
Mitocanic Di- and Triterpenoid Rhodamine B Conjugates



1
Organic Chemistry, Martin-Luther University Halle-Wittenberg, Kurt-Mothes Street 2, D-06120 Halle, Germany
2
Medical and Life Science Faculty, Institute of Precision Medicine, Furtwangen University, Jakob–Kienzle–Street 17, D-78054 Villigen–Schwenningen, Germany
*
Author to whom correspondence should be addressed.
Molecules 2020, 25(22), 5443; https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25225443
Submission received: 29 October 2020
/
Revised: 15 November 2020
/
Accepted: 16 November 2020
/
Published: 20 November 2020
(This article belongs to the Special Issue Anticancer Properties of Natural and Derivative Products)
Abstract
:The combination of the “correct” triterpenoid, the “correct” spacer and rhodamine B (RhoB) seems to be decisive for the ability of the conjugate to accumulate in mitochondria. So far, several triterpenoid rhodamine B conjugates have been prepared and screened for their cytotoxic activity. To obtain cytotoxic compounds with EC50 values in a low nano-molar range combined with good tumor/non-tumor selectivity, the Rho B unit has to be attached via an amine spacer to the terpenoid skeleton. To avoid spirolactamization, secondary amines have to be used. First results indicate that a homopiperazinyl spacer is superior to a piperazinyl spacer. Hybrids derived from maslinic acid or tormentic acid are superior to those from oleanolic, ursolic, glycyrrhetinic or euscaphic acid. Thus, a tormentic acid-derived RhoB conjugate 32, holding a homopiperazinyl spacer can be regarded, at present, as the most promising candidate for further biological studies.
1. Introduction
Cancer remains one of the leading causes of death worldwide, and the incidence is increasing. Cancer is the second-leading cause of death globally, accounting for 9.6 million deaths in 2018 [1]. It is expected that in 2030, 21 million people worldwide will suffer from cancer [2]. Tremendous progress, however, has been made in the treatment of individual cancers [3,4,5,6]. This is due, on one hand, to improved early detection and prophylaxis and, on the other, to the development of highly efficient drugs in a wide range of different substance classes. Thus, the probability of premature death from cancer per year decreased from 8.3% in the year 2000 to 6.9% in 2015. It is expected to be as low as 5.3% in 2030, saving approximately 1.1 million lives per year [7].
Both proteins and small molecules have proven their worth in therapy and are in use in the clinic. However, low-molecular-weight drugs often bear the stigma of reduced selectivity in which the cytotoxic drug not only targets cancerous cells, but it also damages healthy tissue. This increases serious side effects and stress symptoms in patients, including nausea, heart or brain damage, impairments to the central nervous system and damage to cells of the inner ear; losses of fertility, hearing and hair have also been noted [8].
These serious side effects and impairments limit the use and acceptance of a drug, as they reduce patient compliance due to a significantly reduced quality of life. This not only endangers the chances of successful therapy, but it often also leads to discontinuation of the therapy [9,10].
Since the problems of reconversion of cancer cells into normal cells (“reprogramming”, for example, of terminally differentiated cancer cells into cancer cells of benign phenotypes) [11] have only remotely been solved today, cancer cells have to be removed either by surgery or destroyed by radiation or chemotherapy. Cell death by chemotherapy can be induced in many different ways [12,13,14,15,16,17], but the mitochondria play a major role in the life or death of a cell. Thus, agents that target mitochondria and induce a controlled cell death, so called “mitocans”, have received increased attention in recent years [17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38]. This seems even more significant inasmuch as cancer cells are closely linked to dysregulated apoptosis of the cells; as a consequence, drug resistance of the cancer cells can develop [39].
Mitocans (as well as other cytotoxic agents) are often able to induce apoptosis; however, the death of a cell, irrespective of whether this cell is malignant, is not random at all. Triggering of controlled cell death is always preferable to an unselective rupture of membranes following the application of extreme but locally applied heat, freeze–thaw cycles or steep osmotic gradients. Controlled cell death can be triggered on a cellular level from nuclear, reticular, cytoskeletal, lysosomal, membrane or, most important, mitochondrial origins [16].
Usually, the cells in a living organism closely cooperate, and cells are constantly in an equilibrium between life and death. Triggering programmed cell death routines removes damaged, infected and out-of-control cells from the organism. The problem arises from the latter cells, since most cancer cells do not respond to extrinsic apoptotic triggers. Thus, mitochondria present a target of emerging interest for cancer therapy as they can trigger apoptosis through an intrinsic pathway. Apoptosis usually starts with loss of mitochondrial membrane potential, followed by the release of cytochrome c and activation of caspase 3 [40]. Furthermore, permeabilization of the outer mitochondrial membrane and the release of cytochrome c are required in many cell death stimuli [41]. This release of cytochrome c can be regarded as a “point of no return” finally leading to the death of the cell. This highlights the importance of mitochondrial compartmentation and explains the devastating effect following permeabilization of the mitochondrial membrane [42,43,44]. In addition to membrane permeabilization, the opening of the mitochondrial permeability transition pore is also considered an important event resulting in mitochondrial depolarization and the release of apoptotic factors [29].
In recent years, triterpenes have repeatedly and increasingly entered the focus of scientific interest. Extensive studies on their apoptotic and cytotoxic properties have been performed. A major concern in dealing with cancer is the MDR (multiple drug resistance) phenotype [39]. These cancer cells overexpress ATP-dependent transporters that eject toxic compounds from the cell before they cause harm to the cell. Some triterpenes are known inhibitors of the efflux pump MDR1, but they are also known to downregulate the transcription factor NF-κB. For cancer, it is widely accepted [45] that NF-κB promotes tumor migration and tumor proliferation.
2. Results
Mitochondrial membranes of malignant cells hold an increased membrane potential compared to non–malignant cells [46]. This effect fosters the accumulation of cationic molecules [17,47,48], hence inducing high selectivity for mitocans holding a (more or less) lipophilic cation such as a rhodamine scaffold. The same effect applies for triphenylphosphonium cations [49,50,51,52,53,54,55,56,57] and to a small extent for quaternary ammonium ions [58,59,60], zwitterionic N-oxides [60,61] and triterpenes substituted with BODIPYs [62,63,64,65,66] or a safirinium moiety [67]. However, the presence of a cationic center is not alone decisive for achieving high cytotoxic effects [60].
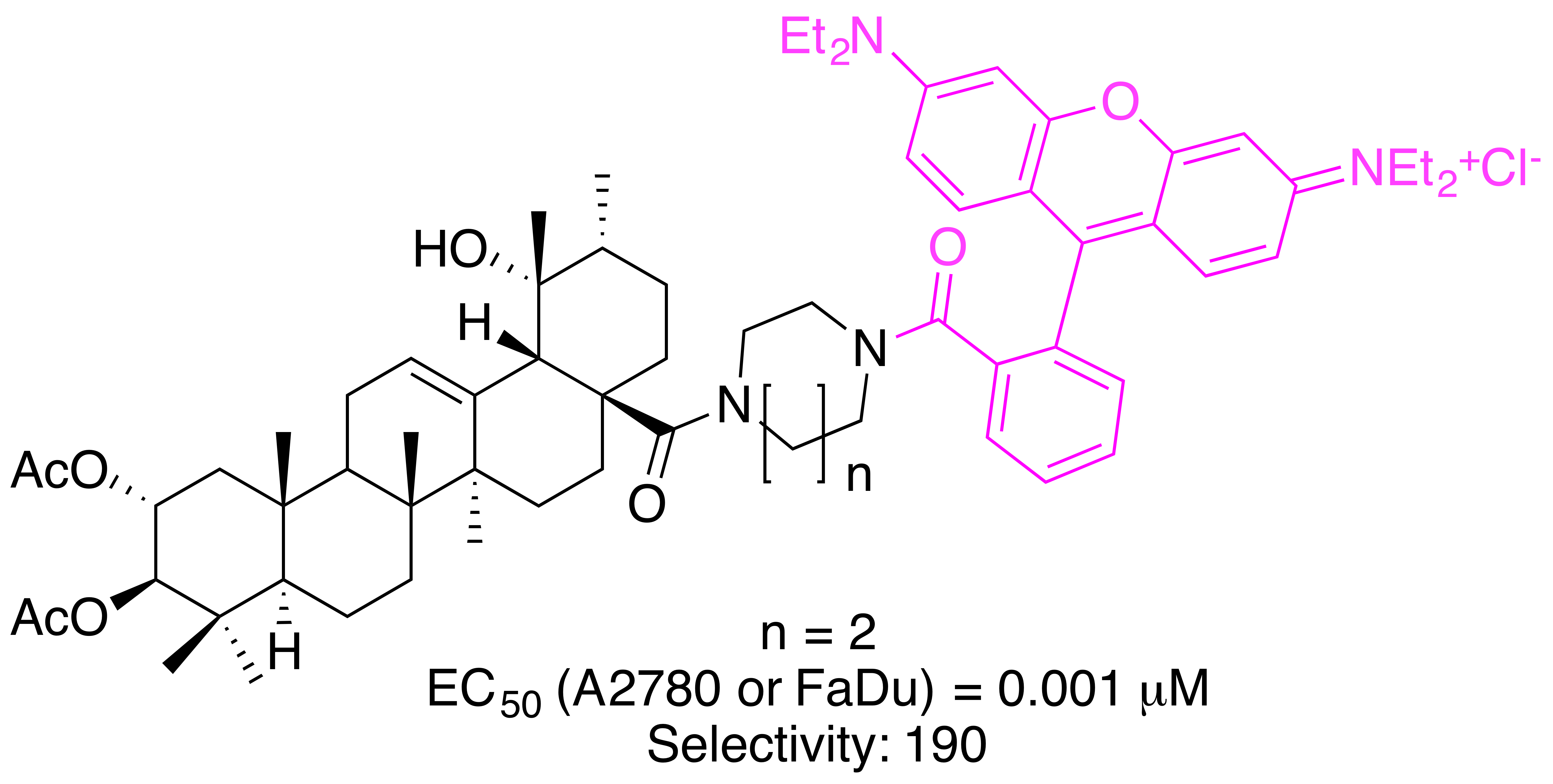
Rhodamine B (RhoB) seems to be a privileged scaffold. This fluorescent dye, also known as rhodamine 610, C.I. Pigment violet 1, basic violet 10, and C.I. 45170 [68], was invented in 1888 (“Tetraethyl-rhodamine”) by M. Cérésole [69,70], and since then it has been widely used in biology, biotechnology and as a biosensor [71,72]. RhoB exists in an equilibrium [73,74,75,76,77] between an “open” positively charged form A (Figure 1) that is fluorescent and a “closed”, non–fluorescent form B. Under acidic conditions, pink-colored A dominates, while colorless B dominates under basic conditions. Further, in less polar organic solvents, the zwitterionic form C undergoes a rapid reversible conversion to B [78,79,80,81].
RhoB is suspected to be carcinogenic [82,83,84,85]. The LD50 value for orally administered RhoB in rats is >500 mg/kg, and an older report classified RhoB (as well as Rho6G) as possibly carcinogenic in rats [85]. RhoB, however, seems not to be mutagenic in Chinese hamster ovary cells [86], but it presents a genotoxic hazard for mammalian organisms [87]. As far as the RhoB–triterpene conjugates are concerned, two types of compounds have been accessed so far: triterpenes with a RhoB moiety directly attached to the skeleton of the triterpene, and compounds wherein these two units are separated by a suitable spacer.
