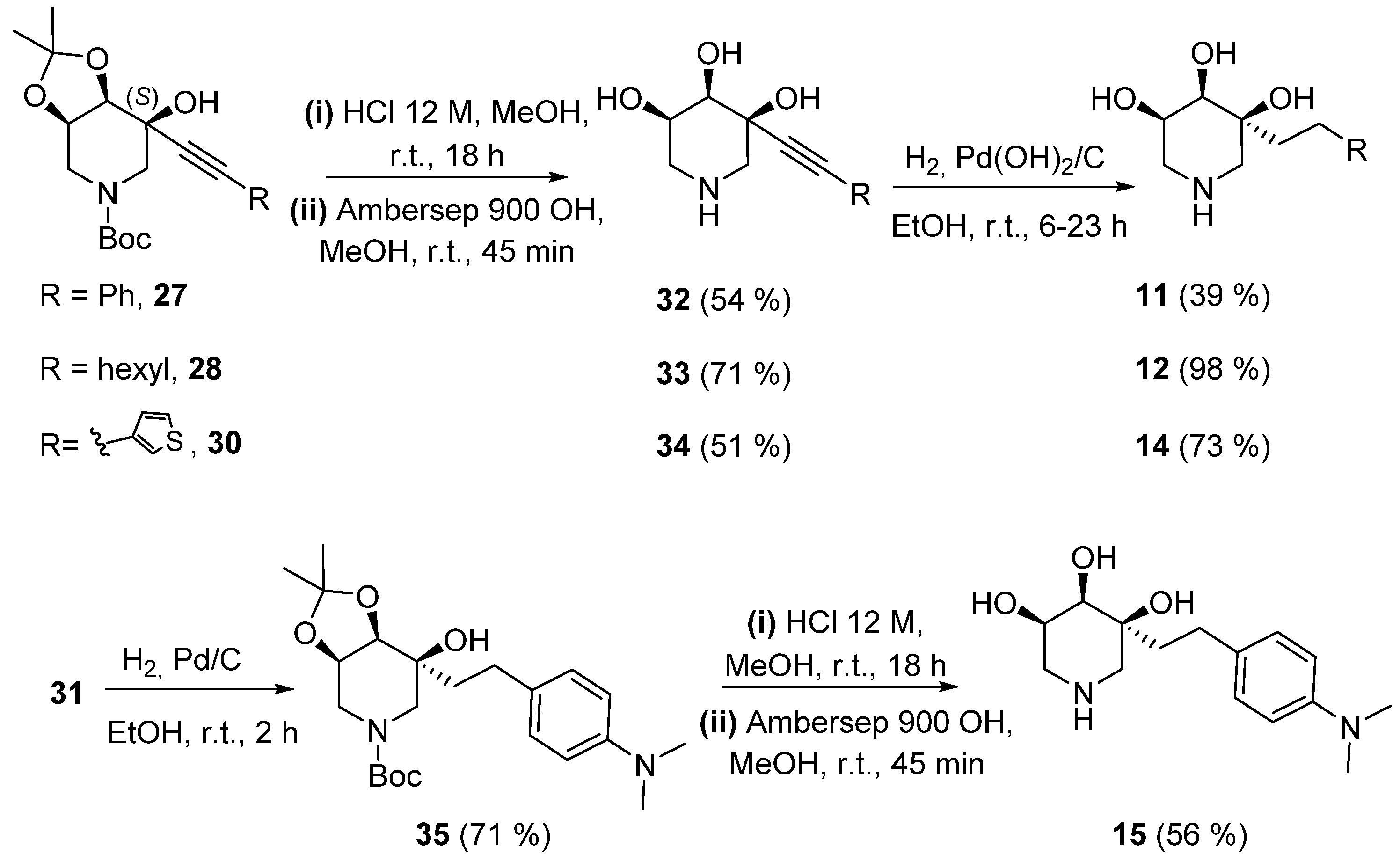
2.1. Synthesis
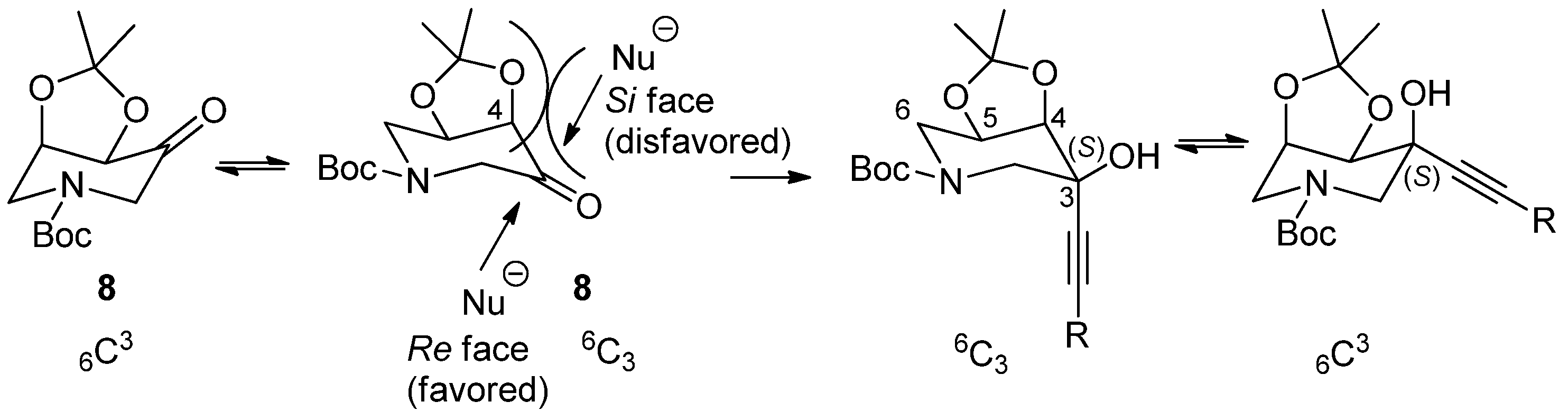
2.2. Configuration Assignment
2.3. Biological Screening
3. Materials and Methods
3.1. General Experimental Procedures for the Syntheses
3.1.1. Synthesis of (3R, 4S, 5S)-3, 4-O-(1-Methylethylidene)-5-Nonyloxy-N-Boc-Piperidine (19)
3.1.2. Synthesis of (3S, 4R, 5R)-4, 5-Dihydroxy-3-(Nonyloxy) Piperidine (9)
3.1.3. Synthesis of (3R, 4S, 5R)-3, 4-O-(1-Methylethylidene)-5-Nonyloxy-N-Boc-Piperidine (20)
3.1.4. Synthesis of (3R, 4R, 5R)-4, 5-Dihydroxy-3-(Nonyloxy) Piperidine (21)
3.1.5. Synthesis of (3R, 4S, 5S)-5-Dodecylamino-3, 4-O-(1-Methylethylidene)-N-Boc-Piperidine (22)
3.1.6. Synthesis of (3R, 4S, 5S)-3, 4-Dihydroxy-5-(Dodecylamino) Piperidine (10)
3.1.7. Synthesis of (3S, 4R, 5R)-3-Hydroxy-4, 5-O-(1-Methylethylidene)-3-Vinyl-N-Boc-Piperidine (26)
3.1.8. Synthesis of (3S, 4R, 5R)-3-Ethyl-3, 4, 5-Trihydroxypiperidine (13)
3.1.9. General Procedure for the Addition of Lithium Acetylides to Ketone 8
3.1.10. Synthesis of (3S, 4R, 5R)-3-Hydroxy-4, 5-O-(1-Methylethylidene)-3-(Phenylethynyl)-N-Boc-Piperidine (27)
3.1.11. Synthesis of (3S, 4R, 5R)-3-Hydroxy-4, 5-O-(1-Methylethylidene)-3-(oct-1-yn-1-yl)-N-Boc-Piperidine (28)
3.1.12. Synthesis of (3S, 4R, 5R)-3-Hydroxy-4, 5-O-(1-Methylethylidene)-3-(3, 3-Diethoxyprop-1-yn-1-yl)-N-Boc-Piperidine (29)
3.1.13. Synthesis of (3S, 4R, 5R)-3-Hydroxy-4, 5-O-(1-Methylethylidene)-3-(3-Thienylethynyl)-N-Boc-Piperidine (30)
3.1.14. Synthesis of (3S, 4R, 5R)-3-Hydroxy-4, 5-O-(1-Methylethylidene)-3-((4-(Dimethylamino) Phenyl) Ethynyl)-N-Boc-Piperidine (31)
3.1.15. General Procedure for the Synthesis of Trihydroxypiperidines 32, 33, and 34
3.1.16. Synthesis of (3S, 4R, 5R)-3, 4, 5-Trihydroxy-3-(Phenylethynyl)-Piperidine (32)
3.1.17. Synthesis of (3S, 4R, 5R)-3, 4, 5-Trihydroxy-3-(oct-1-yn-1-yl)-Piperidine (33)
3.1.18. Synthesis of (3S, 4R, 5R)-3, 4, 5-Trihydroxy-3-(3-Thienylethynyl))-Piperidine (34)
3.1.19. General Procedure for the Reduction of Triple Bond to Piperidines 11, 12, 14
3.1.20. Synthesis of (3S, 4R, 5R)-3, 4, 5-Trihydroxy-3-(2-Phenylethyl)-Piperidine (11)
3.1.21. Synthesis of (3S, 4R, 5R)-3, 4, 5-Trihydroxy-3-Octyl-Piperidine (12)
3.1.22. Synthesis of (3S, 4R, 5R)-3, 4, 5-Trihydroxy-3-((2-(3-Thienyl) Ethyl))-Piperidine (14)
