
Recent Advances in Nanomedicine for the Diagnosis and Treatment of Prostate Cancer Bone Metastasis

 ,
,
Abstract
:1. Introduction
2. Nanomedicine and Prostate Cancer Bone Metastasis
2.1. Preclinical Models
2.1.1. In Vitro Bone Metastatic PCa Models
2.1.2. In Vivo Bone Metastatic PCa Models
2.2. Diagnostics
2.2.1. Current Clinical Diagnostic Modalities

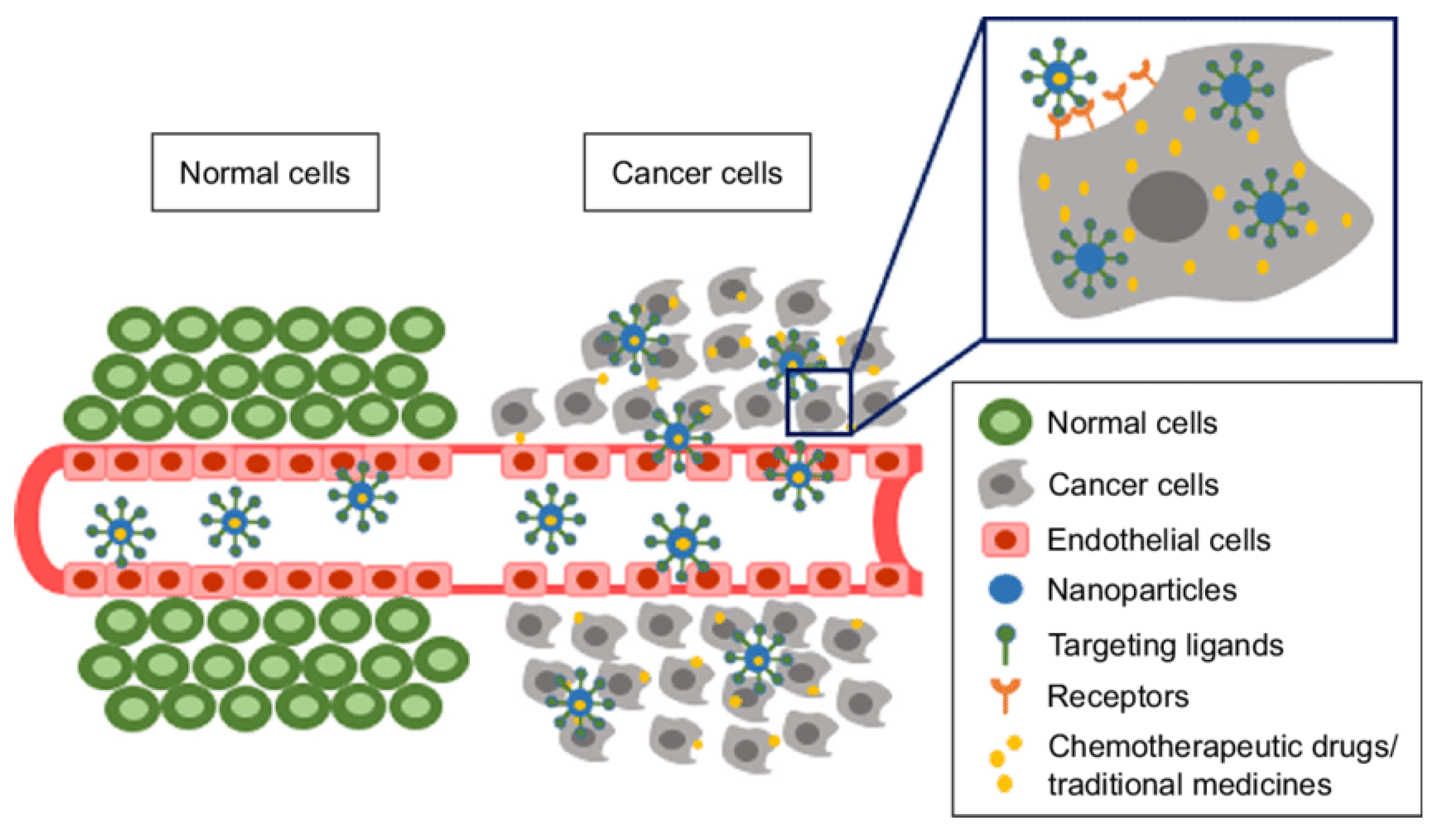{kind=link}
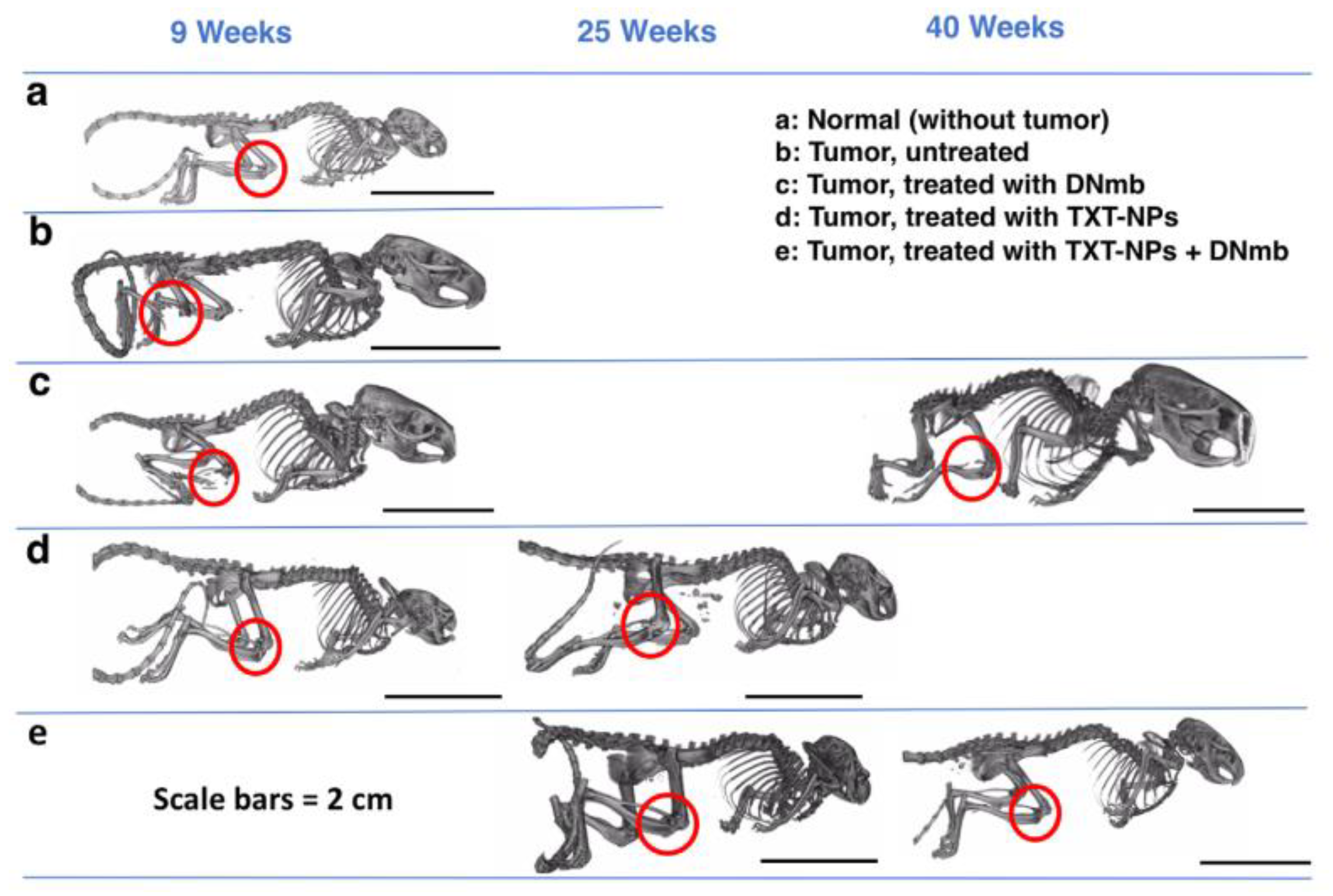{kind=link}
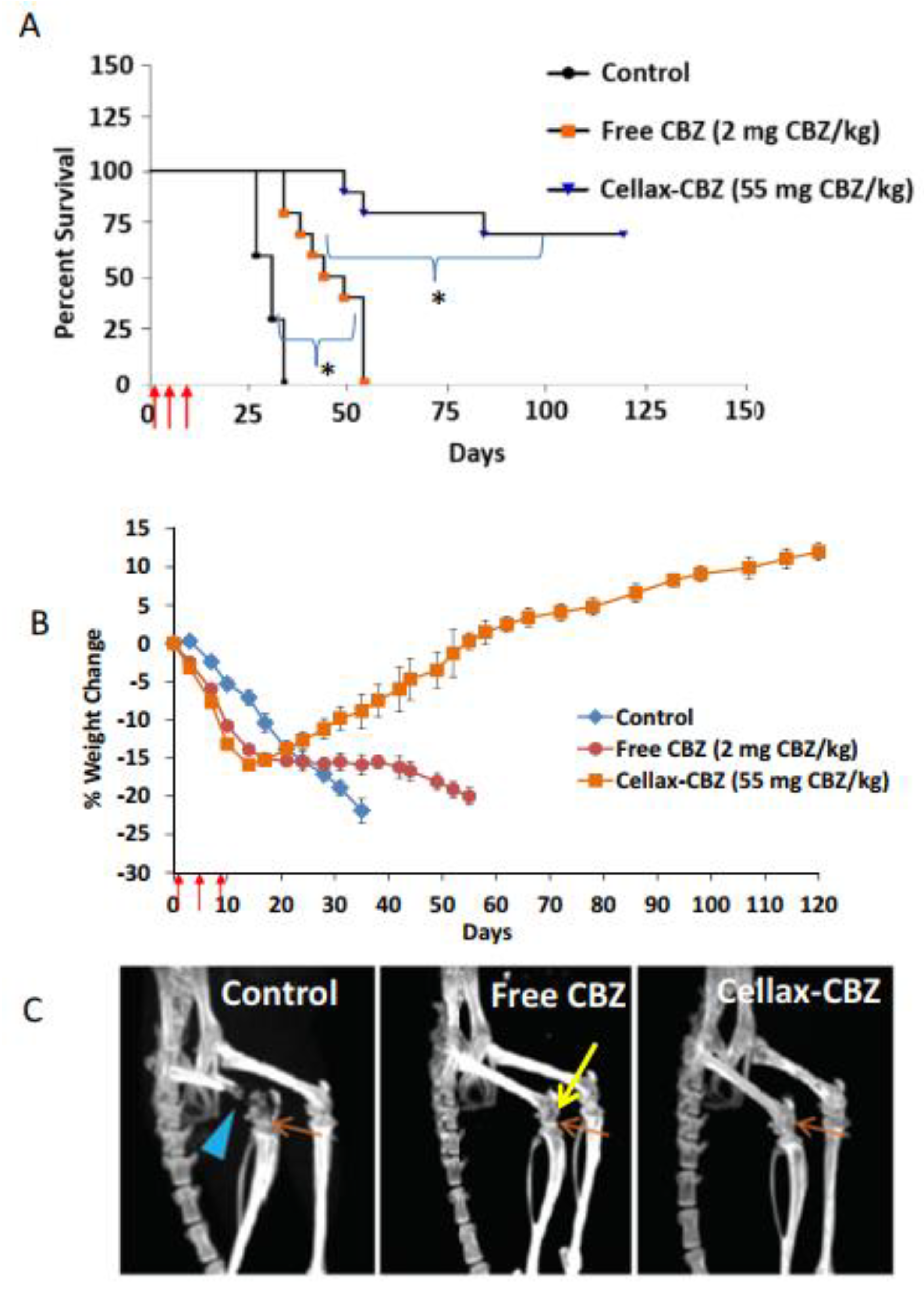{kind=link}
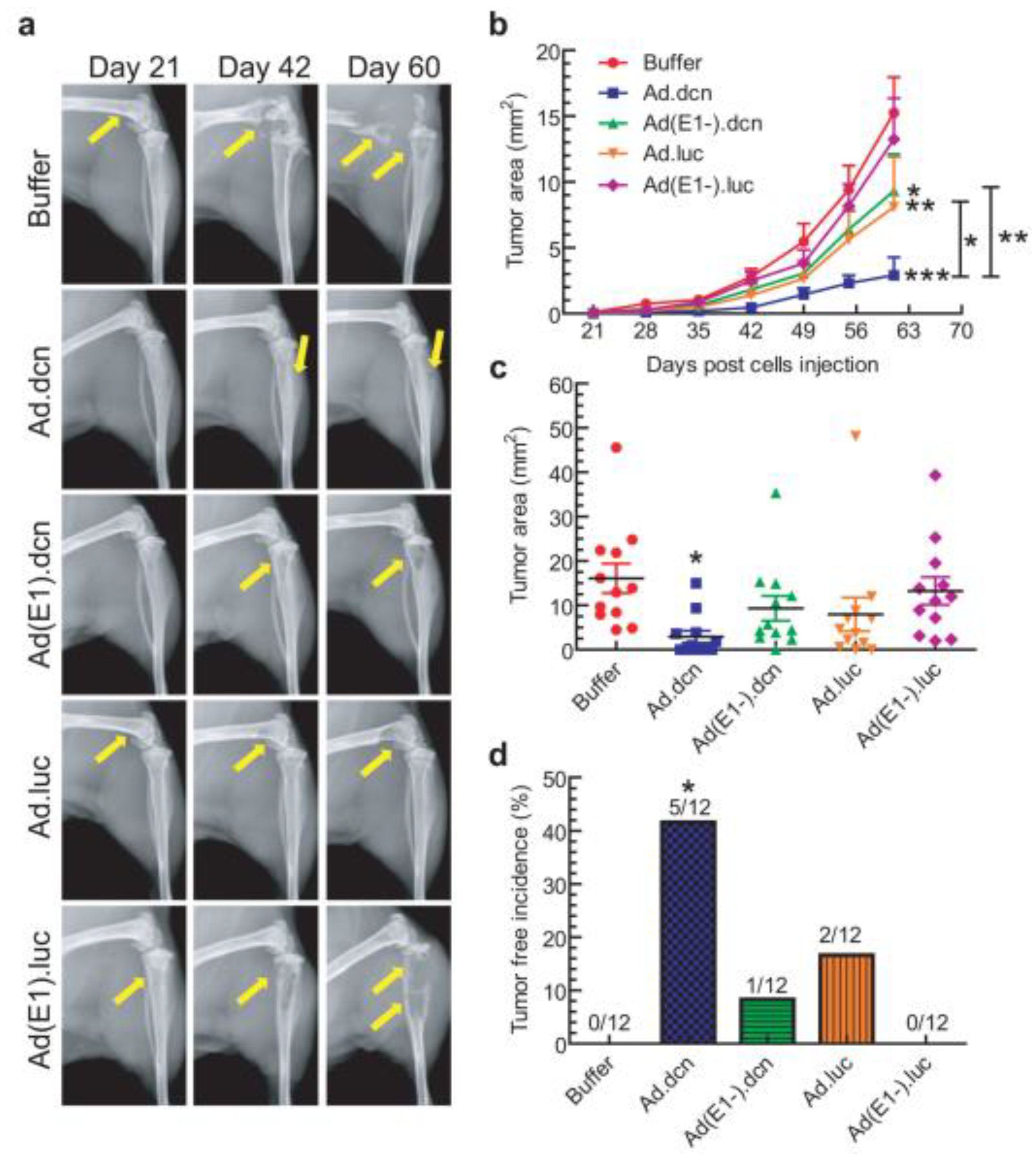{kind=link}
| Name | Core Material | Surface Functionalization | Imaging Modality | Current Status | Refs |
|---|---|---|---|---|---|
| Ferumoxtran-10 | SPIO | Dextran | MRI | Phase 3 Clinical Trial—Europe Ferrotran® | [87] |
| Ferumoxytol | SPIO | Carboxymethyl dextran | MRI | FDA approved (2018) for iron deficiency anemia | [88] |
| GNPs | Gold | Aptamer | CT | Pre-Clinical | [89] |
| Immunolabled QDs | ZnS capped CdSe | Antibody | Optical | Commercially available antibody labeling kits | [90,91] |
| Radiolabled IONPs | SPIO | RGD peptide, Poly(aspartic acid), Radionuclide chelator | PET and MRI | Pre-Clinical Investigation | [92] |
| Radiolabled QDs | [64Cu]CuInS/ZnS | PEG | PET and Optical | Pre-Clinical Investigation | [93] |
| IONPs/QDs | Liposomal SPIO and CdSe QDs | PEG, RGD peptide | MRI and Optical | Pre-Clinical Investigation | [94] |
| C’ dots | Amorphous Silica (SiO2) | PEG, PSMA targeting peptide, radionuclide chelator | PET and Optical | Phase 1—Guided Surgical Treatment of Prostate Cancer Phase 1—Head and Neck Melanoma; Phase 2—Colorectal Cancer | [95] |
2.2.2. Nanoparticles for Bone Metastatic PCa Diagnostics
- A.
- MRI
- B.
- Computed tomography
- C.
- Optical
- D.
- Combination/Multimodal
2.3. Therapeutics
2.3.1. Current Therapies for Bone Metastatic PCa
- A.
- Radiopharmaceuticals
- B.
- Bisphosphonates
- C.
- Chemotherapeutics
- D.
- Denosumab
2.3.2. Nanotechnology-Based Bone Metastatic PCa Therapy
- A.
- Liposomes
- B.
- Polymeric Nanoparticles
- C.
- Polymer-Drug Conjugates
- D.
- 3D-Printed Scaffolds
- E.
- Nanodelivery platforms for gene therapy
3. Future Outlook
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020. [Google Scholar] [CrossRef] [PubMed]
- Howlader, N.; Noone, A.M.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; et al. SEER Cancer Statistics Review, 1975–2017; National Cancer Institute: Bethesda, MD, USA, 2020.
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huggins, C. Studies on prostatic cancer. Arch. Surg. 1941, 43, 209. [Google Scholar] [CrossRef]
- Lee, R.J.; Saylor, P.J.; Smith, M.R. Treatment and prevention of bone complications from prostate cancer. Bone 2011, 48, 88–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bubendorf, L.; Schöpfer, A.; Wagner, U.; Sauter, G.; Moch, H.; Willi, N.; Gasser, T.C.; Mihatsch, M.J. Metastatic patterns of prostate cancer: An autopsy study of 1589 patients. Hum. Pathol. 2000, 31, 578–583. [Google Scholar] [CrossRef] [PubMed]
- Hess, K.R.; Varadhachary, G.R.; Taylor, S.H.; Wei, W.; Raber, M.N.; Lenzi, R.; Abbruzzese, J.L. Metastatic patterns in adenocarcinoma. Cancer 2006, 106, 1624–1633. [Google Scholar] [CrossRef]
- von Moos, R.; Costa, L.; Ripamonti, C.I.; Niepel, D.; Santini, D. Improving quality of life in patients with advanced cancer: Targeting metastatic bone pain. Eur. J. Cancer 2017, 71, 80–94. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, S.C. Spread of prostatic cancer to bone. Urology 1983, 21, 337–344. [Google Scholar] [CrossRef]
- Nieder, C.; Haukland, E.; Pawinski, A.; Dalhaug, A. Anaemia and thrombocytopenia in patients with prostate cancer and bone metastases. BMC Cancer 2010, 10. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.K.; Dayyani, F.; Gallick, G.E. Steps in prostate cancer progression that lead to bone metastasis. Int. J. Cancer 2011, 128, 2545–2561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bussard, K.M.; Gay, C.V.; Mastro, A.M. The bone microenvironment in metastasis; what is special about bone? Cancer Metastasis Rev. 2008, 27, 41–55. [Google Scholar] [CrossRef] [PubMed]
- Pinero, G.J.; Farach-Carson, M.C.; Devoll, R.E.; Aubin, J.E.; Brunn, J.C.; Butler, W.T. Bone matrix proteins in osteogenesis and remodelling in the neonatal rat mandible as studied by immunolocalization of osteopontin, bone sialoprotein, α2HS-glycoprotein and alkaline phosphatase. Arch. Oral Biol. 1995, 40, 145–155. [Google Scholar] [CrossRef]
- Romanowski, R.; Jundt, G.; Termine, J.D.; von der Mark, K.; Schulz, A. Immunoelectron microscopy of osteonectin and type I collagen in osteoblasts and bone matrix. Calcif. Tissue Int. 1990, 46, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Virk, M.S.; Lieberman, J.R. Tumor metastasis to bone. Arthritis Res. Ther. 2007, 9, S5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Logothetis, C.J.; Lin, S.H. Osteoblasts in prostate cancer metastasis to bone. Nat. Rev. Cancer 2005, 5, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Idowu, B.M. Prostate carcinoma presenting with diffuse osteolytic metastases and supraclavicular lymphadenopathy mimicking multiple myeloma. Clin. Case Rep. 2018, 6, 253–257. [Google Scholar] [CrossRef] [Green Version]
- Urwin, G.H.; Percival, R.C.; Harris, S.; Beneton, M.N.; Williams, J.L.; Kanis, J.A. Generalised increase in bone resorption in carcinoma of the prostate. Br. J. Urol. 1985, 57, 721–723. [Google Scholar] [CrossRef]
- Ibrahim, T.; Flamini, E.; Mercatali, L.; Sacanna, E.; Serra, P.; Amadori, D. Pathogenesis of osteoblastic bone metastases from prostate cancer. Cancer 2010, 116, 1406–1418. [Google Scholar] [CrossRef]
- Yang, J.; Fizazi, K.; Peleg, S.; Sikes, C.R.; Raymond, A.K.; Jamal, N.; Hu, M.; Olive, M.; Martinez, L.A.; Wood, C.G.; et al. Prostate cancer cells induce osteoblast differentiation through a Cbfa1-dependent pathway. Cancer Res. 2001, 61, 5652–5659. [Google Scholar]
- Lang, S.H.; Miller, W.R.; Habib, F.K. Stimulation of human prostate cancer cell lines by factors present in human osteoblast-like cells but not in bone marrow. Prostate 1995, 27, 287–293. [Google Scholar] [CrossRef]