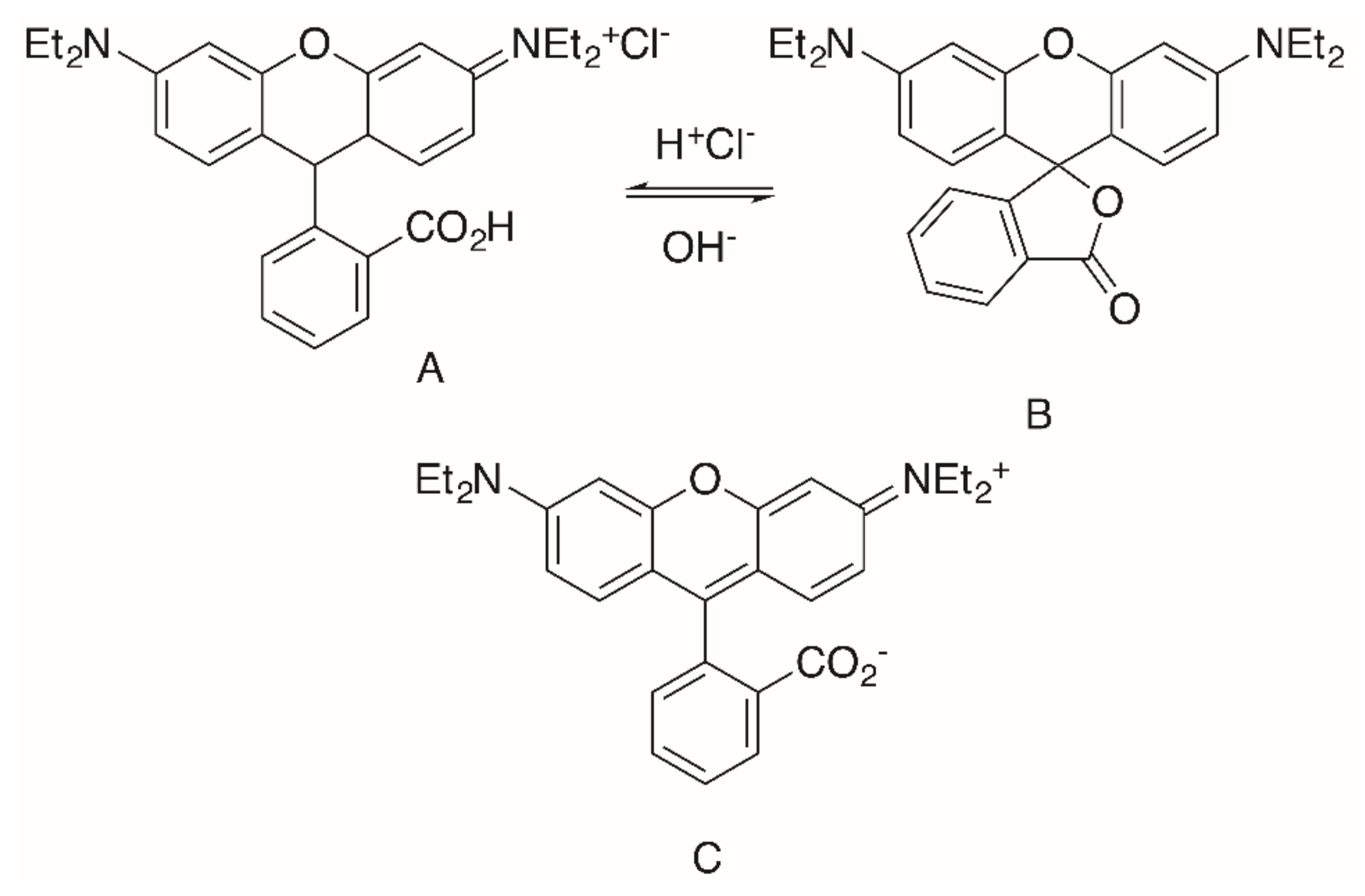
To date, hybrid molecules have been prepared from oleanolic acid (OA, Figure 2), ursolic acid (UA), glycyrrhetinic acid (GA), betulinic acid (BA), maslinic acid (MA), augustic acid (AU), 11-keto-β-boswellic acid (KBA), asiatic acid (AA), tormentic acid (TA) and euscaphic acid (EA).
By means of suitable double-staining experiments, it could be shown that these hybrids are actually effective as mitocans [88], and preliminary molecular modeling studies suggest these compounds might target the mitochondrial NADH dehydrogenase and mitochondrial succinate dehydrogenase [89]. Both enzymes are part of the mitochondrial electron transport chain; this also suggests an increased production of reactive oxygen species (ROS). An increased production of ROS would lead to an oxidative damage of the cell and trigger apoptosis through an intrinsic pathway. Therefore, the integrity of the RhoB basic structure seems to be of crucial importance. It has been shown that derivatives from the triphenylmethane dye malachite green still exhibit increased cytotoxicity as compared to their parent compounds [90]. The cytotoxicity, however, of these hybrids was much lower than those observed for the RhoB derivatives (vide infra).
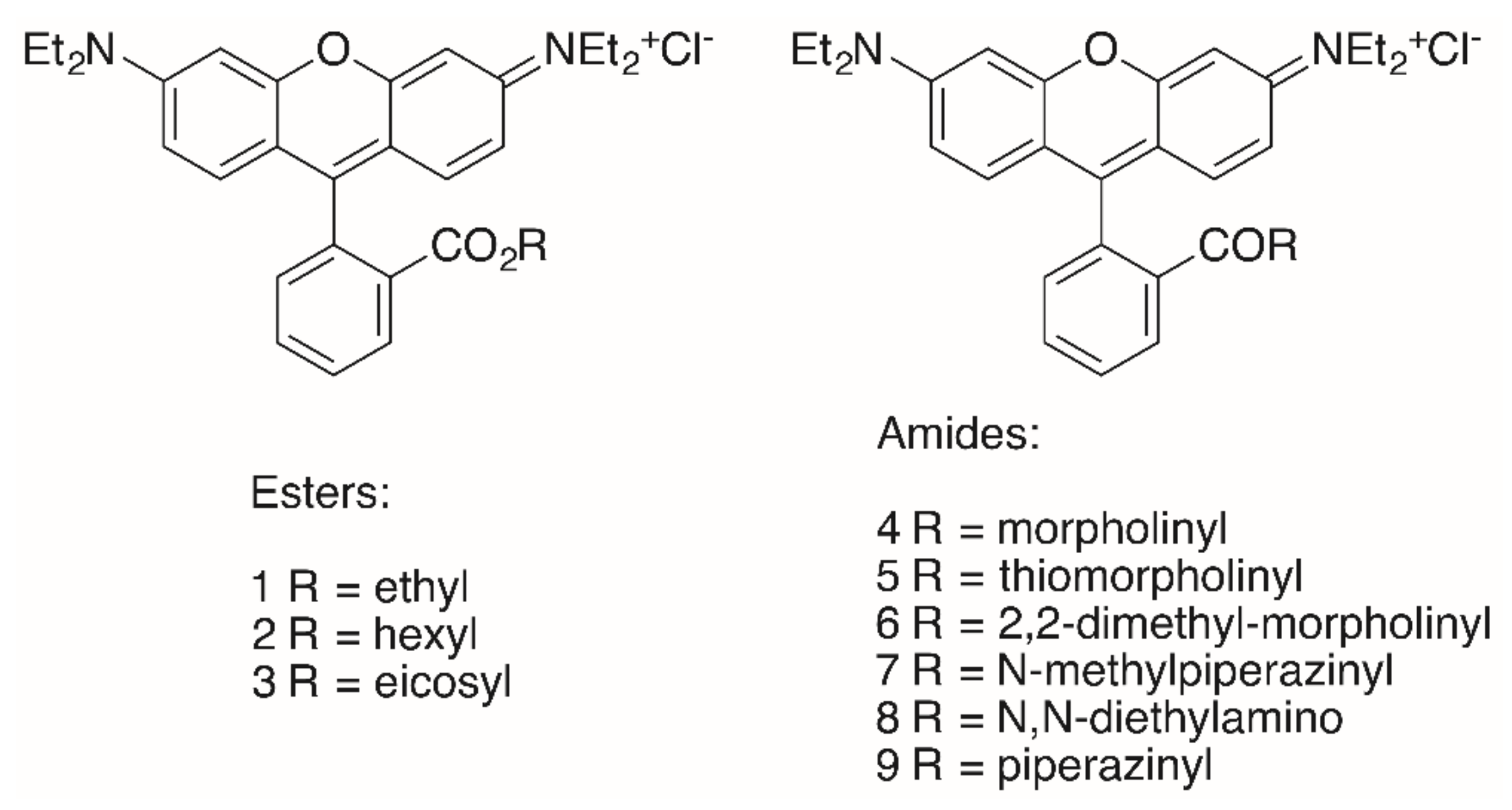
The triterpenoid skeleton is equally important. Here, too, it was shown that “simple” RhoB conjugates 1–9 (Figure 3) also had lower cytotoxicity than the corresponding triterpenoid analogs, but their tumor cell/non-tumor cell selectivity was also diminished (Table 1) [91].
Of special interest seems the morpholinyl derivative 4 inasmuch as this compound held the highest selectivity of this series with respect to MCF-7 carcinoma cells (S = (EC50, NIH 3T3 / EC50, MCF-7) > 19.5) and A2780 ovarian cancer cells (S = (EC50, NIH 3T3 / EC50, A27807) > 18.1) [90].
The highest cytotoxicity was observed for the hexyl ester 2 (EC50 = 0.15–0.19 μM) for the different tumor cell lines. Interestingly, an eicosyl ester 3 with a lipophilicity similar to that of triterpenoids did not show even moderate cytotoxicity [90], while hydroxycinnamic acid rhodamine B conjugates displayed good cytotoxicity in the low μM range [92].