3.1.23. Synthesis of (3S, 4R, 5R)-3-hydroxy-4, 5-O-(1-methylethylidene)-3-(2-((4-(dimethylamino) phenyl) ethyl)-N-Boc-piperidine (35)
3.1.24. Synthesis of (3S, 4R, 5R)-3, 4, 5-Trihydroxy-3-(2-((4-(Dimethylamino) Phenyl) Ethyl)-Piperidine (15)
3.2. Preliminary Biological Screening towards Commercial Glycosidases
3.3. Biological Screening towards Human Lysosomal β-Galactosidase (β-Gal) and β-Glucosidase (GCase)
3.3.1. Human Lysosomal β-Galactosidase (β-Gal) Activity
3.3.2. Human Lysosomal β-Glucosidase (GCase) Activity
3.4. Pharmacological Chaperoning Activity of Compound 10
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Marques, A.R.A.; Saftig, P. Lysosomal storage disorders—challenges, concepts and avenues for therapy: Beyond rare diseases. J. Cell Sci. 2019, 132, jcs221739. [Google Scholar] [CrossRef] [PubMed]
- Platt, F.M.; D’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal storage diseases. Nat. Rev. Dis. Prim. 2018, 4, 27. [Google Scholar] [CrossRef] [PubMed]
- Parenti, G.; Andria, G.; Ballabio, A. Lysosomal Storage Diseases: From Pathophysiology to Therapy. Annu. Rev. Med. 2015, 66, 471–486. [Google Scholar] [CrossRef] [PubMed]
- Ortolano, S.; Viéitez, I.; Navarro, C.; Spuch, C. Treatment of lysosomal storage diseases: Recent patents and future strategies. Recent Patents Endocr. Metab. Immune Drug Discov. 2014, 8, 9–25. [Google Scholar] [CrossRef] [PubMed]
- Caciotti, A.; Garman, S.C.; Rivera-Colón, Y.; Procopio, E.; Catarzi, S.; Ferri, L.; Guido, C.; Martelli, P.; Parini, R.; Antuzzi, D.; et al. GM1 gangliosidosis and Morquio B disease: An update on genetic alterations and clinical findings. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2011, 1812, 782–790. [Google Scholar] [CrossRef] [PubMed]
- Brunetti-Pierri, N.; Scaglia, F.F. GM1 gangliosidosis: Review of clinical, molecular, and therapeutic aspects. Mol. Genet. Metab. 2008, 94, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Pereira, D.M.; Valentão, P.; Andrade, P.B. Tuning protein folding in lysosomal storage diseases: The chemistry behind pharmacological chaperones. Chem. Sci. 2018, 9, 1740–1752. [Google Scholar] [CrossRef] [Green Version]
- Convertino, M.; Das, J.; Dokholyan, N.V. Pharmacological Chaperones: Design and Development of New Therapeutic Strategies for the Treatment of Conformational Diseases. ACS Chem. Boil. 2016, 11, 1471–1489. [Google Scholar] [CrossRef]
