
Targeting Beta-Blocker Drug–Drug Interactions with Fibrinogen Blood Plasma Protein: A Computational and Experimental Study

 , , , , and
, , , , and 
Abstract
:
1. Introduction
2. Results and Discussion
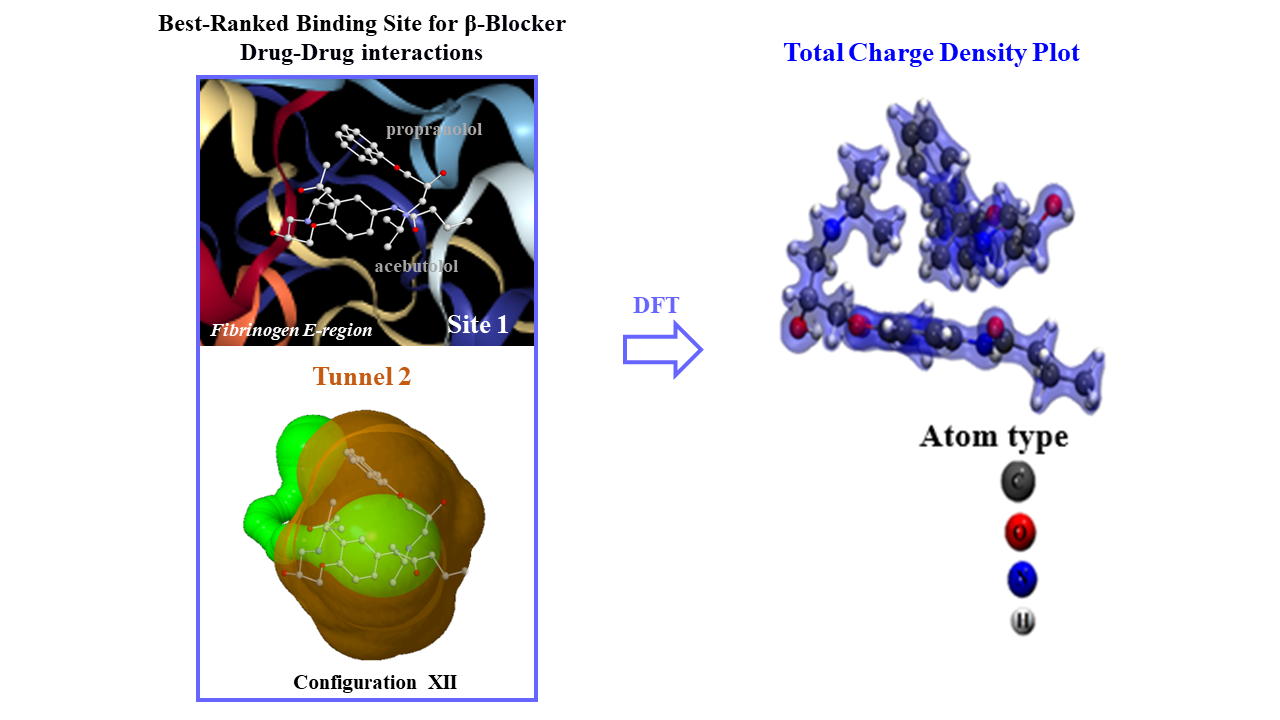
