
In Search for Multi-Target Ligands as Potential Agents for Diabetes Mellitus and Its Complications—A Structure-Activity Relationship Study on Inhibitors of Aldose Reductase and Protein Tyrosine Phosphatase 1B

 , , , ,
, , , ,  , , ,
, , ,
Abstract
:1. Introduction
2. Results and Discussion
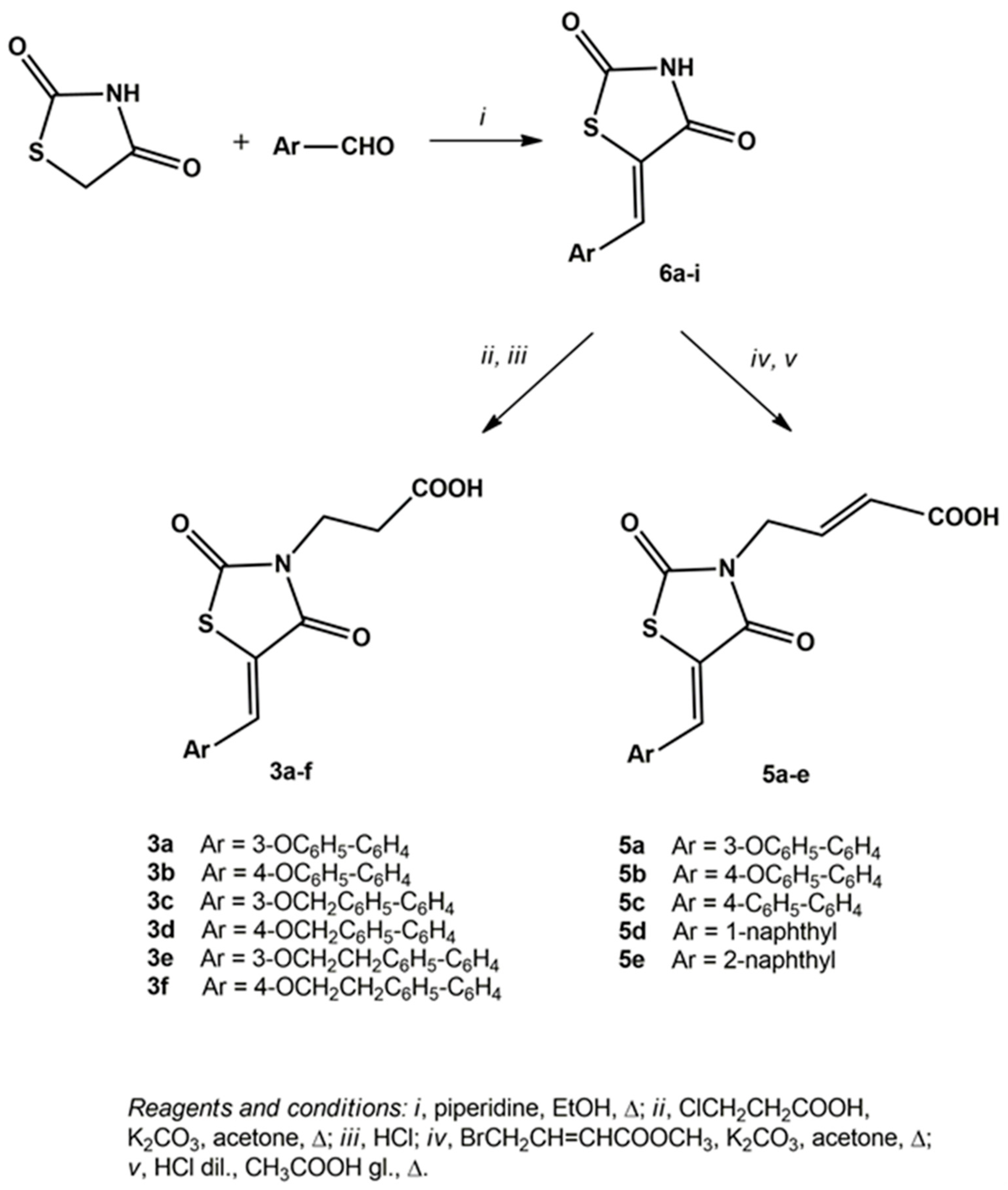
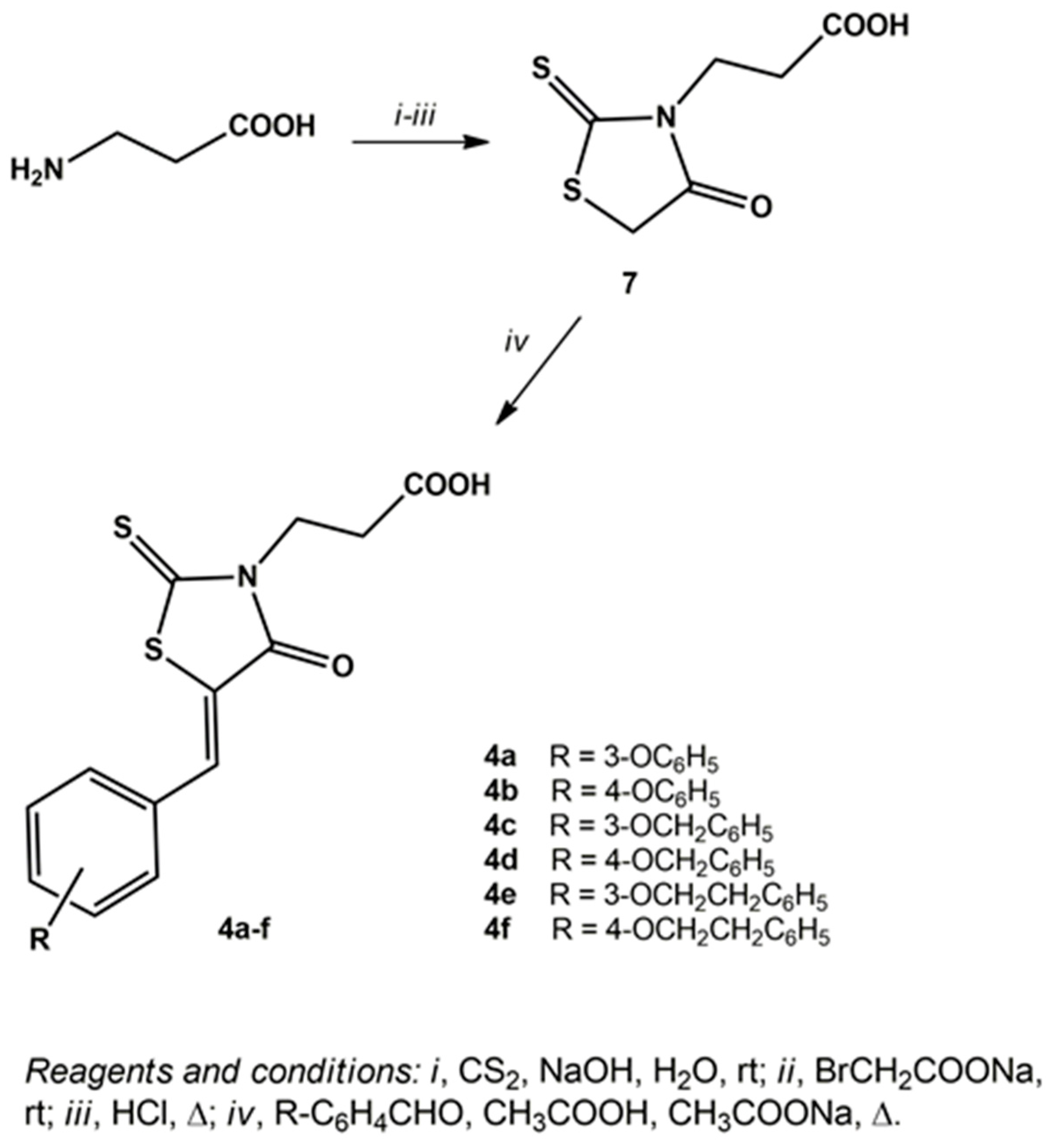
2.1. Chemistry
2.2. AR and PTP1B Inhibition
2.3. Kinetic Studies
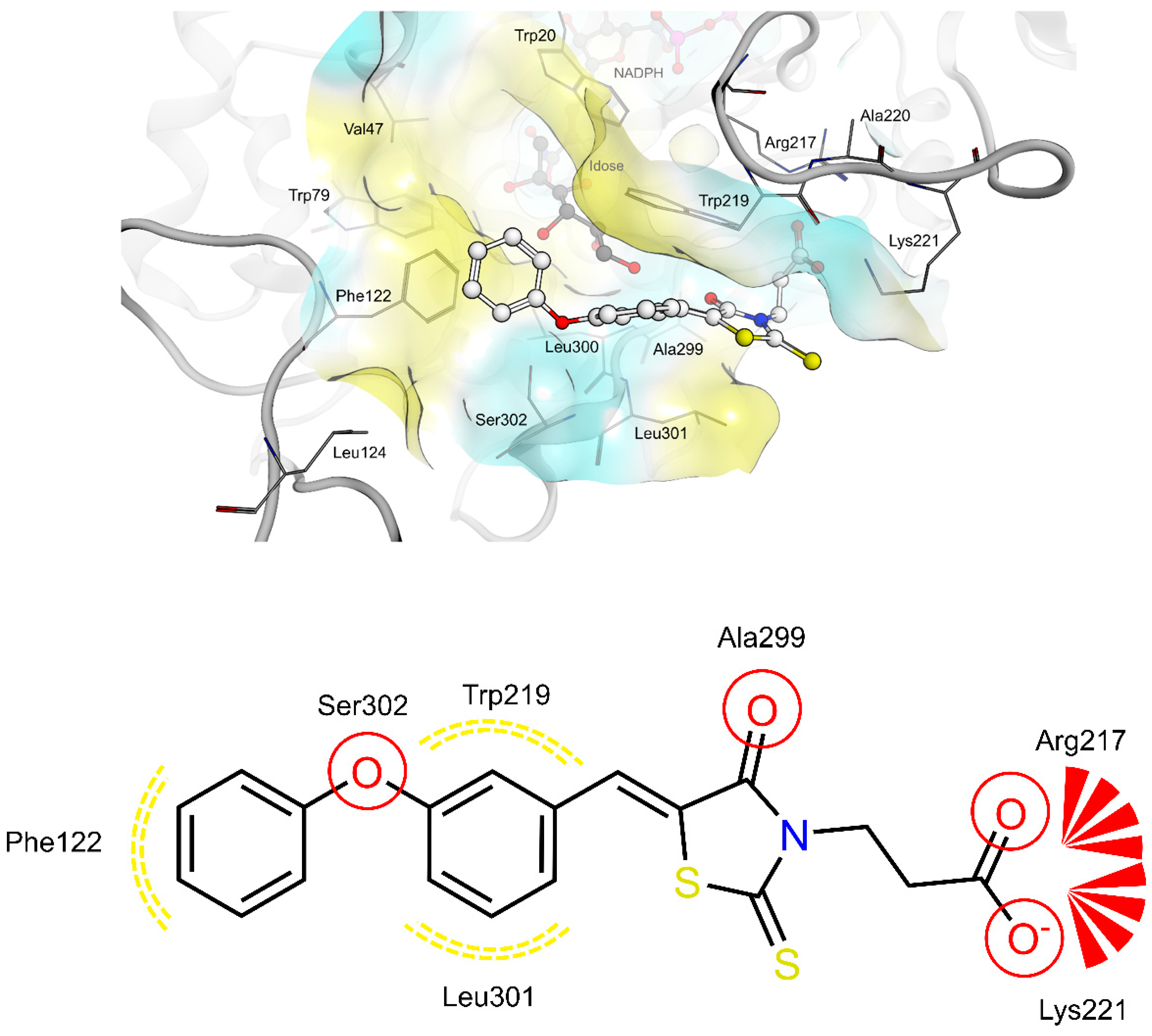
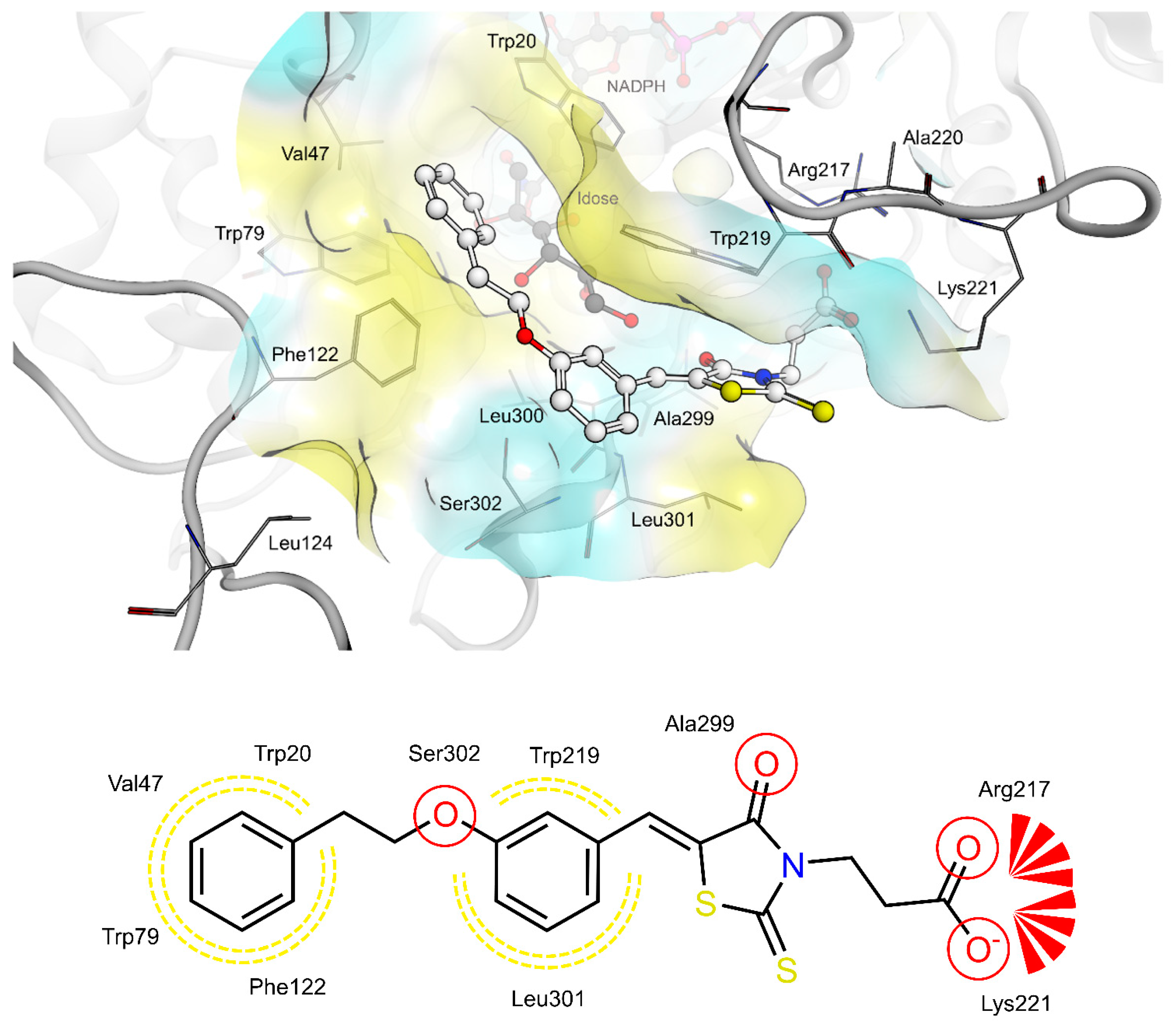
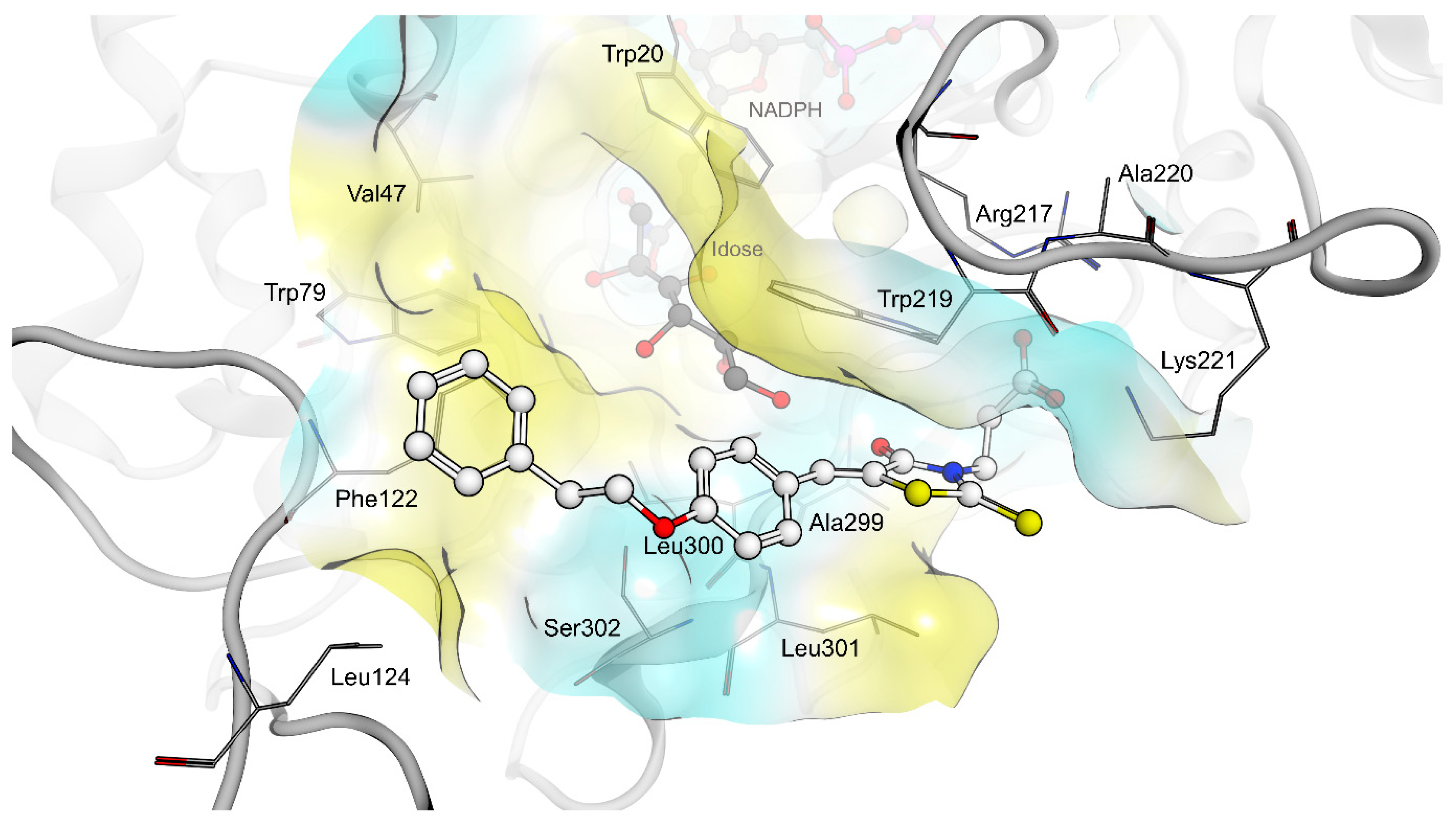
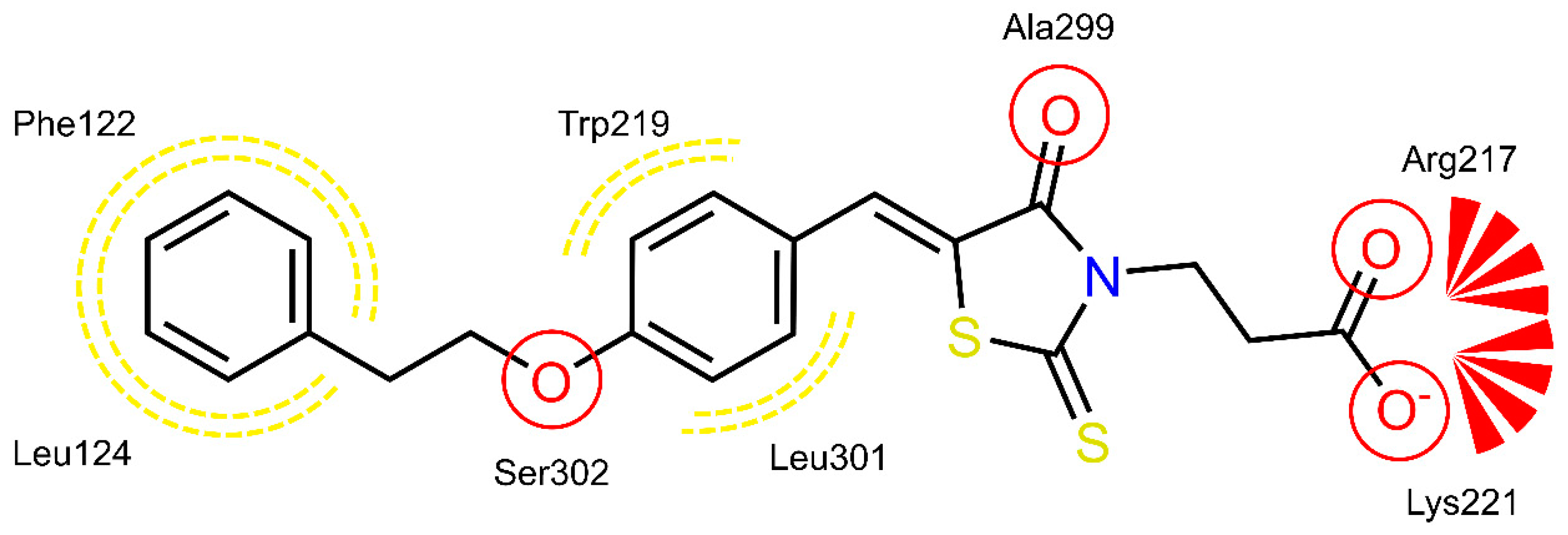
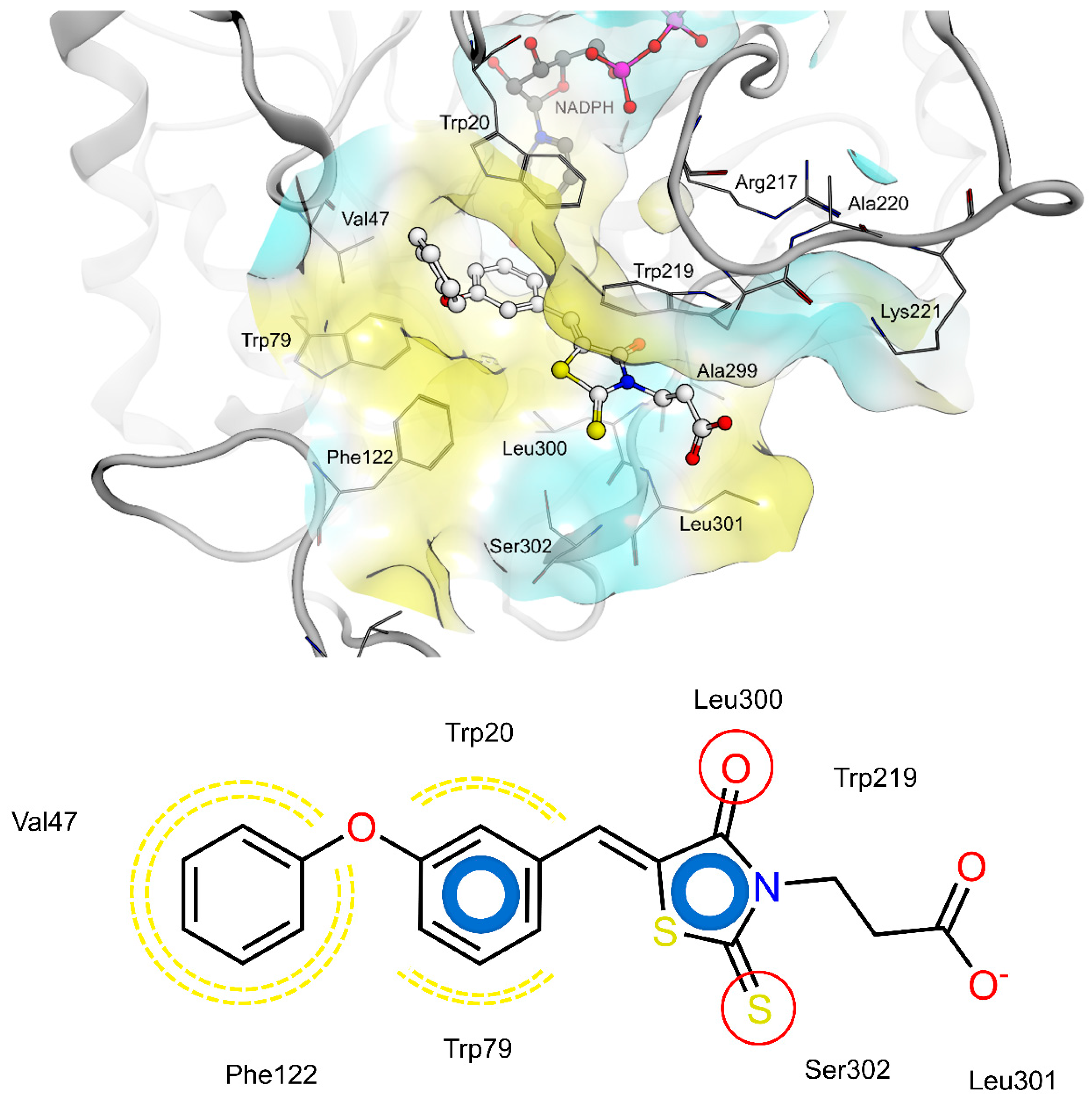
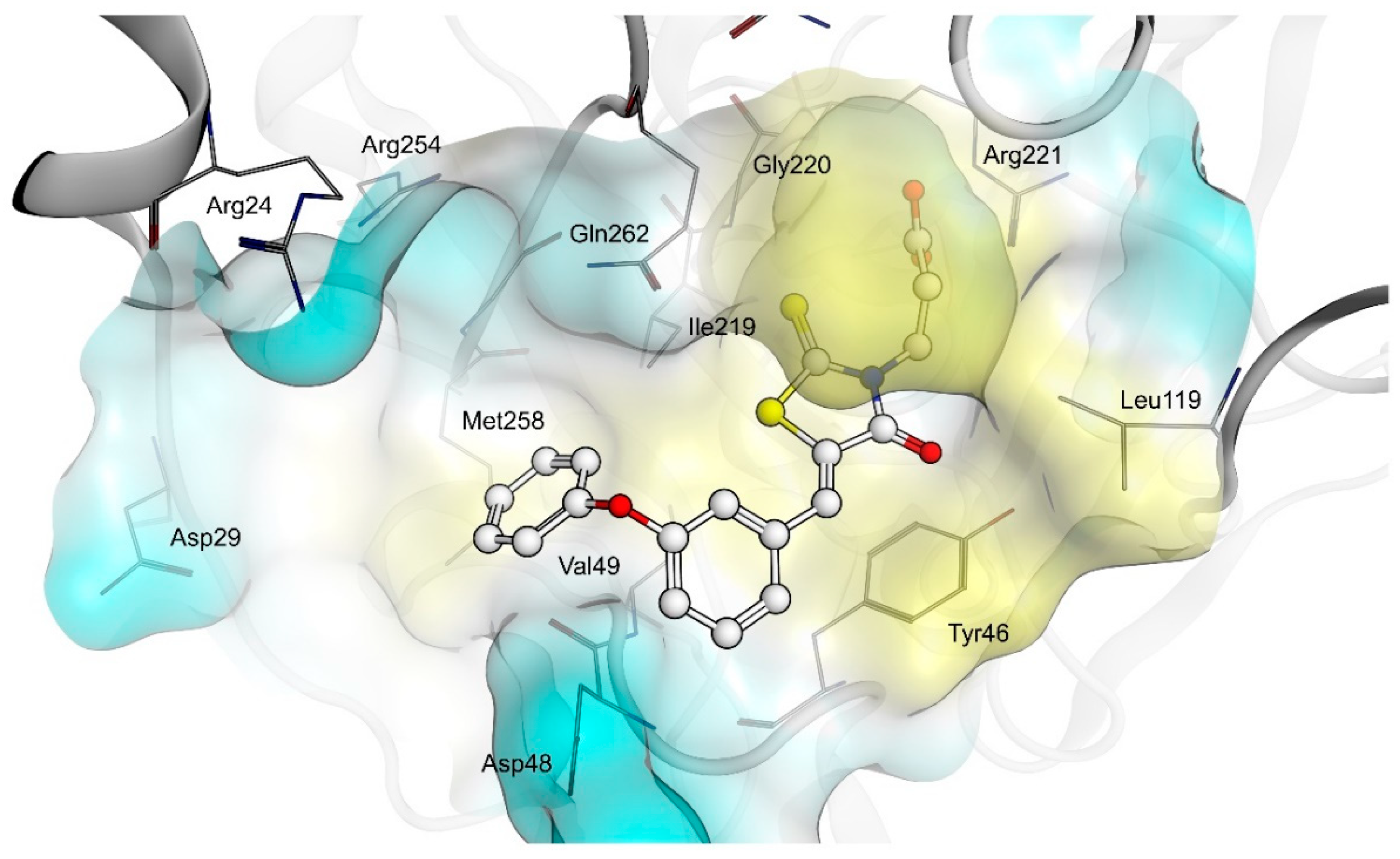
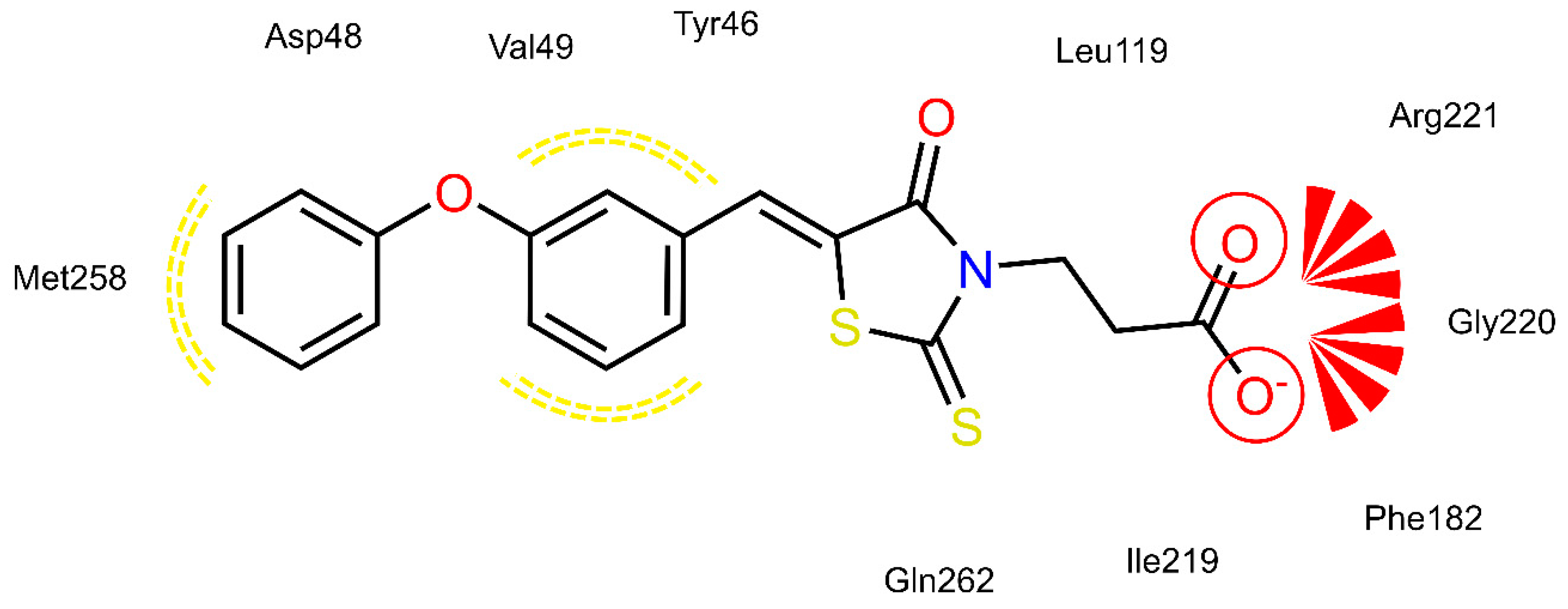
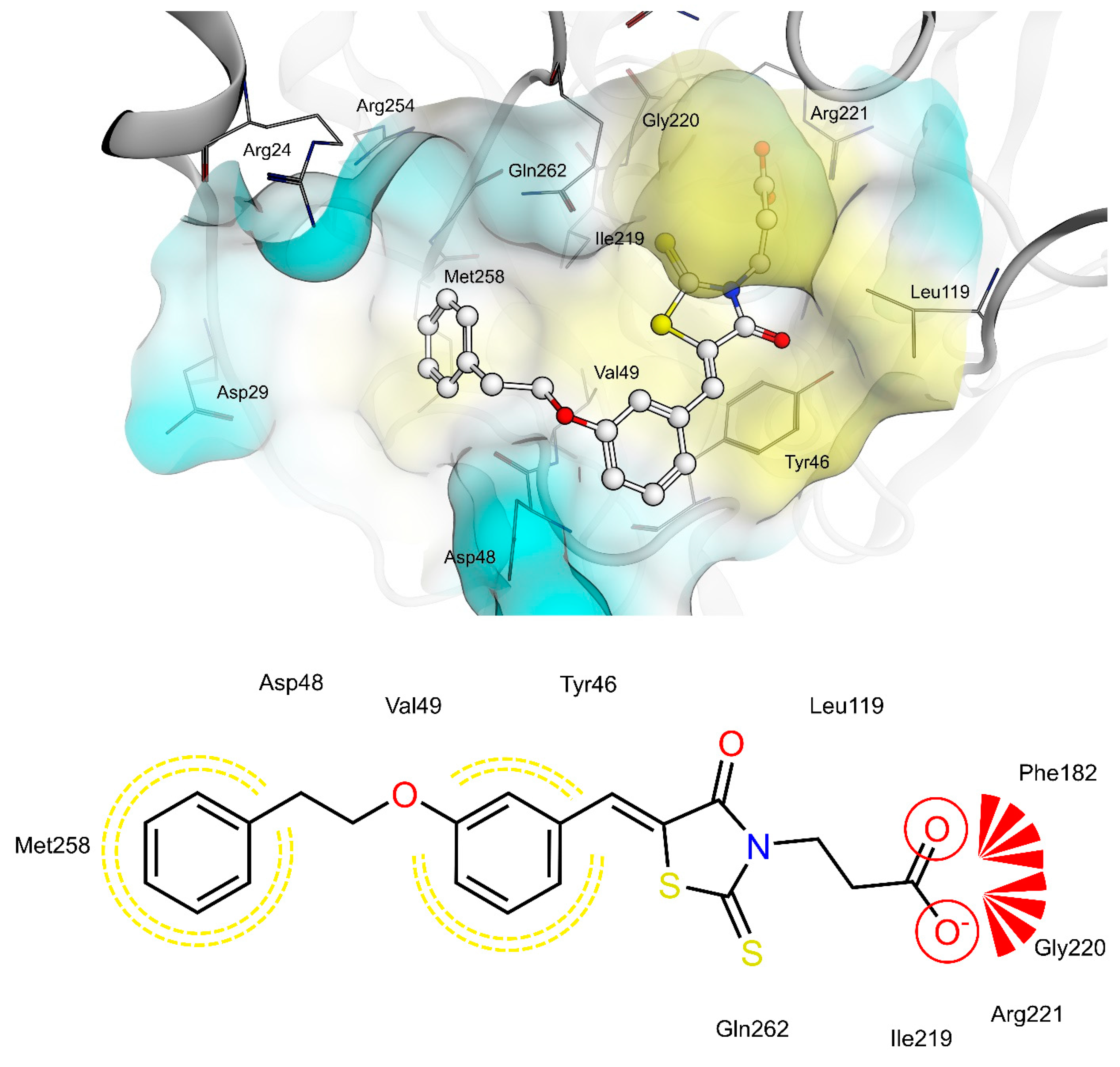
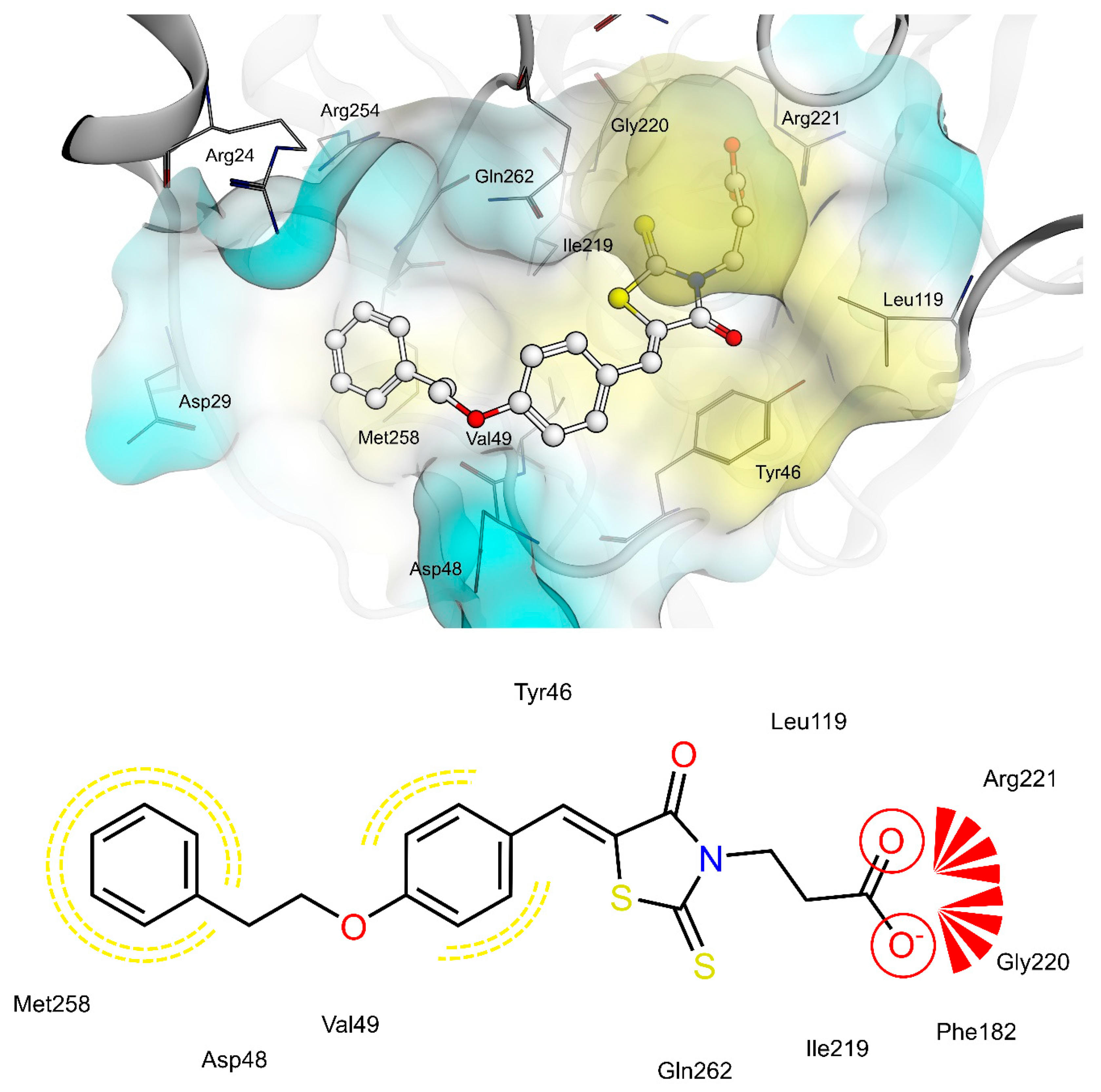
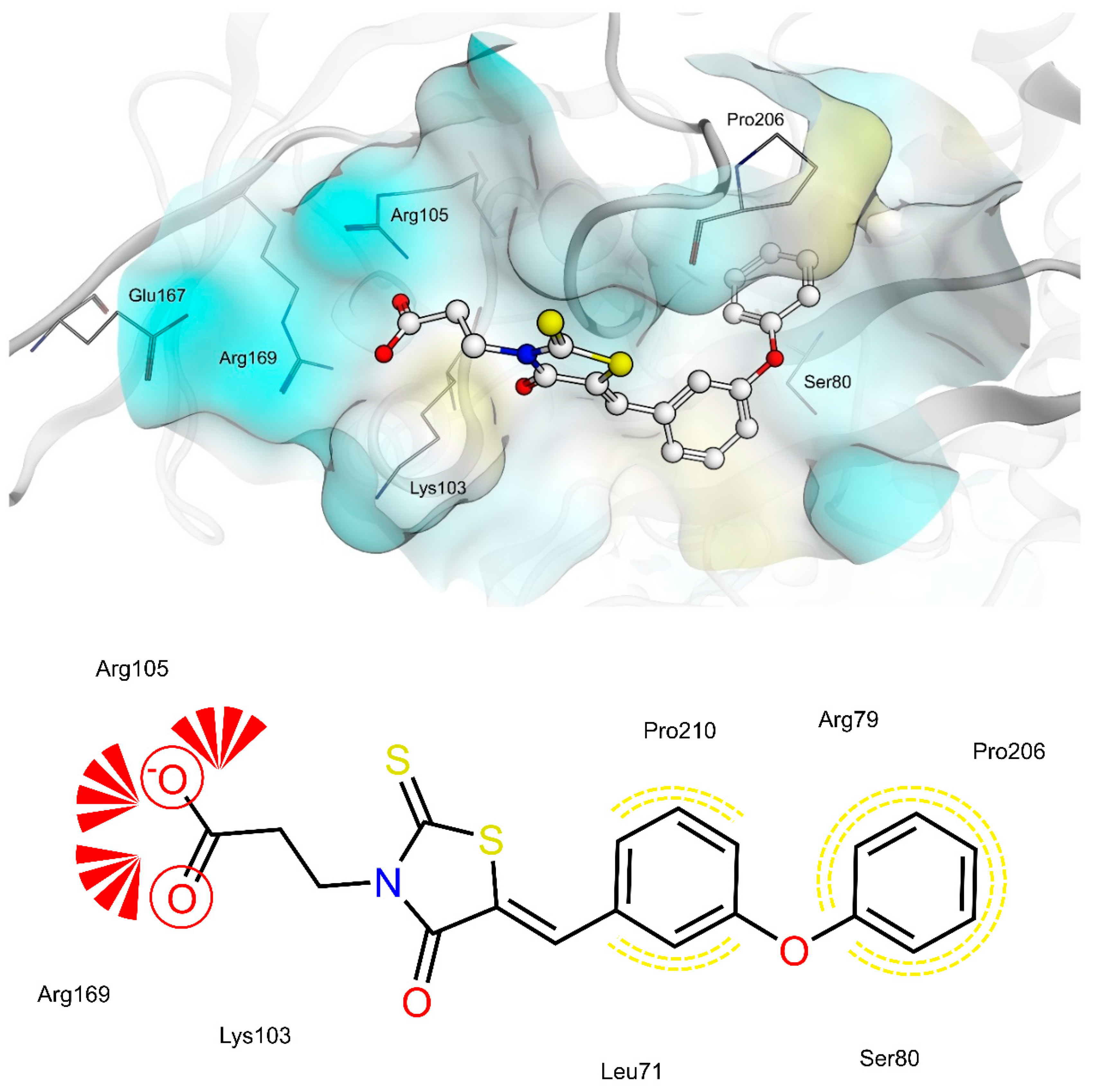
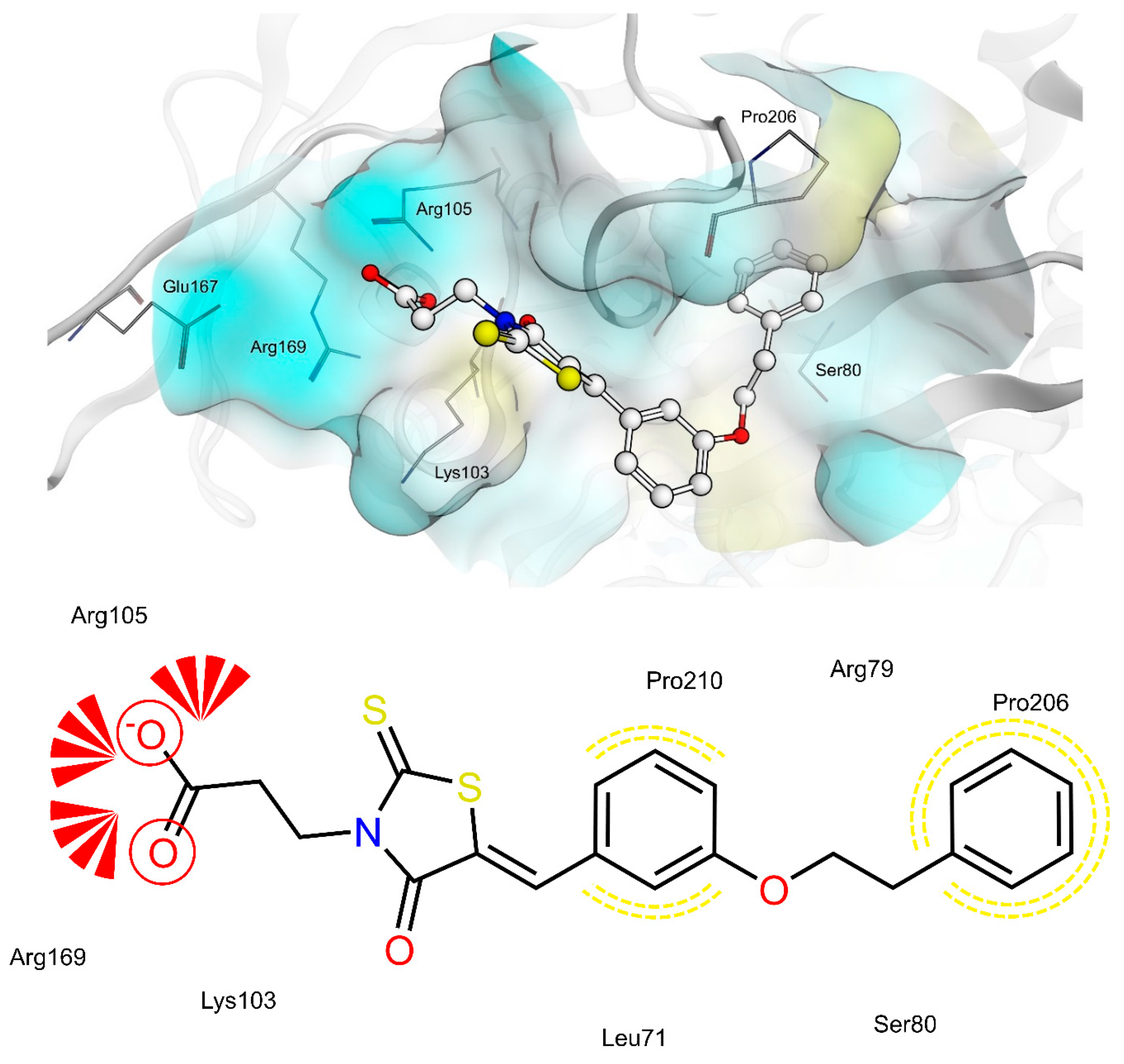
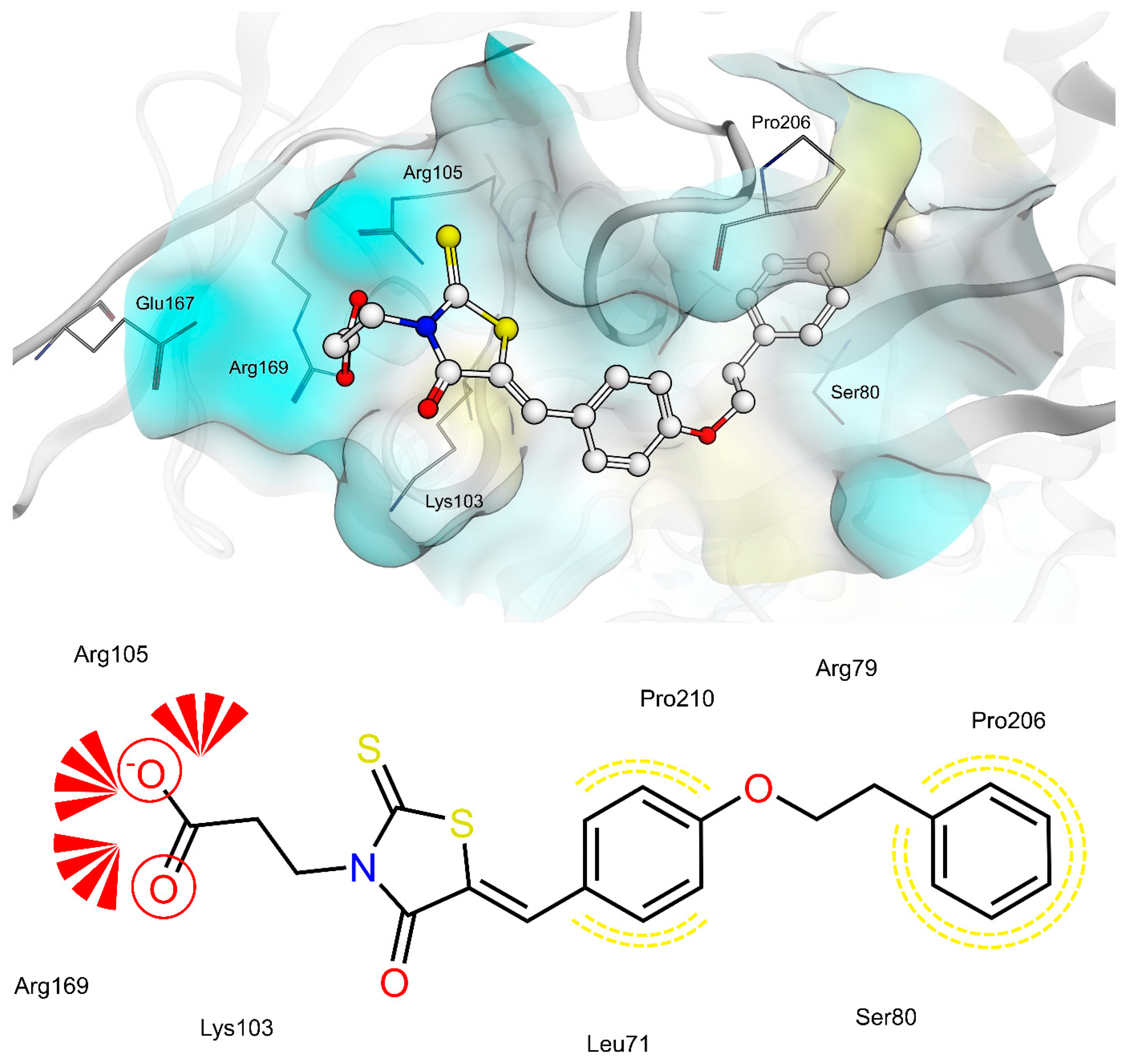
2.4. AR and PTP1B Docking Experiments
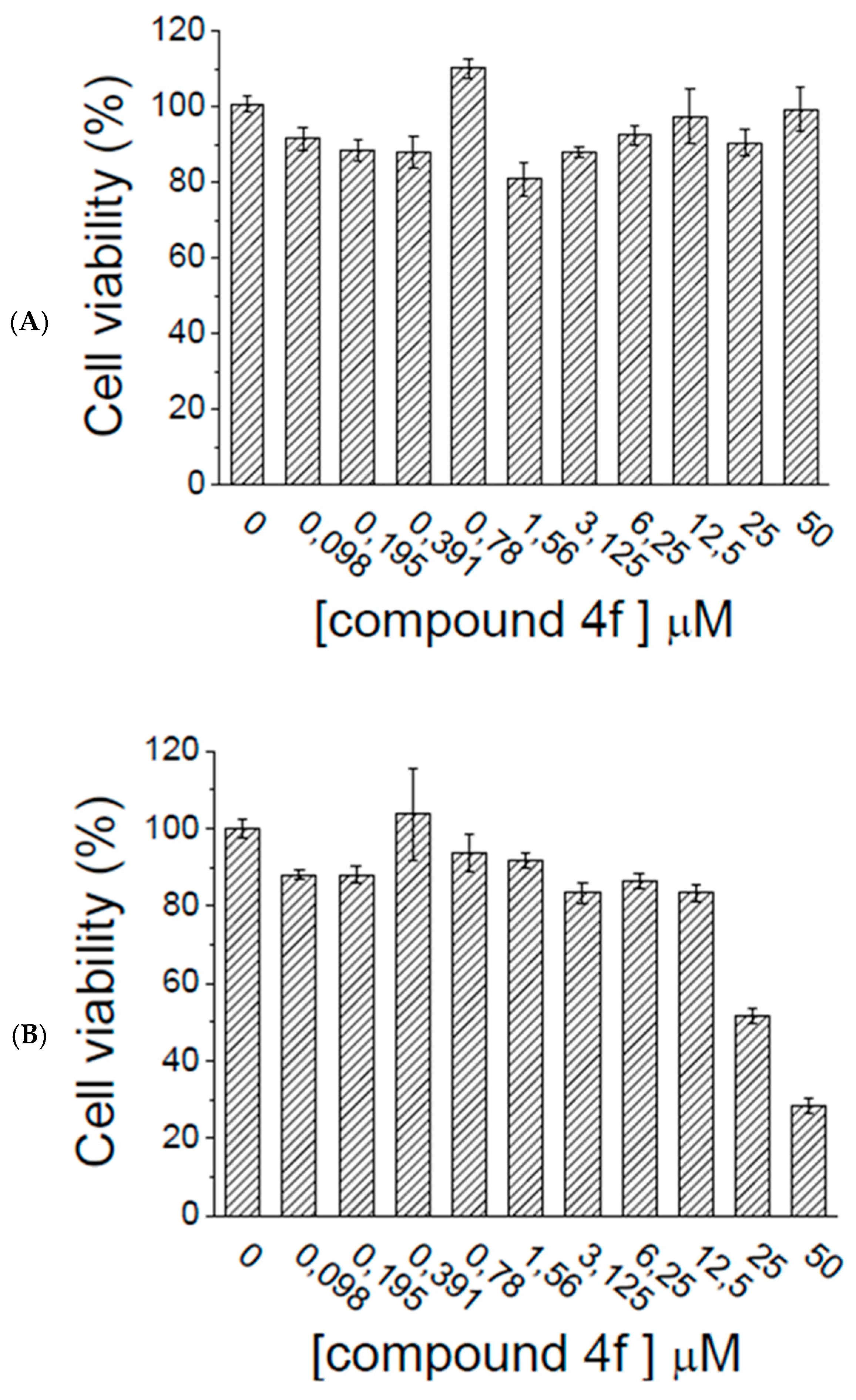
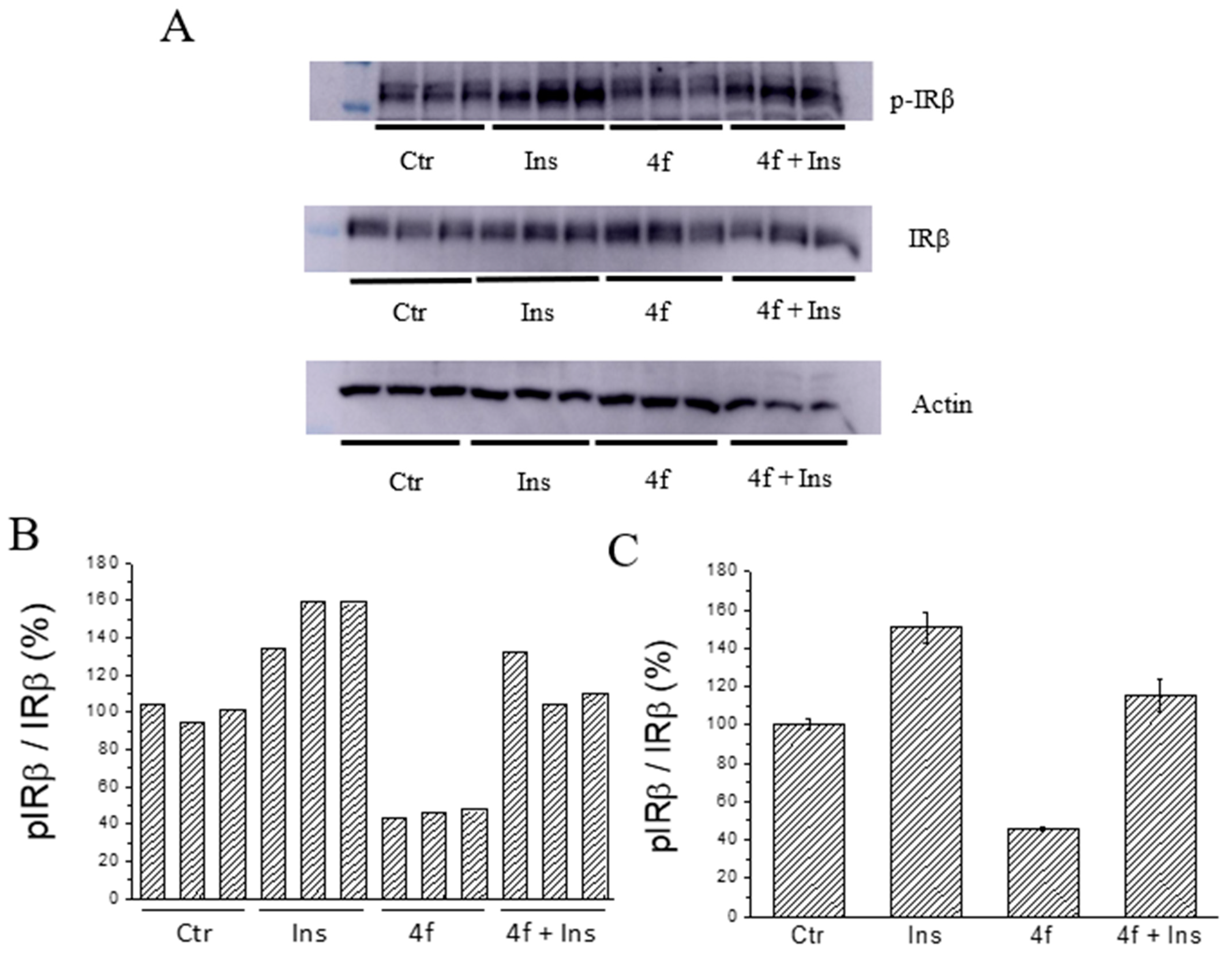
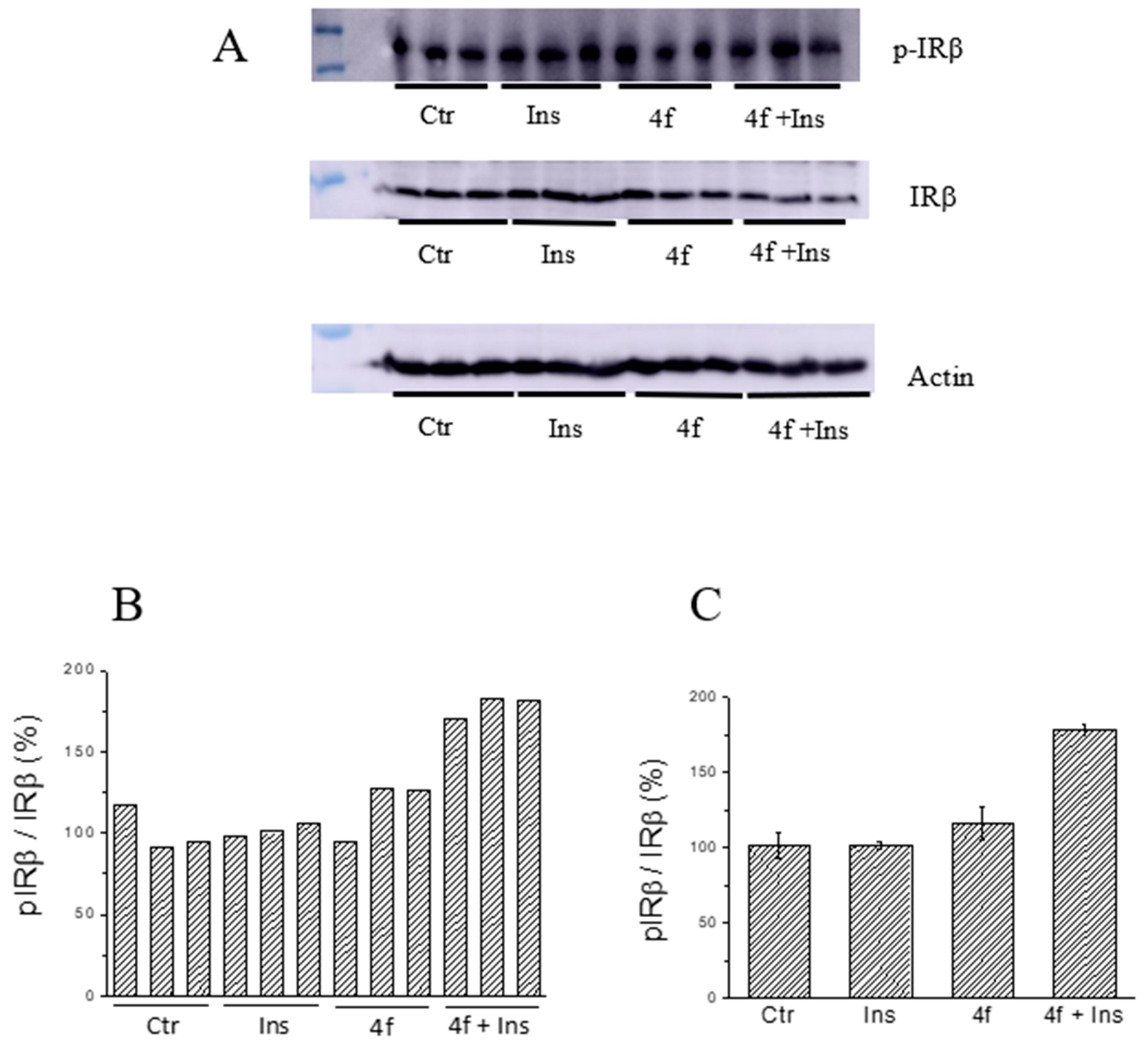
2.5. Ex Vivo Assays
3. Materials and Methods
3.1. Chemistry
3.1.1. General Procedure for the Synthesis of 5-arylidene-2,4-dioxothiazolidinones 6a–i
3.1.2. General Procedure for the Synthesis of 3-(5-arylidene-2,4-dioxothiazolidin-3-yl)propanoic Acids 3a–f
3.1.3. General Procedure for the Synthesis of 3-(5-arylidene-4-oxo-2-thioxothiazolidin-3-yl)propanoic Acids 4a–f
3.1.4. General Procedure for the Synthesis of 4-(5-arylidene-2,4-dioxothiazolidin-3-yl)-2-butenoic Acids 5a–e
3.2. Enzymatic Assays
3.2.1. AR Enzymatic Assay, Expression and Purification
3.2.2. Inhibition Studies on AR
3.2.3. Enzymatic Assays with PTP1B
3.2.4. Reversibility Assay (Dilution Test) with PTP1B
3.2.5. Evaluation of Action Mechanism of Inhibitors
3.3. Ex Vivo Assay
3.3.1. Cell Cultures
3.3.2. Cell Viability Assay
3.3.3. Evaluation of Insulin Mimetic/Sensibilizing Activity of Inhibitors
3.4. Molecular Docking
Virtual Screening
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Morphy, R.; Rankovic, Z. Designed Multiple Ligands. An Emerging Drug Discovery Paradigm. J. Med. Chem. 2005, 48, 6523–6543. [Google Scholar] [CrossRef]
- Csermely, P.; Agoston, V.; Pongor, S. The efficiency of multi-target drugs: The network approach might help drug design. Trends Pharmacol. Sci. 2005, 26, 178–182. [Google Scholar] [CrossRef] [Green Version]
- Hopkins, A.L. Network pharmacology: The next paradigm in drug discovery. Nat. Chem. Biol. 2008, 4, 682–690. [Google Scholar] [CrossRef]