- Parenti, G.; Andria, G.; Valenzano, K.J. Pharmacological Chaperone Therapy: Preclinical Development, Clinical Translation, and Prospects for the Treatment of Lysosomal Storage Disorders. Mol. Ther. 2015, 23, 1138–1148. [Google Scholar] [CrossRef] [Green Version]
- Boyd, R.E.; Lee, G.; Rybczynski, P.; Benjamin, E.R.; Khanna, R.; Wustman, B.A.; Valenzano, K.J. Pharmacological Chaperones as Therapeutics for Lysosomal Storage Diseases. J. Med. Chem. 2013, 56, 2705–2725. [Google Scholar] [CrossRef]
- Aguilar-Moncayo, M.; Takai, T.; Higaki, K.; Mena-Barragán, T.; Hirano, Y.; Yura, K.; Li, L.; Yu, Y.; Ninomiya, H.; Garcıa-Moreno, M.; et al. Tuning glycosidase inhibition through aglycone interactions: Pharmacological chaperones for Fabry disease and GM1 gangliosidosis. Chem. Commun. 2012, 48, 6514–6516. [Google Scholar] [CrossRef] [PubMed]
- Compain, P.; Martin, O.R. (Eds.) Iminosugars: From Synthesis to Therapeutic Applications; John Wiley & Sons Ltd.: Chichester, UK, 2007; ISBN 978-0-470-03391-3. [Google Scholar] [CrossRef]
- Sánchez-Fernández, E.M.; Fernández, J.M.G.; Mellet, C.O. Glycomimetic-based pharmacological chaperones for lysosomal storage disorders: Lessons from Gaucher, GM1-gangliosidosis and Fabry diseases. Chem. Commun. 2016, 52, 5497–5515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCafferty, E.H.; Scott, L.J. Migalastat: A Review in Fabry Disease. Drugs 2019, 79, 543–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuda, J.; Suzuki, O.; Oshima, A.; Yamamoto, Y.; Noguchi, A.; Takimoto, K.; Itoh, M.; Matsuzaki, Y.; Yasuda, Y.; Ogawa, S.; et al. Chemical chaperone therapy for brain pathology in GM1-gangliosidosis. Proc. Natl. Acad. Sci. USA 2003, 100, 15912–15917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rigat, B.A.; Tropak, M.B.; Buttner, J.; Crushell, E.; Benedict, D.; Callahan, J.W.; Martin, D.R.; Mahuran, D.J. Evaluation of N-nonyl-deoxygalactonojirimycin as a pharmacological chaperone for human GM1 gangliosidosis leads to identification of a feline model suitable for testing enzyme enhancement therapy. Mol. Genet. Metab. 2012, 107, 203–212. [Google Scholar] [CrossRef] [Green Version]
