
Effect of the Supporting Electrolyte on Chloroform Reduction at a Silver Electrode in Aqueous Solutions



1
Faculty of Advanced Technology and Chemistry, Institute of Materials Science and Engineering, Military University of Technology, Kaliskiego 2, 00908 Warszawa, Poland
2
Department of Physical Chemistry and Electrochemistry, Faculty of Chemistry, Jagiellonian University, Gronostajowa 2, 30387 Krakow, Poland
*
Author to whom correspondence should be addressed.
Molecules 2021, 26(3), 525; https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26030525
Submission received: 30 December 2020
/
Revised: 14 January 2021
/
Accepted: 19 January 2021
/
Published: 20 January 2021
(This article belongs to the Special Issue Electrochemistry of Thin Films and Nanostructured Materials)
Abstract
:Herein, we report, for the first time, a comparative study on the electrocatalytic reduction of chloroform on silver in different aqueous supporting electrolytes. Cyclic voltammetry measurements were performed at a wide range of scan rates and concentrations of CHCl3 using 0.05 M NaClO4, NaH2PO4, and Na2HPO4 as supporting electrolytes. We observed that a type of supporting electrolyte anion strongly influences both the potential as well as the current density of the chloroform reduction peak, mainly due to the presence of OH− in an alkaline Na2HPO4 solution, which is a specifically interacting anion. Moreover, the highest sensitivity of the Ag electrode toward CHCl3 reduction was observed in a neutral NaClO4 aqueous solution. It was found that the electroreduction of chloroform at the silver surface occurs via a concerted mechanism regardless of the type of the studied supporting electrolyte.
1. Introduction
Chloroform and other trihalomethanes (THMs) are known to be environmental pollutants [1], toxic, and possibly carcinogenic agents to humans [2]. The use of chloroform in consumer products (surgical anesthetic, cough syrups, toothpaste, etc.) was banned in the USA in 1976, as it was found to be carcinogenic in animal tests [3]. The issue of water contamination with THMs is reflected by the EU [4] and USA [5] regulations with the total trihalomethanes level in drinking water at maximum allowable averages of 100 and 80 μg L−1, respectively. The known sources of chloroform pollution are either environmental (volcanic emission and marine algae) or anthropogenic [6]. The CHCl3 can be formed during waste incineration or as a water chlorination by-product. Moreover, chloroform is still widely used as a solvent in the pharmaceutical industry, dyes and pesticides synthesis, fire extinguishers, rubber industry, etc. and, therefore, it can be emitted to the environment in a form of exhaust gas and wastewater.
Catalytic cathodic reduction at silver electrodes, aiming to convert organic halides to the corresponding de-halogenated hydrocarbons, has been reported as a promising approach toward the reduced abatement of such environmental pollutants [7]. At this point, most of the reported studies on the electrocatalytic reduction of organic halides were oriented to explore the mechanism of bond-braking reactions in nonaqueous solutions [8,9,10]. The aqueous solutions were also studied, regardless of the fact that the electrochemical reduction of many organic halides takes place at very negative potentials, at which a simultaneous hydrogen evolution reaction (HER) occurs, significantly increasing the energy consumption of the process [11,12,13,14,15,16,17,18,19]. Nonetheless, further studies on principal aspects of the chloroform electrocatalytic reduction at silver electrodes in aqueous solutions is a crucial step toward environmental applications.
To date, Rondinini et al. [20] studied the effect of the supporting electrolyte on the electrocatalytic activity of silver in nonaqueous solutions. They explored a large set of alkylammonium salts, and showed that the reduction potentials were spread widely in a range of about 300 mV. Authors reported that the major factor determining the reduction potential is the nature of the anion, while the cations exert a finer modulating effect. Moreover, the current density of the peak was also dependent on the supporting electrolyte. Fiori et al. [8] and Rondinini et al. [21] later confirmed the significant influence of the supporting electrolyte on the reduction potential of organic halides at Ag electrodes in a nonaqueous environment. For the chloroform reduction at silver electrodes in aqueous solutions, tetraethylammonium benzoate [22], KClO4 [14,17,23,24], NaClO4 [12], and NaOH [16] were reported as supporting electrolytes. NaClO4 and KClO4 are reportedly used due to negligible specific interactions of silver with perchlorate anions and sodium/potassium cations [25]. Taking into consideration the effect of the supporting electrolyte clearly demonstrated for nonaqueous solutions, it is surprising that no comparative study was reported to date for aqueous solutions. Therefore, in this work, aqueous NaClO4, NaH2PO4, and Na2HPO4 solutions were selected and studied as supporting electrolytes for the electrochemical reduction of chloroform at Ag.
2. Results and Discussion
During the reduction of chloroform at silver electrodes in nonaqueous solvents, 2 or 3 reduction peaks are observed [8,9,10,21,26], which can be ascribed to a gradual dehalogenation of the molecule, i.e., successive removal of halogen atoms [9]. In aqueous solutions, typically one wide peak of chloroform reduction, shifted toward less negative potential values as compared to nonaqueous solvents (up to 0.44 V), was observed [27]. The shift of the chloroform reduction peak toward less negative values, as compared to nonaqueous solvents, was attributed to the ability of water to enhance the turnover frequency of catalytic sites, as it favors the desorption processes [13]. It is also assumed that the electrocatalytic reduction of CHCl3 in an aqueous solvent is a complex phenomenon. Consequently, a superimposition of different peaks, corresponding to subsequent steps in the reaction mechanism, occurs [23], hindering the correct analysis of the process. Table 1 shows chloroform reduction peak potentials (Ep), scan rates (ν), and concentrations of CHCl3 (c) reported in the literature.