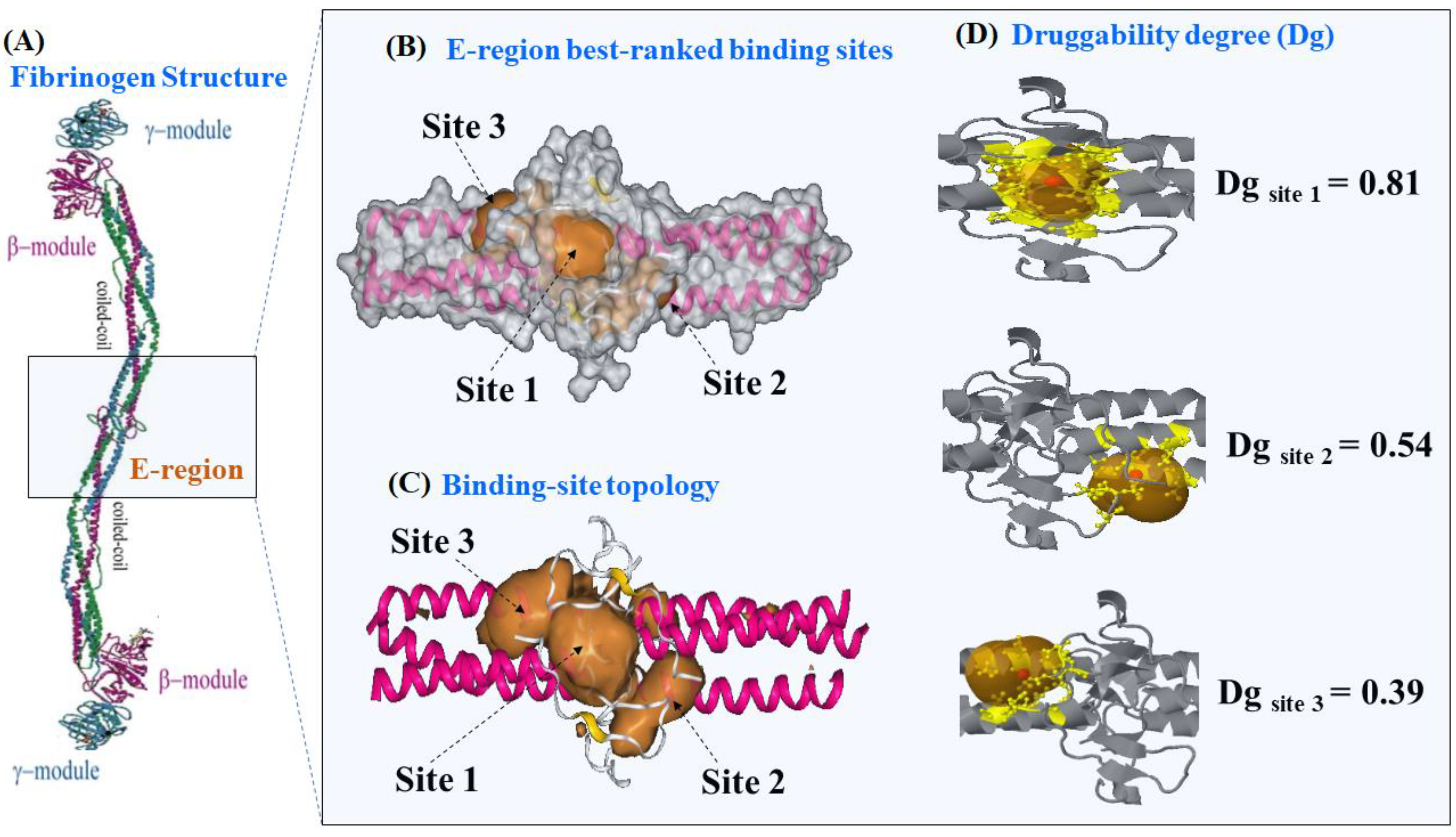
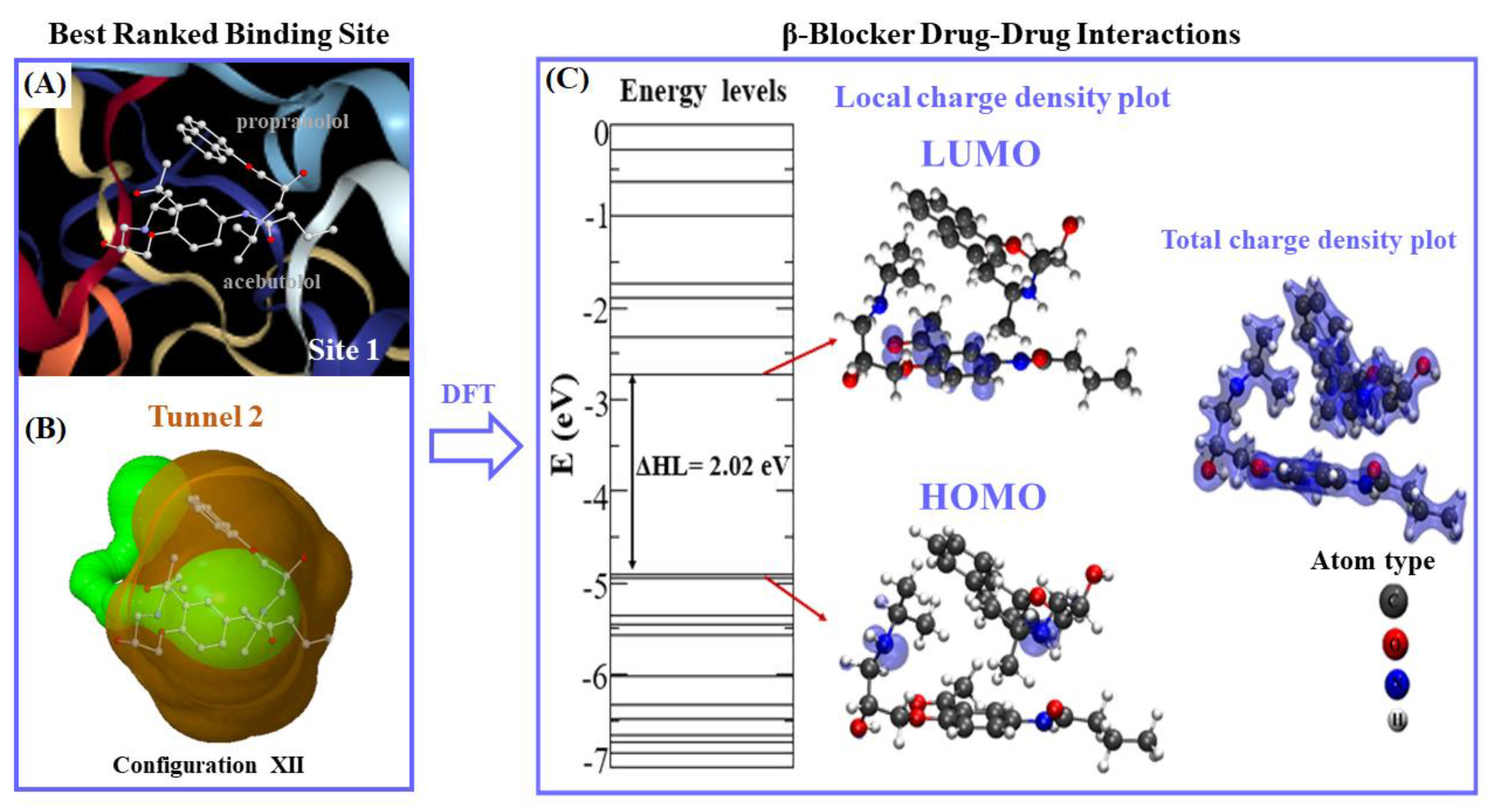
2.1. Prediction of the Binding Sites for Fibrinogen
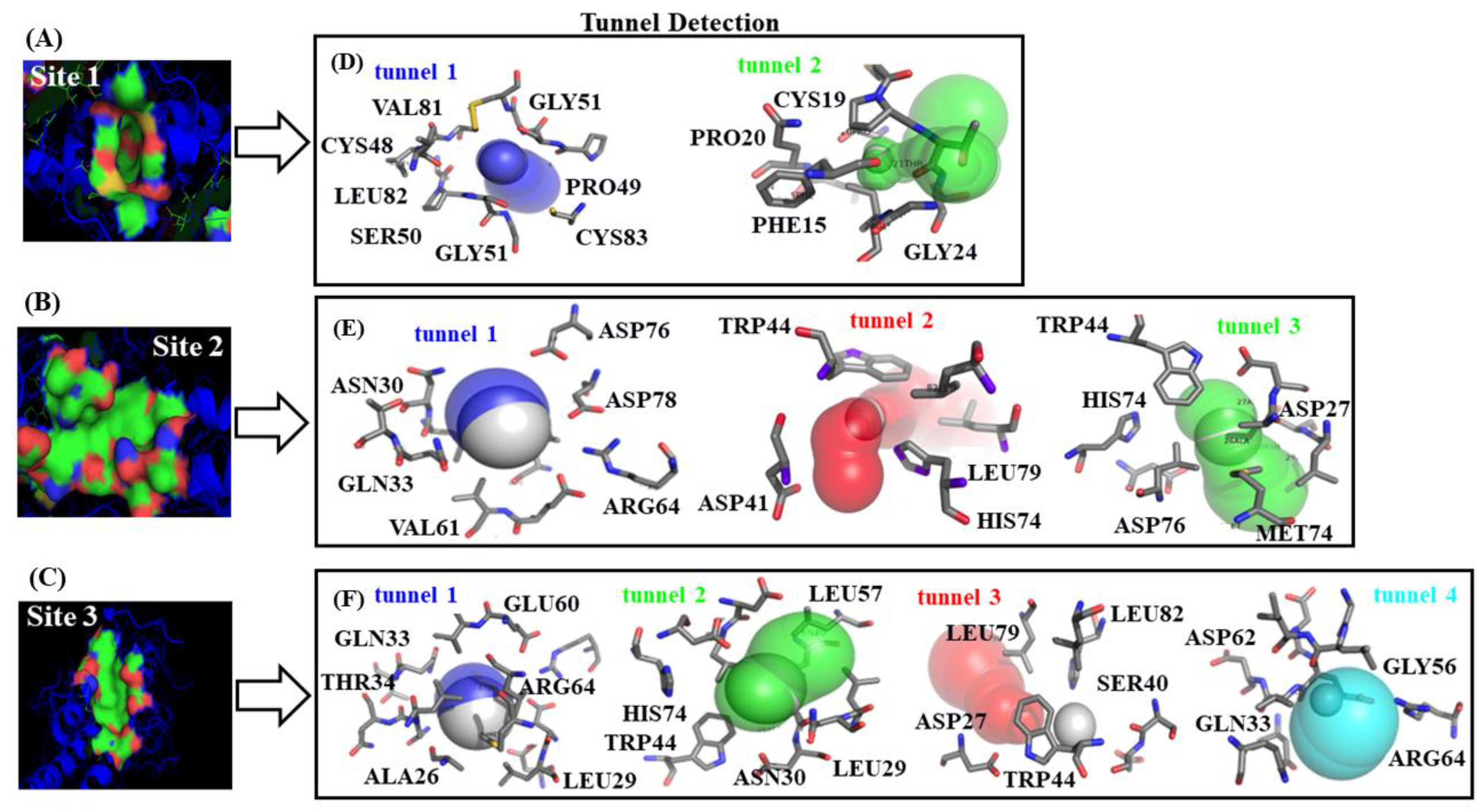
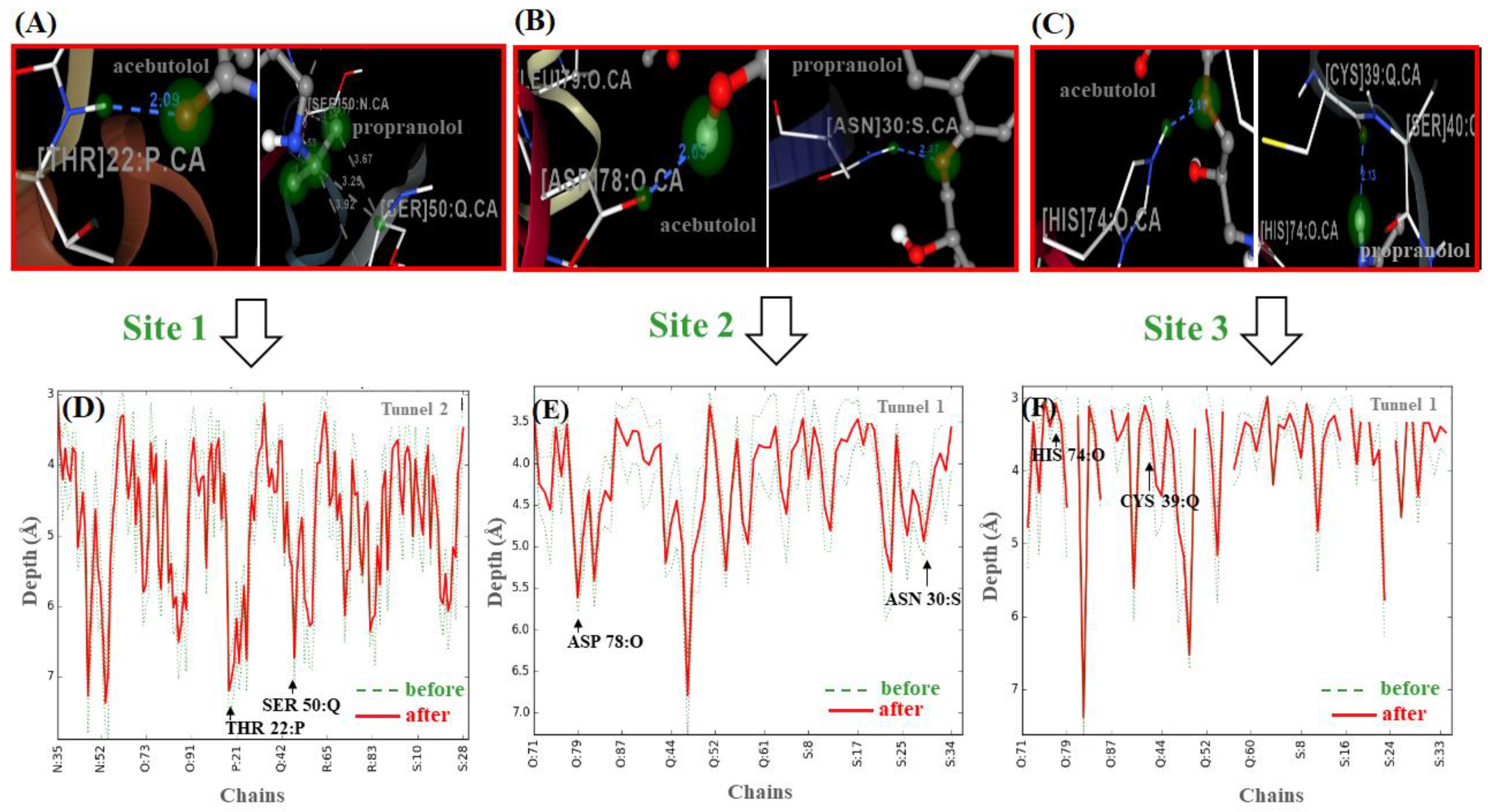
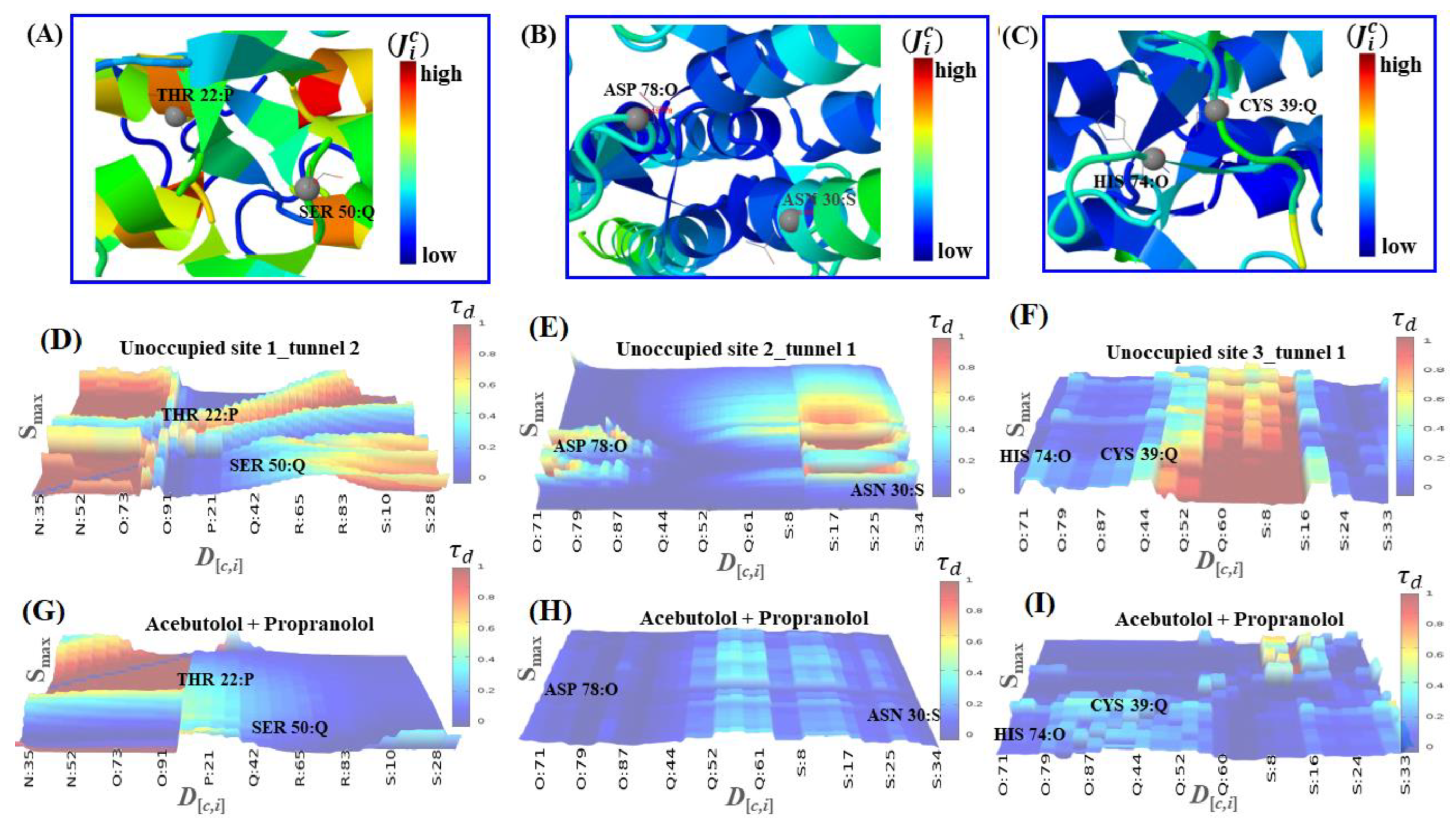