- Prichard, K.; Campkin, D.; O’Brien, N.; Kato, A.; Fleet, G.W.J.; Simone, M.I. Biological activities of 3,4,5-trihydroxypiperidines and their N- and O- derivatives. Chem. Boil. Drug Des. 2018, 92, 1171–1197. [Google Scholar] [CrossRef] [PubMed]
- Biela-Banaś, A.; Oulaïdi, F.; Front, S.; Gallienne, E.; Ikeda-Obatake, K.; Asano, N.; Wenger, D.A.; Martin, O.R. Iminosugar-Based Galactoside Mimics as Inhibitors of Galactocerebrosidase: SAR Studies and Comparison with Other Lysosomal Galactosidases. ChemMedChem 2014, 9, 2647–2652. [Google Scholar] [CrossRef]
- Front, S.; Biela-Banaś, A.; Burda, P.; Ballhausen, D.; Higaki, K.; Caciotti, A.; Morrone, A.; Charollais-Thoenig, J.; Gallienne, E.; Demotz, S.; et al. (5aR)-5a-C-Pentyl-4-epi-isofagomine: A powerful inhibitor of lysosomal β-galactosidase and a remarkable chaperone for mutations associated with GM1-gangliosidosis and Morquio disease type B. Eur. J. Med. Chem. 2017, 126, 160–170. [Google Scholar] [CrossRef]
- Front, S.; Almeida, S.; Zoete, V.; Charollais-Thoenig, J.; Gallienne, E.; Marmy, C.; Pilloud, V.; Marti, R.; Wood, T.; Martin, O.R.; et al. 4-epi-Isofagomine derivatives as pharmacological chaperones for the treatment of lysosomal diseases linked to β-galactosidase mutations: Improved synthesis and biological investigations. Bioorg. Med. Chem. 2018, 26, 5462–5469. [Google Scholar] [CrossRef]
- Siriwardena, A.; Sonawane, D.P.; Bande, O.P.; Markad, P.R.; Yonekawa, S.; Tropak, M.B.; Ghosh, S.; Chopade, B.A.; Mahuran, N.J.; Dhavale, D.D. Synthesis of 1,5-Dideoxy-1,5-iminoribitol C-Glycosides through a Nitrone–Olefin Cycloaddition Domino Strategy: Identification of Pharmacological Chaperones of Mutant Human Lysosomal β-Galactosidase. J. Org. Chem. 2014, 79, 4398–4404. [Google Scholar] [CrossRef]
- Parmeggiani, C.; Catarzi, S.; Matassini, C.; D’Adamio, G.; Morrone, A.; Goti, A.; Paoli, P.; Cardona, F. Human Acid β-Glucosidase Inhibition by Carbohydrate Derived Iminosugars: Towards New Pharmacological Chaperones for Gaucher Disease. ChemBioChem 2015, 16, 2054–2064. [Google Scholar] [CrossRef] [PubMed]