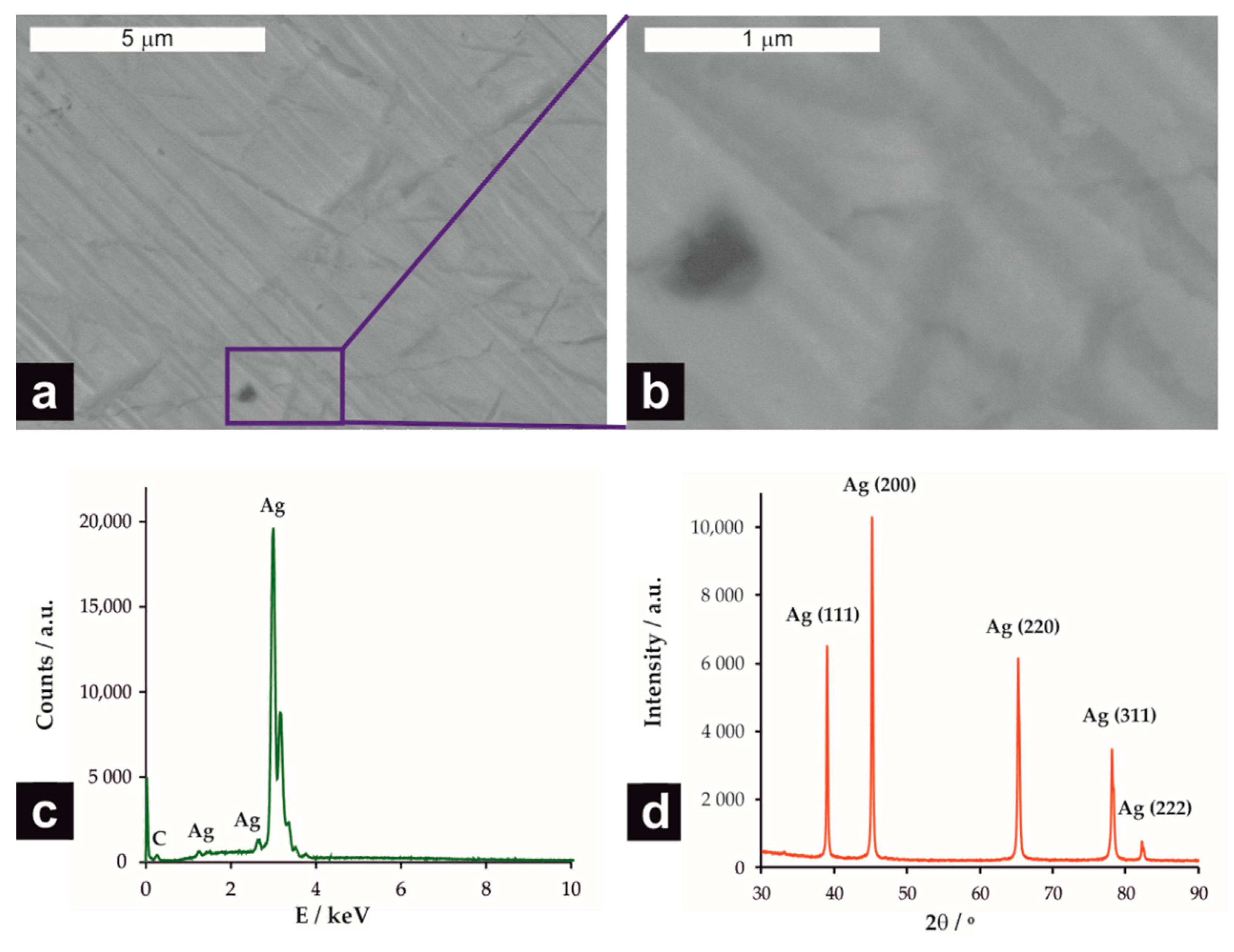
From the data gathered in Table 1, one can conclude that the direct comparison of the influence of different electrode materials from the literature reports is not possible, as the scan rate, concentration of chloroform, as well as type and concentration of the supporting electrolyte, strongly differ. It was previously shown in cyclic voltammetry (CV) measurements that both the potential and current density of the chloroform reduction peak strongly depend on the concentration of CHCl3 [17]. The CV measurements were performed at the silver electrode (Figure 1) in order to assess the effect of the supporting electrolyte on the potential and current density of the chloroform reduction peak. As expected, the EDS spectrum of the Ag electrode (Figure 1c) confirmed its chemical composition and, consequently, the planes (111), (200), (220), (1311), and (222) indexed to face-centered-cubic silver (JCPDS No. 04-0783) were detected in the XRD pattern.
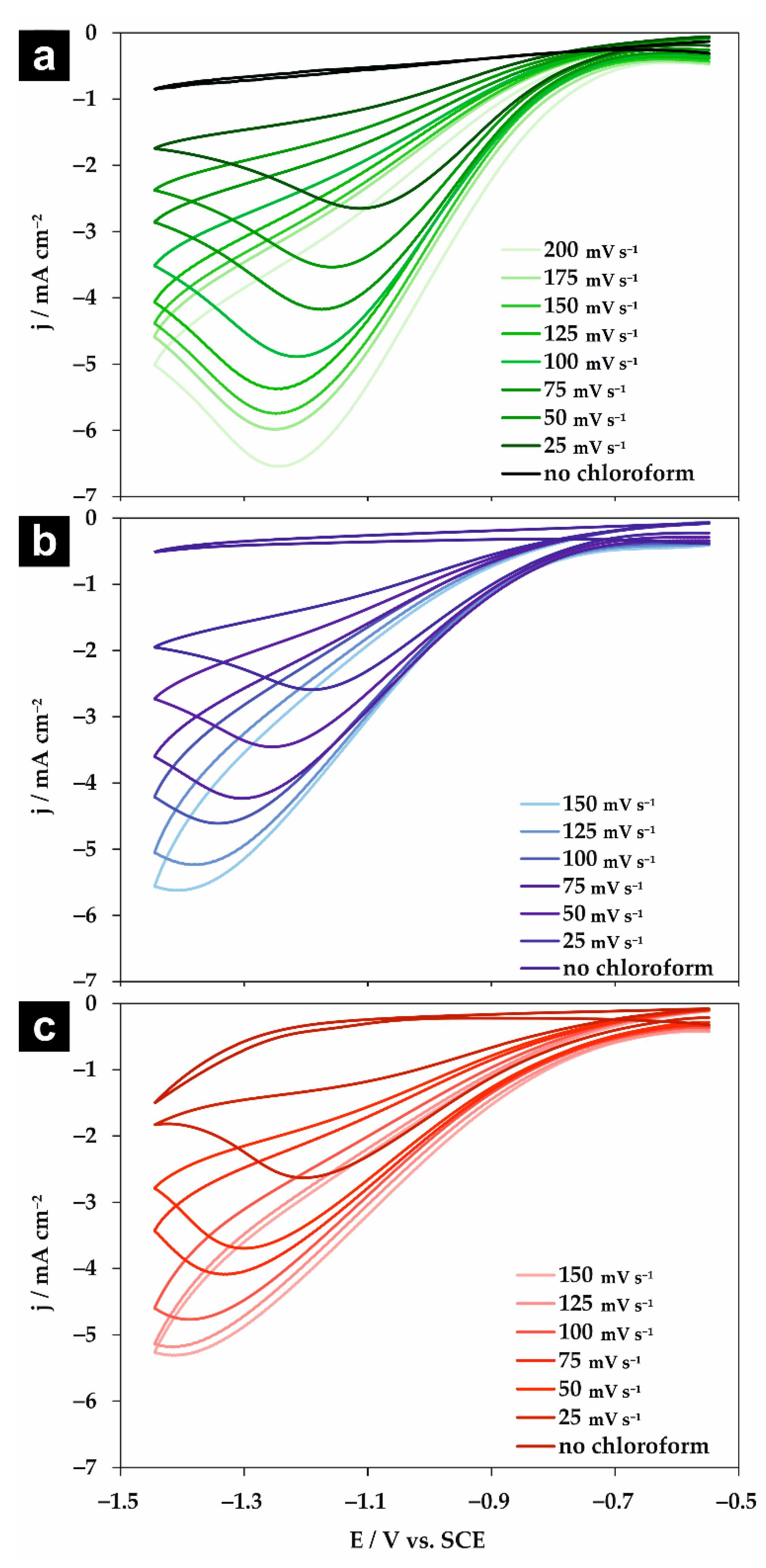
The voltammograms obtained at the Ag electrode in the absence and presence of 8.26 mM chloroform for selected scan rates are shown in Figure 2 for 0.05 M Na2HPO4, NaClO4, and NaH2PO4 solutions. The potentials are referenced to the saturated calomel electrode (SCE). In all studied electrolytes in the potential range of −0.547 to −1.447 V vs. SCE, a competitive reaction (i.e., decomposition of water or supporting electrolyte) was not observed. Only small capacitive currents were observed in the absence of chloroform. Conversely, independently of the studied electrolyte, after the addition of chloroform, one irreversible peak was observed similarly to previous reports (Table 1). Both the potential as well as the current density of the chloroform reduction peak shifted with the scan rate. Such an effect was previously reported for the chloroform reduction in aqueous solutions at Ag [17] and during benzyl chloride reduction in acetonitrile at Ag [29] and AgPd [30] electrodes. It is important to note that the magnitude of the shift strongly depends on the type of studied supporting electrolyte—especially when the results are compared with previously reported data for the chloroform reduction in 0.05 M KClO4 in the same setup [17]. The most significant shift in the reduction potential and highest current density were observed for Na2HPO4. This fact will be discussed later.