2.2. Identification of Tunnels for the Fibrinogen Binding Sites
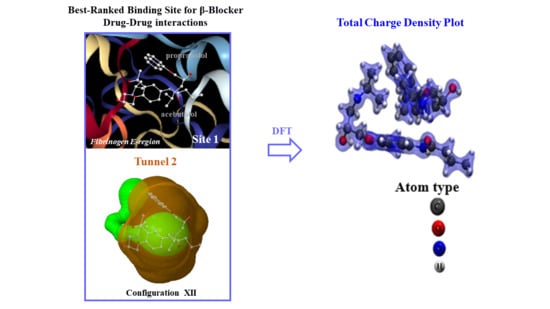
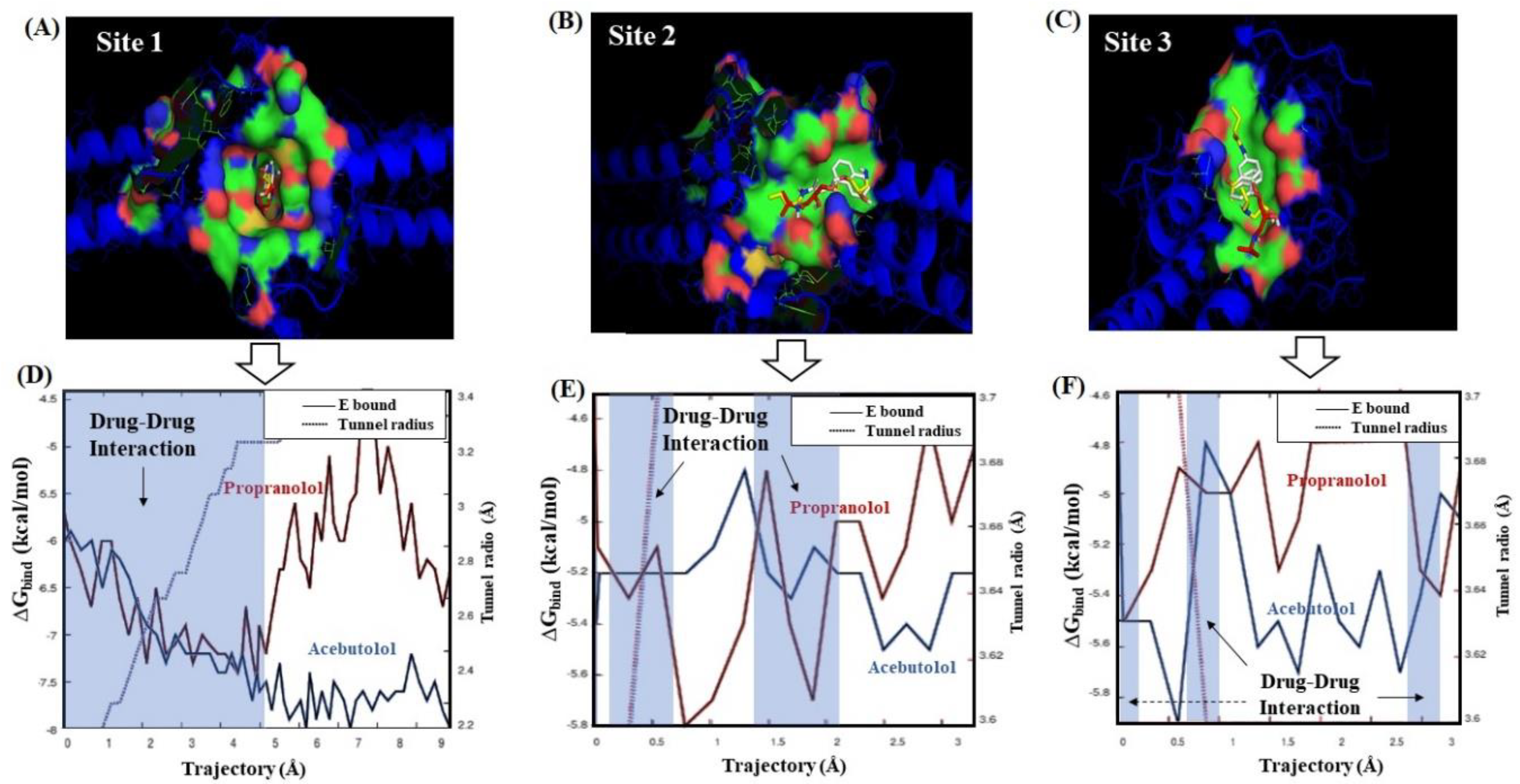
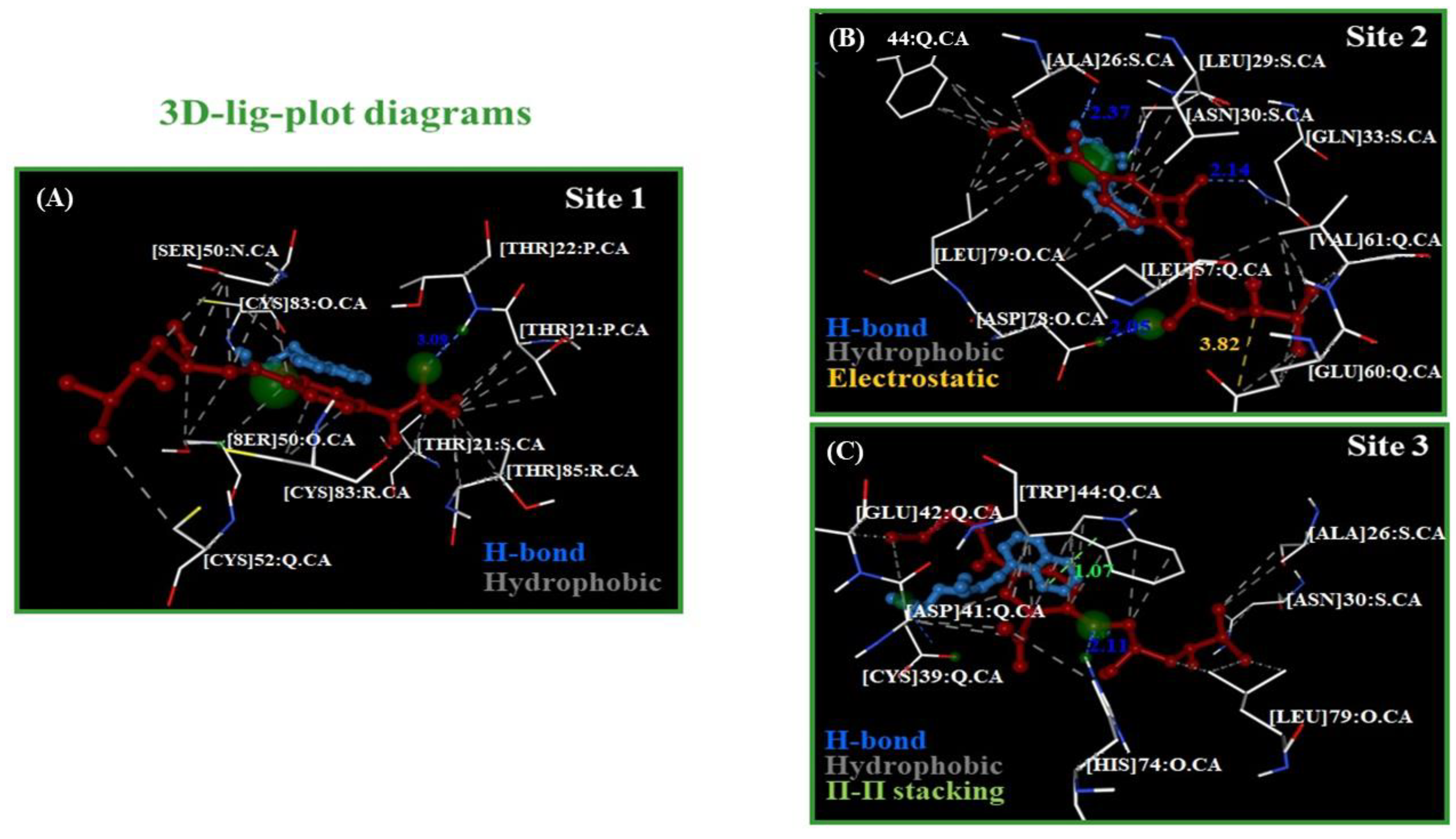
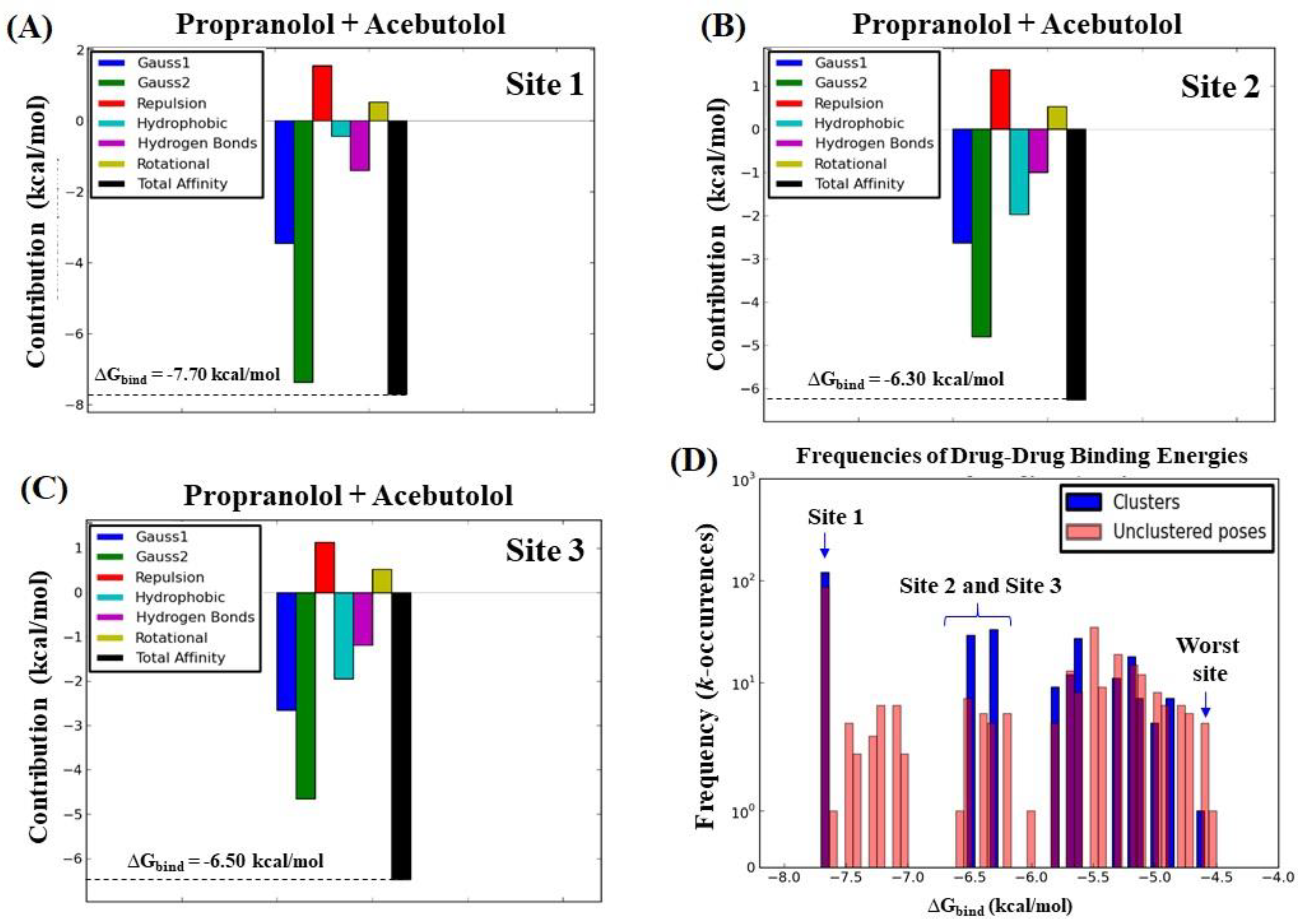
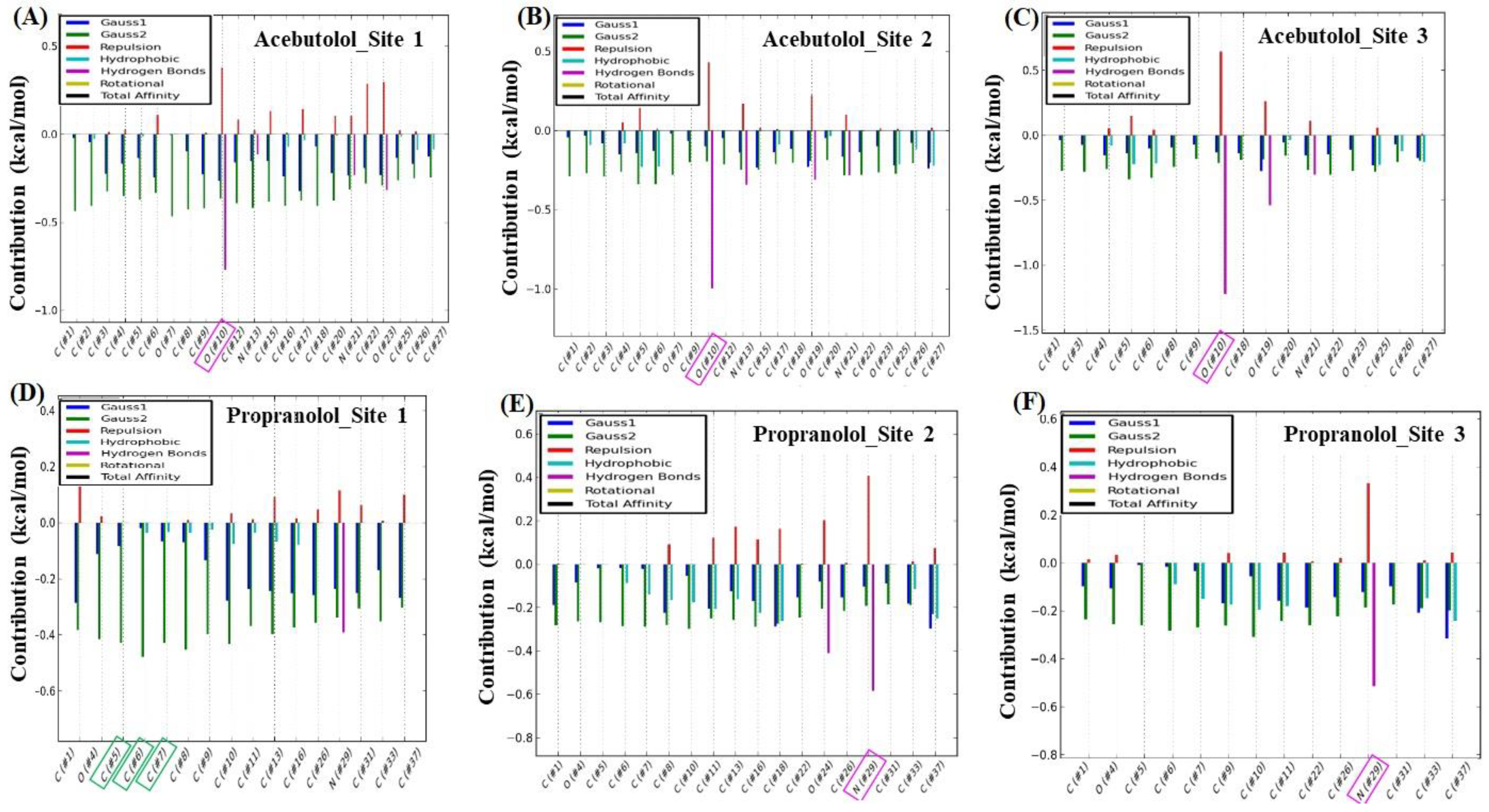