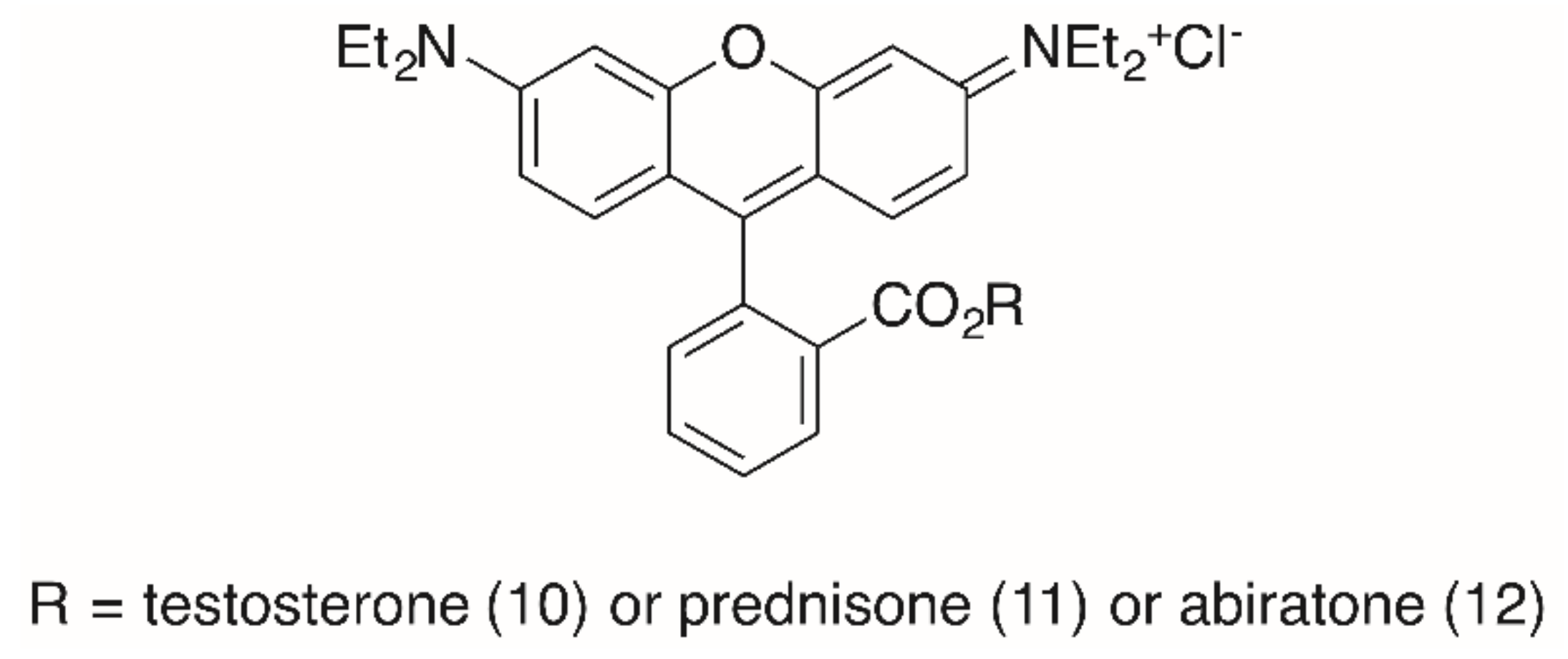
The importance of the presence of a triterpenoid backbone is also evident from studies concerning RhoB steroid conjugates (Figure 4) [93]. In these studies, the reaction of the steroids cholesterol, testosterone, prednisone and abiraterone with an activated RhoB chloride furnished ester conjugates holding low EC50 values (SRB assays with several human tumor cell lines, Table 2). Thus, a testosterone conjugate 10 held EC50 = 60 nm for MCF-7 cells, but acted by necrosis (20%, A2780 cells). A prednisone conjugate 11 was less cytotoxic (0.2 μM for MCF-7 cells) but acted in A2780 cells mainly by apoptosis (48%) and late apoptosis (14%). In addition, this compound showed a higher selectivity for the A2780 tumor cells (S = 73) than for NIH 3T3 fibroblasts. For comparison, an abiratone conjugate 12 was less cytotoxic and also less selective [93].
A closer look at the cell cycle by FACS (with A2780 cells) showed a decrease of the G1 and G2/M peak with an increase of cells in the S phase. For cells treated with 11, the S phase peak and the subG1/apoptosis peak increased significantly. However, for all compounds the selectivity between tumor cells and non-malignant fibroblasts NIH 3T3 was small and never exceeded 7.3 (11, for MCF-7 cells) [93].
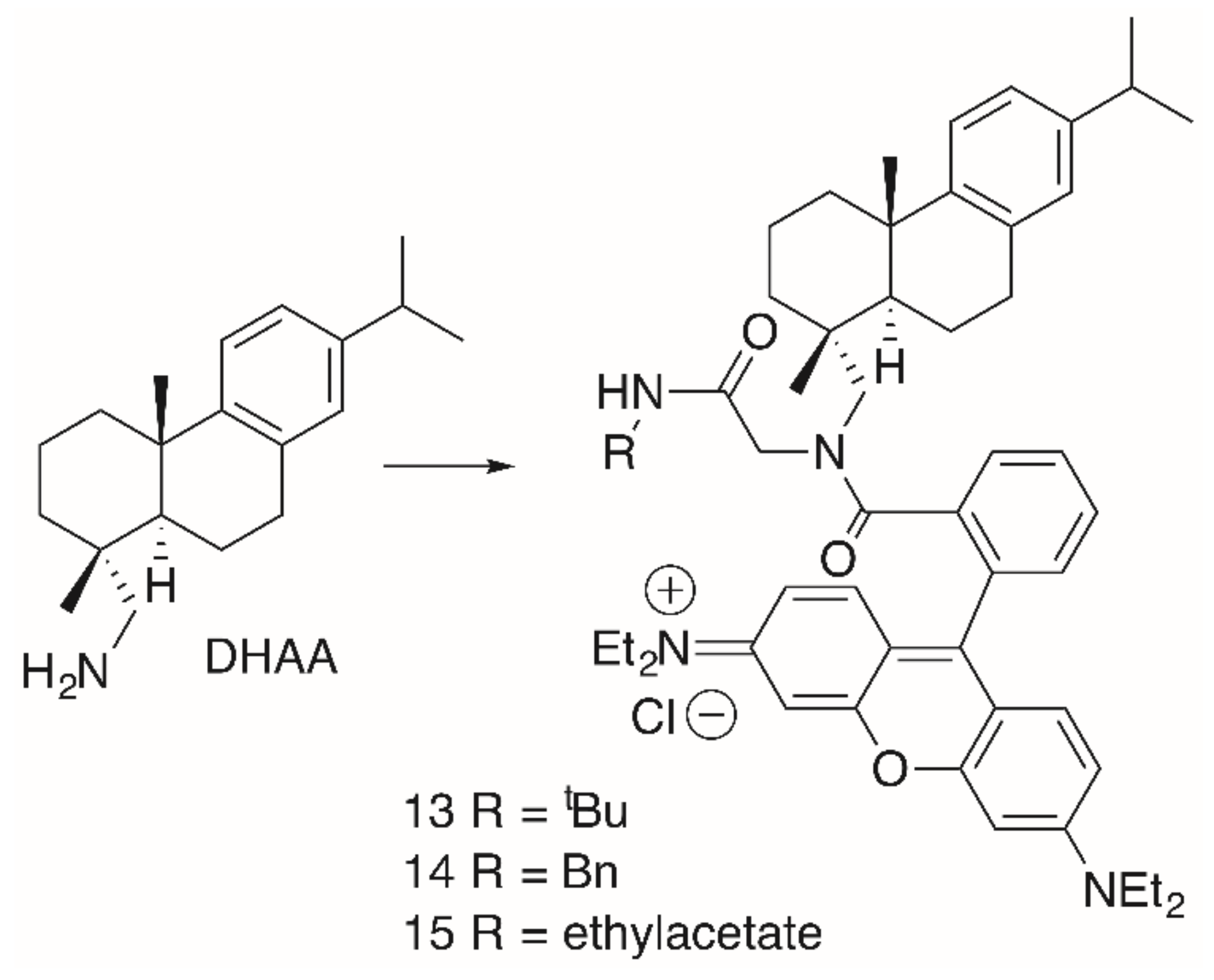
A similar behavior was observed for dehydroabietylamine (DHAA) derivatives 13–16 (Figure 5, Table 3). These products were easily obtained from dehydroabietylamine by the microwave-assisted multicomponent Ugi reaction using paraformaldehyde, an isocyanide and RhoB with yields between 47 and 50% [94].
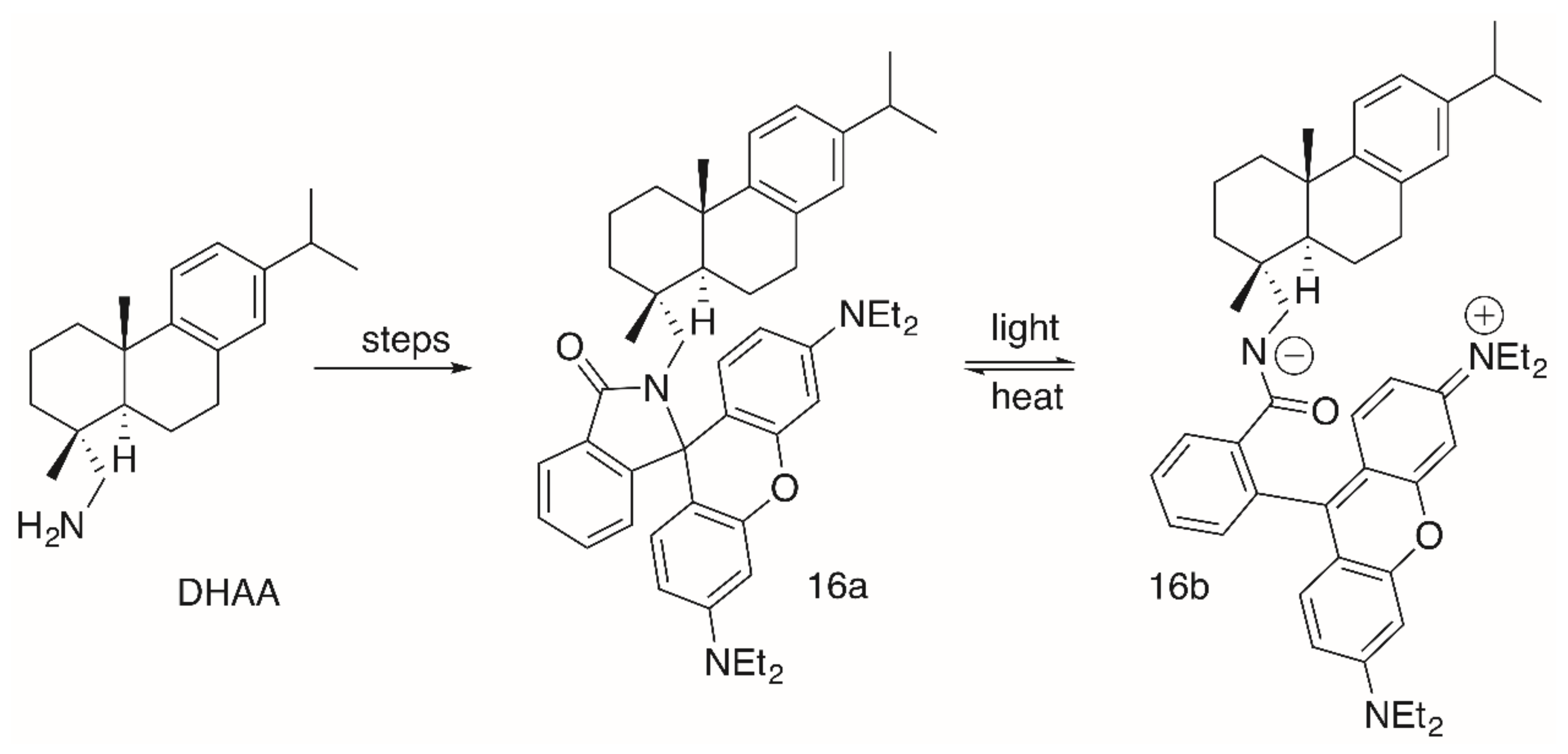
Although the cytotoxicity of these compounds was good, their pharmacological potential was restricted by low selectivity values. Interestingly enough, products 16a/16b (Figure 6), having been obtained from a simple Schotten–Baumann reaction with DHAA and RhoB, were not cytotoxic at all [94]. As mentioned above, RhoB conjugates derived from primary amines are able to form intramolecular non-fluorescent spirolactams (here 16a). From a photo-induced ring opening reaction, 16b was obtained from 16a very quickly within 10 s of irradiation either with visible light or with UV light (λ = 254 or 366 nm). This equilibrium is also strongly influenced by changes in temperature, and at room temperature 16a dominates the equilibrium [94].