- Chackal-Roy, M.; Niemeyer, C.; Moore, M.; Zetter, B.R. Stimulation of human prostatic carcinoma cell growth by factors present in human bone marrow. J. Clin. Investig. 1989, 84, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nature 2003, 423, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Udagawa, N.; Takahashi, N.; Yasuda, H.; Mizuno, A.; Itoh, K.; Ueno, Y.; Shinki, T.; Gillespie, M.T.; Martin, T.J.; Higashio, K.; et al. Osteoprotegerin produced by osteoblasts is an important regulator in osteoclast development and function. Endocrinology 2000, 141, 3478–3484. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M.; Vessella, R.L.; Kostenuik, P.J.; Dunstan, C.R.; Lange, P.H.; Corey, E. Serum osteoprotegerin levels are increased in patients with advanced prostate cancer. Clin Cancer Res 2001, 7, 2977–2983. [Google Scholar] [PubMed]
- Jung, K.; Lein, M.; Stephan, C.; Von Hösslin, K.; Semjonow, A.; Sinha, P.; Loening, S.A.; Schnorr, D. Comparison of 10 serum bone turnover markers in prostate carcinoma patients with bone metastatic spread: Diagnostic and prognostic implications. Int. J. Cancer 2004, 111, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Saad, F.; Gleason, D.M.; Murray, R.; Tchekmedyian, S.; Venner, P.; Lacombe, L.; Chin, J.L.; Vinholes, J.J.; Goas, J.A.; Chen, B. A randomized, placebo-controlled trial of zoledronic acid in patients with hormone-refractory metastatic prostate carcinoma. J. Natl. Cancer Inst. 2002, 94, 1458–1468. [Google Scholar] [CrossRef] [PubMed]
- Quilty, P.M.; Kirk, D.; Bolger, J.J.; Dearnaley, D.P.; Lewington, V.J.; Masone, M.D.; Reed, N.S.E.; Russell, J.M.; Yardley, J. A comparison of the palliative effects of strontium-89 and external beam radiotherapy in metastatic prostate cancer. Radiother. Oncol. 1994, 31, 33–40. [Google Scholar] [CrossRef]
- Sartor, O.; Reid, R.H.; Hoskin, P.J.; Quick, D.P.; Ell, P.J.; Coleman, R.E.; Kotler, J.A.; Freeman, L.M.; Olivier, P. Samarium-153-lexidronam complex for treatment of painful bone metastases in hormone-refractory prostate cancer. Urology 2004, 63, 940–945. [Google Scholar] [CrossRef] [PubMed]
- Han, S.H.; De Klerk, J.M.H.; Tan, S.; Van het Schip, A.D.; Derksen, B.H.; Van Dijk, A.; Kruitwagen, C.L.J.J.; Blijham, G.H.; Van Rijk, P.P.; Zonnenberg, B.A. The placorhen study: A double-blind, placebo-controlled, randomized radionuclide study with 186re-etidronate in hormone-resistant prostate cancer patients with painful bone metastases. J. Nucl. Med. 2002, 43, 1150–1156. [Google Scholar] [PubMed]
- Tu, S.-M.; Millikan, R.E.; Mengistu, B.; Delpassand, E.S.; Amato, R.J.; Pagliaro, L.C.; Daliani, D.; Papandreou, C.N.; Smith, T.L.; Kim, J.; et al. Bone-targeted therapy for advanced androgen-independent carcinoma of the prostate: A randomised phase II trial. Lancet 2001, 357, 336–341. [Google Scholar] [CrossRef]
- Carducci, M.A.; Padley, R.J.; Breul, J.; Vogelzang, N.J.; Zonnenberg, B.A.; Daliani, D.D.; Schulman, C.C.; Nabulsi, A.A.; Humerickhouse, R.A.; Weinberg, M.A.; et al. Effect of endothelin-A receptor blockade with atrasentan on tumor progression in men with hormone-refractory prostate cancer: A randomized, phase II, placebo-controlled trial. J. Clin. Oncol. 2003, 21, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Webster, T.J. Nanomedicine: What’s in a definition? Int. J. Nanomedicine 2006, 1, 115–116. [Google Scholar] [CrossRef] [PubMed]
- Baetke, S.C.; Lammers, T.; Kiessling, F. Applications of nanoparticles for diagnosis and therapy of cancer. Br. J. Radiol. 2015, 88. [Google Scholar] [CrossRef] [PubMed]
- Tran, S.; DeGiovanni, P.-J.; Piel, B.; Rai, P. Cancer nanomedicine: A review of recent success in drug delivery. Clin. Transl. Med. 2017, 6, 10143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melancon, M.P.; Zhou, M.; Li, C. Cancer Theranostics with Near-Infrared Light-Activatable Multimodal Nanoparticles. Acc. Chem. Res. 2011, 44, 947–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, J.; Nakamura, H.; Maeda, H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv. Drug Deliv. Rev. 2011, 63, 136–151. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H. Toward a full understanding of the EPR effect in primary and metastatic tumors as well as issues related to its heterogeneity. Adv. Drug Deliv. Rev. 2015, 91, 3–6. [Google Scholar] [CrossRef]
- Clemons, T.D.; Singh, R.; Sorolla, A.; Chaudhari, N.; Hubbard, A.; Iyer, K.S. Distinction between Active and Passive Targeting of Nanoparticles Dictate Their Overall Therapeutic Efficacy. Langmuir 2018, 34, 15343–15349. [Google Scholar] [CrossRef] [Green Version]
- Attia, M.F.; Anton, N.; Wallyn, J.; Omran, Z.; Vandamme, T.F. An overview of active and passive targeting strategies to improve the nanocarriers efficiency to tumour sites. J. Pharm. Pharmacol. 2019, 71, 1185–1198. [Google Scholar] [CrossRef] [Green Version]
- Muhamad, N.; Plengsuriyakarn, T.; Na-Bangchang, K. Application of active targeting nanoparticle delivery system for chemotherapeutic drugs and traditional/herbal medicines in cancer therapy: A systematic review. Int. J. Nanomed. 2018, 13, 3921–3935. [Google Scholar] [CrossRef] [Green Version]
- Park, S.H.; Eber, M.R.; Shiozawa, Y. Models of Prostate Cancer Bone Metastasis. In Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2019; pp. 295–308. [Google Scholar]
- Tennant, T.R.; Kim, H.; Sokoloff, M.; Rinker-Schaeffer, C.W. The Dunning model. Prostate 2000. [Google Scholar] [CrossRef]
- Rosol, T.J.; Tannehill-Gregg, S.H.; LeRoy, B.E.; Mandl, S.; Contag, C.H. Animal models of bone metastasis. Interdiscip. Int. J. Am. Cancer Soc. 2003, 97, 748–757. [Google Scholar] [CrossRef] [PubMed]
- LeRoy, B.E.; Thudi, N.K.; Nadella, M.V.P.; Toribio, R.E.; Tannehill-Gregg, S.H.; Van Bokhoven, A.; Davis, D.; Corn, S.; Rosol, T.J. New bone formation and osteolysis by a metastatic, highly invasive canine prostate carcinoma xenograft. Prostate 2006. [Google Scholar] [CrossRef] [PubMed]
- Zhi, G.L.; Mathew, P.; Yang, J.; Starbuck, M.W.; Zurita, A.J.; Liu, J.; Sikes, C.; Multani, A.S.; Efstathiou, E.; Lopez, A.; et al. Androgen receptor-negative human prostate cancer cells induce osteogenesis in mice through FGF9-mediated mechanisms. J. Clin. Investig. 2008. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.C.; Lee, Y.C.; Yu, G.; Cheng, C.J.; Zhou, X.; Chu, K.; Murshed, M.; Le, N.T.; Baseler, L.; Abe, J.-I.; et al. Endothelial-to-Osteoblast Conversion Generates Osteoblastic Metastasis of Prostate Cancer. Dev. Cell 2017. [Google Scholar] [CrossRef] [Green Version]
- Astashkina, A.; Mann, B.; Grainger, D.W. A critical evaluation of in vitro cell culture models for high-throughput drug screening and toxicity. Pharmacol. Ther. 2012, 134, 82–106. [Google Scholar] [CrossRef]
- Ivanov, D.P.; Parker, T.L.; Walker, D.A.; Alexander, C.; Ashford, M.B.; Gellert, P.R.; Garnett, M.C. In vitro co-culture model of medulloblastoma and human neural stem cells for drug delivery assessment. J. Biotechnol. 2015, 205, 3–13. [Google Scholar] [CrossRef] [Green Version]