2.3. Calculation of Energetic Contributions for Binding Affinity
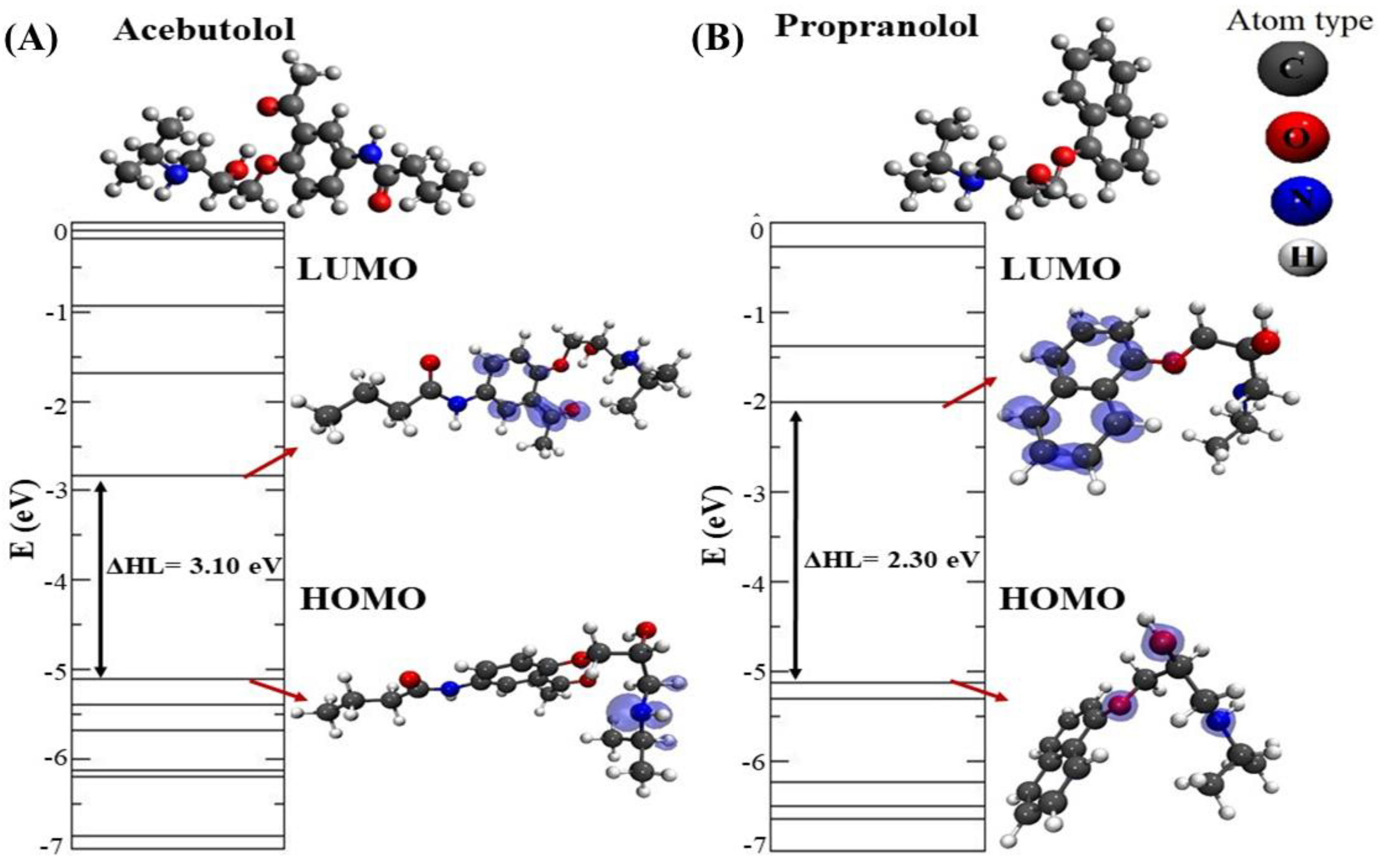
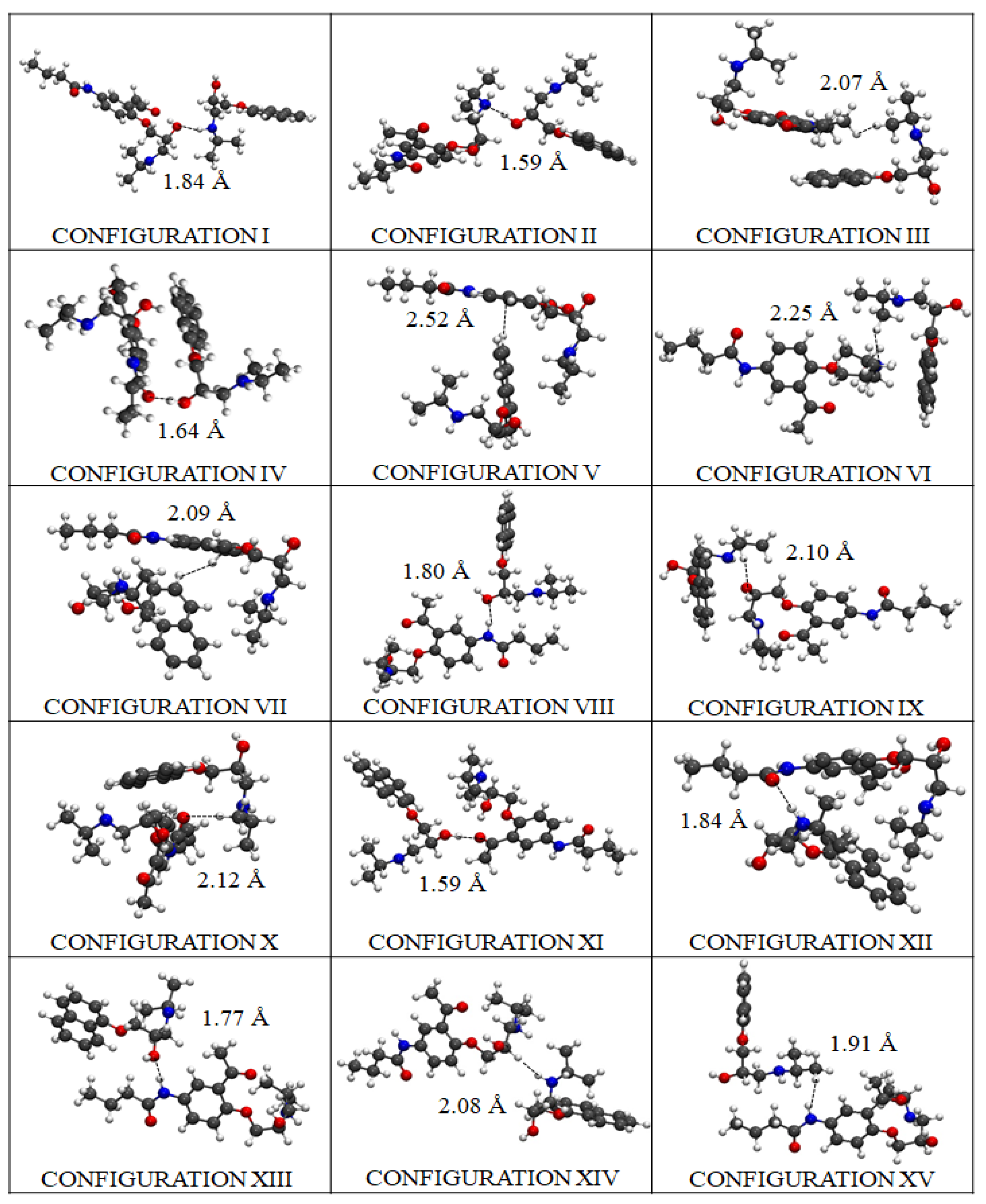
2.4. DFT Modeling of Beta-Blocker Drug–Drug Binding Systems
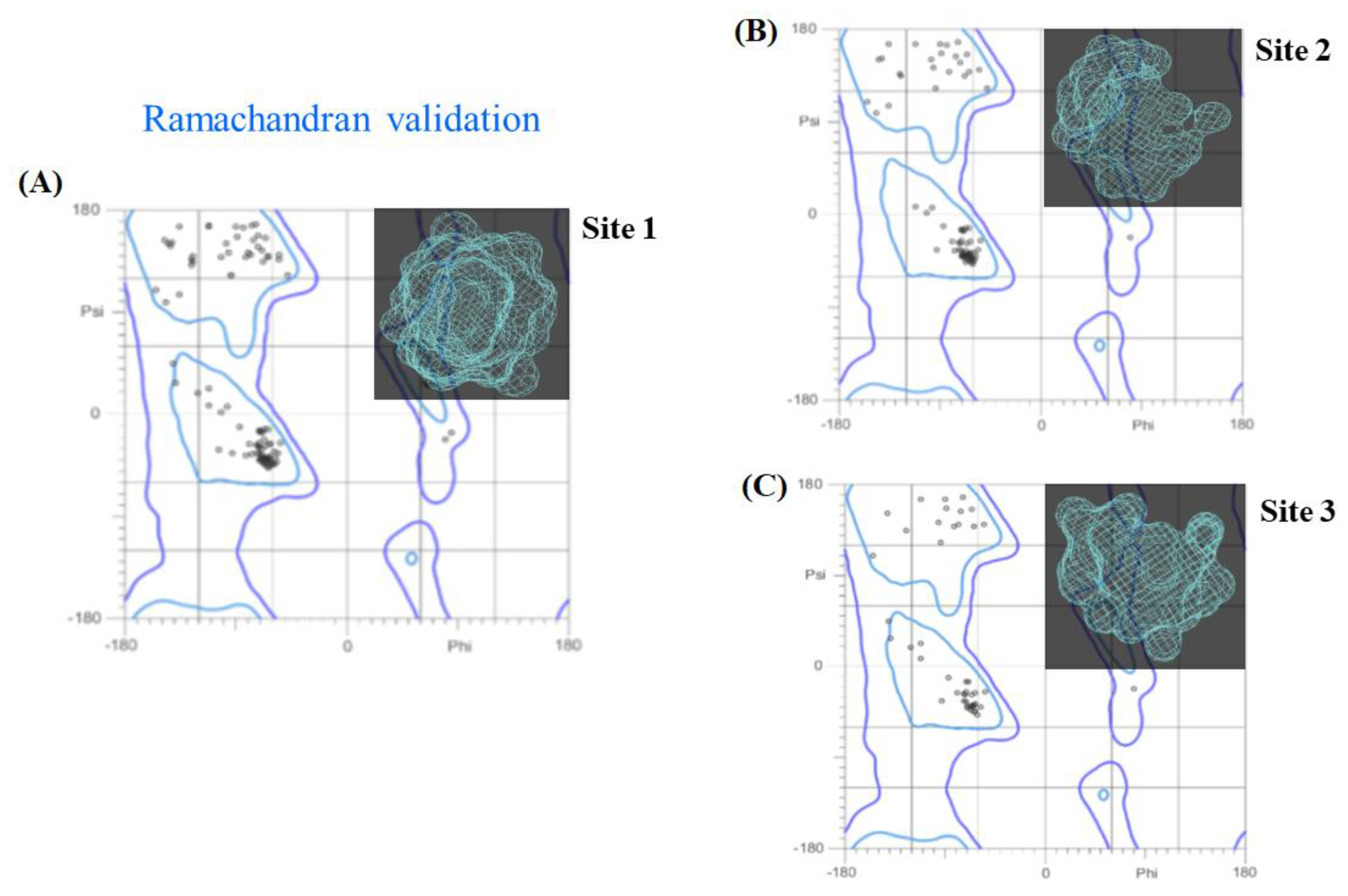
2.5. Experimental Validation