- Proschak, E.; Stark, H.; Merk, D. Polypharmacology by Design: A Medicinal Chemist’s Perspective on Multitargeting Compounds. J. Med. Chem. 2019, 62, 420–444. [Google Scholar] [CrossRef]
- Maccari, R.; Del Corso, A.; Paoli, P.; Adornato, I.; Lori, G.; Balestri, F.; Cappiello, M.; Naß, A.; Wolber, G.; Ottanà, R. An investigation on 4-thiazolidinone derivatives as dual inhibitors of aldose reductase and protein tyrosine phosphatase 1B, in the search for potential agents for the treatment of type 2 diabetes mellitus and its complications. Bioorganic Med. Chem. Lett. 2018, 28, 3712–3720. [Google Scholar] [CrossRef] [PubMed]
- Cho, N.H.; Shaw, J.E.; Karuranga, S.; Huang, Y.; Fernandes, J.D.R.; Ohlrogge, A.; Malanda, B. IDF Diabetes Atlas: Global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res. Clin. Pract. 2018, 138, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gattrell, W.; Johnstone, C.; Patel, S.; Smith, C.S.; Scheel, A.; Schindler, M. Designed multiple ligands in metabolic disease research: From concept to platform. Drug Discov. Today 2013, 18, 692–696. [Google Scholar] [CrossRef] [PubMed]
- Dowarah, J.; Singh, V.P. Anti-diabetic drugs recent approaches and advancements. Bioorganic Med. Chem. 2020, 28, 115263. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.K.; Ramana, K.V.; Bhatnagar, A. Role of Aldose Reductase and Oxidative Damage in Diabetes and the Consequent Potential for Therapeutic Options. Endocr. Rev. 2005, 26, 380–392. [Google Scholar] [CrossRef]
- Maccari, R.; Ottanà, R. Targeting Aldose Reductase for the Treatment of Diabetes Complications and Inflammatory Diseases: New Insights and Future Directions. J. Med. Chem. 2015, 58, 2047–2067. [Google Scholar] [CrossRef] [PubMed]
- Ramana, K.V.; Srivastava, S.K. Aldose reductase: A novel therapeutic target for inflammatory pathologies. Int. J. Biochem. Cell Biol. 2010, 42, 17–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frohnert, B.I.; Long, E.K.; Hahn, W.S.; Bernlohr, D.A. Glutathionylated Lipid Aldehydes Are Products of Adipocyte Oxidative Stress and Activators of Macrophage Inflammation. Diabetes 2013, 63, 89–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Paola, R.; Frittitta, L.; Miscio, G.; Bozzali, M.; Baratta, R.; Centra, M.; Spampinato, D.; Santagati, M.G.; Ercolino, T.; Cisternino, C.; et al. A Variation in 3′ UTR of hPTP1B Increases Specific Gene Expression and Associates with Insulin Resistance. Am. J. Hum. Genet. 