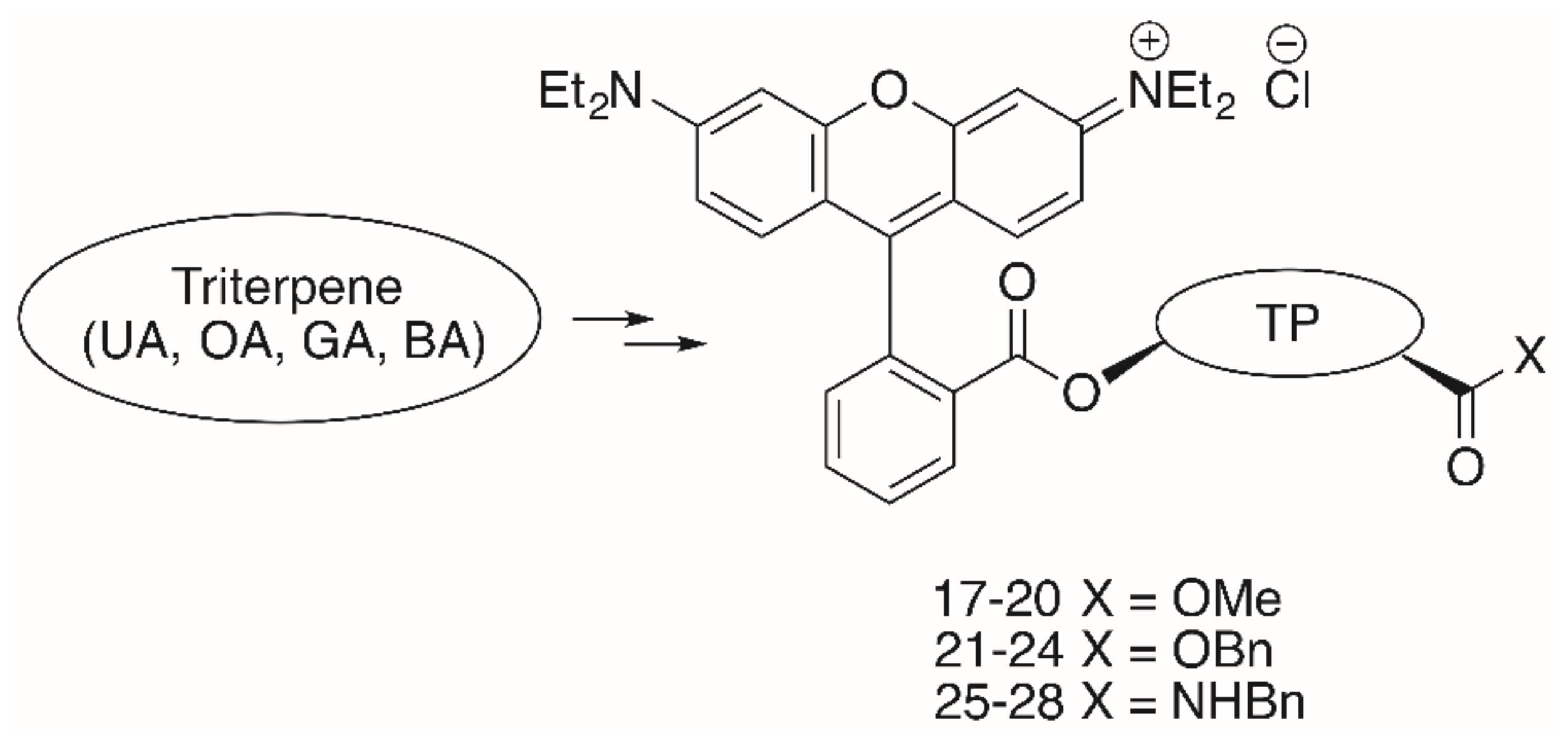
As far as the triterpene RhoB conjugates are concerned, the RhoB moiety can be attached to the triterpenoid scaffold either directly (e.g., in form of a triterpene RhoB ester) or with the aid of a suitable spacer. Pentacyclic triterpenoic acids (Figure 2) holding an RhoB moiety without an extra spacer have been prepared by esterification of UA, OA, GA and BA with RhoB, respectively, (Figure 7) [95].
All of these compounds had EC50 values between 0.02 and 15.8 μM (Table 4); thereby, the cytotoxicity of benzyl esters 21–24 was lower than the cytotoxicity of the methyl esters 17–20, while the benzyl amides 25–28 were the most cytotoxic compounds of this series. The presence of a benzyl ester group as in 21–24 seems to be disadvantageous, while the opposite is true for the benzyl amides 25–28. Compound 27 was the most cytotoxic compound (EC50 = 0.02–0.08 μM), but it was not selective for human tumor cells. Extra staining experiments showed this compound to be accumulated in the mitochondria of A2780 cells and to act mainly by apoptosis [95].
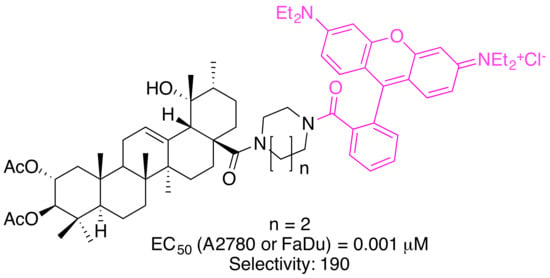
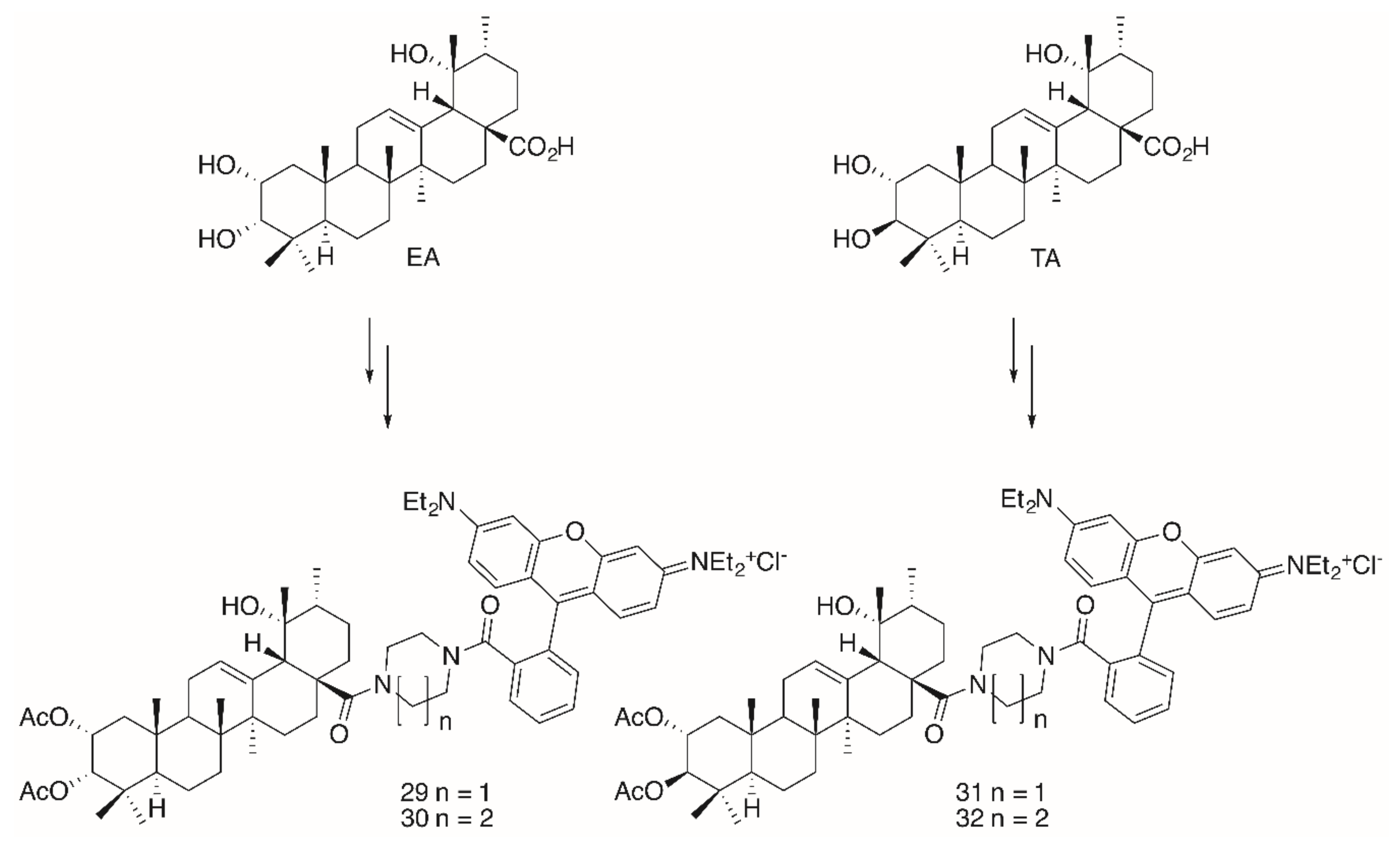
Noteworthy in this context is the higher cytotoxicity of the glycyrrhetinic acid derivatives as compared to analogs derived from OA, UA or BA. Extensions in the design of these compounds led to the synthesis of triterpene conjugates with further modifications in the backbone (→ tormentic acid (TA) and euscaphic acid (EA)) as well as to changes in the ring size of the heterocyclic spacer between the backbone of the triterpene and the RhoB moiety (Figure 8).
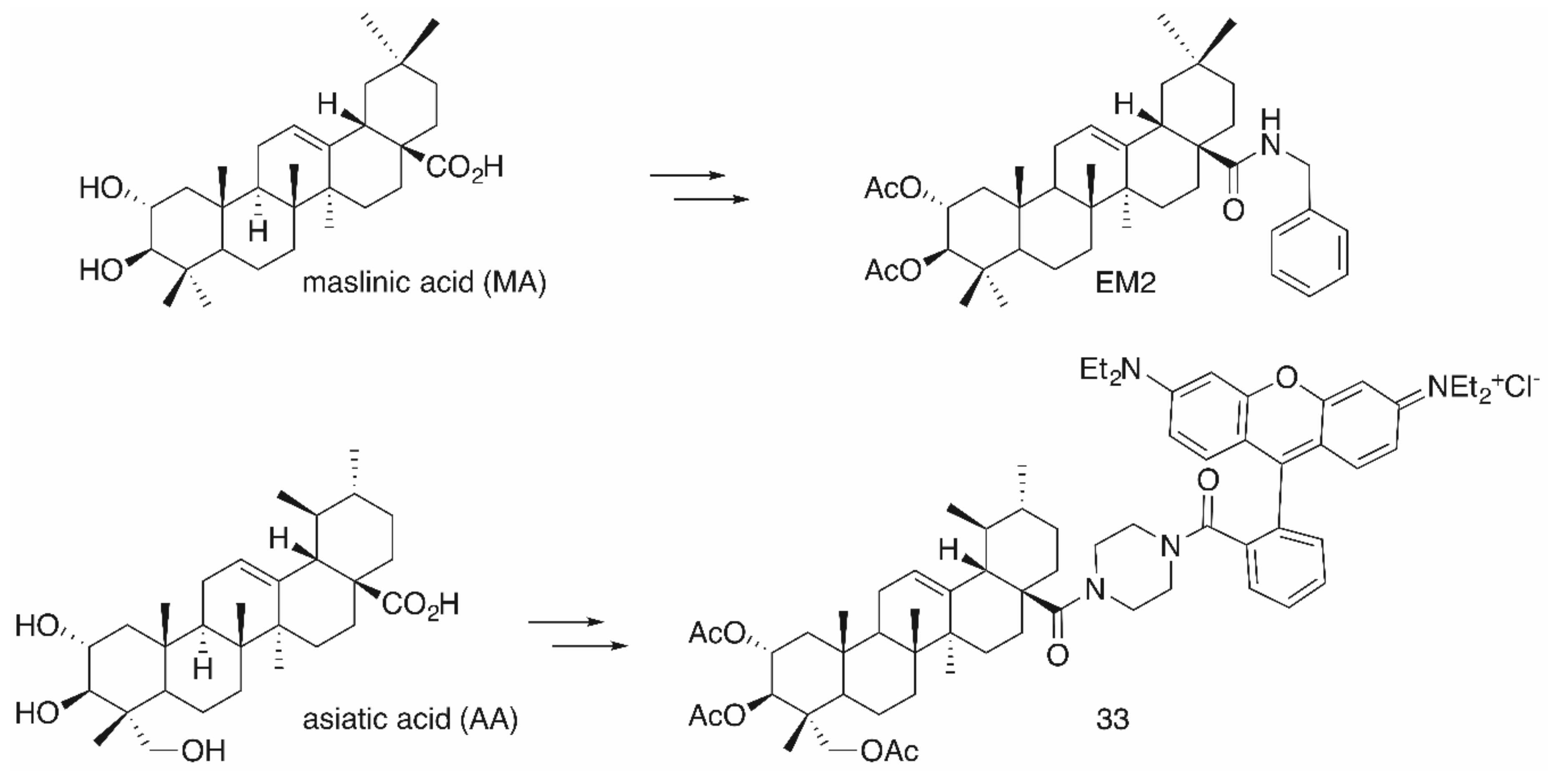
The significantly higher cytotoxicity (Table 5) of TA-derived 32 seems particularly noteworthy when comparing the different spacers: Thereby, the presence of a homopiperazinyl spacer [96] (as in 32) proved to be clearly superior to the piperazinyl moiety (as in 31). A similar trend was also noted for EA-derived compounds 29 and 30. On the other hand, TA-derived compounds were more cytotoxic than the corresponding EA derivatives. Interestingly, the absolute configuration at C–2 and C–3 in TA corresponds exactly to the configuration found in maslinic acid (MA). Several MA derivatives (for example [97,98], a diacetylated benzylamide EM2, Figure 9) were of higher cytotoxicity and better selectivity than their corresponding OA or UA derivatives.