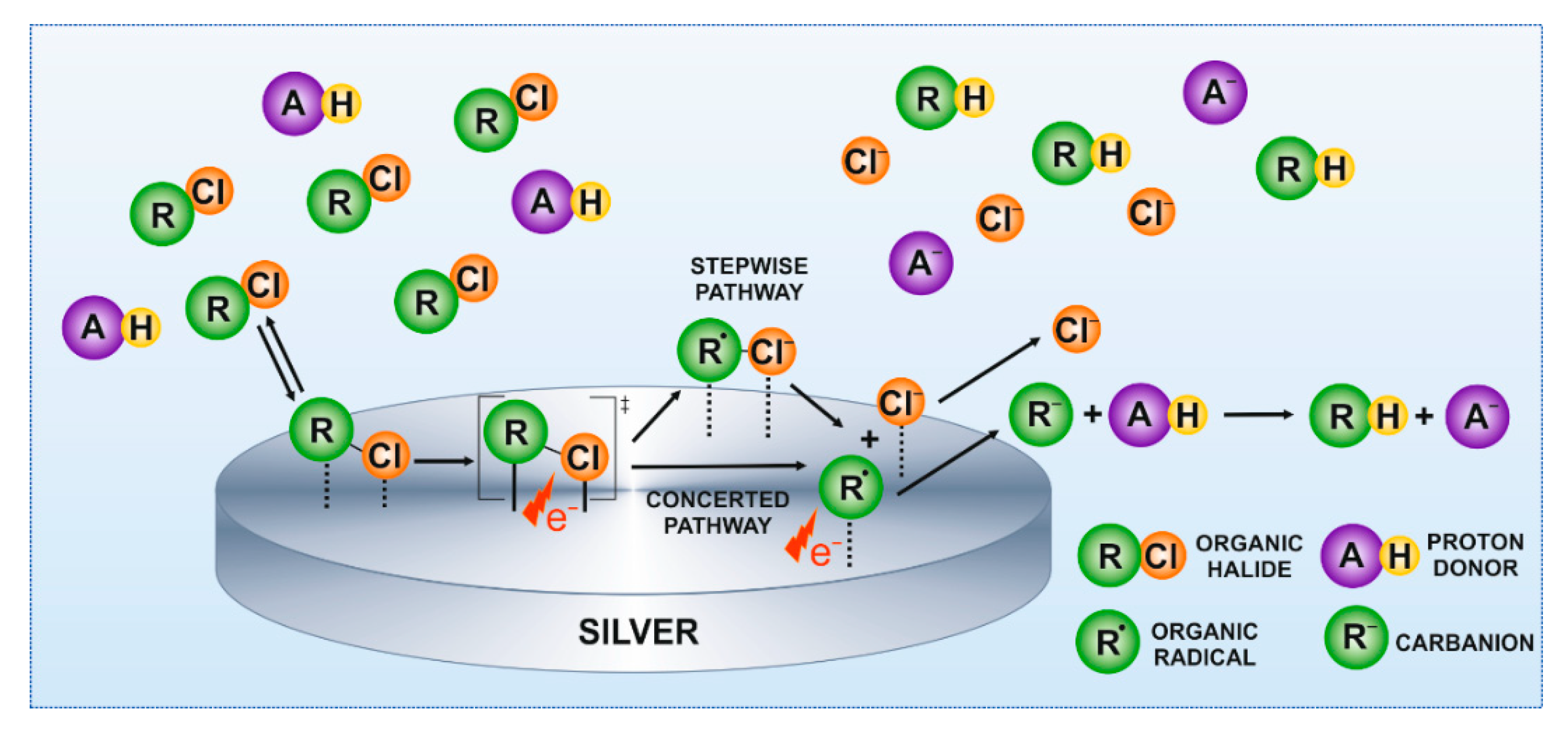
The reduction of organic halides occurs according to a stepwise or concerted pathway (Figure 3).
In the stepwise mechanism, an intermediate radical anion is formed before the C–X bond cleavage. In the concerted mechanism, the electron transfer and the C–X bond cleavage occur simultaneously. Those reactions are considered the slowest step of the reduction. The second electron transfer to the formed radical leads to the formation of organic anions, and is recognized as a very fast step occurring at less negative potentials. The created carbanion is protonated rapidly by a proton donor [27]. The cleavage mechanism of organic halide R–X bonds depends mainly on the type of reduced molecule and electrode material [31]. A kinetic indicator κ can be a useful tool to discriminate between the mechanisms involved in the R–X bond cleavage [32], and can be determined by voltammetric analysis using Equations (1) and (2) [26]:
where: κ—electron transfer coefficient; R—universal gas constant, R = 8.3145 J mol−1 K−1; T—temperature, K; ν—scan rate, V s−1; Ep—reduction potential of chloroform, V; Ep/2—potential corresponding to half of the peak current, V; F—Faraday constant, F = 9.6485 · 104 C mol−1.