- Fitzgerald, K.A.; Malhotra, M.; Curtin, C.M.; O’Brien, F.J.; O’Driscoll, C.M. Life in 3D is never flat: 3D models to optimise drug delivery. J. Control. Release 2015, 215, 39–54. [Google Scholar] [CrossRef]
- Fitzgerald, K.A.; Guo, J.; Raftery, R.M.; Castaño, I.M.; Curtin, C.M.; Gooding, M.; Darcy, R.; O’Brien, F.J.; O’Driscoll, C.M. Nanoparticle-mediated siRNA delivery assessed in a 3D co-culture model simulating prostate cancer bone metastasis. Int. J. Pharm. 2016, 511, 1058–1069. [Google Scholar] [CrossRef]
- Lynch, C.C. Matrix metalloproteinases as master regulators of the vicious cycle of bone metastasis. Bone 2011, 48, 44–53. [Google Scholar] [CrossRef]
- Chinni, S.R.; Sivalogan, S.; Dong, Z.; Trindade Filho, J.C.; Deng, X.; Bonfil, R.D.; Cher, M.L. CXCL12/CXCR4 signaling activates Akt-1 and MMP-9 expression in prostate cancer cells: The role of bone microenvironment-associated CXCL12. Prostate 2006, 66, 32–48. [Google Scholar] [CrossRef]
- Evans, J.C.; McCarthy, J.; Torres-Fuentes, C.; Cryan, J.F.; Ogier, J.; Darcy, R.; Watson, R.W.; O’Driscoll, C.M. Cyclodextrin mediated delivery of NF-κB and SRF siRNA reduces the invasion potential of prostate cancer cells in vitro. Gene Ther. 2015, 22, 802–810. [Google Scholar] [CrossRef] [PubMed]
- Bock, N.; Shokoohmand, A.; Kryza, T.; Röhl, J.; Meijer, J.; Tran, P.A.; Nelson, C.C.; Clements, J.A.; Hutmacher, D.W. Engineering osteoblastic metastases to delineate the adaptive response of androgen-deprived prostate cancer in the bone metastatic microenvironment. Bone Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Berish, R.B.; Ali, A.N.; Telmer, P.G.; Ronald, J.A.; Leong, H.S. Translational models of prostate cancer bone metastasis. Nat. Rev. Urol. 2018, 15, 403–421. [Google Scholar] [CrossRef] [PubMed]
- Corey, E.; Quinn, J.E.; Bladou, F.; Brown, L.G.; Roudier, M.P.; Brown, J.M.; Buhler, K.R.; Vessella, R.L. Establishment and characterization of osseous prostate cancer models: Intra-tibial injection of human prostate cancer cells. Prostate 2002, 52, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.; Shiozawa, Y.; Wang, J.; Mcgregor, N.; Dai, J.; Park, S.I.; Berry, J.E.; Havens, A.M.; Joseph, J.; Kim, J.K.; et al. Prevalence of prostate cancer metastases after intravenous inoculation provides clues into the molecular basis of dormancy in the bone marrow microenvironment. Neoplasia (USA) 2012, 14, 429–439. [Google Scholar] [CrossRef] [Green Version]
- Arguello, F.; Duerst, R.E.; McQueen, K.; Frantz, C.N.; Baggs, R.B.; Johnstone, L. Pathogenesis of vertebral metastasis and epidural spinal cord compression. Cancer 1990, 65, 98–106. [Google Scholar] [CrossRef]
- Bonfil, R.D.; Dong, Z.; Trindade Filho, J.C.; Sabbota, A.; Osenkowski, P.; Nabha, S.; Yamamoto, H.; Chinni, S.R.; Zhao, H.; Mobashery, S.; et al. Prostate cancer-associated membrane type 1-matrix metalloproteinase: A pivotal role in bone response and intraosseous tumor growth. Am. J. Pathol. 2007, 170, 2100–2111. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.T.; Sikes, R.A.; Cui, Q.; Thalmann, G.N.; Kao, C.; Murphy, C.F.; Yang, H.; Zhau, H.E.; Balian, G.; Chung, L.W.K. Establishing human prostate cancer cell xenografts in bone: Induction of osteoblastic reaction by prostate-specific antigen-producing tumors in athymic and SCID/bg mice using LNCaP and lineage-derived metastatic sublines. Int. J. Cancer 1998, 77, 887–894. [Google Scholar] [CrossRef]
- Yonou, H.; Ochiai, A.; Goya, M.; Kanomata, N.; Hokama, S.; Morozumi, M.; Sugaya, K.; Hatano, T.; Ogawa, Y. Intraosseous Growth of Human Prostate Cancer in Implanted Adult Human Bone: Relationship of Prostate Cancer Cell to Osteoclasts in Osteoblastic Metastatic Lesions. Prostate 2004, 58, 406–413. [Google Scholar] [CrossRef]
- Nemeth, J.A.; Harb, J.F.; Barroso, U.; He, Z.; Grignon, D.J.; Cher, M.L. Severe Combined Immunodeficient-hu Model of Human Prostate Cancer Metastasis to Human Bone. Cancer Res. 1999, 59, 1987–1993. [Google Scholar] [PubMed]
- Shiozawa, Y.; Pedersen, E.A.; Havens, A.M.; Jung, Y.; Mishra, A.; Joseph, J.; Kim, J.K.; Patel, L.R.; Ying, C.; Ziegler, A.M.; et al. Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J. Clin. Investig. 2011, 121, 1298–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaiselbuh, S.R.; Edelman, M.; Lipton, J.M.; Liu, J.M. Ectopic human mesenchymal stem cell-coated scaffolds in NOD/SCID mice: An in vivo model of the leukemia niche. Tissue Eng.—Part C Methods 2010, 16, 1523–1531. [Google Scholar] [CrossRef]
- Hesami, P.; Holzapfel, B.M.; Taubenberger, A.; Roudier, M.; Fazli, L.; Sieh, S.; Thibaudeau, L.; Gregory, L.S.; Hutmacher, D.W.; Clements, J.A. A humanized tissue-engineered in vivo model to dissect interactions between human prostate cancer cells and human bone. Clin. Exp. Metastasis 2014, 31, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Landgraf, M.; Lahr, C.A.; Sanchez-Herrero, A.; Meinert, C.; Shokoohmand, A.; Pollock, P.M.; Hutmacher, D.W.; Shafiee, A.; McGovern, J.A. Humanized bone facilitates prostate cancer metastasis and recapitulates therapeutic effects of zoledronic acid in vivo. Bone Res. 2019, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Heindel, W. The diagnostic imaging of bone metastases. Dtsch. Arztebl. Int. 2014, 111, 741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isaac, A.; Dalili, D.; Dalili, D.; Weber, M.-A. State-of-the-art imaging for diagnosis of metastatic bone diseaseModernste Bildgebung zur Diagnose von Knochenmetastasen. Radiologe 2020, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanishi, D. 99mTc-MDP accumulation mechanisms in bone. Oral Surg. Oral Med. Oral Pathol. 1993, 75, 239–246. [Google Scholar] [CrossRef]
- Okamoto, Y. Accumulation of technetium-99m methylene diphosphonate. Conditions affecting adsorption to hydroxyapatite. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 1995, 80, 115–119. [Google Scholar] [CrossRef]
- Fogelman, I.; Cook, G.; Israel, O.; Van Der Wall, H. Positron emission tomography and bone metastases. Semin. Nucl. Med. 2005. [Google Scholar] [CrossRef] [PubMed]
- Langsteger, W.; Heinisch, M.; Fogelman, I. The role of fluorodeoxyglucose, 18F-dihydroxyphenylalanine, 18F-choline, and 18F-fluoride in bone imaging with emphasis on prostate and breast. Semin. Nucl. Med. 2006. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Kosaka, N.; Kishi, H. PET imaging of prostate cancer using carbon-11-choline. J. Nucl. Med. 1998. [Google Scholar]