The same configuration is found in asiatic acid (AA). Again, its acetylated piperazinyl-rhodamine B conjugate 33 was most cytotoxic to many human tumor cell lines, being accumulated in the mitochondria, and it also acted as a mitocan [99]. However, for this compound an unusual non-linear rate of growth was detected for some human tumor cell lines (e.g., colorectal carcinoma HT29 and melanoma 518A2). In a bimodal manner at two different concentrations the tumor cells were killed, a phenomenon that might be due to an accelerated recovery of the mitochondrial membrane potential or due to a modulation of the mitochondrial permeability pores. However, at present a concentration-triggered activation of a metabolizing enzyme cannot completely be ruled out [99].
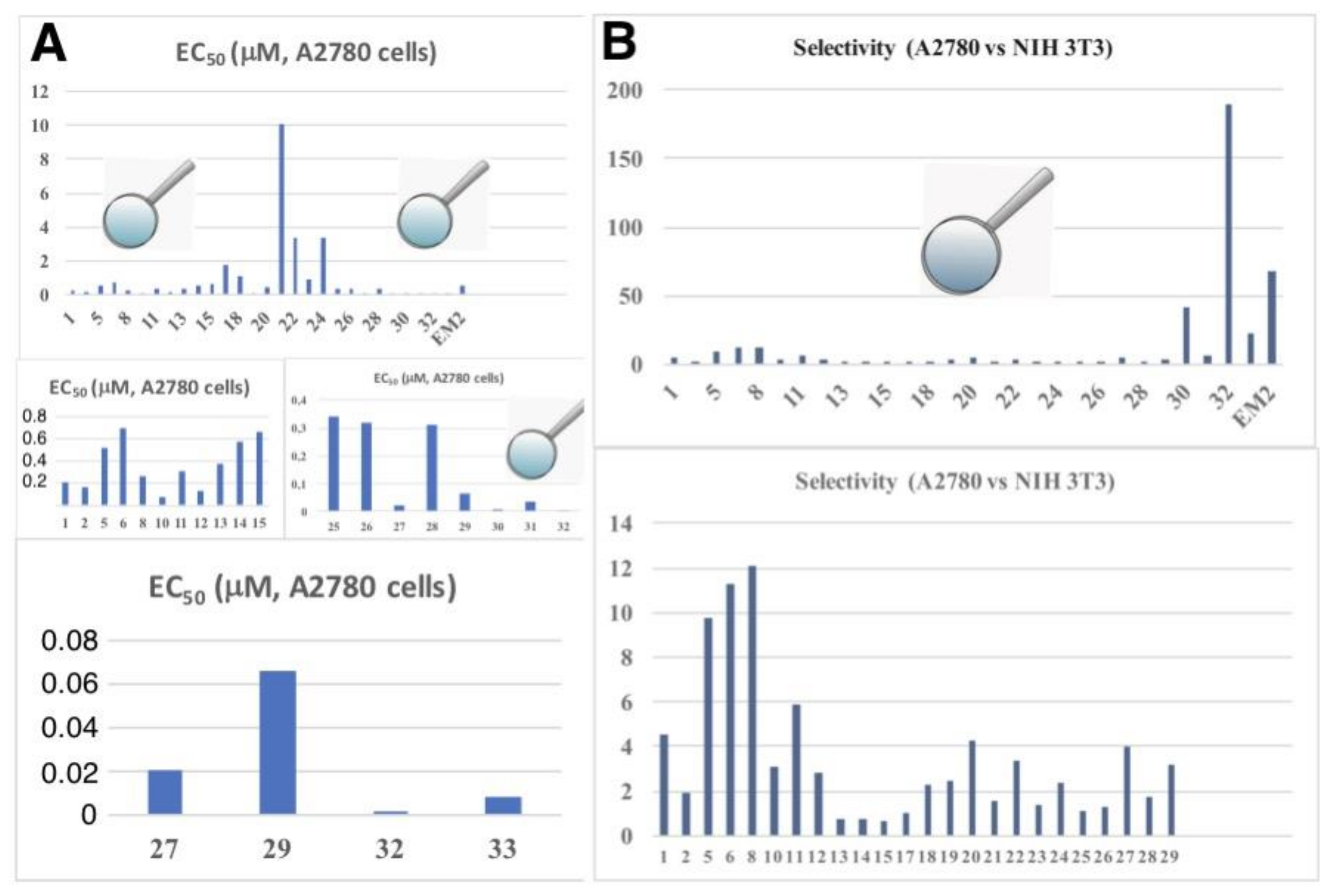
A graphical comparison of all derivatives (using the target line A2780 as an example) is given in Figure 10 including a comparison of tumor cell/non-tumor cell selectivity (A2780 vs. NIH 3T3) of all compounds.
From Figure 10 the high potential of compound 32 (selectivity for A2780 or FaDu cells, ca. 190) becomes clearly visible, making this compound an interesting drug for advanced testing and biological screening.
3. Conclusions
OA-derived RhoB conjugates appear to be superior to analog UA-derived compounds in the majority of cases with respect to their cytotoxicity. Although AKBA-derived derivatives have good cytotoxicity properties, they were found to be less cytotoxic compared to other triterpene carboxylic acid derivatives, but they often showed better tumor cell/non-tumor cell selectivity. So far, the best cytotoxicity properties have been found for MA-, EA- and TA-derived derivatives. These allowed the transition to compounds of nano-molar activity, while many other triterpene carboxylic acid derivatives were cytotoxic only on a micro-molar concentration range. MA- derived derivatives seem to be approximately equivalent to EA-derived compounds. They are currently only surpassed in many tumor cell lines only by the analogous derivatives from TA. From results available so far, it can be concluded that compounds holding a homopiperazinyl spacer are superior to those with a piperazinyl spacer. This underlines the importance of the spacer for obtaining good cytotoxicity properties. Replacement of the secondary amide derived spacer by a primary amine like ethylenediamine has invariably led to RhoB conjugates of insignificant cytotoxicity (EC50 > 30 μM) due to the formation of a spirolactam holding no positive charge in the RhoB part.
However, the presence of a distal cation is not sufficient to obtain compounds with excellent cytotoxicity, as has been shown for several quaternary ammonium compounds or compounds where the RhoB part has been replaced by, for example, malachite green, a BODIPY residue or a safirinium group. In addition, the latter compounds do not act as mitocans, since their primary target is the endoplasmic reticulum.
A statement on the extent to which the replacement of the RhoB group with another rhodamine has a positive effect on biological activity cannot be made at present. The cytotoxic properties of these compounds, other spacers and other triterpene carboxylic acids are currently the subject of further investigation. The combination of the “correct” parent structure, the “correct” spacer and the “correct” RhoB seems to be decisive for the ability of the conjugate to accumulate in mitochondria. So far, a tormentic acid acid-derived RhoB conjugate 32 holding a homopiperazinyl spacer can be regarded as the most promising candidate for further biological studies. At present, no extended investigations have been carried out on the precise mode of action of these molecules.
Author Contributions
Conceptualization, R.C. and H.-P.D.; validation, S.H., I.S. and R.C.; investigation, S.H.; writing—original draft preparation, R.C. writing—review and editing, R.C., S.H., I.S., H.-P.D. All authors have read and agreed to the published version of the manuscript.
Funding
We acknowledge the financial support within the funding program Open Access Publishing by the German Research Foundation (DFG).
Acknowledgments
We like to thank all members of our groups for their continuing support and skillful work in the laboratories.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Available online: https://www.who.int/health-topics/cancer-tab=tab_1 (accessed on 20 October 2020).
- Bray, F.; Jemal, A.; Grey, N.; Ferlay, J.; Forman, D. Global cancer transitions according to the Human Development Index (2008–2030): A population-based study. Lancet Oncol. 2012, 13, 790–801. [Google Scholar] [CrossRef]
- Siegel, R.L.; Mph, K.D.M.; Sauer, A.G.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2020. CA: A Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markham, M.J.; Wachter, K.; Agarwal, N.; Bertagnolli, M.M.; Chang, S.M.; Dale, W.; Diefenbach, C.S.M.; Rodriguez-Galindo, C.; George, D.J.; Gilligan, T.D.; et al. Clinical Cancer Advances 2020: Annual Report on Progress Against Cancer From the American Society of Clinical Oncology. J. Clin. Oncol. 2020, 38, 1081. [Google Scholar] [CrossRef] [PubMed]
- Oostveen, M.; Pritchard-Jones, K. Pharmacotherapeutic Management of Wilms Tumor: An Update. Pediatr. Drugs 2019, 21, 1–13. [Google Scholar] [CrossRef]
- Di Paolo, A.; Arrigoni, E.; Luci, G.; Cucchiara, F.; Danesi, R.; Galimberti, S. Precision Medicine in Lymphoma by Innovative Instrumental Platforms. Front. Oncol. 2019, 9, 1417. [Google Scholar] [CrossRef]
- Available online: https://www.paho.org/hq/index.php?option=com_docman&view=download&category_slug=4-cancer-country-profiles-2020&alias=51561-global-cancer-profile-2020&Itemid=270&lang=fr (accessed on 20 October 2020).