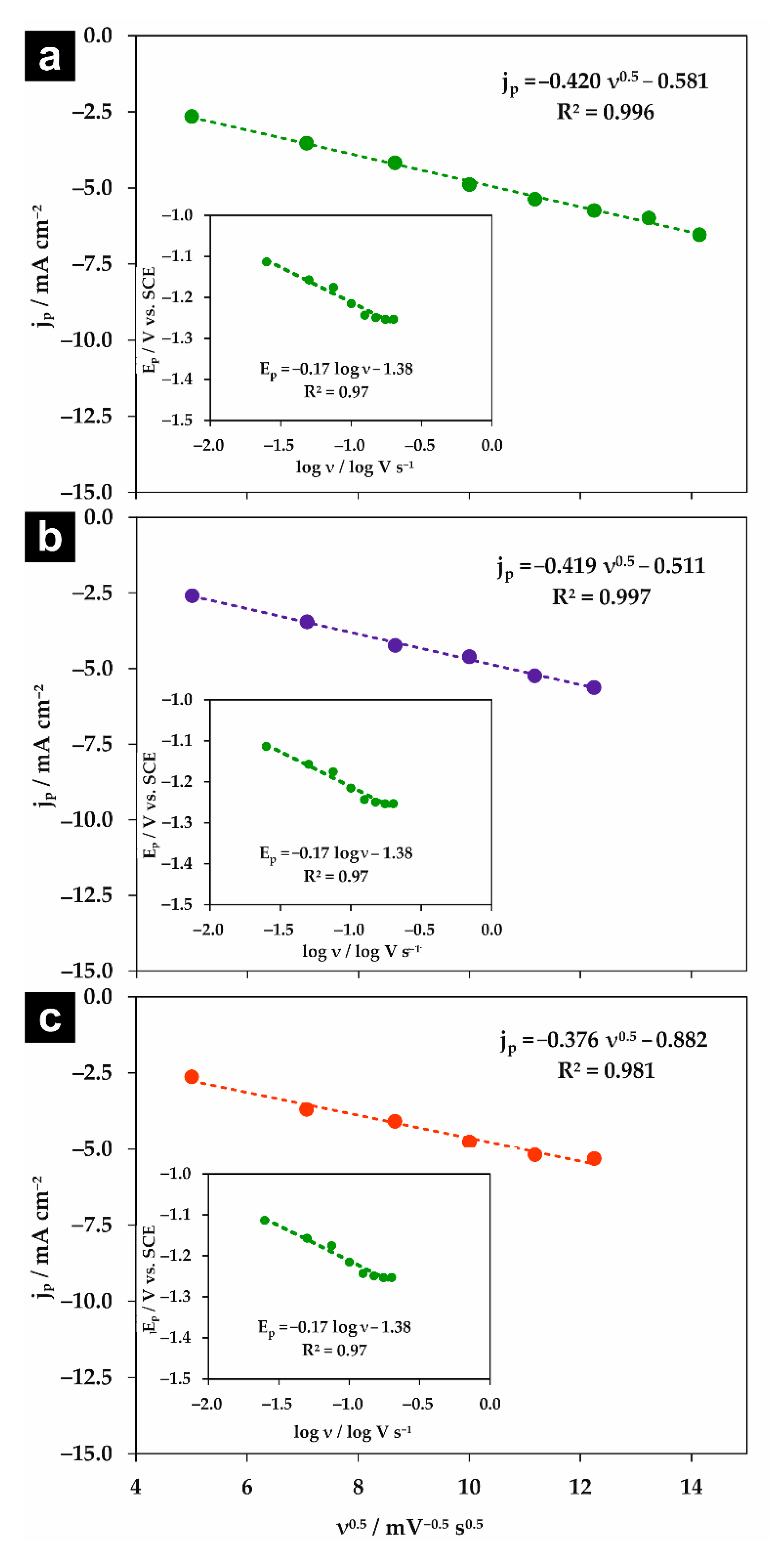
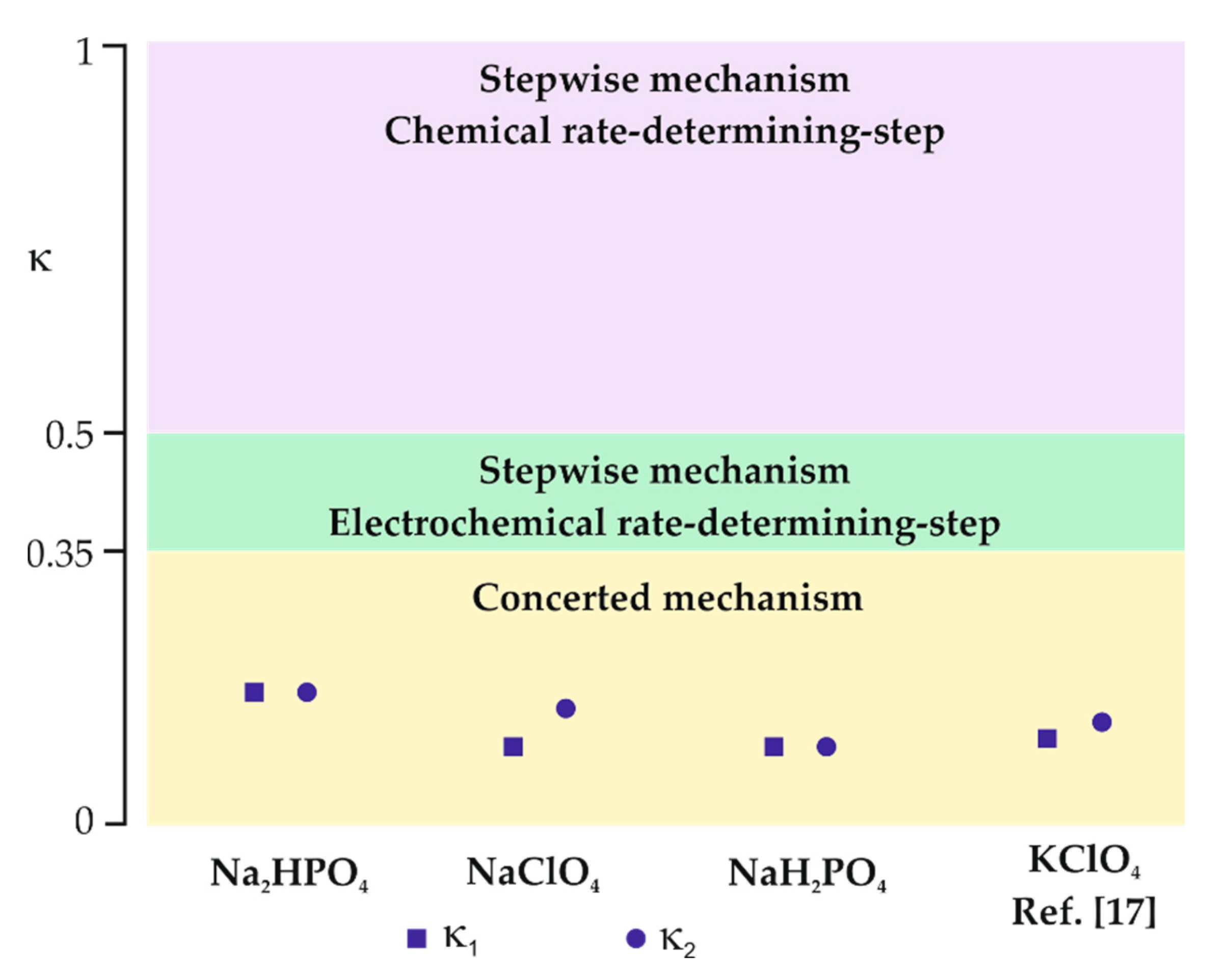
If κ is in the range between 0.35 and 0.5, the process occurs via the stepwise mechanism with an electrochemical rate-determining step. For κ < 0.35, the reaction follows the concerted mechanism and κ values can be considered as a transfer coefficient [17]. Moreover, Equation (1) clearly shows that the observed shift in the reduction potential with scan rate is correlated with the electron transfer coefficient. As can be seen in Figure 4, the peak current density (jp) is linearly dependent on the square root of the scan rate (ν0.5) for all used supporting electrolytes, which indicates that the reaction is diffusion-controlled [31]. Since Ep vs. log ν dependencies (insets in Figure 4) are linear with negative slopes, it was possible to estimate κ values (Table 2) from Equations (1) and (2).