- Pomykala, K.L.; Czernin, J.; Grogan, T.R.; Armstrong, W.R.; Williams, J.; Calais, J. Total-Body 68Ga-PSMA-11 PET/CT for Bone Metastasis Detection in Prostate Cancer Patients: Potential Impact on Bone Scan Guidelines. J. Nucl. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Paolillo, V.; Ta, R.T.; Damasco, J.; Rojo, R.D.; Carl, J.C.; Melancon, M.P.; Ravizzini, G.C.; Le, D.B.; Santos, E.B. Fully automated preparation of 68Ga-PSMA-11atcurie level quantity using cyclotron-produced 68Ga for clinical applications. Appl. Radiat. Isot. 2020. [Google Scholar] [CrossRef] [PubMed]
- Bihan, D. Le Apparent diffusion coefficient and beyond: What diffusion mr imaging can tell us about tissue structure. Radiology 2013, 268, 318–322. [Google Scholar] [CrossRef] [PubMed]
- Galbán, C.J.; Hoff, B.A.; Chenevert, T.L.; Ross, B.D. Diffusion MRI in early cancer therapeutic response assessment. NMR Biomed. 2017, 30, 3458. [Google Scholar] [CrossRef]
- Chilla, G.S.; Tan, C.H.; Xu, C.; Poh, C.L. Diffusion weighted magnetic resonance imaging and its recent trend-a survey. Quant. Imaging Med. Surg. 2015, 5, 407–422. [Google Scholar] [CrossRef]
- Thoeny, H.C.; Ross, B.D. Predicting and monitoring cancer treatment response with diffusion-weighted MRI. J. Magn. Reson. Imaging 2010, 32, 2–16. [Google Scholar] [CrossRef] [Green Version]
- Padhani, A.R.; Koh, D.M. Diffusion MR imaging for monitoring of treatment response. Magn. Reson. Imaging Clin. N. Am. 2011, 19, 181–209. [Google Scholar] [CrossRef]
- Tabotta, F.; Jreige, M.; Schaefer, N.; Becce, F.; Prior, J.O.; Nicod Lalonde, M. Quantitative bone SPECT/CT: High specificity for identification of prostate cancer bone metastases. BMC Musculoskelet. Disord. 2019, 20, 619. [Google Scholar] [CrossRef] [Green Version]
- Moffat, B.A.; Chenevert, T.L.; Lawrence, T.S.; Meyer, C.R.; Johnson, T.D.; Dong, Q.; Tsien, C.; Mukherji, S.; Quint, D.J.; Gebarski, S.S.; et al. Functional diffusion map: A noninvasive MRI biomarker for early stratification of clinical brain tumor response. Proc. Natl. Acad. Sci. USA. 2005, 102, 5524–5529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moffat, B.A.; Chenevert, T.L.; Meyer, C.R.; Mckeevery, P.E.; Hall, D.E.; Hoff, B.A.; Johnson, T.D.; Rehemtulla, A.; Ross, B.D. The functional diffusion map: An imaging biomarker for the early prediction of cancer treatment outcome. Neoplasia 2006, 8, 259–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamstra, D.A.; Galbán, C.J.; Meyer, C.R.; Johnson, T.D.; Sundgren, P.C.; Tsien, C.; Lawrence, T.S.; Junck, L.; Ross, D.J.; Rehemtulla, A.; et al. Functional diffusion map as an early imaging biomarker for high-grade glioma: Correlation with conventional radiologic response and overall survival. J. Clin. Oncol. 2008, 26, 3387–3394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brisset, J.-C.; Hoff, B.A.; Chenevert, T.L.; Jacobson, J.A.; Boes, J.L.; Galbán, S.; Rehemtulla, A.; Johnson, T.D.; Pienta, K.J.; Galbán, C.J.; et al. Integrated Multimodal Imaging of Dynamic Bone-Tumor Alterations Associated with Metastatic Prostate Cancer. PLoS ONE 2015, 10, e0123877. [Google Scholar] [CrossRef] [PubMed]
- Turkbey, B.; Agarwal, H.K.; Shih, J.; Bernardo, M.; McKinney, Y.L.; Daar, D.; Griffiths, G.L.; Sankineni, S.; Johnson, L.; Grant, K.B.; et al. A Phase I Dosing Study of Ferumoxytol for MR Lymphography at 3 T in Patients With Prostate Cancer. Am. J. Roentgenol. 2015, 205, 64–69. [Google Scholar] [CrossRef]
- Mahan, M.M.; Doiron, A.L. Gold Nanoparticles as X-Ray, CT, and Multimodal Imaging Contrast Agents: Formulation, Targeting, and Methodology. J. Nanomater. 2018, 2018. [Google Scholar] [CrossRef]
- Ruan, Y.; Yu, W.; Cheng, F.; Zhang, X.; Larré, S. Detection of prostate stem cell antigen expression in human prostate cancer using quantum-dot-based technology. Sensors 2012, 12, 5461–5470. [Google Scholar] [CrossRef] [Green Version]
- Reiter, R.E.; Gu, Z.; Watabe, T.; Thomas, G.; Szigeti, K.; Davis, E.; Wahl, M.; Nisitani, S.; Yamashiro, J.; Le Beau, M.M.; et al. Prostate stem cell antigen: A cell surface marker overexpressed in prostate cancer. Proc. Natl. Acad. Sci. USA 1998, 95, 1735–1740. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.Y.; Li, Z.; Chen, K.; Hsu, A.R.; Xu, C.; Xie, J.; Sun, S.; Chen, X. PET/MRI dual-modality tumor imaging using arginine-glycine-aspartic (RGD)-conjugated radiolabeled iron oxide nanoparticles. J. Nucl. Med. 2008, 49, 1371–1379. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Sun, X.; Jacobson, O.; Yan, X.; Min, K.; Srivatsan, A.; Niu, G.; Kiesewetter, D.O.; Chang, J.; Chen, X. Intrinsically radioactive [64Cu]CuInS/ZnS quantum dots for pet and optical imaging: Improved radiochemical stability and controllable cerenkov luminescence. ACS Nano 2015, 9, 488–495. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Chen, Z.; Zhu, L. cRGD-conjugated magnetic-fluorescent liposomes for targeted dual-modality imaging of bone metastasis from prostate cancer. J. Liposome Res. 2015, 25, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Ma, K.; Benezra, M.; Zhang, L.; Cheal, S.M.; Phillips, E.; Yoo, B.; Pauliah, M.; Overholtzer, M.; Zanzonico, P.; et al. Cancer-Targeting Ultrasmall Silica Nanoparticles for Clinical Translation: Physicochemical Structure and Biological Property Correlations. Chem. Mater. 2017, 29, 8766–8779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, L.; Bergstralh, E.J.; Cheville, J.C.; Slezak, J.; Corica, F.A.; Zincke, H.; Blute, M.L.; Bostwick, D.G. Cancer Volume of Lymph Node Metastasis Predicts Progression in Prostate Cancer. Am. J. Surg. Pathol. 1998, 22, 1491–1500. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; Greco, F.A.; Pavlidis, N.; Daugaard, G.; Oien, K.; Pentheroudakis, G. Cancers of unknown primary site: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2015. [Google Scholar] [CrossRef]
- Yin, M.; Dhir, R.; Parwani, A.V. Diagnostic utility of p501s (prostein) in comparison to prostate specific antigen (PSA) for the detection of metastatic prostatic adenocarcinoma. Diagn. Pathol. 2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genega, E.M.; Hutchinson, B.; Reuter, V.E.; Gaudin, P.B. Immunophenotype of high-grade prostatic adenocarcinoma and urothelial carcinoma. Mod. Pathol. 2000. [Google Scholar] [CrossRef]
- Mhawech, P.; Uchida, T.; Pelte, M.F. Immunohistochemical profile of high-grade urothelial bladder carcinoma and prostate adenocarcinoma. Hum. Pathol. 2002. [Google Scholar] [CrossRef]