- Green, D.R. Cancer and Apoptosis: Who Is Built to Last? Cancer Cell 2017, 31, 2–4. [Google Scholar] [CrossRef] [Green Version]
- Pretta, A.; Trevisi, E.; Bregni, G.; Deleporte, A.; Hendlisz, A.; Sclafani, F. Treatment compliance in early-stage anal cancer. Ann. Oncol. 2020, 31, 1282–1284. [Google Scholar] [CrossRef]
- Muzumder, S.; Srikantia, N.; Vashishta, G.D.; Udayashankar, A.H.; Raj, J.M.; Sebastian, M.G.J.; Kainthaje, P.B. Compliance, toxicity and efficacy in weekly versus 3-weekly cisplatin concurrent chemoradiation in locally advanced head and neck cancer. J. Radiother. Pr. 2018, 18, 21–25. [Google Scholar] [CrossRef]
- Gong, L.; Yan, Q.; Zhang, Y.; Fang, X.; Liu, B.; Guan, X.-Y. Cancer cell reprogramming: A promising therapy converting malignancy to benignity. Cancer Commun. 2019, 39, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, Y.; Steller, H. Live to die another way: Modes of programmed cell death and the signals emanating from dying cells. Nat. Rev. Mol. Cell Biol. 2015, 16, 329–344. [Google Scholar] [CrossRef]
- Wang, C.X.; Youle, R.J. The Role of Mitochondria in Apoptosis. Annu. Rev. Genet. 2009, 43, 95–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnstone, R.W.; Ruefli, A.A.; Lowe, S.W. Apoptosis: A link between cancer genetics and chemotherapy. Cell 2002, 108, 153–164. [Google Scholar] [CrossRef] [Green Version]
- Fiers, W.; Beyaert, R.; Declercq, W.; Vandenabeele, P. More than one way to die: Apoptosis, necrosis and reactive oxygen damage. Oncogene 1999, 18, 7719–7730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Pedro, J.M.B.-S.; Kroemer, G. Organelle-specific initiation of cell death. Nat. Cell Biol. 2014, 16, 728–736. [Google Scholar] [CrossRef] [PubMed]
- Bing, Y.; Dong, L.; Neuzil, J. Mitochondria: An intriguing target for killing tumour-initiating cells. Mitochondrion 2016, 26, 86–93. [Google Scholar] [CrossRef]
- Pei, J.; Panina, S.B.; Kirienko, N.V. An Automated Differential Nuclear Staining Assay for Accurate Determination of Mitocan Cytotoxicity. J. Vis. Exp. 2020, 159, e61295. [Google Scholar] [CrossRef]
- Panina, S.B.; Pei, J.; Baran, N.; Konopleva, M.; Kirienko, N.V. Utilizing Synergistic Potential of Mitochondria-Targeting Drugs for Leukemia Therapy. Front. Oncol. 2020, 10, 435. [Google Scholar] [CrossRef] [Green Version]
- Ralph, S.J.; Neuzil, J. Mitochondria as Targets for Cancer Therapy. In Mitochondria and Cancer; Costello, L., Singh, K., Eds.; Springer: New York, NY, USA, 2009; pp. 211–249. [Google Scholar]
- Ralph, S.J.; Rodríguez-Enríquez, S.; Neuzil, J.; Moreno-Sánchez, R. Bioenergetic pathways in tumor mitochondria as targets for cancer therapy and the importance of the ROS-induced apoptotic trigger. Mol. Asp. Med. 2010, 31, 29–59. [Google Scholar] [CrossRef] [Green Version]
- Ralph, S.J.; Rodríguez-Enríquez, S.; Neuzil, J.; Saavedra, E.; Moreno-Sánchez, R. The causes of cancer revisited: “Mitochondrial malignancy” and ROS-induced oncogenic transformation – Why mitochondria are targets for cancer therapy. Mol. Asp. Med. 2010, 31, 145–170. [Google Scholar] [CrossRef] [PubMed]
- Chiu, H.Y.; Tay, E.X.Y.; Ong, D.S.T.; Taneja, R. Mitochondrial Dysfunction at the Center of Cancer Therapy. Antioxidants Redox Signal. 2020, 32, 309–330. [Google Scholar] [CrossRef]
- Singh, Y.; Viswanadham, K.K.D.R.; Pawar, V.K.; Meher, J.; Jajoriya, A.K.; Omer, A.; Jaiswal, S.; Dewangan, J.; Bora, H.K.; Singh, P.; et al. Induction of Mitochondrial Cell Death and Reversal of Anticancer Drug Resistance via Nanocarriers Composed of a Triphenylphosphonium Derivative of Tocopheryl Polyethylene Glycol Succinate. Mol. Pharm. 2019, 16, 3744–3759. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, C.; Pandey, S. Exploiting Mitochondrial Vulnerabilities to Trigger Apoptosis Selectively in Cancer Cells. Cancers 2019, 11, 916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olivas-Aguirre, M.; Pottosin, I.; Dobrovinskaya, O. Mitochondria as emerging targets for therapies against T cell acute lymphoblastic leukemia. J. Leukoc. Biol. 2019, 105, 935–946. [Google Scholar] [CrossRef]
- Sassi, N.; Biasutto, L.; Mattarei, A.; Carraro, M.; Giorgio, V.; Citta, A.; Bernardi, P.; Garbisa, S.; Szabò, I.; Paradisi, C.; et al. Cytotoxicity of a mitochondriotropic quercetin derivative: Mechanisms. Biochim. et Biophys. Acta (BBA) - Gen. Subj. 2012, 1817, 1095–1106. [Google Scholar] [CrossRef] [PubMed]
- Sassi, N.; Mattarei, A.; Azzolini, M.; Bernardi, P.; Szabò, I.; Paradisi, C.; Zoratti, M.; Biasutto, L. Mitochondria-targeted resveratrol derivatives act as cytotoxic pro-oxidants. Curr. Pharm. Des. 2014, 20, 172–179. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Wang, F.; Trachootham, D.; Huang, P. Preferential killing of cancer cells with mitochondrial dysfunction by natural compounds. Mitochondrion 2010, 10, 614–625. [Google Scholar] [CrossRef] [Green Version]
- Guzman-Villanueva, D.; Weissig, V. Mitochondria-Targeted Agents: Mitochondriotropics, Mitochondriotoxics, and Mitocans. Muscarinic Receptors 2016, 240, 423–438. [Google Scholar] [CrossRef]
- Rohlena, J.; Dong, L.-F.; Neuzil, J. Targeting the mitochondrial electron transport chain complexes for the induction of apoptosis and cancer treatment. Curr. Pharm. Biotechnol. 2013, 14, 377–389. [Google Scholar] [CrossRef]
- Rohlena, J.; Dong, L.; Ralph, S.J.; Neuzil, J. Anticancer Drugs Targeting the Mitochondrial Electron Transport Chain. Antioxidants Redox Signal. 2011, 15, 2951–2974. [Google Scholar] [CrossRef]
- Kornblihtt, L.I.; Carreras, M.C.A.; Blanco, G. Targeting Mitophagy in Combined Therapies of Haematological Malignancies. Autophagy Curr. Trends Cell. Physiol. Pathol. 2016, 19, 411–431. [Google Scholar] [CrossRef] [Green Version]
- Neuzil, J.; Dong, L.-F.; Rohlena, J.; Truksa, J.; Ralph, S.J. Classification of mitocans, anti-cancer drugs acting on mitochondria. Mitochondrion 2013, 13, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Neuzil, J.; Dyason, J.C.; Freeman, R.; Dong, L.; Prochazka, L.; Wang, X.-F.; Scheffler, I.; Ralph, S.J. Mitocans as anti-cancer agents targeting mitochondria: Lessons from studies with vitamin E analogues, inhibitors of complex II. J. Bioenerg. Biomembr. 2007, 39, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Neuzil, J.; Wang, X.-F.; Dong, L.-F.; Low, P.; Ralph, S.J. Molecular mechanism of ‘mitocan’-induced apoptosis in cancer cells epitomizes the multiple roles of reactive oxygen species and Bcl-2 family proteins. FEBS Lett. 2006, 580, 5125–5129. [Google Scholar] [CrossRef] [PubMed]
- Morrison, B.J.; Andera, L.; Reynolds, B.A.; Ralph, S.J.; Neuzil, J. Future use of mitocans against tumour-initiating cells? Mol. Nutr. Food Res. 2009, 53, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Hahn, T.; Polanczyk, M.J.; Borodovsky, A.; Ramanathapuram, L.V.; Akporiaye, E.T.; Ralph, S.J. Use of anti-cancer drugs, mitocans, to enhance the immune responses against tumors. Curr. Pharm. Biotechnol. 2013, 14, 357–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaral, L.; Spengler, G.; Molnar, J. Identification of Important Compounds Isolated from Natural Sources that Have Activity Against Multidrug-resistant Cancer Cell Lines: Effects on Proliferation, Apoptotic Mechanism and the Efflux Pump Responsible for Multi-resistance Phenotype. Anticancer. Res. 2016, 36, 5665–5672. [Google Scholar] [CrossRef] [Green Version]
- Abdelmageed, N.; Morad, S.A.; Elghoneimy, A.A.; Syrovets, T.; Simmet, T.; El-Zorba, H.; El-Banna, H.A.; Cabot, M.; Abdel-Aziz, M.I. Oleanolic acid methyl ester, a novel cytotoxic mitocan, induces cell cycle arrest and ROS-Mediated cell death in castration-resistant prostate cancer PC-3 cells. Biomed. Pharmacother. 2017, 96, 417–425. [Google Scholar] [CrossRef]
- Bhola, P.D.; Letai, A. Mitochondria—Judges and Executioners of Cell Death Sentences. Mol. Cell 2016, 61, 695–704. [Google Scholar] [CrossRef] [Green Version]
- Jacotot, E.E.; Ferri, K.K.; El Hamel, C.C.; Brenner, C.C.; Druillennec, S.S.; Hoebeke, J.; Rustin, P.P.; Métivier, D.D.; Lenoir, C.C.; Geuskens, M.; et al. Control of Mitochondrial Membrane Permeabilization by Adenine Nucleotide Translocator Interacting with HIV-1 Viral Protein R and Bcl-2. J. Exp. Med. 2001, 193, 509–520. [Google Scholar] [CrossRef]
- Zamzami, N.; Kroemer, G. The mitochondrion in apoptosis: How Pandora’s box opens. Nat. Rev. Mol. Cell Biol. 2001, 2, 67–71. [Google Scholar] [CrossRef]
- Zamzami, N.; Maisse, C.; Métivier, D.; Kroemer, G. Chapter 8 Measurement of membrane permeability and permeability transition of mitochondria. Echinoderms Part B 2001, 65, 147–158. [Google Scholar] [CrossRef]
- Yang, H.; Dou, Q.P. Targeting apoptosis pathway with natural terpenoids: Implications for treatment of breast and prostate cancer. Curr. Drug Targets 2010, 11, 733–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulda, S.; Kroemer, G. Mitochondria as Therapeutic Targets for the Treatment of Malignant Disease. Antioxidants Redox Signal. 2011, 15, 2937–2949. [Google Scholar] [CrossRef] [PubMed]
- Serafim, T.L.; Carvalho, F.S.; Pereira, G.; Greene, A.; Perkins, E.; Holy, J.; Krasutsky, D.A.; Kolomitsyna, O.N.; Krasutsky, P.A.; Oliveira, P.J. Lupane triterpenoids as mitocans against breast cancer cells. Eur. J. Clin. Invest. 2013, 43, 48–49. [Google Scholar]
- Serafim, T.L.; Carvalho, F.S.; Bernardo, T.C.; Pereira, G.C.; Perkins, E.; Holy, J.; Krasutsky, D.A.; Kolomitsyna, O.N.; Krasutsky, P.A.; Oliveira, P.J. New derivatives of lupine triterpenoids disturb breast cancer mitochondria and induce cell death. Bioorg. Med. Chem. 2014, 22, 6270–6287. [Google Scholar] [CrossRef] [PubMed]
- Grymel, M.; Zawojak, M.; Adamek, J. Triphenylphosphonium Analogues of Betulin and Betulinic Acid with Biological Activity: A Comprehensive Review. J. Nat. Prod. 2019, 82, 1719–1730. [Google Scholar] [CrossRef]
- Kolevzon, N.; Kuflik, U.; Shmuel, M.; Benhamron, S.; Ringel, I.; Yavin, E. Multiple Triphenylphosphonium Cations as a Platform for the Delivery of a Pro-Apoptotic Peptide. Pharm. Res. 2011, 28, 2780–2789. [Google Scholar] [CrossRef]
- Spivak, A.Y.; Nedopekina, D.A.; Khalitova, R.R.; Gubaidullin, R.; Odinokov, V.N.; Bel’Skii, Y.P.; Bel’Skaya, N.V.; Khazanov, V.A. Triphenylphosphonium cations of betulinic acid derivatives: Synthesis and antitumor activity. Med. Chem. Res. 2017, 26, 518–531. [Google Scholar] [CrossRef]