For all studied supporting electrolytes, the observed current density of the chloroform reduction peak is similar; however, the peak potential strongly differs. Moreover, the calculated κ values are significantly lower than 0.35 and are in good agreement for both estimation methods. It indicates that the chloroform reduction in studied supporting electrolytes takes place via the concerted mechanism at the surface of the polycrystalline silver electrode (Figure 5). Such a mechanism is often observed for cases when a proton donor is present in the solution [31]. For instance, the transfer coefficient reported for the chloroform reduction at the Ag electrode was in the range of 0.11–0.13 for 8.26 mM CHCl3 in a 0.05 M aqueous solution of KClO4 [17]. The charge transfer coefficient depends on the peak potential of analyte reduction [33,34]; therefore, observed discrepancies are expected.
Similarly, as in the case of the electrocatalytic reduction of organic halides in nonaqueous solutions, the selected aqueous supporting electrolytes influence the reduction potential of the studied compound [20]. Importantly, the nucleophilic character of the hydrogen phosphate ion (HPO42–) has to be considered, which is about twice as great as for the hydroxyl ion, leading to hydrolysis-like reactions and providing easier removal of Cl− ions [35]. This could lead to (i) lowering of the concentration of chloroform in the electrolyte and, therefore, a less negative potential of the chloroform reduction, and (ii) a slightly higher cathodic current density of the reduction peak due to a higher turnover frequency in the case of Na2HPO4. However, further extensive studies are necessary for the better understanding of this phenomenon. Moreover, in the aqueous solutions, additional hydrolysis of chloroform can occur, according to a neutral mechanism (Equation (3)) or an alkaline-catalyzed mechanism (Equation (4)) [35]:
CHCl3 + 5H2O → HCOOH + 3H3O+ + 3Cl−
CHCl3 + 4OH− → HCOO− + 2H2O + 3Cl−
The neutral hydrolysis is the dominant pathway at pH < 4. Between pH 4 and 8, none of the hydrolysis mechanisms are predominant, and at pH > 8, the alkaline-catalyzed hydrolysis pathway becomes important due to the strong neutrophilic character of the hydroxyl ion [35]. The presence of OH−, specifically interacting ion in a solution, is therefore another reason for the less negative potential of chloroform reduction observed in an alkaline solution due to a lower concentration of chloroform. The hydrolysis half-life of chloroform at room temperature is 1000 years at pH = 7, and even longer for more acidic solutions; therefore, the influence of hydrolysis could be neglected.
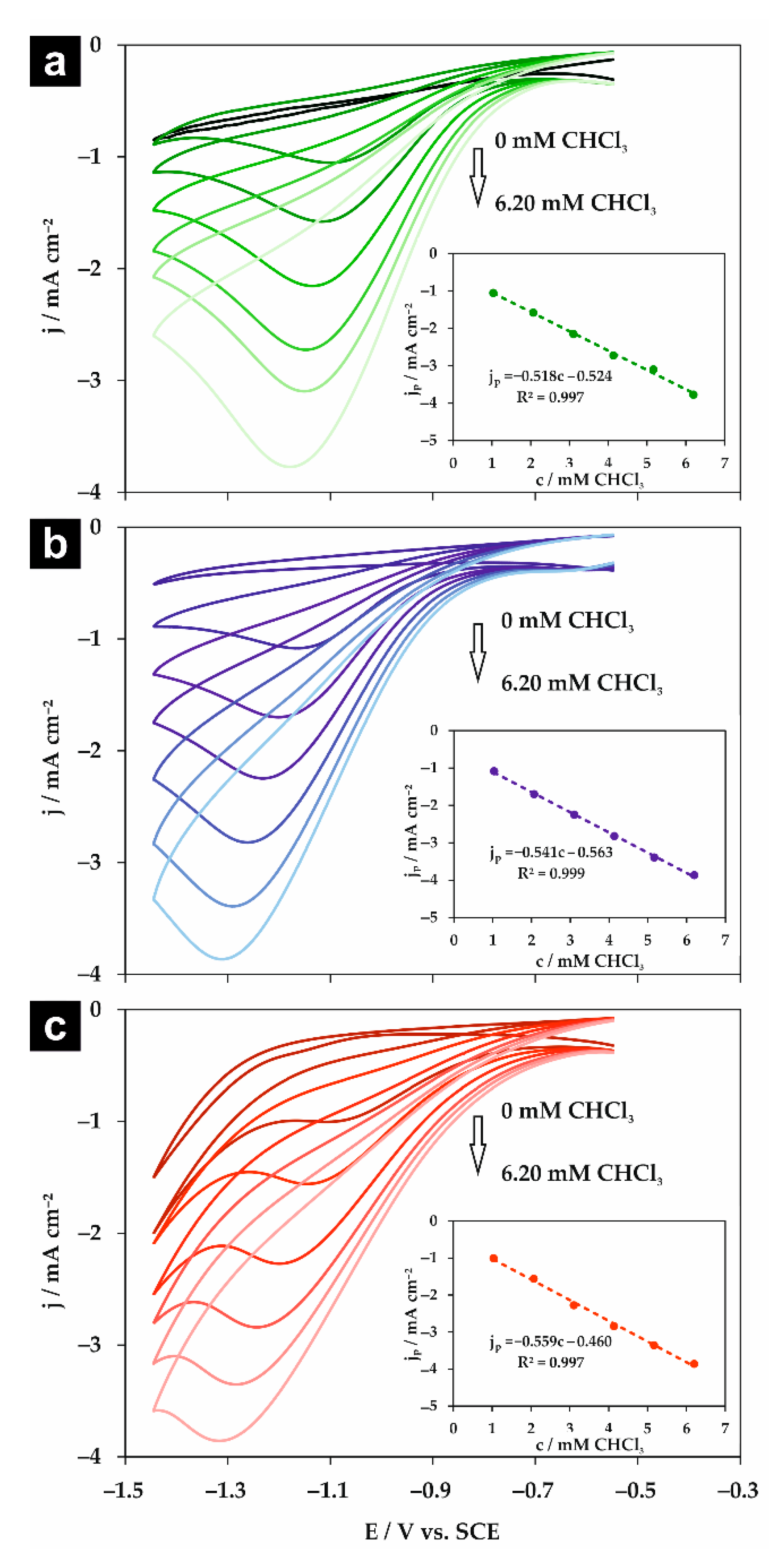
Subsequently, the influence of chloroform concentration on the peak potential and current density for all studied electrolytes was explored. The CV measurements were performed in a 0.05 M solution of supporting electrolyte at the scan rate of 100 mV s−1. The registered voltammograms are shown in Figure 6. For all studied supporting electrolytes, a linear correlation between the peak current density and CHCl3 concentration was observed for the concentration range of 1.03–6.20 mM (see insets in Figure 6). Similarly to previous reports [10,17], the potential of chloroform reduction shifts toward more negative values with its concentration. Therefore, it is important to consider the concentration of organic halides, when the catalytic activity of different electrodes is compared. The calculated sensitivity, limit of detection (LOD, calculated as 3σb/a), and limit of quantification (LOQ, calculated as 10σb/a) are gathered in Table 3. Clearly, the lowest LOD and LOQ values, as well as the highest sensitivity, were obtained for NaClO4, which implies an inert character of the supporting electrolyte and justifies the claim that this supporting electrolyte is the best choice among those studied here.