3. Materials and Methods
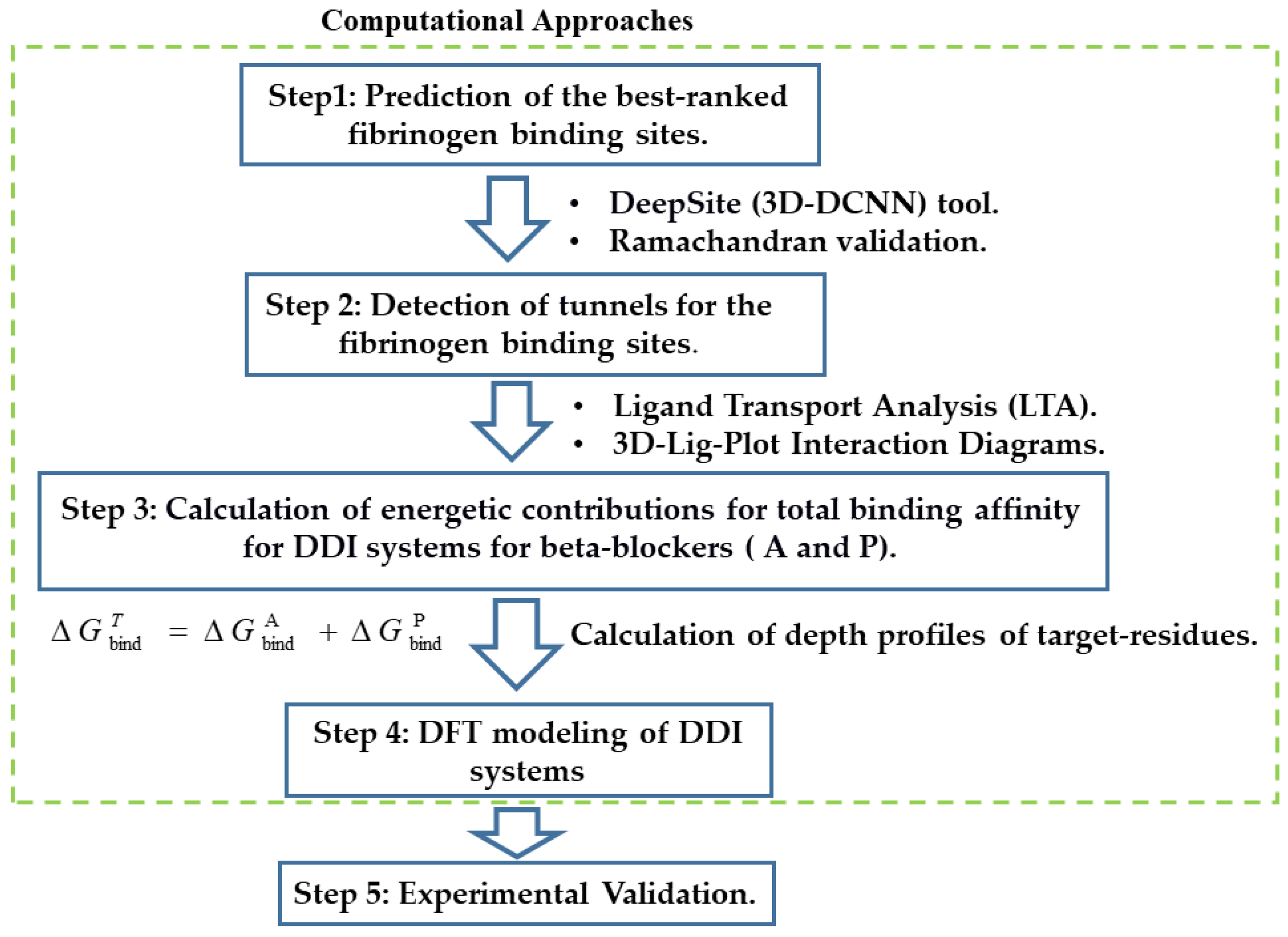
General Workflow
- (i)
- Predict the binding sites for the fibrinogen E-region by applying an appropriate machine learning framework.
- (ii)
- Select from the fibrinogen E-region binding sites found, the three best-ranked ones based on their highest druggability. For comparison purposes, ascertain as well the undruggable or “worst fibrinogen E-region binding site” to be used as reference control denoting absence of drug–drug interactions.