- Akhmetova, V.R.; Nedopekina, D.A.; Shakurova, E.R.; Khalitova, R.R.; Gubaidullin, R.R.; Odinokov, V.N.; Dzhemilev, U.M.; Bel’Skii, Y.P.; Bel’Skaya, N.V.; Stankevich, S.A.; et al. Synthesis of lupane triterpenoids with triphenylphosphonium substituents and studies of their antitumor activity. Russ. Chem. Bull. 2013, 62, 188–198. [Google Scholar] [CrossRef]
- Huang, H.; Wu, H.; Huang, Y.; Zhang, S.; Lam, Y.-W.; Ao, N. Antitumor activity and antitumor mechanism of triphenylphosphonium chitosan against liver carcinoma. J. Mater. Res. 2018, 33, 2586–2597. [Google Scholar] [CrossRef]
- Strobykina, I.Y.; Andreeva, O.V.; Belenok, M.G.; Semenova, M.N.; Semenov, V.V.; Chuprov-Netochin, R.N.; Sapunova, A.S.; Voloshina, A.D.; Dobrynin, A.B.; Semenov, V.E.; et al. Triphenylphosphonium conjugates of 1,2,3-triazolyl nucleoside analogues. Synthesis and cytotoxicity evaluation. Med. Chem. Res. 2020, 29, 1–15. [Google Scholar] [CrossRef]
- Jara, J.A.; Castro-Castillo, V.; Saavedra-Olavarría, J.; Peredo, L.; Pavanni, M.; Jaña, F.; Letelier, M.E.; Parra, E.; Becker, M.I.; Morello, A.; et al. Antiproliferative and Uncoupling Effects of Delocalized, Lipophilic, Cationic Gallic Acid Derivatives on Cancer Cell Lines. Validation in Vivo in Singenic Mice. J. Med. Chem. 2014, 57, 2440–2454. [Google Scholar] [CrossRef] [PubMed]
- Kafkova, A.; Trnka, J. Mitochondria-targeted compounds in the treatment of cancer. Neoplasma 2020, 67, 450–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Y.; Zhang, T.; Yuan, H.; Li, D.; Lou, H.; Fan, P. Mitochondria-Targeted Lupane Triterpenoid Derivatives and Their Selective Apoptosis-Inducing Anticancer Mechanisms. J. Med. Chem. 2017, 60, 6353–6363. [Google Scholar] [CrossRef]
- Sarek, J.; Biedermann, D.; Eignerova, B.; Hajduch, M. Synthesis and Evaluation of Biological Activity of the Quaternary Ammonium Salts of Lupane-, Oleanane-, and Ursane-Type Acids. Synthesis 2010, 22, 3839–3848. [Google Scholar] [CrossRef]
- Kataev, V.E.; Strobykina, I.Y.; Zakharova, L.Y. Quaternary ammonium derivatives of natural terpenoids. Synthesis and properties. Russ. Chem. Bull. 2014, 63, 1884–1900. [Google Scholar] [CrossRef]
- Brandes, B.; Koch, L.; Hoenke, S.; Deigner, H.-P.; Csuk, R. The presence of a cationic center is not alone decisive for the cytotoxicity of triterpene carboxylic acid amides. Steroids 2020, 163, 108713. [Google Scholar] [CrossRef]
- Perreault, M.; Maltais, R.; Dutour, R.; Poirier, D. Explorative study on the anticancer activity, selectivity and metabolic stability of related analogs of aminosteroid RM-133. Steroids 2016, 115, 105–113. [Google Scholar] [CrossRef]
- Brandes, B.; Hoenke, S.; Fischer, L.; Csuk, R. Design, synthesis and cytotoxicity of BODIPY FL labelled triterpenoids. Eur. J. Med. Chem. 2020, 185, 111858. [Google Scholar] [CrossRef]
- Barut, B.; Çoban, Ö.; Özgür, Y.C.; Baş, H.; Sari, S.; Biyiklioglu, Z.; Demirbaş, Ü.; Özel, A. Synthesis, DNA interaction, in vitro/in silico topoisomerase II inhibition and photodynamic therapy activities of two cationic BODIPY derivatives. Dye. Pigment. 2020, 174, 108072. [Google Scholar] [CrossRef]
- Barut, B.; Özgür, Y.C.; Sari, S.; Çoban, Ö.; Keleş, T.; Biyiklioglu, Z.; Abudayyak, M.; Demirbaş, Ü.; Özel, A. Novel water soluble BODIPY compounds: Synthesis, photochemical, DNA interaction, topoisomerases inhibition and photodynamic activity properties. Eur. J. Med. Chem. 2019, 183, 11685. [Google Scholar] [CrossRef] [PubMed]
- Taki, S.; Ardestani, M.S. Novel nanosized AS1411-chitosan-BODIPY conjugate for molecular fluorescent imaging. Int. J. Nanomed. 2019, 14, 3543–3555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Li, X.; Cao, Z.; Wu, Y.; Chen, J.-A.; Gao, J.; Wang, Z.; Guo, W.; Gu, X. Mitochondria-targeting BODIPY-loaded micelles as novel class of photosensitizer for photodynamic therapy. Eur. J. Med. Chem. 2018, 157, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Kraft, O.; Kozubek, M.; Hoenke, S.; Serbian, I.; Major, D.; Csuk, R. Cytotoxic triterpenoid–safirinium conjugates target the endoplasmic reticulum. Eur. J. Med. Chem. 2020, 112920. [Google Scholar] [CrossRef] [PubMed]
- Cooksey, C. Quirks of dye nomenclature. 5. Rhodamines. Biotech. Histochem. 2015, 91, 1–6. [Google Scholar] [CrossRef]
- Ceresole, M. Rhodamin Dye. U.S. Patent 516589 A 18940313, 13 March 1894. [Google Scholar]
- Ceresole, M. Red Dyestuff. U.S. Patent 456081 18910714, 14 July 1891. [Google Scholar]
- Kim, H.N.; Lee, M.H.; Kim, H.J.; Kim, J.S.; Yoon, J. A new trend in rhodamine-based chemosensors: Application of spirolactam ring-opening to sensing ions. Chem. Soc. Rev. 2008, 37, 1465–1472. [Google Scholar] [CrossRef]
- Wang, D.; Wang, Z.; Li, Y.; Song, Y.; Song, Y.; Zhang, M.; Yu, H. A single rhodamine spirolactam probe for localization and pH monitoring of mitochondrion/lysosome in living cells. New J. Chem. 2018, 42, 11102–11108. [Google Scholar] [CrossRef]
- Adamovich, L.P.; Mel’nik, V.V.; McHedlov-Petrosyan, N.O. Rhodamine B equilibriums in water-salt solutions. Zh. Fiz. Khim. 1979, 53, 356–359. [Google Scholar]
- Hinckley, D.A.; Seybold, P.G. Thermodynamics of the rhodamine B lactone zwitterion equilibrium: An undergraduate laboratory experiment. J. Chem. Educ. 1987, 64, 362. [Google Scholar] [CrossRef]
- Ramart-Lucas, P. Structure of rhodamine from its absorption spectrum. Compt. Rend. 1938, 207, 1416–1418. [Google Scholar]
- Ramette, R.W.; Blackburn, T.R. Specific effects of cations on rhodamine B equilibriums. J. Phys. Chem. 1958, 62, 1601–1603. [Google Scholar] [CrossRef]
- Ramette, R.W.; Sandell, E.B. Rhodamine B equilibriums. J. Am. Chem. Soc. 1956, 78, 4872–4878. [Google Scholar] [CrossRef]
- Hinckley, D.A.; Seybold, P.G.; Borris, D.P. Solvatochromism and thermochromism of rhodamine solutions. Spectrochim. Acta Part A Mol. Spectrosc. 1986, 42, 747–754. [Google Scholar] [CrossRef]
- Beija, M.; Afonso, C.A.M.; Martinho, J.M.G. Synthesis and applications of Rhodamine derivatives as fluorescent probes. Chem. Soc. Rev. 2009, 38, 2410–2433. [Google Scholar] [CrossRef] [Green Version]
- Arbeloa, F.L.; Estevez, M.J.T. Photophysics of rhodamines: Molecular structure and solvent effects. J. Phys. Chem. 1991, 95, 2203–2208. [Google Scholar] [CrossRef]
- Arbeloa, T.L.; Estévez, M.T.; López-Arbeloa, F.; Aguirresacona, I.U.; Arbeloa, I.L. Luminescence properties of rhodamines in water/ethanol mixtures. J. Lumin 1991, 48, 400–404. [Google Scholar] [CrossRef]
- Milvy, P.; Kay, K. Mutagenicity of 19 major graphic arts and printing dyes. J. Toxicol. Environ. Heal. Part A 1978, 4, 31–36. [Google Scholar] [CrossRef]
- Smart, P.L. A review of the toxicity of twelve fluorescent dyes used for water tracing. NSS Bull. 1984, 46, 21–33. [Google Scholar]
- Umeda, M. Experimental production of sarcoma in rats by injection of rhodamine B. I. Gann 1952, 43, 120–122. [Google Scholar]
- Umeda, M. Experimental study of xanthene dyes as carcinogenic agents. Gann 1956, 47, 51–78. [Google Scholar]
- Nestmann, E.R.; Douglas, G.R.I.; Matula, T.E.; Grant, C.; Kowbel, D.J. Mutagenic activity of rhodamine dyes and their impurities as detected by mutation induction in Salmonella and DNA damage in Chinese hamster ovary cells. Cancer Res. 1979, 39, 4412–4417. [Google Scholar] [PubMed]
- Elliott, G.S.; Mason, R.W.; Edwards, I.R. Studies on the pharmacokinetics and mutagenic potential of rhodamine b. J. Toxicol. Clin. Toxicol. 1990, 28, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Sommerwerk, S.; Heller, L.; Kerzig, C.; Kramell, A.E.; Csuk, R. Rhodamine B conjugates of triterpenoic acids are cytotoxic mitocans even at nanomolar concentrations. Eur. J. Med. Chem. 2017, 127, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Macasoi, I.; Mioc, M.; Vaduva, D.B.; Ghiulai, R.; Mioc, A.; Soica, C.; Muntean, D.; Dumitrascu, V. In silico Evaluation of the Antiproliferative Mithocondrial Targeted Mechanism of Action of Some Pentacyclic Triterpene Derivatives. Rev. Chim. 2019, 69, 3361–3363. [Google Scholar] [CrossRef]
- Friedrich, S.; Serbian, I.; Hoenke, S.; Wolfram, R.K.; Csuk, R. Synthesis and cytotoxic evaluation of malachite green derived oleanolic and ursolic acid piperazineamides. Med. Chem. Res. 2020, 29, 926–933. [Google Scholar] [CrossRef] [Green Version]
- Serbian, I.; Hoenke, S.; Kraft, O.; Csuk, R. Ester and amide derivatives of rhodamine B exert cytotoxic effects on different human tumor cell lines. Med. Chem. Res. 2020, 29, 1655–1661. [Google Scholar] [CrossRef]
- Kozubek, M.; Serbian, I.; Hoenke, S.; Kraft, O.; Csuk, R. Synthesis and cytotoxic evaluation of hydroxycinnamic acid rhodamine B conjugates. Results Chem. 2020, 2, 100057. [Google Scholar] [CrossRef]
- Serbian, I.; Hoenke, S.; Csuk, R. Synthesis of some steroidal mitocans of nanomolar cytotoxicity acting by apoptosis. Eur. J. Med. Chem. 2020, 199, 112425. [Google Scholar] [CrossRef]
- Wiemann, J.; Fischer, L.; Kessler, J.; Ströhl, D.; Csuk, R. Ugi Multicomponent-reaction: Syntheses of cytotoxic dehydroabietylamine derivatives. Bioorganic Chem. 2018, 81, 567–576. [Google Scholar] [CrossRef]
- Wolfram, R.K.; Heller, L.; Csuk, R. Targeting mitochondria: Esters of rhodamine B with triterpenoids are mitocanic triggers of apoptosis. Eur. J. Med. Chem. 2018, 152, 21–30. [Google Scholar] [CrossRef]
- Wolfram, R.K.; Fischer, L.; Kluge, R.; Ströhl, D.; Al-Harrasi, A.; Csuk, R. Homopiperazine-rhodamine B adducts of triterpenoic acids are strong mitocans. Eur. J. Med. Chem. 2018, 155, 869–879. [Google Scholar] [CrossRef]
- Siewert, B.; Pianowski, E.; Obernauer, A.; Csuk, R. Towards cytotoxic and selective derivatives of maslinic acid. Bioorg. Med. Chem. 2014, 22, 594–615. [Google Scholar] [CrossRef]
- Sommerwerk, S.; Heller, L.; Kuhfs, J.; Csuk, R. Urea derivates of ursolic, oleanolic and maslinic acid induce apoptosis and are selective cytotoxic for several human tumor cell lines. Eur. J. Med. Chem. 2016, 119, 1–16. [Google Scholar] [CrossRef]
- Kahnt, M.; Wiemann, J.; Fischer, L.; Sommerwerk, S.; Csuk, R. Transformation of asiatic acid into a mitocanic, bimodal-acting rhodamine B conjugate of nanomolar cytotoxicity. Eur. J. Med. Chem. 2018, 159, 143–148. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Structure of Rhodamine B (RhoB) in its “open” form A, “closed” lactone form B and the zwitterion C.