3. Materials and Methods
Pure disodium phosphate (Na2HPO4·2H2O), sodium phosphate (NaH2PO4·H2O), and sodium perchlorate (NaClO4·H2O) were purchased from Sigma-Aldrich (St. Louis, MO, USA), and chloroform (CHCl3) was acquired from Chempur® (Piekary Śląskie, Poland). The polishing Al2O3 fine abrasive powder was purchased from Alfa Aesar® (Thermo Fisher Scientific, Lancashire, United Kingdom). Highly deionized water (18.2 MΩcm) was obtained using the MilliQ® Millipore System (Merck KGaA, Darmstadt, Germany).
All electrochemical measurements were performed in a custom-made, conventional three-electrode setup with the Gamry Instrument Reference 3000 potentiostat/galvanostat (Warminster, PA, USA). For cyclic voltammetry measurements, Teflon-coated polycrystalline silver (Ag, 7 mm diameter, 99.999% Specpure, Johnson Matthey Chemicals Ltd., England) served as a working electrode, the Ag|AgCl|Cl− (3 M NaCl) electrode in a Luggin capillary (G0100 Vycor, BASi, West Lafayette, IN, USA) filled with a 0.1 M solution of the selected supporting electrolyte was used as a reference electrode, and a Pt wire coiled on a Teflon® frame was used as a counter electrode [17]. The CV measurements were recorded at the potential range from −0.547 to −1.447 V at selected scan rates (25–200 mV s−1) and chloroform concentrations (0–6.02 mM). All used electrolytes, i.e., NaClO4, NaH2PO4, Na2HPO4, were prepared using deionized water. Before each series of experiments, the surface of the silver electrode was polished with the Al2O3 powder, and electrochemically cleaned between each measurement in the 0.05 M supporting electrolyte by CV cycling in abovementioned potential range. Due to the fact that in the literature, the reduction potentials for organic halides are consistently provided versus the SCE, all potentials measured in this study were also recalculated to this reference.
The surface morphology of Ag electrode and its composition was studied using a field emission scanning electron microscope (FE-SEM, Hitachi S-4700, Hitachi High-Tech Corporation, Tokyo, Japan) equipped with an energy-dispersive X-ray spectrometer (EDS, Noran System 7, Thermo Electron Scientific Instruments LLC, Madison, WI, USA). The crystallinity of the Ag electrode was examined using a Rigaku Mini Flex II diffractometer with Cu K α radiation (1.54060 Å) at 30–90° 2θ with a step size of 0.005° and at a rate of 5° min.−1.
4. Conclusions
In this work, for the first time, the influence of the supporting electrolyte on the potential of chloroform reduction in aqueous environment at the surface of the silver electrode was reported. Similarly to previously reported nonaqueous supporting electrolytes, we observed that both the peak current density and peak potential of the chloroform reduction depend on the type of the used supporting electrolyte. On the other hand, it was found that the electroreduction of chloroform at the silver surface occurs via a concerted mechanism regardless of the type of studied supporting electrolyte. The highest sensitivity of the Ag electrode toward CHCl3 reduction was observed in a neutral NaClO4 aqueous solution. NaClO4 is widely reported in the literature as an inert aqueous supporting electrolyte for the electrocatalytic reduction of organic halides at Ag. However, the influence of other supporting electrolytes and noninert additives affecting the solution pH is often ignored. This is particularly important for the electrocatalytic reduction of organic halides in real water samples, where multiple anions and cations can be present.
Author Contributions
Conceptualization, A.M.B. and A.B.; methodology, A.M.B. and A.B.; formal analysis, A.M.B.; investigation, A.M.B. and A.B.; writing—original draft preparation, A.M.B.; writing—review and editing, A.B. and G.D.S.; visualization, A.M.B. and G.D.S.; supervision, G.D.S.; project administration, A.M.B.; funding acquisition, A.M.B. and G.D.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the National Science Centre, Poland (Grant No. 2015/19/N/ST4/00313).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data sharing not applicable.
Acknowledgments
The FE-SEM imaging was performed in the Laboratory of Field Emission Scanning Electron Microscopy and Microanalysis at the Institute of Geological Sciences, Jagiellonian University, Poland. We would like to acknowledge Monika Sołtys-Mróz for performing the XRD measurement.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Sample Availability
Samples of the compounds are not available from the authors.
References
- Trihalomethanes in Drinking-Water. In Background Document for Development of WHO Guidelines for Drinking-Water Quality, WHO/SDE/WSH/03.04/64; World Health Organization: Geneva, Switzerland, 2004; pp. 1–18.
- Chloroform. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; IARC: Lyon, France, 1999; Volume 73, pp. 131–182. [Google Scholar]
- Huang, B.; Lei, C.; Wei, C.; Zeng, G. Chlorinated volatile organic compounds (Cl-VOCs) in environment—sources, potential human health impacts, and current remediation technologies. Environ. Int. 2014, 71, 118–138. [Google Scholar] [CrossRef]
- Council Directive 98/83/EC of 3 November 1998 on the Quality of Water Intended for Human Consumption. Off. J. L 1998, 330, 32–54.
- Stage 2 Disinfectants and Disinfection Byproducts Rule (Stage 2 DBPR) 71 FR 388; United States Environmental Protection: Washington, DC, USA, 2006; Volume 71, pp. 1–2.
- Ivahnenko, T.; Zogorski, J.S. Sources and Occurrence of Chloroform and Other Trihalomethanes in Drinking-Water Supply Wells in the United States. 1986–2001; Scientific Investigations Report 2006–5015; U.S. Geological Survey: Storrs, CT, USA, 2006.