- Mhawech-Fauceglia, P.; Zhang, S.; Terracciano, L.; Sauter, G.; Chadhuri, A.; Herrmann, F.R.; Penetrante, R. Prostate-specific membrane antigen (PSMA) protein expression in normal and neoplastic tissues and its sensitivity and specificity in prostate adenocarcinoma: An immunohistochemical study using mutiple tumour tissue microarray technique. Histopathology 2007. [Google Scholar] [CrossRef] [PubMed]
- Kalos, M.; Askaa, J.; Hylander, B.L.; Repasky, E.A.; Cai, F.; Vedvick, T.; Reed, S.G.; Wright, G.L.; Fanger, G.R. Prostein expression is highly restricted to normal and malignant prostate tissues. Prostate 2004. [Google Scholar] [CrossRef]
- Braun, M.; Goltz, D.; Shaikhibrahim, Z.; Vogel, W.; Böhm, D.; Scheble, V.; Sotlar, K.; Fend, F.; Tan, S.H.; Dobi, A.; et al. ERG protein expression and genomic rearrangement status in primary and metastatic prostate cancer - A comparative study of two monoclonal antibodies. Prostate Cancer Prostatic Dis. 2012. [Google Scholar] [CrossRef] [Green Version]
- Downes, M.R.; Torlakovic, E.E.; Aldaoud, N.; Zlotta, A.R.; Evans, A.J.; Van Der Kwast, T.H. Diagnostic utility of androgen receptor expression in discriminating poorly differentiated urothelial and prostate carcinoma. J. Clin. Pathol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Visakorpi, T.; Hyytinen, E.; Koivisto, P.; Tanner, M.; Keinänen, R.; Palmberg, C.; Palotie, A.; Tammela, T.; Isola, J.; Kallioniemi, O.P. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat. Genet. 1995. [Google Scholar] [CrossRef] [PubMed]
- Queisser, A.; Hagedorn, S.A.; Braun, M.; Vogel, W.; Duensing, S.; Perner, S. Comparison of different prostatic markers in lymph node and distant metastases of prostate cancer. Mod. Pathol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Islam, T.; Wolf, G. The pharmacokinetics of the lymphotropic nanoparticle MRI contrast agent ferumoxtran-10. Cancer Biomark. 2009, 5, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.W. Surface properties of superparamagnetic iron oxide MR contrast agents: Ferumoxides, ferumoxtran, ferumoxsil. Magn. Reson. Imaging 1995, 13, 675–691. [Google Scholar] [CrossRef]
- Hudgins, P.A.; Anzai, Y.; Morris, M.R.; Lucas, M.A. Ferumoxtran-10, a Superparamagnetic Iron Oxide as a Magnetic Resonance Enhancement Agent for Imaging Lymph Nodes: A Phase 2 Dose Study. Am. J. Neuroradiol. 2002, 23, 649–656. [Google Scholar]
- Harisinghani, M.; Ross, R.W.; Guimaraes, A.R.; Weissleder, R. Utility of a new bolus-injectable nanoparticle for clinical cancer staging. Neoplasia 2007, 9, 1160–1165. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.; Liao, F.; Molesa, S.; Redinger, D.; Subramanian, V. Plastic-Compatible Low Resistance Printable Gold Nanoparticle Conductors for Flexible Electronics. J. Electrochem. Soc. 2003, 150, G412. [Google Scholar] [CrossRef] [Green Version]
- Ali, M.E.; Hashim, U.; Mustafa, S.; Che Man, Y.B.; Islam, K.N. Gold nanoparticle sensor for the visual detection of pork adulteration in meatball formulation. J. Nanomater. 2012, 2012. [Google Scholar] [CrossRef]
- Carabineiro, S.A.C. Supported Gold Nanoparticles as Catalysts for the Oxidation of Alcohols and Alkanes. Front. Chem. 2019, 7, 702. [Google Scholar] [CrossRef] [Green Version]
- Brown, S.D.; Nativo, P.; Smith, J.A.; Stirling, D.; Edwards, P.R.; Venugopal, B.; Flint, D.J.; Plumb, J.A.; Graham, D.; Wheate, N.J. Gold nanoparticles for the improved anticancer drug delivery of the active component of oxaliplatin. J. Am. Chem. Soc. 2010, 132, 4678–4684. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.; Tisch, U.; Adams, O.; Hakim, M.; Shehada, N.; Broza, Y.Y.; Billan, S.; Abdah-Bortnyak, R.; Kuten, A.; Haick, H. Diagnosing lung cancer in exhaled breath using gold nanoparticles. Nat. Nanotechnol. 2009, 4, 669–673. [Google Scholar] [CrossRef] [PubMed]
- Alivisatos, A.P. Semiconductor Clusters, Nanocrystals, and Quantum Dots. Science 1996, 271, 933–937. [Google Scholar] [CrossRef] [Green Version]
- Lauber, D.T.; Fülöp, A.; Kovács, T.; Szigeti, K.; Máthé, D.; Szijártó, A. State of the art in vivo imaging techniques for laboratory animals. Lab. Anim. 2017, 51, 465–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.P.; Zhang, Y.; Zhang, S.; Xia, J.G.; Liu, J.W.; Xu, K.; Gu, N. Preparation and characterization of water-soluble monodisperse magnetic iron oxide nanoparticles via surface double-exchange with DMSA. Colloids Surf. A Physicochem. Eng. Asp. 2008, 316, 210–216. [Google Scholar] [CrossRef]
- Liu, P.; Qin, L.; Wang, Q.; Sun, Y.; Zhu, M.; Shen, M.; Duan, Y. CRGD-functionalized mPEG-PLGA-PLL nanoparticles for imaging and therapy of breast cancer. Biomaterials 2012, 33, 6739–6747. [Google Scholar] [CrossRef]
- Touijer, K.; Vargas, A. The Use of Nanoparticles to Guide the Surgical Treatment of Prostate Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT04167969?term=nanoparticle&cond=prostate+cancer&draw=1&rank=2 (accessed on 28 December 2020).
- El-Amm, J.; Patel, N.; Freeman, A.; Aragon-Ching, J.B. Metastatic Castration-Resistant Prostate Cancer: Critical Review of Enzalutamide. Clin. Med. Insights Oncol. 2013, 7, CMO.S11670. [Google Scholar] [CrossRef] [Green Version]
- Gdowski, A.S.; Ranjan, A.; Vishwanatha, J.K. Current concepts in bone metastasis, contemporary therapeutic strategies and ongoing clinical trials. J. Exp. Clin. Cancer Res. 2017, 36. [Google Scholar] [CrossRef] [Green Version]
- Vengalil, S.; O’Sullivan, J.M.; Parker, C.C. Use of radionuclides in metastatic prostate cancer: Pain relief and beyond. Curr. Opin. Support. Palliat. Care 2012, 6, 310–315. [Google Scholar] [CrossRef]
- Longo, J.; Lutz, S.; Johnstone, C. Samarium-153-ethylene diamine tetramethylene phosphonate, a beta-emitting bone-targeted radiopharmaceutical, useful for patients with osteoblastic bone metastases. Cancer Manag. Res. 2013, 5, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Croke, J.; Leung, E.; Segal, R.; Malone, S. Clinical benefits of alpharadin in castrate-chemotherapy-resistant prostate cancer: Case report and literature review. BMJ Case Rep. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Parker, C.; Nilsson, D.; Heinrich, S.; Helle, S.I.; O’Sullivan, J.M.; Fosså, S.D.; Chodacki, A.; Wiechno, P.; Logue, J.; Seke, M.; et al. Alpha emitter radium-223 and survival in metastatic prostate cancer. N. Engl. J. Med. 2013, 369, 213–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- European Medicines Agency. EMA Restricts use of Prostate Cancer Medicine Xofigo; European Medicines Agency: Amsterdam, The Netherlands, 2018.