Figure 1.
Structure of Rhodamine B (RhoB) in its “open” form A, “closed” lactone form B and the zwitterion C.

Figure 2.
Structure of some important pentacyclic triterpenoic acids.

Figure 3.
Structures of “simple” RhoB conjugates 1–9.

Figure 4.
RhoB steroid conjugates from the esterification of RhoB with testosterone (→ 10), prednisone (→ 11) or abiratone (→ 12), respectively.
Figure 4.
RhoB steroid conjugates from the esterification of RhoB with testosterone (→ 10), prednisone (→ 11) or abiratone (→ 12), respectively.

Figure 5.
Dehydroabiethylamine (DHAA)-derived RhoB conjugates 13–15 obtained by Ugi-multi component reactions.
Figure 5.
Dehydroabiethylamine (DHAA)-derived RhoB conjugates 13–15 obtained by Ugi-multi component reactions.

Figure 6.
Synthesis of DHAA-derived RhoB conjugates 16a/16b and their equilibrium.

Figure 7.
Un-spacered UA-, OA-, GA- and BA-derived esters 17–24 and amides 25–28.

Figure 8.
Synthesis of euscaphic (EA)- or tormentic acid (TA)-derived RhoB conjugates 29–32.

Figure 9.
Synthesis of maslinic acid (MA)-derived EM2 holding the same absolute configuration of hydroxyl groups in ring A as asiatic acid (AA)-derived conjugate 33.
Figure 9.
Synthesis of maslinic acid (MA)-derived EM2 holding the same absolute configuration of hydroxyl groups in ring A as asiatic acid (AA)-derived conjugate 33.

Figure 10.
Graphical representation of the cytotoxicity of all compounds (EC50 in μM) for the cell line A2780 (A) combined with a comparison of tumor cell/non-tumor cell selectivity (A2780 vs. NIH 3T3, selectivity = (EC50, NIH3T3/EC50, A2780)) of all compounds (B).
Figure 10.
Graphical representation of the cytotoxicity of all compounds (EC50 in μM) for the cell line A2780 (A) combined with a comparison of tumor cell/non-tumor cell selectivity (A2780 vs. NIH 3T3, selectivity = (EC50, NIH3T3/EC50, A2780)) of all compounds (B).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Cytotoxicity of selected “simple” RhoB conjugates.
| Compound | A375 | HT29 | MCF-7 | A2780 | FaDu | NIH 3T3 |
|---|---|---|---|---|---|---|
| RhoB | >30 | >30 | >30 | >30 | >30 | >30 |
| 1 | 0.38 | 0.41 | 0.23 | 0.21 | 0.30 | 0.96 |
| 2 | 0.19 | 0.19 | 0.14 | 0.17 | 0.15 | 0.32 |
| 3 | >30 | >30 | >30 | >30 | >30 | >30 |
| 4 | 7.09 | 5.46 | 1.54 | 1.66 | 4.53 | >30 |
| 5 | 1.79 | 1.54 | 0.44 | 0.52 | 1.12 | 5.09 |
| 6 | 3.05 | 1.74 | 0.49 | 0.70 | 1.52 | 7.92 |
| 7 | 16.05 | 17.34 | 3.74 | 3.62 | 11.78 | >30 |
| 8 | 1.03 | 0.54 | 0.32 | 0.27 | 0.64 | 3.27 |
| 9 | >30 | >30 | 17.80 | 26.40 | >30 | >30 |
EC50 in μM from SRB assays; cut-off 30 μM.
Table 2.
Cytotoxicity of selected steroidal RhoB conjugates.
| Compound | A375 | HT29 | MCF-7 | A2780 | FaDu | NIH 3T3 |
|---|---|---|---|---|---|---|
| 10 | 0.16 | 0.12 | 0.06 | 0.08 | 0.26 | 0.25 |
| 11 | 0.11 | 0.64 | 0.21 | 0.31 | 0.40 | 1.81 |
| 12 | 0.22 | 0.21 | 0.23 | 0.13 | 0.24 | 0.37 |
EC50 in μM from SRB assays.
Table 3.
Cytotoxicity of selected DHAA-derived RhoB conjugates.
| Compound | A375 | HT29 | MCF-7 | A2780 | FaDu | NIH 3T3 |
|---|---|---|---|---|---|---|
| 13 | 3.2 | 0.18 | 0.10 | 0.37 | 0.23 | 0.28 |
| 14 | 0.23 | 0.32 | 0.16 | 0.57 | 0.35 | 0.41 |
| 15 | 0.20 | 0.28 | 0.12 | 0.66 | 0.32 | 0.44 |
| 16a/16b | >30 | >30 | >30 | >30 | >30 | >30 |
EC50 in μM from SRB assays.
Table 4.
Cytotoxicity of un-spacered esters UA-, OA-, GA- and BA-derived esters 17–24 and amides 25–28.
Table 4.
Cytotoxicity of un-spacered esters UA-, OA-, GA- and BA-derived esters 17–24 and amides 25–28.
| Compound | TP | R | FaDu | A2780 | HT29 | MCF-7 | SW1736 | NIH 3T3 |
|---|---|---|---|---|---|---|---|---|
| 17 | UA | OMe | 1.96 | 1.75 | 1.85 | 1.83 | 1.72 | 1.84 |
| 18 | OA | OMe | 1.99 | 1.14 | 2.75 | 2.31 | 1.76 | 2.63 |
| 19 | GA | OMe | 0.19 | 0.08 | 0.15 | 0.18 | 0.15 | 0.20 |
| 20 | BA | OMe | 1.29 | 0.42 | 0.61 | 0.81 | 0.74 | 1.77 |
| 21 | UA | OBn | 15.79 | 10.10 | 11.41 | 13.75 | 12.66 | 15.42 |
| 22 | OA | OBn | 9.12 | 3.35 | 8.90 | 9.40 | 9.05 | 11.25 |
| 23 | GA | OBn | 1.54 | 0.90 | 1.42 | 1.47 | 1.13 | 1.28 |
| 24 | BA | OBn | 7.59 | 3.36 | 5.33 | 5.05 | 6.43 | 8.04 |
| 25 | UA | NBn | 0.44 | 0.34 | 0.45 | 0.30 | 0.24 | 0.37 |
| 26 | OA | NBn | 0.50 | 0.32 | 0.46 | 0.36 | 0.27 | 0.40 |
| 27 | GA | NBn | 0.06 | 0.02 | 0.06 | 0.04 | 0.04 | 0.08 |
| 28 | BA | NBn | 0.54 | 0.31 | 0.53 | 0.47 | 0.45 | 0.54 |
EC50 in μM from SRB assays.
Table 5.
Cytotoxicity of euscaphic (EA)- or tormentic acid (TA)-derived RhoB conjugates 29–32, asiatic acid (AA)-derived 33 and maslinic acid (MA)-derived amide EM2.
Table 5.
Cytotoxicity of euscaphic (EA)- or tormentic acid (TA)-derived RhoB conjugates 29–32, asiatic acid (AA)-derived 33 and maslinic acid (MA)-derived amide EM2.
| Compound | A375 | HT29 | MCF-7 | A2780 | FaDu | NIH 3T3 |
|---|---|---|---|---|---|---|
| 29 | 0.19 | 0,19 | 0.094 | 0.066 | 0.074 | 0.21 |
| 30 | 0.012 | 0.012 | 0.022 | 0.004 | 0.004 | 0.164 |
| 31 | 0.14 | 0.16 | 0.0084 | 0.037 | 0.041 | 0.25 |
| 32 | 0.06 | 0.005 | 0.008 | 0.001 | 0.001 | 0.19 |
| 33 | n.d. | 0.017 | 0.012 | 0.008 | n.d. | 0.178 |
| EM2 | n.d. | 4.70 | 7.70 | 0.50 | n.d. | 33.8 |
EC50 in μM from SRB assays.
Sample Availability: Samples of the compounds are not available from the authors. | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hoenke, S.; Serbian, I.; Deigner, H.-P.; Csuk, R. Mitocanic Di- and Triterpenoid Rhodamine B Conjugates. Molecules 2020, 25, 5443. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25225443
AMA Style
Hoenke S, Serbian I, Deigner H-P, Csuk R. Mitocanic Di- and Triterpenoid Rhodamine B Conjugates. Molecules. 2020; 25(22):5443. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25225443
Chicago/Turabian StyleHoenke, Sophie, Immo Serbian, Hans-Peter Deigner, and René Csuk. 2020. "Mitocanic Di- and Triterpenoid Rhodamine B Conjugates" Molecules 25, no. 22: 5443. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules25225443