2002, 70, 806–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, A.; Uetani, N.; Simoncic, P.D.; Chaubey, V.P.; Lee-Loy, A.; McGlade, C.J.; Kennedy, B.P.; Tremblay, M.L. Attenuation of leptin action and regulation of obesity by protein tyrosine phosphatase 1B. Dev. Cell 2002, 2, 497–503. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.-Y.; Dodd, G.T.; Tiganis, T. Protein Tyrosine Phosphatases in Hypothalamic Insulin and Leptin Signaling. Trends Pharmacol. Sci. 2015, 36, 661–674. [Google Scholar] [CrossRef] [Green Version]
- Zabolotny, J.M.; Kim, Y.-B.; Welsh, L.A.; Kershaw, E.E.; Neel, B.G.; Kahn, B.B. Protein-tyrosine Phosphatase 1B Expression Is Induced by Inflammation in Vivo. J. Biol. Chem. 2008, 283, 14230–14241. [Google Scholar] [CrossRef] [Green Version]
- Elchebly, M. Increased Insulin Sensitivity and Obesity Resistance in Mice Lacking the Protein Tyrosine Phosphatase-1B Gene. Science 1999, 283, 1544–1548. [Google Scholar] [CrossRef]
- Klaman, L.D.; Boss, O.; Peroni, O.D.; Kim, J.K.; Martino, J.L.; Zabolotny, J.M.; Moghal, N.; Lubkin, M.; Kim, Y.-B.; Sharpe, A.H.; et al. Increased Energy Expenditure, Decreased Adiposity and Tissue-Specific Insulin Sensitivity in Protein-Tyrosine Phosphatase 1B-Deficient Mice. Mol. Cell. Biol. 2000, 20, 5479–5489. [Google Scholar] [CrossRef] [Green Version]
- Combs, A.P. Recent Advances in the Discovery of Competitive Protein Tyrosine Phosphatase 1B Inhibitors for the Treatment of Diabetes, Obesity and Cancer. J. Med. Chem. 2010, 53, 2333–2344. [Google Scholar] [CrossRef]
- Lantz, K.A.; Hart, S.G.E.; Planey, S.L.; Roitman, M.F.; Ruiz-White, I.A.; Wolfe, H.R.; McLane, M.P. Inhibition of PTP1B by Trodusquemine (MSI-1436) Causes Fat-specific Weight Loss in Diet-induced Obese Mice. Obesity 2010, 18, 1516–1523. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo-Figueroa, S.; Estrada-Soto, S.; Ramírez-Espinosa, J.J.; Paoli, P.; Lori, G.; León-Rivera, I.; Navarrete-Vazquez, G. Synthesis and evaluation of thiazolidine-2,4-dione/benzazole derivatives as inhibitors of protein tyrosine phosphatase 1B (PTP-1B): Antihyperglycemic activity with molecular docking study. Biomed. Pharmacother. 2018, 107, 1302–1310. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Silakari, O.; Kaur, M.; Singh, B. Integrated pharmacophore and docking-based designing of dual inhibitors of aldose reductase (ALR2) and protein tyrosine phosphatase 1B (PTP1B) as novel therapeutics for insulin-resistant diabetes and its complications. J. Chemom. 2015, 29, 109–125. [Google Scholar] [CrossRef]