- Sedhom, R.; Antonarakis, E.S. Radium-223 plus abiraterone in metastatic castration-resistant prostate cancer: A cautionary tale. Transl. Androl. Urol. 2019, 8, S341–S345. [Google Scholar] [CrossRef] [PubMed]
- Fazil, M.; Baboota, S.; Sahni, J.K.; Ameeduzzafar; Ali, J. Bisphosphonates: Therapeutics potential and recent advances in drug delivery. Drug Deliv. 2015, 22, 1–9. [Google Scholar] [CrossRef]
- Cole, L.E.; Vargo-Gogola, T.; Roeder, R.K. Targeted delivery to bone and mineral deposits using bisphosphonate ligands. Adv. Drug Deliv. Rev. 2016, 99, 12–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, R.G.G.; Xia, Z.; Dunford, J.E.; Oppermann, U.; Kwaasi, A.; Hulley, P.A.; Kavanagh, K.L.; Triffitt, J.T.; Lundy, M.W.; Phipps, R.J.; et al. Bisphosphonates: An update on mechanisms of action and how these relate to clinical efficacy. Ann. N. Y. Acad. Sci. 2007, 1117, 209–257. [Google Scholar] [CrossRef] [PubMed]
- Body, J.-J. Breast Cancer: Bisphosphonate Therapy for Metastatic Bone Disease. Clin. Cancer Res. 2006, 12, 6258s–6263s. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, A.; Scher, N.; Williams, G.; Sridhara, R.; Li, N.; Chen, G.; Leighton, J.; Booth, B.; Gobburu, J.V.S.; Rahman, A.; et al. Approval summary for zoledronic acid for treatment of multiple myeloma and cancer bone metastases. Clin. Cancer Res. 2003, 9, 2394–2399. [Google Scholar]
- Hengst, V.; Oussoren, C.; Kissel, T.; Storm, G. Bone targeting potential of bisphosphonate-targeted liposomes. Preparation, characterization and hydroxyapatite binding in vitro. Int. J. Pharm. 2007, 331, 224–227. [Google Scholar] [CrossRef]
- Stapleton, M.; Sawamoto, K.; Alméciga-Díaz, C.J.; Mackenzie, W.G.; Mason, R.W.; Orii, T.; Tomatsu, S. Development of bone targeting drugs. Int. J. Mol. Sci. 2017, 18, 1345. [Google Scholar] [CrossRef]
- Tannock, I.F.; De Wit, R.; Berry, W.R.; Horti, J.; Pluzanska, A.; Chi, K.N.; Oudard, S.; Théodore, C.; James, N.D.; Turesson, I.; et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N. Engl. J. Med. 2004, 351, 1502–1512. [Google Scholar] [CrossRef] [Green Version]
- Shelley, M.; Mason, M.D. Docetaxel plus prednisone improves survival in men with advanced prostate cancer. Cancer Treat. Rev. 2005, 31, 403–407. [Google Scholar] [CrossRef] [PubMed]
- De Bono, J.S.; Oudard, S.; Ozguroglu, M.; Hansen, S.; MacHiels, J.P.; Kocak, I.; Gravis, G.; Bodrogi, I.; MacKenzie, M.J.; Shen, L.; et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: A randomised open-label trial. Lancet 2010, 376, 1147–1154. [Google Scholar] [CrossRef]
- Richards, L. Prostate cancer: Cabazitaxel boosts post-docetaxel survival. Nat. Rev. Urol. 2010, 7, 645. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, E.M.; Ritchlin, C.T. Clinical development of anti-RANKL therapy. Arthritis Res. Ther. 2007, 9, 2171. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.R.; Saad, F.; Coleman, R.; Shore, N.; Fizazi, K.; Tombal, B.; Miller, K.; Sieber, P.; Karsh, L.; Damião, R.; et al. Denosumab and bone-metastasis-free survival in men with castration-resistant prostate cancer: Results of a phase 3, randomised, placebo-controlled trial. Lancet 2012, 379, 39–46. [Google Scholar] [CrossRef] [Green Version]
- Tallman, M.S. Monoclonal antibody therapies in leukemias. Semin. Hematol. 2002, 39, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Ohaegbulam, K.C.; Assal, A.; Lazar-Molnar, E.; Yao, Y.; Zang, X. Human cancer immunotherapy with antibodies to the PD-1 and PD-L1 pathway. Trends Mol. Med. 2015, 21, 24–33. [Google Scholar] [CrossRef] [Green Version]
- Sui, H.; Ma, N.; Wang, Y.; Li, H.; Liu, X.; Su, Y.; Yang, J. Anti-PD-1/PD-L1 Therapy for Non-Small-Cell Lung Cancer: Toward Personalized Medicine and Combination Strategies. J. Immunol. Res. 2018, 2018, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Peggs, K.S.; Quezada, S.A.; Korman, A.J.; Allison, J.P. Principles and use of anti-CTLA4 antibody in human cancer immunotherapy. Curr. Opin. Immunol. 2006, 18, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Conte, P.; Coleman, R. Bisphosphonates in the treatment of skeletal metastases. Semin. Oncol. 2004, 31, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.E. Bisphosphonates: Clinical Experience. Oncologist 2004, 9, 14–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroon, J.; Buijs, J.T.; Van Der Horst, G.; Cheung, H.; Van Der Mark, M.; Van Bloois, L.; Rizzo, L.Y.; Lammers, T.; Pelger, R.C.; Storm, G.; et al. Liposomal delivery of dexamethasone attenuates prostate cancer bone metastatic tumor growth in Vivo. Prostate 2015, 75, 815–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, P.; Mathur, V.; Kumar, A.; Khedgikar, V.; Teja, B.V.; Chaudhary, D.; Kushwaha, P.; Bora, H.K.; Konwar, R.; Trivedi, R.; et al. Nanoemulsion based concomitant delivery of curcumin and etoposide: Impact on cross talk between prostate cancer cells and osteoblast during metastasis. J. Biomed. Nanotechnol. 2014, 10, 3381–3391. [Google Scholar] [CrossRef] [PubMed]
- Gdowski, A.S.; Ranjan, A.; Sarker, M.R.; Vishwanatha, J.K. Bone-targeted cabazitaxel nanoparticles for metastatic prostate cancer skeletal lesions and pain. Nanomedicine 2017, 12, 2083–2095. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.K.; Thareja, S. In vitro and in vivo characterization of pharmaceutical nanocarriers used for drug delivery. Artif. Cells, Nanomedicine, Biotechnol. 2019, 47, 524–539. [Google Scholar] [CrossRef] [Green Version]
- Yoo, J.; Won, Y.-Y. Phenomenology of the Initial Burst Release of Drugs from PLGA Microparticles. ACS Biomater. Sci. Eng. 2020. [Google Scholar] [CrossRef]
- Kopeček, J.; Kopečková, P. HPMA copolymers: Origins, early developments, present, and future. Adv. Drug Deliv. Rev. 2010, 62, 122–149. [Google Scholar] [CrossRef] [Green Version]
- Ekladious, I.; Colson, Y.L.; Grinstaff, M.W. Polymer–drug conjugate therapeutics: Advances, insights and prospects. Nat. Rev. Drug Discov. 2019, 18, 273–294. [Google Scholar] [CrossRef]
- Adjei, I.M.; Sharma, B.; Peetla, C.; Labhasetwar, V. Inhibition of bone loss with surface-modulated, drug-loaded nanoparticles in an intraosseous model of prostate cancer. J. Control. Release 2016, 232, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Vijayaraghavalu, S.; Gao, Y.; Rahman, M.T.; Rozic, R.; Sharifi, N.; Midura, R.J.; Labhasetwar, V. Synergistic combination treatment to break cross talk between cancer cells and bone cells to inhibit progression of bone metastasis. Biomaterials 2020, 227, 119558. [Google Scholar] [CrossRef] [PubMed]
- Gaur, S.; Wen, Y.; Song, J.H.; Parikh, N.U.; Mangala, L.S.; Blessing, A.M.; Ivan, C.; Wu, S.Y.; Varkaris, A.; Shi, Y.; et al. Chitosan nanoparticle-mediated delivery of miRNA-34a decreases prostate tumor growth in the bone and its expression induces non-canonical autophagy. Oncotarget 2015, 6, 29161–29177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, Z.; Fan, W.; Hao, J.; Wu, X.; Zeng, G.Q.; Zhang, L.J.; Nie, S.F.; Wang, X.D. Efficient delivery of micro RNA to bone-metastatic prostate tumors by using aptamer-conjugated atelocollagen in vitro and in vivo. Drug Deliv. 2016, 23, 874–881. [Google Scholar] [CrossRef] [PubMed]
- Hoang, B.; Ernsting, M.J.; Murakami, M.; Undzys, E.; Li, S.D. Docetaxel-carboxymethylcellulose nanoparticles display enhanced anti-tumor activity in murine models of castration-resistant prostate cancer. Int. J. Pharm. 2014, 471, 224–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoang, B.; Ernsting, M.J.; Tang, W.H.S.; Bteich, J.; Undzys, E.; Kiyota, T.; Li, S.D. Cabazitaxel-conjugated nanoparticles for docetaxel-resistant and bone metastatic prostate cancer. Cancer Lett. 2017, 410, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Dozono, H.; Yanazume, S.; Nakamura, H.; Etrych, T.; Chytil, P.; Ulbrich, K.; Fang, J.; Arimura, T.; Douchi, T.; Kobayashi, H.; et al. HPMA Copolymer-Conjugated Pirarubicin in Multimodal Treatment of a Patient with Stage IV Prostate Cancer and Extensive Lung and Bone Metastases. Target. Oncol. 2016, 11, 101–106. [Google Scholar] [CrossRef]
- Ahangar, P.; Akoury, E.; Ramirez Garcia Luna, A.; Nour, A.; Weber, M.; Rosenzweig, D. Nanoporous 3D-Printed Scaffolds for Local Doxorubicin Delivery in Bone Metastases Secondary to Prostate Cancer. Materials 2018, 11, 1485. [Google Scholar] [CrossRef] [Green Version]
- Swenson, S.D.; Silva-Hirschberg, C.; Markland, F.S. Methods for evaluation of a snake venom-derived disintegrin in animal models of human cancer. Methods Mol. Biol. 2020, 2068, 185–204. [Google Scholar] [CrossRef]