- (iii)
- Validate the three best-ranked binding sites found by examining their Ramachandran plots.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Niu, J.; Straubinger, R.M.; Mager, D.E. Pharmacodynamic drug–drug interactions. Clin. Pharmacol. Ther. 2019, 105, 1395–1406. [Google Scholar] [CrossRef] [PubMed]
- Boran, A.D.; Iyengar, R. Systems approaches to polypharmacology and drug discovery. Curr. Opin. Drug Discov. Devel. 2010, 13, 297–309. [Google Scholar] [PubMed]
- Mestres, J.; Gregori-Puigjané, E. Conciliating binding efficiency and polypharmacology. Trends Pharmacol. Sci. 2009, 30, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.-U. Polypharmacology—Foe or Friend? J. Med. Chem. 2013, 56, 8955–8971. [Google Scholar] [CrossRef]
- Hu, Y.; Bajorath, J. Compound promiscuity: What can we learn from current data? Drug Discov. Today 2013, 18, 644–650. [Google Scholar] [CrossRef]
- Simon, Z.; Peragovics, A.; Vigh-Smeller, M.; Csukly, G.; Tombor, L.; Yang, Z.; Zahoránszky-Kőhalmi, G.; Végner, L.; Jelinek, B.; Hari, P. Drug effect prediction by polypharmacology-based interaction profiling. J. Chem. Inf. Model. 2011, 52, 134–145. [Google Scholar] [CrossRef]
- Chaudhari, R.; Tan, Z.; Huang, B.; Zhang, S. Computational polypharmacology: A new paradigm for drug discovery. Expert Opin. Drug Discov. 2017, 12, 279–291. [Google Scholar] [CrossRef]
- Ruso, J.M.; González-Pérez, A.; Prieto, G.; Sarmiento, F. A volumetric study of two related amphiphilic beta-blockers as a function of temperature and electrolyte concentration. Colloids Surf. B 2004, 33, 165–175. [Google Scholar]
- Johnsson, G.; Regardh, C.G. Clinical pharmacokinetics of beta-adrenoreceptor blocking drugs. Clin. Pharmacokinet. 1976, 1, 233–263. [Google Scholar] [CrossRef]
- Borchard, U. Pharmacokinetics of beta-adrenoceptor blocking agents: Clinical significance of hepatic and/or renal clearance. Clin. Physiol. Bioch. 1990, 8, 28–34. [Google Scholar]
- Lounkine, E.; Keiser, M.J.; Whitebread, S.; Mikhailov, D.; Hamon, J.; Jenkins, J.L.; Lavan, P.; Weber, E.; Doak, A.K.; Côté, S. Large-scale prediction and testing of drug activity on side-effect targets. Nature 2012, 486, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Zitnik, M.; Agrawal, M.; Leskovec, J. Modeling polypharmacy side effects with graph convolutional networks. Bioinformatics 2018, 34, i457–i466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moya-García, A.; Adeyelu, T.; Kruger, F.A.; Dawson, N.L.; Lees, J.G.; Overington, J.P.; Orengo, C.; Ranea, J.A. Structural and functional view of polypharmacology. Sci. Rep. 2017, 7, 10102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmer, A.; Katzir, I.; Dekel, E.; Mayo, A.E.; Alon, U. Prediction of multidimensional drug dose responses based on measurements of drug pairs. Proc. Natl. Acad. Sci. USA 2016, 113, 10442–10447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loewe, S. Effect of combinations: Mathematical basis of problem. Arch. Exp. Pathol. Pharmakol. 1926, 114, 313–326. [Google Scholar] [CrossRef]
- Lehár, J.; Zimmermann, G.R.; Krueger, A.S.; Molnar, R.A.; Ledell, J.T.; Heilbut, A.M.; Short, G.F.; Giusti, L.C.; Nolan, G.P.; Magid, O.A. Chemical combination effects predict connectivity in biological systems. Mol. Syst. Biol. 2007, 3, 80. [Google Scholar] [CrossRef]
- Ryu, J.Y.; Kim, H.U.; Lee, S.Y. Deep learning improves prediction of drug-drug and drug-food interactions. Proc. Natl. Acad. Sci. USA 2018, 115, E4304–E4311. [Google Scholar] [CrossRef] [Green Version]
- Owens, J. Target validation: Determining druggability. Nat. Rev. Drug Discov. 2007, 6, 187. [Google Scholar] [CrossRef]
- Cheng, A.C.; Coleman, R.G.; Smyth, K.T.; Cao, Q.; Soulard, P.; Caffrey, D.R.; Salzberg, A.C.; Huang, E.S. Structure-based maximal affinity model predicts small-molecule druggability. Nat. Biotechnol. 2007, 25, 71. [Google Scholar] [CrossRef]
- Bakan, A.; Nevins, N.; Lakdawala, A.S.; Bahar, I. Druggability assessment of allosteric proteins by dynamics simulations in the presence of probe molecules. J. Chem. Theory Comput. 2012, 8, 2435–2447. [Google Scholar] [CrossRef]
- Pasznik, P.; Rutkowska, E.; Niewieczerzal, S.; Cielecka-Piontek, J.; Latek, D. Potential off-target effects of beta-blockers on gut hormone receptors: In silico study including GUT-DOCK—A web service for small-molecule docking. PLoS ONE 2019, 14, e0210705. [Google Scholar] [CrossRef] [PubMed]
- Hassan, N.; Maldonado-Valderrama, J.; Gunning, A.P.; Morris, V.J.; Ruso, J.M. Surface characterization and AFM imaging of mixed fibrinogen—Surfactant films. J. Phys. Chem. B 2011, 115, 6304–6311. [Google Scholar] [CrossRef] [PubMed]
- Hassan, N.; Ruso, J.M.; Somasundaran, P. Mechanisms of fibrinogen–acebutolol interactions: Insights from DSC, CD and LS. Colloids Surf. B 2011, 82, 581–587. [Google Scholar] [CrossRef]
- Pucci, G.; Ranalli, M.G.; Battista, F.; Schillaci, G. Effects of β-blockers with and without vasodilating properties on central blood pressure: Systematic review and meta-analysis of randomized trials in hypertension. Hypertension 2016, 67, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Bratek-Skicki, A.; Żeliszewska, P.; Ruso, J.M. Fibrinogen: A journey into biotechnology. Soft Matter 2016, 12, 8639–8653. [Google Scholar] [CrossRef]
- Madrazo, J.; Brown, J.H.; Litvinovich, S.; Dominguez, R.; Yakovlev, S.; Medved, L.; Cohen, C. Crystal structure of the central region of bovine fibrinogen (E5 fragment) at 1.4-Å resolution. Proc. Natl. Acad. Sci. USA 2001, 98, 11967–11972. [Google Scholar] [CrossRef] [Green Version]
- Kollman, J.M.; Pandi, L.; Sawaya, M.R.; Riley, M.; Doolittle, R.F. Crystal structure of human fibrinogen. Biochemistry 2009, 48, 3877–3886. [Google Scholar] [CrossRef]
- Pechik, I.; Yakovlev, S.; Mosesson, M.W.; Gilliland, G.L.; Medved, L. Structural basis for sequential cleavage of fibrinopeptides upon fibrin assembly. Biochemistry 2006, 45, 3588–3597. [Google Scholar] [CrossRef] [Green Version]
- Weisel, J.W.; Phillips, J.G.; Cohen, C. The structure of fibrinogen and fibrin: II. Architecture of the fibrin clot. Ann. N.Y. Acad. Sci. 1983, 408, 367–379. [Google Scholar] [CrossRef]
- Brown, J.H.; Volkmann, N.; Jun, G.; Henschen-Edman, A.H.; Cohen, C. The crystal structure of modified bovine fibrinogen. Proc. Natl. Acad. Sci. USA 2000, 97, 85–90. [Google Scholar] [CrossRef] [Green Version]
- Hassan, N.; Barbosa, L.R.; Itri, R.; Ruso, J.M. Fibrinogen stability under surfactant interaction. J. Colloid Interface Sci. 2011, 362, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, J.; Doerr, S.; Martínez-Rosell, G.; Rose, A.S.; De Fabritiis, G. DeepSite: Protein-binding site predictor using 3D-convolutional neural networks. Bioinformatics 2017, 33, 3036–3042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rarey, M.; Kramer, B.; Lengauer, T. Multiple automatic base selection: Protein–ligand docking based on incremental construction without manual intervention. J. Comput. Aided Mol. 1997, 11, 369–384. [Google Scholar] [CrossRef] [PubMed]
- Seeliger, D.; De Groot, B.L. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J. Comput. Aided Mol. 2010, 24, 417–422. [Google Scholar] [CrossRef] [Green Version]
- Shoichet, B.K. Virtual screening of chemical libraries. Nature 2004, 432, 862. [Google Scholar] [CrossRef]
- Stourac, J.; Vavra, O.; Kokkonen, P.; Filipovic, J.; Pinto, G.; Brezovsky, J.; Damborsky, J.; Bednar, D. Caver Web 1.0: Identification of tunnels and channels in proteins and analysis of ligand transport. Nucleic Acids Res. 2019, 47, W414–W422. [Google Scholar] [CrossRef] [Green Version]
- Le Guilloux, V.; Schmidtke, P.; Tuffery, P. Fpocket: An open source platform for ligand pocket detection. BMC Bioinformatics 2009, 10, 168. [Google Scholar] [CrossRef] [Green Version]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Tan, K.P.; Nguyen, T.B.; Patel, S.; Varadarajan, R.; Madhusudhan, M.S. Depth: A web server to compute depth, cavity sizes, detect potential small-molecule ligand-binding cavities and predict the pKa of ionizable residues in proteins. Nucleic Acids Res. 2013, 41, W314–W321. [Google Scholar] [CrossRef]
- Tan, K.P.; Varadarajan, R.; Madhusudhan, M.S. DEPTH: A web server to compute depth and predict small-molecule binding cavities in proteins. Nucleic Acids Res. 2011, 29, W242–W248. [Google Scholar] [CrossRef]
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational protein–ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016, 11, 905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef] [Green Version]
- González-Durruthy, M.; Scanavachi, G.; Rial, R.; Liu, Z.; Cordeiro, M.N.D.S.; Itri, R.; Ruso, J.M. Structural and energetic evolution of fibrinogen toward to the betablocker interactions. Int. J. Biol. Macromol. 2019, 137, 405–419. [Google Scholar] [CrossRef] [PubMed]