- Kong, W.-J.; Vernierib, C.; Foiani, M.; Jiang, J. Berberine in the treatment of metabolism-related chronic diseases: A drug cloud (dCloud) effect to target multifactorial disorders. Pharmacol. Ther. 2020, 209, 107496. [Google Scholar] [CrossRef] [PubMed]
- Bruno, G.; Costantino, L.; Curinga, C.; Maccari, R.; Monforte, F.; Nicolò, F.; Ottanà, R.; Vigorita, M. Synthesis and aldose reductase inhibitory activity of 5-arylidene-2,4-thiazolidinediones. Bioorganic Med. Chem. 2002, 10, 1077–1084. [Google Scholar] [CrossRef]
- Maccari, R.; Ottanà, R.; Curinga, C.; Vigorita, M.G.; Rakowitz, D.; Steindl, T.; Langer, T. Structure–activity relationships and molecular modelling of 5-arylidene-2,4-thiazolidinediones active as aldose reductase inhibitors. Bioorganic Med. Chem. 2005, 13, 2809–2823. [Google Scholar] [CrossRef] [PubMed]
- Maccari, R.; Ottanà, R.; Ciurleo, R.; Vigorita, M.; Rakowitz, D.; Steindl, T.; Langer, T. Evaluation of in vitro aldose redutase inhibitory activity of 5-arylidene-2,4-thiazolidinediones. Bioorganic Med. Chem. Lett. 2007, 17, 3886–3893. [Google Scholar] [CrossRef]
- Maccari, R.; Ottanà, R.; Ciurleo, R.; Rakowitz, D.; Matuszczak, B.; Laggner, C.; Langer, T. Synthesis, induced-fit docking investigations and in vitro aldose reductase inhibitory activity of non-carboxylic acid containing 2,4-thiazolidinedione derivatives. Bioorganic Med. Chem. 2008, 16, 5840–5852. [Google Scholar] [CrossRef]
- Maccari, R.; Ciurleo, R.; Giglio, M.; Cappiello, M.; Moschini, R.; Del Corso, A.; Mura, U.; Ottanà, R. Identification of new non-carboxylic acid containing inhibitors of aldose reductase. Bioorganic Med. Chem. 2010, 18, 4049–4055. [Google Scholar] [CrossRef]
- Ottanà, R.; Maccari, R.; Giglio, M.; Del Corso, A.; Cappiello, M.; Mura, U.; Cosconati, S.; Marinelli, L.; Novellino, E.; Sartini, S.; et al. Identification of 5-arylidene-4-thiazolidinone derivatives endowed with dual activity as aldose reductase inhibitors and antioxidant agents for the treatment of diabetic complications. Eur. J. Med. Chem. 2011, 46, 2797–2806. [Google Scholar] [CrossRef]
- Maccari, R.; Del Corso, A.; Giglio, M.; Moschini, R.; Mura, U.; Ottanà, R. In vitro evaluation of 5-arylidene-2-thioxo-4-thiazolidinones active as aldose reductase inhibitors. Bioorganic Med. Chem. Lett. 2011, 21, 200–203. [Google Scholar] [CrossRef] [PubMed]