- Clemente, F.; Matassini, C.; Goti, A.; Morrone, A.; Paoli, P.; Cardona, F. Stereoselective Synthesis of C-2 Alkylated Trihydroxypiperidines: Novel Pharmacological Chaperones for Gaucher Disease. ACS Med. Chem. Lett. 2019, 10, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Clemente, F.; Matassini, C.; Faggi, C.; Giachetti, S.; Cresti, C.; Morrone, A.; Paoli, P.; Goti, A.; Martínez-Bailén, M.; Cardona, F. Glucocerebrosidase (GCase) activity modulation by 2-alkyl trihydroxypiperidines: Inhibition and pharmacological chaperoning. Bioorg. Chem. 2020, 98, 103740. [Google Scholar] [CrossRef] [PubMed]
- Matassini, C.; Mirabella, S.; Goti, A.; Cardona, F. Double Reductive Amination and Selective Strecker Reaction of a D-Lyxaric Aldehyde: Synthesis of Diversely Functionalized 3,4,5-Trihydroxypiperidines. Eur. J. Org. Chem. 2012, 3920–3924. [Google Scholar] [CrossRef]
- Matassini, C.; Clemente, F.; Cardona, F. The double reductive amination approach to the synthesis of polyhydroxypiperidines. In Targets in Heterocyclic Systems—Chemistry and Properties (THS); Attanasi, O.A., Merino, P., Spinelli, D., Eds.; Societa’ Chimica Italiana: Rome, Italy, 2019; Volume 23, pp. 283–301. [Google Scholar] [CrossRef]
- Clemente, F.; Matassini, C.; Cardona, F. Reductive Amination Routes in the Synthesis of Piperidine IminoSugars. Eur. J. Org. Chem. 2020, 4447–4462. [Google Scholar] [CrossRef]
- Santos, C.; Stauffert, F.; Ballereau, S.; Dehoux, C.; Rodriguez, F.; Bodlenner, A.; Compain, P.; Génisson, Y. Iminosugar-based ceramide mimicry for the design of new CERT START domain ligands. Bioorganic Med. Chem. 2017, 25, 1984–1989. [Google Scholar] [CrossRef]
- Matassini, C.; Mirabella, S.; Ferhati, X.; Faggi, C.; Robina, I.; Goti, A.; Clavijo, E.M.; Moreno-Vargas, A.J.; Cardona, F. Polyhydroxyamino-Piperidine-Type Iminosugars and Pipecolic Acid Analogues from aD-Mannose-Derived Aldehyde. Eur. J. Org. Chem. 2014, 5419–5432. [Google Scholar] [CrossRef]
- Mirabella, S.; Fibbi, G.; Matassini, C.; Faggi, C.; Goti, A.; Cardona, F. Accessing 2-substituted piperidine iminosugars by organometallic addition/intramolecular reductive amination: Aldehyde vs. nitrone route. Org. Biomol. Chem. 2017, 15, 9121–9126. [Google Scholar] [CrossRef]
- Kankanamalage, A.C.G.; Kim, Y.; Damalanka, V.C.; Rathnayake, A.D.; Fehr, A.R.; Mehzabeen, N.; Battaile, K.P.; Lovell, S.; Lushington, G.H.; Perlman, S.; et al. Structure-guided design of potent and permeable inhibitors of MERS coronavirus 3CL protease that utilize a piperidine moiety as a novel design element. Eur. J. Med. Chem. 2018, 150, 334–346. [Google Scholar] [CrossRef]