- Xu, W.; Neill, T.; Yang, Y.; Hu, Z.; Cleveland, E.; Wu, Y.; Hutten, R.; Xiao, X.; Stock, S.R.; Shevrin, D.; et al. The systemic delivery of an oncolytic adenovirus expressing decorin inhibits bone metastasis in a mouse model of human prostate cancer. Gene Ther. 2015, 22, 247–256. [Google Scholar] [CrossRef] [Green Version]
- Malam, Y.; Loizidou, M.; Seifalian, A.M. Liposomes and nanoparticles: Nanosized vehicles for drug delivery in cancer. Trends Pharmacol. Sci. 2009, 30, 592–599. [Google Scholar] [CrossRef]
- Maranhão, R.C.; Vital, C.G.; Tavoni, T.M.; Graziani, S.R. Clinical experience with drug delivery systems as tools to decrease the toxicity of anticancer chemotherapeutic agents. Expert Opin. Drug Deliv. 2017, 14, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Herrero, E.; Fernández-Medarde, A. Advanced targeted therapies in cancer: Drug nanocarriers, the future of chemotherapy. Eur. J. Pharm. Biopharm. 2015, 93, 52–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLane, M.A.; Joerger, T.; Mahmoud, A. Disintegrins in health and disease. Front. Biosci. 2008, 13, 6617–6637. [Google Scholar] [CrossRef] [PubMed]
- Gould, R.J.; Polokoff, M.A.; Friedman, P.A.; Huang, T.F.; Holt, J.C.; Cook, J.J.; Niewiarowski, S. Disintegrins: A Family of Integrin Inhibitory Proteins from Viper Venoms. Proc. Soc. Exp. Biol. Med. 1990, 195, 168–171. [Google Scholar] [CrossRef] [PubMed]
- Minea, R.O.; Helchowski, C.M.; Zidovetzki, S.J.; Costa, F.K.; Swenson, S.D.; Markland, F.S. Vicrostatin - An anti-invasive multi-integrin targeting chimeric disintegrin with tumor anti-angiogenic and pro-apoptotic activities. PLoS ONE 2010, 5. [Google Scholar] [CrossRef]
- Minea, R.; Helchowski, C.; Rubino, B.; Brodmann, K.; Swenson, S.; Markland, F. Development of a chimeric recombinant disintegrin as a cost-effective anti-cancer agent with promising translational potential. Toxicon 2012, 59, 472–486. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Oyamada, S.; Wu, T.; Robich, M.P.; Wu, H.; Wang, X.; Buchholz, B.; McCarthy, S.; Bianchi, C.F.; Sellke, F.W.; et al. In vitro and in vivo degradation of poly(D, L-lactide-co-glycolide)/amorphous calcium phosphate copolymer coated on metal stents. J. Biomed. Mater. Res. A 2011, 96, 632–638. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Giottonini, K.Y.; Rodríguez-Córdova, R.J.; Gutiérrez-Valenzuela, C.A.; Peñuñuri-Miranda, O.; Zavala-Rivera, P.; Guerrero-Germán, P.; Lucero-Acuña, A. PLGA nanoparticle preparations by emulsification and nanoprecipitation techniques: Effects of formulation parameters. RSC Adv. 2020, 10, 4218–4231. [Google Scholar] [CrossRef] [Green Version]
- Sarin, H. Physiologic upper limits of pore size of different blood capillary types and another perspective on the dual pore theory of microvascular permeability. J. Angiogenes. Res. 2010, 2. [Google Scholar] [CrossRef] [Green Version]
- Hegemann, M.; Bedke, J.; Stenzl, A.; Todenhöfer, T. Denosumab treatment in the management of patients with advanced prostate cancer: Clinical evidence and experience. Ther. Adv. Urol. 2017, 9, 81–88. [Google Scholar] [CrossRef] [Green Version]
- Arvizo, R.R.; Miranda, O.R.; Moyano, D.F.; Walden, C.A.; Giri, K.; Bhattacharya, R.; Robertson, J.D.; Rotello, V.M.; Reid, J.M.; Mukherjee, P. Modulating Pharmacokinetics, Tumor Uptake and Biodistribution by Engineered Nanoparticles. PLoS ONE 2011, 6, e24374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miele, E.; Spinelli, G.P.; Miele, E.; Tomao, F.; Tomao, S. Albumin-bound formulation of paclitaxel (Abraxane® ABI-007) in the treatment of breast cancer. Int. J. Nanomedicine 2009, 4, 99–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibrahim, N.K.; Desai, N.; Legha, S.; Soon-Shiong, P.; Theriault, R.L.; Rivera, E.; Esmaeli, B.; Ring, S.E.; Bedikian, A.; Hortobagyi, G.N.; et al. Phase I and Pharmacokinetic Study of ABI-007, a Cremophor-free, Protein-stabilized, Nanoparticle Formulation of Paclitaxel. Clin. Cancer Res. 2002, 8, 1038–1044. [Google Scholar] [PubMed]
- Datta-Mannan, A.; Choi, H.; Stokell, D.; Tang, J.; Murphy, A.; Wrobleski, A.; Feng, Y. The Properties of Cysteine-Conjugated Antibody-Drug Conjugates Are Impacted by the IgG Subclass. AAPS J. 2018, 20, 103. [Google Scholar] [CrossRef] [PubMed]
- Takakura, Y.; Hashida, M. Macromolecular carrier systems for targeted drug delivery: Pharmacokinetic considerations on biodistribution. Pharm. Res. 1996, 13, 820–831. [Google Scholar] [CrossRef] [PubMed]
- Sezaki, H.; Hashida, M. Macromolecule-drug conjugates in targeted cancer chemotherapy. Crit. Rev. Ther. Drug Carrier Syst. 1984, 1, 1–38. [Google Scholar]
- Ernsting, M.J.; Tang, W.L.; Maccallum, N.; Li, S.D. Synthetic modification of carboxymethylcellulose and use thereof to prepare a nanoparticle forming conjugate of docetaxel for enhanced cytotoxicity against cancer cells. Bioconjug. Chem. 2011, 22, 2474–2486. [Google Scholar] [CrossRef]
- Bteich, J.; McManus, S.A.; Ernsting, M.J.; Mohammed, M.Z.; Prud’Homme, R.K.; Sokoll, K.K. Using Flash Nanoprecipitation to Produce Highly Potent and Stable Cellax Nanoparticles from Amphiphilic Polymers Derived from Carboxymethyl Cellulose, Polyethylene Glycol, and Cabazitaxel. Mol. Pharm. 2017, 14, 3998–4007. [Google Scholar] [CrossRef]
- Seruga, B.; Tannock, I.F. Chemotherapy-based treatment for castration-resistant prostate cancer. J. Clin. Oncol. 2011, 29, 3686–3694. [Google Scholar] [CrossRef]
- Harrington, J.A.; Jones, R.J. Management of metastatic castration-resistant prostate cancer after first-line docetaxel. Eur. J. Cancer 2011, 47, 2133–2142. [Google Scholar] [CrossRef]
- Shih, Y.-C.T.; Halpern, M.T. Economic Evaluations of Medical Care Interventions for Cancer Patients: How, Why, and What Does it Mean? CA Cancer J. Clin. 2008, 58, 231–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuelsson, E.; Shen, H.; Blanco, E.; Ferrari, M.; Wolfram, J. Contribution of Kupffer cells to liposome accumulation in the liver. Colloids Surfaces B Biointerfaces 2017, 158, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.N.; Poon, W.; Tavares, A.J.; McGilvray, I.D.; Chan, W.C.W. Nanoparticle–liver interactions: Cellular uptake and hepatobiliary elimination. J. Control. Release 2016, 240, 332–348. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Kopeček, J. The light at the end of the tunnel—second generation HPMA conjugates for cancer treatment. Curr. Opin. Colloid Interface Sci. 2017, 31, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Sonawane, S.J.; Kalhapure, R.S.; Govender, T. Hydrazone linkages in pH responsive drug delivery systems. Eur. J. Pharm. Sci. 2017, 99, 45–65. [Google Scholar] [CrossRef] [PubMed]
- Tsukigawa, K.; Liao, L.; Nakamura, H.; Fang, J.; Greish, K.; Otagiri, M.; Maeda, H. Synthesis and therapeutic effect of styrene-maleic acid copolymer-conjugated pirarubicin. Cancer Sci. 2015, 106, 270–278. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, H.; Etrych, T.; Chytil, P.; Ohkubo, M.; Fang, J.; Ulbrich, K.; Maeda, H. Two step mechanisms of tumor selective delivery of N-(2-hydroxypropyl) methacrylamide copolymer conjugated with pirarubicin via an acid-cleavable linkage. J. Control. Release 2014, 174, 81–87. [Google Scholar] [CrossRef]
- Jakus, A.E.; Rutz, A.L.; Shah, R.N. Advancing the field of 3D biomaterial printing. Biomed. Mater. 2016, 11. [Google Scholar] [CrossRef]
- Murphy, S.V.; Atala, A. 3D bioprinting of tissues and organs. Nat. Biotechnol. 2014, 32, 773–785. [Google Scholar] [CrossRef]
- Yan, Y.; Chen, H.; Zhang, H.; Guo, C.; Yang, K.; Chen, K.; Cheng, R.; Qian, N.; Sandler, N.; Zhang, Y.S.; et al. Vascularized 3D printed scaffolds for promoting bone regeneration. Biomaterials 2019, 190–191, 97–110. [Google Scholar] [CrossRef]
- Park, J.H.; Jang, J.; Lee, J.S.; Cho, D.W. Three-Dimensional Printing of Tissue/Organ Analogues Containing Living Cells. Ann. Biomed. Eng. 2017, 45, 180–194. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Mangadlao, J.D.; Wallat, J.; De Leon, A.; Pokorski, J.K.; Advincula, R.C. 3D printing biocompatible polyurethane/poly(lactic acid)/graphene oxide nanocomposites: Anisotropic properties. ACS Appl. Mater. Interfaces 2017, 9, 4015–4023. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food & Drug Administration Fact Sheet: Breakthrough Therapies|FDA. Available online: https://www.fda.gov/regulatory-information/food-and-drug-administration-safety-and-innovation-act-fdasia/fact-sheet-breakthrough-therapies (accessed on 19 May 2020).