- Maccari, R.; Vitale, R.M.; Ottanà, R.; Rocchiccioli, M.; Marrazzo, A.; Cardile, V.; Graziano, A.C.E.; Amodeo, P.; Mura, U.; Del Corso, A. Structure–activity relationships and molecular modelling of new 5-arylidene-4-thiazolidinone derivatives as aldose reductase inhibitors and potential anti-inflammatory agents. Eur. J. Med. Chem. 2014, 81, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Maccari, R.; Paoli, P.; Ottanà, R.; Jacomelli, M.; Ciurleo, R.; Manao, G.; Steindl, T.; Langer, T.; Vigorita, M.G.; Camici, G. 5-Arylidene-2,4-thiazolidinediones as inhibitors of protein tyrosine phosphatases. Bioorganic Med. Chem. 2007, 15, 5137–5149. [Google Scholar] [CrossRef] [PubMed]
- Ottanà, R.; Maccari, R.; Ciurleo, R.; Paoli, P.; Jacomelli, M.; Manao, G.; Camici, G.; Laggner, C.; Langer, T. 5-Arylidene-2-phenylimino-4-thiazolidinones as PTP1B and LMW-PTP inhibitors. Bioorganic Med. Chem. 2009, 17, 1928–1937. [Google Scholar] [CrossRef] [PubMed]
- Maccari, R.; Ottanà, R.; Ciurleo, R.; Paoli, P.; Manao, G.; Camici, G.; Laggner, C.; Langer, T. Structure-Based Optimization of Benzoic Acids as Inhibitors of Protein Tyrosine Phosphatase 1B and Low Molecular Weight Protein Tyrosine Phosphatase. ChemMedChem 2009, 4, 957–962. [Google Scholar] [CrossRef]
- Ottanà, R.; Maccari, R.; Amuso, S.; Wolber, G.; Schuster, D.; Herdlinger, S.; Manao, G.; Camici, G.; Paoli, P. New 4-[(5-arylidene-2-arylimino-4-oxo-3-thiazolidinyl)methyl]benzoic acids active as protein tyrosine phosphatase inhibitors endowed with insulinomimetic effect on mouse C2C12 skeletal muscle cells. Eur. J. Med. Chem. 2012, 50, 332–343. [Google Scholar] [CrossRef]
- Ottanà, R.; Maccari, R.; Mortier, J.; Caselli, A.; Amuso, S.; Camici, G.; Rotondo, A.; Wolber, G.; Paoli, P. Synthesis, biological activity and structure–activity relationships of new benzoic acid-based protein tyrosine phosphatase inhibitors endowed with insulinomimetic effects in mouse C2C12 skeletal muscle cells. Eur. J. Med. Chem. 2014, 71, 112–127. [Google Scholar] [CrossRef]
- Ottanà, R.; Paoli, P.; Naß, A.; Lori, G.; Cardile, V.; Adornato, I.; Rotondo, A.; Graziano, A.C.E.; Wolber, G.; Maccari, R. Discovery of 4-[(5-arylidene-4-oxothiazolidin-3-yl)methyl]benzoic acid derivatives active as novel potent allosteric inhibitors of protein tyrosine phosphatase 1B: In silico studies and in vitro evaluation as insulinomimetic and anti-inflammatory agents. Eur. J. Med. Chem. 2017, 127, 840–858. [Google Scholar] [CrossRef]
- Park, H.; Yu, K.R.; Ku, A.B.; Kim, B.-Y.; Kim, S.J. Identification of novel PTPRQ phosphatase inhibitors based on the virtual screening with docking simulations. Theor. Biol. Med. Model. 2013, 10, 49. [Google Scholar] [CrossRef] [Green Version]