- Cowan, D.O.; Mosher, H.S. Comparison of the Reactions of Grignard Reagents and Dialkylmagnesium Compounds in Addition, Reduction, and Enolization Reactions1. J. Org. Chem. 1962, 27, 1–5. [Google Scholar] [CrossRef]
- Sassian, M.; Tuulmets, A. Solvation Effects in theGrignard Reaction with Carbonyl Compounds. Helv. Chim. Acta 2003, 86, 82–90. [Google Scholar] [CrossRef]
- Engel, D.A.; Dudley, G.B. Olefination of Ketones Using a Gold(III)-Catalyzed Meyer-Schuster Rearrangement. Org. Lett. 2006, 8, 4027–4029. [Google Scholar] [CrossRef] [PubMed]
- Lamb, N.; Abrams, S.R. Synthesis of optically active cyclohexanone analogs of the plant hormone abscisic acid. Can. J. Chem. 1990, 68, 1151–1162. [Google Scholar] [CrossRef]
- Rocquet, F.; Battioni, J.-P.; Capmau, M.-L.; Chodkiewicz, W. Stereochemistry of organometallic compound addition to cyclohexanones monomethylated in the 2, 3, and 4 positions. C. R. Acad. Sci. Ser. C 1969, 268, 1449–1452. [Google Scholar]
- Ando, K.; Houk, K.N.; Busch, J.; Menassé, A.A.; Séquin, U. Experimental and Computational Studies of Nucleophilic Additions of Metal Hydrides and Organometallics to Hindered Cyclohexanones. J. Org. Chem. 1998, 63, 1761–1766. [Google Scholar] [CrossRef]
- Chérest, M.; Felkin, H.; Prudent, N. Torsional strain involving partial bonds. The stereochemistry of the lithium aluminium hydride reduction of some simple open-chain ketones. Tetrahedron Lett. 1968, 9, 2199–2204. [Google Scholar] [CrossRef]
- Chérest, M.; Felkin, H. Torsional strain involving partial bonds. The steric course of the reaction between allyl magnesium bromide and 4-t-butyl-cyclohexanone. Tetrahedron Lett. 1968, 9, 2205–2208. [Google Scholar] [CrossRef]
- Anh, N.T.; Eisenstein, O. Theoretical interpretation of 1-2 asymmetric induction: The importance of antiperiplanarity. Nouv. J. Chim. 1977, 1, 61–70. [Google Scholar]
- Battioni, J.-P.; Chodkiewicz, W. cheminform abstract: Reaction of organometallics with 2-methoxycyclohexanone and cis- and trans-2-methoxy-4-tert-butylcyclohexanones. Chem. Inf. 1978, 9, 320–328. [Google Scholar] [CrossRef]
- Grabowski, G.A.; Gatt, S.; Horowitz, M. Acid β-Glucosidase: Enzymology and molecular biology of Gaucher disease. Crit. Rev. Biochem. Mol. Biol. 1990, 25, 385–414. [Google Scholar] [CrossRef]