- Sonoyama, N.; Seike, S.; Sueoka, T.; Sakata, T. Electrochemical decomposition of ppb level trihalomethane in tap water. J. Appl. Electrochem. 2003, 33, 1049–1055. [Google Scholar] [CrossRef]
- Fiori, G.; Rondinini, S.; Sello, G.; Vertova, A.; Cirja, M.; Conti, L. Electroreduction of volatile organic halides on activated silver cathodes. J. Appl. Electrochem. 2005, 35, 363–368. [Google Scholar] [CrossRef]
- Isse, A.A.; Sandonà, G.; Durante, C.; Gennaro, A. Voltammetric investigation of the dissociative electron transfer to polychloromethanes at catalytic and noncatalytic electrodes. Electrochim. Acta 2009, 54, 3235–3243. [Google Scholar] [CrossRef]
- Vertova, A.; Barhdadi, R.; Cachet-Vivier, C.; Locateli, C.; Minguzzi, A.; Nedelec, J.-Y.; Rondinini, S. Cavity microelectrodes for the voltammetric investigation of electrocatalysts: The electroreduction of volatile organic halides on micro- sized silver powders. J. Appl. Electrochem. 2008, 38, 965–971. [Google Scholar] [CrossRef]
- Xu, Y.; Zhu, Y.; Zhao, F.; Ma, F.C. Electrocatalytic reductive dehalogenation of polyhalogenated phenols in aqueous solution on Ag electrodes. Appl. Catal. A 2007, 324, 83–86. [Google Scholar] [CrossRef]
- Scialdone, O.; Guarisco, C.; Galia, A.; Filardo, G.; Silvestri, G.; Amatore, C.; Sella, C.; Thouin, L. Anodic abatement of organic pollutants in water in micro reactors. J. Electroanal. Chem. 2010, 638, 293–296. [Google Scholar] [CrossRef]
- Scialdone, O.; Guarisco, C.; Galia, A.; Herbois, R. Electroreduction of aliphatic chlorides at silver cathodes in water. J. Electroanal. Chem. 2010, 641, 14–22. [Google Scholar] [CrossRef]
- Rondinini, S.; Mussini, P.R.; Muttini, P.; Sello, G. Silver as a powerful electrocatalyst for organic halide reduction: The critical role of molecular structure. Electrochim. Acta 2001, 46, 3245–3258. [Google Scholar] [CrossRef]
- Sonoyama, N.; Sakata, T. Electrochemical continuous decomposition of chloroform and other volatile chlorinated hydrocarbons in water using a column type metal impregnated carbon fiber electrode. Environ. Sci. Technol. 1999, 33, 3438–3442. [Google Scholar] [CrossRef]
- Brzózka, A.; Jeleń, A.; Brudzisz, A.M.; Marzec, M.M.; Sulka, G.D. Electrocatalytic reduction of chloroform at nanostructured silver electrodes. Electrochim. Acta 2017, 225, 574–583. [Google Scholar] [CrossRef]
- Brudzisz, A.; Sulka, G.D.; Brzózka, A. A facile approach to silver nanowire array electrode preparation and its application for chloroform reduction. Electrochim. Acta 2020, 362, 137110. [Google Scholar] [CrossRef]
- Petro, A.G.C.; Thapa, B.; Karty, J.A.; Raghavachari, K.; Baker, L.A.; Peters, D.G. Direct electrochemical reduction of acetochlor at carbon and silver cathodes in dimethylformamide. J. Electrochem. Soc. 2020, 167, 155517. [Google Scholar] [CrossRef]
- Huang, B.; Li, J.; Cao, X.; Zhu, Y.; Chen, W.; Lei, C. Electrochemical reduction of p-chloronitrobenzene (p-CNB) at silver cathode in dimethylformamide. Electrochim. Acta 2019, 296, 980–988. [Google Scholar] [CrossRef]
- Rondinini, S.B.; Mussini, P.R.; Crippa, F.; Sello, G. Electrocatalytic potentialities of silver as a cathode for organic halide reductions. Electrochem. Comm. 2000, 2, 491–496. [Google Scholar] [CrossRef]
- Rondinini, S.; Vertova, A. Electrocatalysis on silver and silver alloys for dichloromethane and trichloromethane dehalogenation. Electrochim. Acta 2004, 49, 4035–4046. [Google Scholar] [CrossRef]
- Peverly, A.A.; Peters, D.G. Electrochemical determination of trihalomethanes in water by means of stripping analysis. Anal. Chem. 2012, 84, 6110–6115. [Google Scholar] [CrossRef]
- Lugaresi, O.; Preales-Rondon, J.V.; Minguzzi, A.; Solla-Gullon, J.; Rondinini, S.; Feliu, J.M.; Sanchez-Sanchez, C.M. Rapid screening of silver nanoparticles for the catalytic degradation of chlorinated pollutants in water. Appl. Catal. B. 2015, 163, 554–563. [Google Scholar] [CrossRef] [Green Version]
- Minguzzi, A.; Lugaresi, O.; Aricci, G.; Rondinini, S.; Vertova, A. Silver nanoparticles for hydrodehalogenation reduction: Evidence of a synergistic effect between catalyst and support. Electrochem. Commun. 2012, 22, 25–28. [Google Scholar] [CrossRef]
- Ardizzone, S.; Cappelletti, G.; Mussini, P.R.; Rondinini, S.; Doubova, L.M. Adsorption competition effects in the electrocatalytic reduction of organic halides on silver. J. Electroanal. Chem. 2002, 532, 285–293. [Google Scholar] [CrossRef]
- Durante, C.; Perazzolo, V.; Perini, L.; Favaro, M.; Granozzi, G.; Gennaro, A. Electrochemical activation of carbon–halogen bonds: Electrocatalysis at silver/copper nanoparticles. Appl. Catal. B 2014, 158–159, 286–295. [Google Scholar] [CrossRef]
- Durante, C.; Isse, A.A.; Sandona, G.; Gennaro, A. Electrochemical hydrodehalogenation of polychloromethanes at silver and carbon electrodes. Appl. Catal. B 2009, 88, 479–489. [Google Scholar] [CrossRef]
- Rondinini, S.; Aricci, G.; Krpetic, Z.; Locatelli, C.; Minguzzi, A.; Porta, F.; Vertova, A. Electroreductions on silver-based electrocatalysts: The use of Ag nanoparticles for CHCl3 to CH4 conversion. Fuel Cells 2009, 9, 253–263. [Google Scholar] [CrossRef]
- Wang, A.; Huang, Y.-F.; Sur, U.K.; Wu, D.-Y.; Ren, B.; Rondinini, S.; Amatore, C.; Tian, Z.-Q. In situ identification of intermediates of benzyl chloride reduction at a silver electrode by SERS coupled with DFT calculations. J. Am. Chem. Soc. 2010, 132, 9534–9536. [Google Scholar] [CrossRef] [PubMed]
- An, C.; Kuang, Y.; Fu, C.; Zeng, F.; Wang, W.; Zhu, H. Study on Ag–Pd bimetallic nanoparticles for electrocatalytic reduction of benzyl chloride. Electrochem. Commun. 2011, 13, 1413–1416. [Google Scholar] [CrossRef]
- Costentin, C.; Robert, M.; Savéant, J.M. Stepwise and concerted electron- transfer/bond breaking reactions. Solvent control of the existence of unstable πion radicals and of the activation barriers of their heterolytic cleavage. J. Am. Chem. Soc. 2004, 126, 16834–16840. [Google Scholar] [CrossRef] [PubMed]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods: Fundamentals and Applications, 2nd ed.; Wiley: Hoboken, NJ, USA, 2001. [Google Scholar]
- Corrigan, D.A.; Evans, D.H. Cyclic voltammetric study of tertnitrobutane reduction in acetonitrile at mercury and platinum electrodes: Observation of a potential dependent electrochemical transfer coefficient and the influence of the electrolyte cation on the rate constant. J. Electroanal. Chem. Interf. Electrochem. 1980, 106, 287–304. [Google Scholar] [CrossRef]
- Garreau, D.; Saveant, J.M.; Tessier, D. Potential dependence of the electrochemical transfer coefficient. An impedance study of the reduction of aromatic compounds. J. Phys. Chem. 1979, 83, 3003–3007. [Google Scholar] [CrossRef]
- Amonette, J.E.; Jeffers, P.M.; Qafoku, O.; Russell, C.K.; Humphrys, D.R.; Wietsma, T.W.; Truex, M.J. Abiotic Degradation Rates for Carbon Tetrachloride and Chloroform: Final Report; U.S. Department of Energy: Germantown, MD, USA, 2012. [CrossRef] [Green Version]
Figure 1.