- Liang, D.; Robinson, E.; Hom, K.; Yu, W.; Nguyen, N.; Li, Y.; Zong, Q.; Wilks, A.; Xue, F. Structure-based design and biological evaluation of inhibitors of the pseudomonas aeruginosa heme oxygenase (pa-HemO). Bioorganic Med. Chem. Lett. 2018, 28, 1024–1029. [Google Scholar] [CrossRef]
- Dasgupta, R.; Gonsalves, F. Preparation of substituted oxazole derivatives and analogs for use as beta-catenin modulators. PCT Int. Appl. 2009, 2009097113. [Google Scholar]
- Ottanà, R.; Maccari, R.; Barreca, M.L.; Bruno, G.; Rotondo, A.; Rossi, A.; Chiricosta, G.; Di Paola, R.; Sautebin, L.; Cuzzocrea, S.; et al. 5-Arylidene-2-imino-4-thiazolidinones: Design and synthesis of novel anti-inflammatory agents. Bioorganic Med. Chem. 2005, 13, 4243–4252. [Google Scholar] [CrossRef]
- Del-Corso, A.; Balestri, F.; Di Bugno, E.; Moschini, R.; Cappiello, M.; Sartini, S.; La-Motta, C.; Da Settimo, F.; Mura, U. A New Approach to Control the Enigmatic Activity of Aldose Reductase. PLoS ONE 2013, 8, e74076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balestri, F.; Quattrini, L.; Coviello, V.; Sartini, S.; Da Settimo, F.; Cappiello, M.; Moschini, R.; Del Corso, A.; Mura, U.; La Motta, C. Acid Derivatives of Pyrazolo[1,5-a]pyrimidine as Aldose Reductase Differential Inhibitors. Cell Chem. Biol. 2018, 25, 1414–1418.e3. [Google Scholar] [CrossRef] [Green Version]
- Balestri, F.; Poli, G.; Pineschi, C.; Moschini, R.; Cappiello, M.; Mura, U.; Tuccinardi, T.; Del Corso, A. Aldose Reductase Differential Inhibitors in Green Tea. Biomolecules 2020, 10, 1003. [Google Scholar] [CrossRef]
- Puius, Y.A.; Zhao, Y.; Sullivan, M.; Lawrence, D.S.; Almo, S.C.; Zhang, Z.-Y. Identification of a second aryl phosphate-binding site in protein-tyrosine phosphatase 1B: A paradigm for inhibitor design. Proc. Natl. Acad. Sci. USA 1997, 94, 13420–13425. [Google Scholar] [CrossRef] [Green Version]
- Balestri, F.; Cappiello, M.; Moschini, R.; Rotondo, R.; Buggiani, I.; Pelosi, P.; Mura, U.; Del Corso, A. l-Idose: An attractive substrate alternative to d-glucose for measuring aldose reductase activity. Biochem. Biophys. Res. Commun. 2015, 456, 891–895. [Google Scholar] [CrossRef] [Green Version]
- Balestri, F.; Rotondo, R.; Moschini, R.; Pellegrino, M.; Cappiello, M.; Barracco, V.; Misuri, L.; Sorce, C.; Andreucci, A.; Del Corso, A.; et al. Zolfino landrace (Phaseolus vulgaris L.) from Pratomagno: General and specific features of a functional food. Food Nutr. Res. 2016, 60, 31792. [Google Scholar] [CrossRef] [Green Version]