- Advances in Gaucher Disease: Basic and Clinical Perspectives Future Medicine; Future Medicine Ltd.: London, UK, 2013; ISBN 978-1-78084-201-1. [CrossRef]
- Kato, A.; Nakagome, I.; Sato, K.; Yamamoto, A.; Adachi, I.; Nash, R.J.; Fleet, G.W.J.; Natori, Y.; Watanabe, Y.; Imahori, T.; et al. Docking study and biological evaluation of pyrrolidine-based iminosugars as pharmacological chaperones for Gaucher disease. Org. Biomol. Chem. 2016, 14, 1039–1048. [Google Scholar] [CrossRef] [PubMed]
- Compain, P.; Martin, O.R.; Boucheron, C.; Godin, G.; Yu, L.; Ikeda, K.; Asano, N. Design and Synthesis of Highly Potent and Selective Pharmacological Chaperones for the Treatment of Gaucher’s disease. ChemBioChem 2006, 7, 1356–1359. [Google Scholar] [CrossRef] [PubMed]
- Oulaïdi, F.; Front-Deschamps, S.; Gallienne, E.; Lesellier, E.; Ikeda, K.; Asano, N.; Compain, P.; Martin, O.R. Second-Generation Iminoxylitol-Based Pharmacological Chaperones for the Treatment of Gaucher Disease. ChemMedChem 2011, 6, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Mellor, H.R.; Nolan, J.; Pickering, L.; Wormald, M.R.; Platt, F.M.; Dwek, R.A.; Fleet, G.W.; Butters, T.D. Preparation, biochemical characterization and biological properties of radiolabelled N-alkylated deoxynojirimycins. Biochem. J. 2002, 366, 225–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellor, H.R.; Platt, F.M.; Dwek, R.A.; Butters, T.D. Membrane disruption and cytotoxicity of hydrophobic N-alkylated imino sugars is independent of the inhibition of protein and lipid glycosylation. Biochem. J. 2003, 374, 307–314. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Samples of the compounds are not available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry a | Alkyne | Time (h) | Product | Yield (%) |
|---|---|---|---|---|
| 1 | Phenylacetylene | 2.5 | 27 | 77 |
| 2 | 1-octyne | 3 | 28 | 78 |
| 3 | 3,3-diethoxyprop-1-yne | 3 | 29 | 65 |
| 4 | 3-ethynylthiophene | 4 | 30 | 88 |
| 5 | 4-ethynyl-N,N-dimethylaniline | 4 | 31 | 83 |
 | H-4 | H-5 | H-6a | H-6b |
|---|---|---|---|---|
| δ (ppm) | δ (ppm) | δ (ppm) | δ (ppm) | |
| R = vinyl, 26 | 4.07 (d, J = 6.8 Hz) | 4.33 (br s) | 3.95–3.69 (m) | 3.53–3.32 (m) |
R =  , 35 , 35 | 3.98 (d, J = 6.4 Hz) | 4.30 (br s) | 3.68–3.55 (m) | 3.43–3.10 (m) |