- Tao, A.; Huang, Y.; Shinohara, Y.; Caylor, M.L.; Pashikanti, S.; Xu, D. ezCADD: A Rapid 2D/3D visualization-enabled web modeling environment for democratizing computer-aided drug design. J. Chem. Inf. Model. 2018, 59, 18–24. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand–protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Newell, N.E. Mapping side chain interactions at protein helix termini. BMC Bioinformatics 2015, 16, 231. [Google Scholar] [CrossRef] [Green Version]
- Bachmann, A.; Wildemann, D.; Praetorius, F.; Fischer, G.; Kiefhaber, T. Mapping backbone and side-chain interactions in the transition state of a coupled protein folding and binding reaction. Proc. Natl. Acad. Sci. USA 2011, 108, 3952–3957. [Google Scholar] [CrossRef] [Green Version]
- Stefan, M.I.; Edelstein, S.J.; Le Novère, N. Computing phenomenologic Adair-Klotz constants from microscopic MWC parameters. BMC Syst. Biol. 2009, 3, 68. [Google Scholar] [CrossRef] [Green Version]
- Haiech, J.; Gendrault, Y.; Kilhoffer, M.-C.; Ranjeva, R.; Madec, M.; Lallement, C. A general framework improving teaching ligand binding to a macromolecule. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 2348–2355. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, J.E.; Spector, A.A.; Ashbrook, J.D. Analysis of macromolecule-ligand binding by determination of stepwise equilibrium constants. Biochemistry 1970, 9, 4580–4587. [Google Scholar] [CrossRef]
- Konkoli, Z. Safe uses of Hill’s model: An exact comparison with the Adair-Klotz model. Theor. Biol. Medical Model. 2011, 8, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menezes, V.M.; Michelon, E.; Rossato, J.; Zanella, I.; Fagan, S.B. Carbon nanostructures interacting with vitamins A, B3 and C: Ab initio simulations. J. Biomed. Nanotechnol. 2012, 8, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Linares, I.; Pérez-Sánchez, H.; Cecilia, J.M.; García, J.M. High-throughput parallel blind virtual screening using BINDSURF. BMC Bioinformatics 2012, 13, S13. [Google Scholar]
- Chovancova, E.; Pavelka, A.; Benes, P.; Strnad, O.; Brezovsky, J.; Kozlikova, B.; Gora, A.; Sustr, V.; Klvana, M.; Medek, P. CAVER 3.0: A tool for the analysis of transport pathways in dynamic protein structures. PLoS Comput. Biol. 2012, 8, e1002708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Z.-R.; Hwang, M.-J. An interaction-motif-based scoring function for protein-ligand docking. BMC Bioinformatics 2010, 11, 298. [Google Scholar] [CrossRef] [Green Version]
- Feldman, H.A. Mathematical theory of complex ligand-binding systems at equilibrium: Some methods for parameter fitting. Anal. Biochem. 1972, 48, 317–338. [Google Scholar] [CrossRef]
- Soler, J.M.; Artacho, E.; Gale, J.D.; García, A.; Junquera, J.; Ordejón, P.; Sánchez-Portal, D. The SIESTA method for ab initio order-N materials simulation. J. Condens. Matter Phys. 2002, 14, 2745–2779. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Xu, Z.; Moe, Y.N. On the performance of local density approximation in describing the adsorption of electron donating/accepting molecules on graphene. Chem. Phys. 2012, 406, 78–85. [Google Scholar] [CrossRef] [Green Version]
- Arrigoni, M.; Madsen, G.K. Comparing the performance of LDA and GGA functionals in predicting the lattice thermal conductivity of III-V semiconductor materials in the zincblende structure: The cases of AlAs and BAs. Comput. Mater. Sci. 2019, 156, 354–360. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, A.R.; Saleem, H.; Subashchandrabose, S.; Sundaraganesan, N.; Sebastain, S. Molecular structure, vibrational spectroscopic (FT-IR, FT-Raman), UV and NBO analysis of 2-chlorobenzonitrile by density functional method. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2011, 78, 582–589. [Google Scholar] [CrossRef]
- Troullier, N.; Martins, J.L. Efficient pseudopotentials for plane-wave calculations. Phys. Rev. B 1991, 43, 1993–2006. [Google Scholar] [CrossRef]
- Motomura, K.; Yamanaka, M.; Aratono, M. Thermodynamic consideration of the mixed micelle of surfactants. Colloid Polym. Sci. 1984, 262, 948–955. [Google Scholar] [CrossRef]
- Holland, P.M. Mixed Surfactant Systems; ACS Symposium Series; Holland, P.M., Rubingh, D.N., Eds.; American Chemical Society: Washington, DC, USA, 1992; Volume 501, Chapter 2; pp. 31–44. [Google Scholar]
- Landau, L.D.; Lifshitz, E.M. Course of Theoretical Physics, Vol 5: Statistical Physics; Pergamon Press: Oxford, UK, 1987. [Google Scholar]
- Ruso, J.M.; Attwood, D.; Rey, C.; Taboada, P.; Mosquera, V.; Sarmiento, F. Light scattering and NMR studies of the self-association of the amphiphilic molecule propranolol hydrochloride in aqueous electrolyte solutions. J. Phys. Chem. B 1999, 103, 7092–7096. [Google Scholar] [CrossRef]
- Blanco, E.; Verdes, P.V.; Ruso, J.M.; Prieto, G.; Sarmiento, F. Interactions in binary mixed systems involving betablockers with different lipophilicity as a function of temperature and mixed ratios. Colloids Surf. A Physicochem. Eng. Asp. 2009, 334, 116–123. [Google Scholar] [CrossRef]
- Westbrook, J.; Feng, Z.; Chen, L.; Yang, H.; Berman, H.M. The protein data bank and structural genomics. Nucleic Acids Res. 2003, 31, 489–491. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Gerard Martínez-Rosell, G.; Giorgino, T.; De Fabritiis, G. PlayMolecule ProteinPrepare: A Web Application for Protein Preparation for Molecular Dynamics Simulations. J. Chem. Inf. Model. 2017, 57, 1511–1516. [Google Scholar] [CrossRef]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A. PubChem substance and compound databases. Nucleic Acids Res. 2015, 44, D1202–D1213. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminformatics 2012, 4, 17. [Google Scholar] [CrossRef] [Green Version]
- Feinstein, W.P.; Brylinski, M. Calculating an optimal box size for ligand docking and virtual screening against experimental and predicted binding pockets. J. Cheminformatics 2015, 7, 18. [Google Scholar] [CrossRef] [Green Version]
- Quiroga, R.; Villarreal, M.A. Vinardo: A scoring function based on Autodock Vina improves scoring, docking, and virtual screening. PLoS ONE 2016, 11, e0155183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marti-Renom, M.A.; Madhusudhan, M.; Sali, A. Alignment of protein sequences by their profiles. Protein Sci. 2004, 13, 1071–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864. [Google Scholar] [CrossRef] [Green Version]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133. [Google Scholar]
- Sankey, O.F.; Niklewski, D.J. Ab initio multicenter tight-binding model for molecular-dynamics simulations and other applications in covalent systems. Phys. Rev. B 1989, 40, 3979–3995. [Google Scholar] [CrossRef]
- Perdew, J.P.; Zunger, A. Self-interaction correction to density-functional approximations for many-electron systems. Phys. Rev. B 1981, 23, 5048–5079. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Samples of the beta-blocker compounds (propranolol and acebutolol) are available from the authors (Spain). |














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ranking | Configuration | |Eb| (eV) | ΔHL (eV) | dA–P(Å) |
|---|---|---|---|---|
| 1 | XII | 2.40 | 2.02 | 1.84 (O-H) |
| 2 | IV | 2.36 | 2.11 | 1.64 (O-H) |
| 3 | II | 1.99 | 1.87 | 1.59 (N-H) |
| 4 | XIII | 1.99 | 2.26 | 1.77 (H-O) |
| 5 | XIV | 1.99 | 2.15 | 2.08 (N-H) |
| 6 | XI | 1.97 | 1.63 | 1.59 (O-H) |
| 7 | VI | 1.92 | 1.90 | 2.25 (N-H) |
| 8 | VII | 1.88 | 2.13 | 2.09 (H-H) |
| 9 | X | 1.84 | 2.29 | 2.12 (O-H) |
| 10 | VIII | 1.83 | 2.30 | 1.80 (H-O) |
| 11 | III | 1.82 | 2.22 | 2.07 (H-H) |
| 12 | I | 1.76 | 1.47 | 1.84 (O-H) |
| 13 | IX | 1.71 | 2.20 | 2.10 (O-H) |
| 14 | V | 1.67 | 1.97 | 2.52 (C-H) |
| 15 | XV | 1.67 | 2.21 | 1.91 (H-H) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Durruthy, M.; Concu, R.; Vendrame, L.F.O.; Zanella, I.; Ruso, J.M.; Cordeiro, M.N.D.S. Targeting Beta-Blocker Drug–Drug Interactions with Fibrinogen Blood Plasma Protein: A Computational and Experimental Study. Molecules 2020, 25, 5425. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25225425
González-Durruthy M, Concu R, Vendrame LFO, Zanella I, Ruso JM, Cordeiro MNDS. Targeting Beta-Blocker Drug–Drug Interactions with Fibrinogen Blood Plasma Protein: A Computational and Experimental Study. Molecules. 2020; 25(22):5425. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25225425
Chicago/Turabian StyleGonzález-Durruthy, Michael, Riccardo Concu, Laura F. Osmari Vendrame, Ivana Zanella, Juan M. Ruso, and M. Natália D. S. Cordeiro. 2020. "Targeting Beta-Blocker Drug–Drug Interactions with Fibrinogen Blood Plasma Protein: A Computational and Experimental Study" Molecules 25, no. 22: 5425. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25225425