- Misuri, L.; Cappiello, M.; Balestri, F.; Moschini, R.; Barracco, V.; Mura, U.; Del Corso, A. The use of dimethylsulfoxide as a solvent in enzyme inhibition studies: The case of aldose reductase. J. Enzym. Inhib. Med. Chem. 2017, 32, 1152–1158. [Google Scholar] [CrossRef]
- Scapin, G.; Patel, S.B.; Becker, J.W.; Wang, Q.; Desponts, C.; Waddleton, D.; Skorey, K.; Cromlish, W.; Bayly, C.; Therien, M.; et al. The Structural Basis for the Selectivity of Benzotriazole Inhibitors of PTP1B. Biochemistry 2003, 42, 11451–11459. [Google Scholar] [CrossRef]
- Zheng, X.; Zhang, L.; Chen, W.; Chen, Y.; Xie, W.; Hu, X. Partial Inhibition of Aldose Reductase by Nitazoxanide and Its Molecular Basis. ChemMedChem 2012, 7, 1921–1923. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolber, G.; Langer, T. LigandScout: 3-D Pharmacophores Derived from Protein-Bound Ligands and Their Use as Virtual Screening Filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Wolber, G.; Dornhofer, A.A.; Langer, T. Efficient overlay of small organic molecules using 3D pharmacophores. J. Comput. Mol. Des. 2006, 20, 773–788. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck molecular force field. II. MMFF94 van der Waals and electrostatic parameters for intermolecular interactions. J. Comput. Chem. 1996, 17, 520–552. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE), 2019.01; Chemical Computing Group ULC: Montreal, QC, Canada, 2020.
- Hawkins, P.C.D.; Skillman, A.A.G.; Nicholls, A. Comparison of Shape-Matching and Docking as Virtual Screening Tools. J. Med. Chem. 2007, 50, 74–82. [Google Scholar] [CrossRef]
- Wilson, D.P.; Wan, Z.-K.; Xu, W.-X.; Kirincich, S.J.; Follows, B.C.; Joseph-McCarthy, D.; Foreman, K.; Moretto, A.; Wu, J.; Zhu, M.; et al. Structure-Based Optimization of Protein Tyrosine Phosphatase 1B Inhibitors: From the Active Site to the Second Phosphotyrosine Binding Site. J. Med. Chem. 2007, 50, 4681–4698. [Google Scholar] [CrossRef]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| X | R | Ar | AR IC50 (µM) | PTP1B IC50 (µM) | |
|---|---|---|---|---|---|
| 3a | O | (CH2)2COOH | 3-OC6H5-C6H4 | 11.9 ± 0.9 | 79% at 50 µM |
| 3b | O | (CH2)2COOH | 4-OC6H5-C6H4 | 43.8 ± 7.1 | 56% at 50 µM |
| 3c | O | (CH2)2COOH | 3-OCH2C6H5-C6H4 | 14.3 ± 1.0 | 76% at 50 µM |
| 3d | O | (CH2)2COOH | 4-OCH2C6H5-C6H4 | 35.7 ± 3.0 | 77% at 50 µM |
| 3e | O | (CH2)2COOH | 3-OCH2CH2C6H5-C6H4 | 27.9 ± 3.1 | 64% at 50 µM |