FE-SEM micrographs of the silver electrode (a) and a higher magnification of the selected area (b). The EDS analysis (c) and XRD diffraction pattern (d) of the Ag electrode.
Figure 1.
FE-SEM micrographs of the silver electrode (a) and a higher magnification of the selected area (b). The EDS analysis (c) and XRD diffraction pattern (d) of the Ag electrode.

Figure 2.
Cyclic voltammograms obtained at the silver electrode in the absence and presence of 8.26 mM chloroform in 0.05 M (a) Na2HPO4, (b) NaClO4, and (c) NaH2PO4 solutions for selected scan rates.
Figure 2.
Cyclic voltammograms obtained at the silver electrode in the absence and presence of 8.26 mM chloroform in 0.05 M (a) Na2HPO4, (b) NaClO4, and (c) NaH2PO4 solutions for selected scan rates.

Figure 3.
Schematic representation of the mechanism of chloroform reduction at the surface of Ag electrode in aqueous solutions.
Figure 3.
Schematic representation of the mechanism of chloroform reduction at the surface of Ag electrode in aqueous solutions.

Figure 4.
The dependencies of the cathodic current density of the CHCl3 reduction peak (jp) vs. the square root of the scan rate (ν0.5) measured in 0.05 M (a) Na2HPO4, (b) NaClO4, and (c) NaH2PO4. Insets: The corresponding dependencies of the peak potential (Ep) vs. log ν.
Figure 4.
The dependencies of the cathodic current density of the CHCl3 reduction peak (jp) vs. the square root of the scan rate (ν0.5) measured in 0.05 M (a) Na2HPO4, (b) NaClO4, and (c) NaH2PO4. Insets: The corresponding dependencies of the peak potential (Ep) vs. log ν.

Figure 5.
Kinetic indicator κ for identification of the mechanism involved in the R-X bond cleavage together with experimental κ values obtained for the reduction of chloroform in different aqueous supporting electrolytes.
Figure 5.
Kinetic indicator κ for identification of the mechanism involved in the R-X bond cleavage together with experimental κ values obtained for the reduction of chloroform in different aqueous supporting electrolytes.

Figure 6.
Cyclic voltammograms obtained at 100 mV s−1 for the silver electrode in a 0.05 M solution of (a) Na2HPO4, (b) NaClO4, and (c) NaH2PO4 containing trichloromethane in the concentration range of 0–6.20 mM. Insets: The corresponding dependencies of the peak cathodic current density of CHCl3 reduction (jp) vs. chloroform concentration.
Figure 6.
Cyclic voltammograms obtained at 100 mV s−1 for the silver electrode in a 0.05 M solution of (a) Na2HPO4, (b) NaClO4, and (c) NaH2PO4 containing trichloromethane in the concentration range of 0–6.20 mM. Insets: The corresponding dependencies of the peak cathodic current density of CHCl3 reduction (jp) vs. chloroform concentration.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Overview of experimental data for the chloroform reduction in aqueous solutions.
| Electrode | Ep, V vs. SCE | ν, V s−1 | C CHCl3, mM | Supporting Electrolyte | Ref. |
|---|---|---|---|---|---|
| Ag | −1.05 | 0.1 | 5.0 | 0.010 M TEAB | [22] |
| Ag | −1.0 | 0.1 | 5 | 0.035 M NaClO4 | [28] |
| Ag | −1.32 | 0.1 | 8.26 | 0.05 M KClO4 | [17] |
| Ag NWs | −1.15 | 0.1 | 8.26 | 0.05 M KClO4 | [17] |
| Ag NPs @ modified carbon powder | −0.85 | 0.2 | 10 | 0.1 M KClO4 | [15] |
| Ag NPs in C-ME | −0.6–−1 | 0.3 | 10 | 0.1 M KClO4 | [14] |
| Ag | −1.127 | 0.1 | 10.63 | 0.1 M NaOH | [16] |
| Ag NCs | −1.067 | 0.1 | 10.63 | 0.1 M NaOH | [16] |
| Ag NWs | −0.957 | 0.1 | 10.63 | 0.1 M NaOH | [16] |
TEAB—tetraethylammonium benzoate, NWs—nanowires, NPs—nanoparticles, C-ME—microcavity electrode, NCs—nanocones.
Table 2.
Data for the chloroform reduction at the silver electrode in selected aqueous supporting electrolytes.
Table 2.
Data for the chloroform reduction at the silver electrode in selected aqueous supporting electrolytes.
| Electrolyte | pH | Ep, V vs. SCE at 100 mV s−1 | jp, mA cm−2 at 100 mV s−1 | κ1 | κ2 | |
|---|---|---|---|---|---|---|
| Na2HPO4 | 9.4 | −1.215 | −4.89 | −0.171 | 0.17 | 0.17 |
| NaClO4 | 7.0 | −1.344 | −4.61 | −0.284 | 0.10 | 0.15 |
| NaH2PO4 | 4.6 | −1.389 | −4.77 | −0.287 | 0.10 | 0.10 |
Table 3.
The analytical parameters of the chloroform reduction at silver electrode in different aqueous solutions.
Table 3.
The analytical parameters of the chloroform reduction at silver electrode in different aqueous solutions.
| Electrolyte | LOD, mM | LOQ, mM | Sensitivity, mA cm−2 M−1 |
|---|---|---|---|
| Na2HPO4 | 0.348 | 1.16 | 1.04 |
| NaClO4 | 0.204 | 0.68 | 1.10 |
| NaH2PO4 | 0.354 | 1.18 | 1.02 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Brudzisz, A.M.; Brzózka, A.; Sulka, G.D. Effect of the Supporting Electrolyte on Chloroform Reduction at a Silver Electrode in Aqueous Solutions. Molecules 2021, 26, 525. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26030525
AMA Style
Brudzisz AM, Brzózka A, Sulka GD. Effect of the Supporting Electrolyte on Chloroform Reduction at a Silver Electrode in Aqueous Solutions. Molecules. 2021; 26(3):525. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26030525
Chicago/Turabian StyleBrudzisz, Anna M., Agnieszka Brzózka, and Grzegorz D. Sulka. 2021. "Effect of the Supporting Electrolyte on Chloroform Reduction at a Silver Electrode in Aqueous Solutions" Molecules 26, no. 3: 525. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26030525