 | H-4 | H-5 | H-6a | H-6b |
|---|---|---|---|---|
| δ (ppm) | δ (ppm) | δ (ppm) | δ (ppm) | |
R =  , 32 , 32 | 3.89 (br s) | 3.98–3.91 (m) | 2.86–2.76 (m) | |
R =  , 11 , 11 | 3.51 (br d, J = 2.2 Hz) | 3.81 (br s) | 2.96 (dd, J = 13.7, 4.0 Hz) | 2.76–2.70 (m) |
| R = octyl, 12 | 3.44 (br s) | 3.77 (br s) | 2.92 (d, J = 12.6 Hz) | 2.68 (d, J = 12.6 Hz) |
| R = ethyl, 13 | 3.47 (d, J = 3.2 Hz) | 3.81 (br s) | 2.96 (dd, J = 13.6, 3.4 Hz) | 2.72 (dd, J = 13.7, 2.4 Hz) |
R =  , 14 , 14 | 3.53 (br d, J = 2.7 Hz) | 3.86 (br s) | 3.02 (dd, J = 13.6, 3.8 Hz) | 2.79 (br d, J = 13.6 Hz) |
R =  , 15 , 15 | 3.49 (br s) | 3.80 (br s) | 2.95 (d, J = 13.2 Hz) | 2.70 (d, J = 13.8 Hz) |
| Entry | Compound | β-Gal | GCase | |
|---|---|---|---|---|
| Inhibition (%) a | Inhibition (%) a | IC50 (µM) b | ||
| 1 |  | 22 | 98 | 12 ± 6 |
| 2 |  | 0 | 100 | 6.4 ± 0.7 |
| 3 |  | 0 | 36 | n.d. |
| 4 |  | 0 | 93 | 60 ± 23 |
| 5 |  | 3 | 30 | n.d. |
| 6 |  | 16 | 9 | n.d. |
| 7 |  | 6 | 6 | n.d. |
| 8 |  | 0 | 100 | 130 ± 13 |
| 9 |  | 0 | 25 | n.d. |
| 10 |  | 4 | 65 | n.d. |
| 11 |  | 15 | 93 c | 40 ± 3 c |
| 12 |  | 14 d | 100 d | 29 ± 2 d |
| 13 |  | 5 d | 100 d | 1.5 ± 0.1 d |
| 14 |  | 31 d | 80 d | 94 ± 5 d |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Davighi, M.G.; Clemente, F.; Matassini, C.; Morrone, A.; Goti, A.; Martínez-Bailén, M.; Cardona, F. Synthesis of “All-Cis” Trihydroxypiperidines from a Carbohydrate-Derived Ketone: Hints for the Design of New β-Gal and GCase Inhibitors. Molecules 2020, 25, 4526. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25194526
Davighi MG, Clemente F, Matassini C, Morrone A, Goti A, Martínez-Bailén M, Cardona F. Synthesis of “All-Cis” Trihydroxypiperidines from a Carbohydrate-Derived Ketone: Hints for the Design of New β-Gal and GCase Inhibitors. Molecules. 2020; 25(19):4526. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25194526
Chicago/Turabian StyleDavighi, Maria Giulia, Francesca Clemente, Camilla Matassini, Amelia Morrone, Andrea Goti, Macarena Martínez-Bailén, and Francesca Cardona. 2020. "Synthesis of “All-Cis” Trihydroxypiperidines from a Carbohydrate-Derived Ketone: Hints for the Design of New β-Gal and GCase Inhibitors" Molecules 25, no. 19: 4526. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25194526