| 3f | O | (CH2)2COOH | 4-OCH2CH2C6H5-C6H4 | 50.2 ± 4.6 | 46% at 50 µM |
| 4a | S | (CH2)2COOH | 3-OC6H5-C6H4 | 2.2 ± 0.1 | 34.1 ± 0.5 |
| 4b | S | (CH2)2COOH | 4-OC6H5-C6H4 | 7.6 ± 0.6 | 29.5 ± 0.4 |
| 4c | S | (CH2)2COOH | 3-OCH2C6H5-C6H4 | 3.8 ± 0.1 | 42.8 ± 0.7 |
| 4d | S | (CH2)2COOH | 4-OCH2C6H5-C6H4 | 8.4 ± 0.7 | 34.9 ± 0.7 |
| 4e | S | (CH2)2COOH | 3-OCH2CH2C6H5-C6H4 | 2.3 ± 0.1 | 55.5 ± 0.8 |
| 4f | S | (CH2)2COOH | 4-OCH2CH2C6H5-C6H4 | 5.3 ± 0.4 | 12.7 ± 0.3 |
| 5a | O | CH2CH=CHCOOH | 3-OC6H5-C6H4 | 3.9 ± 0.2 | 42.1 ± 0.3 |
| 5b | O | CH2CH=CHCOOH | 4-OC6H5-C6H4 | 84% at 10 µM | 39.7 ± 0.1 |
| 5c | O | CH2CH=CHCOOH | 4-C6H5-C6H4 | 88% at 5 µM | 34.8 ± 0.5 |
| 5d | O | CH2CH=CHCOOH | 1-naphthyl | 3.7 ± 0.2 | 40.3 ± 0.5 |
| 5e | O | CH2CH=CHCOOH | 2-naphthyl | 86% at 10 µM | 37.1 ± 0.4 |
| Epalrestat | 0.102 ± 0.005 | ||||
| Vanadate | 0.4 ± 0.01 | ||||
| AR | PTP1B | |||
|---|---|---|---|---|
| Inhibitor | Ki (µM) | K′i (µM) | Ki (µM) | K′i (µM) |
| 4a | 5.0 ± 0.57 | 1.4 ± 0.022 | 10.6 ± 1.7 | 68.9 ± 1.5 |
| 4e | >8 | 0.66 ± 0.30 | 2.2 ± 0.3 | >12 |
| 4f | >30 | 2.9 ± 0.12 | 0.9 ± 0.1 | 2.3 ± 0.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ottanà, R.; Paoli, P.; Cappiello, M.; Nguyen, T.N.; Adornato, I.; Del Corso, A.; Genovese, M.; Nesi, I.; Moschini, R.; Naß, A.; et al. In Search for Multi-Target Ligands as Potential Agents for Diabetes Mellitus and Its Complications—A Structure-Activity Relationship Study on Inhibitors of Aldose Reductase and Protein Tyrosine Phosphatase 1B. Molecules 2021, 26, 330. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26020330
Ottanà R, Paoli P, Cappiello M, Nguyen TN, Adornato I, Del Corso A, Genovese M, Nesi I, Moschini R, Naß A, et al. In Search for Multi-Target Ligands as Potential Agents for Diabetes Mellitus and Its Complications—A Structure-Activity Relationship Study on Inhibitors of Aldose Reductase and Protein Tyrosine Phosphatase 1B. Molecules. 2021; 26(2):330. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26020330
Chicago/Turabian StyleOttanà, Rosaria, Paolo Paoli, Mario Cappiello, Trung Ngoc Nguyen, Ilenia Adornato, Antonella Del Corso, Massimo Genovese, Ilaria Nesi, Roberta Moschini, Alexandra Naß, and et al. 2021. "In Search for Multi-Target Ligands as Potential Agents for Diabetes Mellitus and Its Complications—A Structure-Activity Relationship Study on Inhibitors of Aldose Reductase and Protein Tyrosine Phosphatase 1B" Molecules 26, no. 2: 330. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26020330