- Fournier, P.G.J.; Juárez, P.; Jiang, G.; Clines, G.A.; Niewolna, M.; Kim, H.S.; Walton, H.W.; Peng, X.H.; Liu, Y.; Mohammad, K.S.; et al. The TGF-β Signaling Regulator PMEPA1 Suppresses Prostate Cancer Metastases to Bone. Cancer Cell 2015, 27, 809–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Hirata, H.; Shiraki, M.; Kamohara, A.; Nishioka, K.; Miyamoto, H.; Kukita, T.; Kukita, A. Prostate transmembrane protein androgen induced 1 is induced by activation of osteoclasts and regulates bone resorption. FASEB J. 2019, 33, 4365–4375. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Chen, X.; Zhang, H.; Wang, H.; Chen, H.; Huang, S.; Xu, Y.; Zhang, Y.; Wu, X.; Chen, J. Study on the cellular internalization mechanisms and in vivo anti-bone metastasis prostate cancer efficiency of the peptide T7-modified polypeptide nanoparticles. Drug Deliv. 2020, 27, 161–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, T.; Yu, X.; Carbone, E.J.; Nelson, C.; Kan, H.M.; Lo, K.W.H. Poly aspartic acid peptide-linked PLGA based nanoscale particles: Potential for bone-targeting drug delivery applications. Int. J. Pharm. 2014, 475, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Jiang, W.; Wu, X.; Huang, S.; Huang, Z.; Shi, Y.; Dai, Q.; Chen, J.; Ren, F.; Gao, S. Peptide T7-modified polypeptide with disulfide bonds for targeted delivery of plasmid DNA for gene therapy of prostate cancer. Int. J. Nanomed. 2018, 13, 6913–6927. [Google Scholar] [CrossRef] [Green Version]
- Edwards, I.J. Proteoglycans in prostate cancer. Nat. Rev. Urol. 2012, 9, 196–206. [Google Scholar] [CrossRef]
- Neill, T.; Painter, H.; Buraschi, S.; Owens, R.T.; Lisanti, M.P.; Schaefer, L.; Iozzo, R.V. Decorin antagonizes the angiogenic network: Concurrent inhibition of met, hypoxia inducible factor 1α, vascular endothelial growth factor A, and induction of thrombospondin-1 and tiMP3. J. Biol. Chem. 2012, 287, 5492–5506. [Google Scholar] [CrossRef] [Green Version]
- Sofeu Feugaing, D.D.; Götte, M.; Viola, M. More than matrix: The multifaceted role of decorin in cancer. Eur. J. Cell Biol. 2013, 92, 1–11. [Google Scholar] [CrossRef]
- Li, X.; Ling, W.; Khan, S.; Yaccoby, S. Therapeutic effects of intrabone and systemic mesenchymal stem cell cytotherapy on myeloma bone disease and tumor growth. J. Bone Miner. Res. 2012, 27, 1635–1648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Pennisi, A.; Yaccoby, S. Role of decorin in the antimyeloma effects of osteoblasts. Blood 2008, 112, 159–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sano, A.; Maeda, M.; Nagahara, S.; Ochiya, T.; Honma, K.; Itoh, H.; Miyata, T.; Fujioka, K. Atelocollagen for protein and gene delivery. Adv. Drug Deliv. Rev. 2003, 55, 1651–1677. [Google Scholar] [CrossRef] [PubMed]
- Ochiya, T.; Takahama, Y.; Nagahara, S.; Sumita, Y.; Hisada, A.; Itoh, H.; Nagai, Y.; Terada, M. New delivery system for plasmid DNA in vivo using atelocollagen as a carrier material: The Minipellet. Nat. Med. 1999, 5, 707–710. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Teply, B.A.; Sherifi, I.; Sung, J.; Luther, G.; Gu, F.X.; Levy-Nissenbaum, E.; Radovic-Moreno, A.F.; Langer, R.; Farokhzad, O.C. Formulation of functionalized PLGA-PEG nanoparticles for in vivo targeted drug delivery. Biomaterials 2007, 28, 869–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhar, S.; Gu, F.X.; Langer, R.; Farokhza, O.C.; Lippard, S.J. Targeted delivery of cisplatin to prostate cancer cells by aptamer functionalized Pt(IV) prodrug-PLGA - PEG nanoparticles. Proc. Natl. Acad. Sci. USA 2008, 105, 17356–17361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonci, D.; Coppola, V.; Musumeci, M.; Addario, A.; Giuffrida, R.; Memeo, L.; D’Urso, L.; Pagliuca, A.; Biffoni, M.; Labbaye, C.; et al. The miR-15a-miR-16-1 cluster controls prostate cancer by targeting multiple oncogenic activities. Nat. Med. 2008, 14, 1271–1277. [Google Scholar] [CrossRef]
- Clevers, H. Wnt/β-Catenin Signaling in Development and Disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef] [Green Version]
- Tsigos, I.; Martinou, A.; Kafetzopoulos, D.; Bouriotis, V. Chitin deacetylases: New, versatile tools in biotechnology. Trends Biotechnol. 2000, 18, 305–312. [Google Scholar] [CrossRef]
- Hintzen, F.; Laffleur, F.; Sarti, F.; Shahnaz, G.; Bernkop-Schnürch, A. Thiomers: Influence of molar mass on in situ gelling properties. Int. J. Pharm. 2012, 436, 120–126. [Google Scholar] [CrossRef]
- Panos, I.; Acosta, N.; Heras, A. New Drug Delivery Systems Based on Chitosan. Curr. Drug Discov. Technol. 2008, 5, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Hauptstein, S.; Bonengel, S.; Griessinger, J.; Bernkop-Schnürch, A. Synthesis and characterization of pH tolerant and mucoadhesive (Thiol-Polyethylene Glycol) chitosan graft polymer for drug delivery. J. Pharm. Sci. 2014, 103, 594–601. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Kelnar, K.; Liu, B.; Chen, X.; Calhoun-Davis, T.; Li, H.; Patrawala, L.; Yan, H.; Jeter, C.; Honorio, S.; et al. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat. Med. 2011, 17, 211–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamura, S.; Saini, S.; Majid, S.; Hirata, H.; Ueno, K.; Deng, G.; Dahiya, R. Microrna-34a modulates c-Myc transcriptional complexes to suppress malignancy in human prostate cancer cells. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.H.; Yin, A.A.; Cheng, J.X.; Huang, H.Y.; Li, X.M.; Zhang, Y.Q.; Han, N.; Zhang, X. TRIM24 promotes glioma progression and enhances chemoresistance through activation of the PI3K/Akt signaling pathway. Oncogene 2015, 34, 600–610. [Google Scholar] [CrossRef] [PubMed]
- Groner, A.C.; Cato, L.; de Tribolet-Hardy, J.; Bernasocchi, T.; Janouskova, H.; Melchers, D.; Houtman, R.; Cato, A.C.B.; Tschopp, P.; Gu, L.; et al. TRIM24 Is an Oncogenic Transcriptional Activator in Prostate Cancer. Cancer Cell 2016, 29, 846–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fong, K.; Zhao, J.C.; Song, B.; Zheng, B.; Yu, J. TRIM28 protects TRIM24 from SPOP-mediated degradation and promotes prostate cancer progression. Nat. Commun. 2018, 9, 5007. [Google Scholar] [CrossRef]
- Shi, S.J.; Wang, L.J.; Han, D.H.; Wu, J.H.; Jiao, D.; Zhang, K.L.; Chen, J.W.; Li, Y.; Yang, F.; Zhang, J.L.; et al. Therapeutic effects of human monoclonal PSMA antibody-mediated TRIM24 siRNA delivery in PSMA-positive castration-resistant prostate cancer. Theranostics 2019, 9, 1247–1263. [Google Scholar] [CrossRef]
- Brown, J.E.; Sim, S. Evolving role of bone biomarkers in castration-resistant prostate cancer. Neoplasia 2010. [Google Scholar] [CrossRef] [Green Version]
- Koizumi, M.; Yonese, J.; Fukui, I.; Ogata, E. The serum level of the amino-terminal propeptide of type I procollagen is a sensitive marker for prostate cancer metastasis to bone. BJU Int. 2001. [Google Scholar] [CrossRef]
- Bergmann, P.; Body, J.J.; Boonen, S.; Boutsen, Y.; Devogelaer, J.P.; Goemaere, S.; Kaufman, J.M.; Reginster, J.Y.; Gangji, V. Evidence-based guidelines for the use of biochemical markers of bone turnover in the selection and monitoring of bisphosphonate treatment in osteoporosis: A consensus document of the Belgian Bone Club. Int. J. Clin. Pract. 2009, 63, 19–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Sircar, K.; Aprikian, A.; Potti, A.; Goltzman, D.; Rabbani, S.A. Expression of RANKL/RANK/OPG in primary and metastatic human prostate cancer as markers of disease stage and functional regulation. Cancer 2006. [Google Scholar] [CrossRef] [PubMed]
- Damasco, J.A.; Ravi, S.; Perez, J.D.; Hagaman, D.E.; Melancon, M.P. Understanding nanoparticle toxicity to direct a safe-by-design approach in cancer nanomedicine. Nanomaterials 2020, 10, 2186. [Google Scholar] [CrossRef] [PubMed]




| Drug Delivery Vehicle | Material | Therapeutic Agent | Results | Refs |
|---|---|---|---|---|
| Polymeric Nanoparticles | Poly(d,l-lactide-co-glycolide) | Paclitaxel | ~50% lower tumor burden at 5 weeks post-treatment compared to saline (p < 0.05). No bone loss compared to >50% bone resorption in control. | [154] |
| Docetaxel | Median survival for untreated group = 10 weeks vs. >48 weeks for the treated group, which is beyond the study end point | [155] | ||
| Cabizataxel | Significant tumor weight reduction compared to the free Cabizataxel treatment group (p < 0.05) | [149] | ||
| Chitosan | miRNA | Successfully delivered tumor suppressive microRNA to PCa cells | [156] | |
| Atellocollagen | miRNA | PSMA targeting atellocollagen enhanced transfection efficiency of microRNA in both in vitro/in vivo models | [157] | |
| Polymer-Drug Conjugates | Pegylated carboxymethyl cellulose (Cellax) | Docetaxel | Mean survival times 2X that of free docetaxel treated groups (p < 0.05) | [158] |
| Cabizataxel | Approximately 4X increase in overall survival time compared to saline and free cabizataxel treatment groups | [159] | ||
| HPMA | Pirarubicin | A single human case study reported complete tumor regression for this treatment in combination with proton beam radiotherapy | [160] | |
| 3D-printed scaffold | Poly(urethane)/poly(vinyl alcohol) copolymer | Doxorubicin | Exhibits sustained drug release (>7 days) and reduced proliferation of LAPC4 cells in vitro | [161] |
| Liposome | Not published | Vicrostatin | Decreased metastatic potential and inhibited tumor growth in androgen dependent in vivo model | [162] |
| DSPE-PEG-2000/DPPC/cholesterol | Dexamethasone | Passively targeted bone lesions and significantly inhibited growth up to 26 days compared to empty vehicle (p < 0.001) | [147] | |
| Tocopherol acetate/Labrasol | Etoposide and Curcumin | Confirmed intracellular delivery to PC3 cells and 1.5-fold enhancement in cytotoxicity compared to free drug (p < 0.05) | [148] | |
| Viral Vector | Recombinant oncolytic adenovirus | Decorin gene | Decorin expression inhibited tumor cell migration and significantly reduced skeletal metastases (p < 0.05) | [163] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hagaman, D.E.; Damasco, J.A.; Perez, J.V.D.; Rojo, R.D.; Melancon, M.P. Recent Advances in Nanomedicine for the Diagnosis and Treatment of Prostate Cancer Bone Metastasis. Molecules 2021, 26, 384. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26020384
Hagaman DE, Damasco JA, Perez JVD, Rojo RD, Melancon MP. Recent Advances in Nanomedicine for the Diagnosis and Treatment of Prostate Cancer Bone Metastasis. Molecules. 2021; 26(2):384. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26020384
Chicago/Turabian StyleHagaman, Daniel E., Jossana A. Damasco, Joy Vanessa D. Perez, Raniv D. Rojo, and Marites P. Melancon. 2021. "Recent Advances in Nanomedicine for the Diagnosis and Treatment of Prostate Cancer Bone Metastasis" Molecules 26, no. 2: 384. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26020384