
New Photochromic α-Methylchalcones Are Highly Photostable, Even under Singlet Oxygen Conditions: Breaking the α-Methyl Michael-System Reactivity by Reversible Peroxybiradical Formation


Abstract
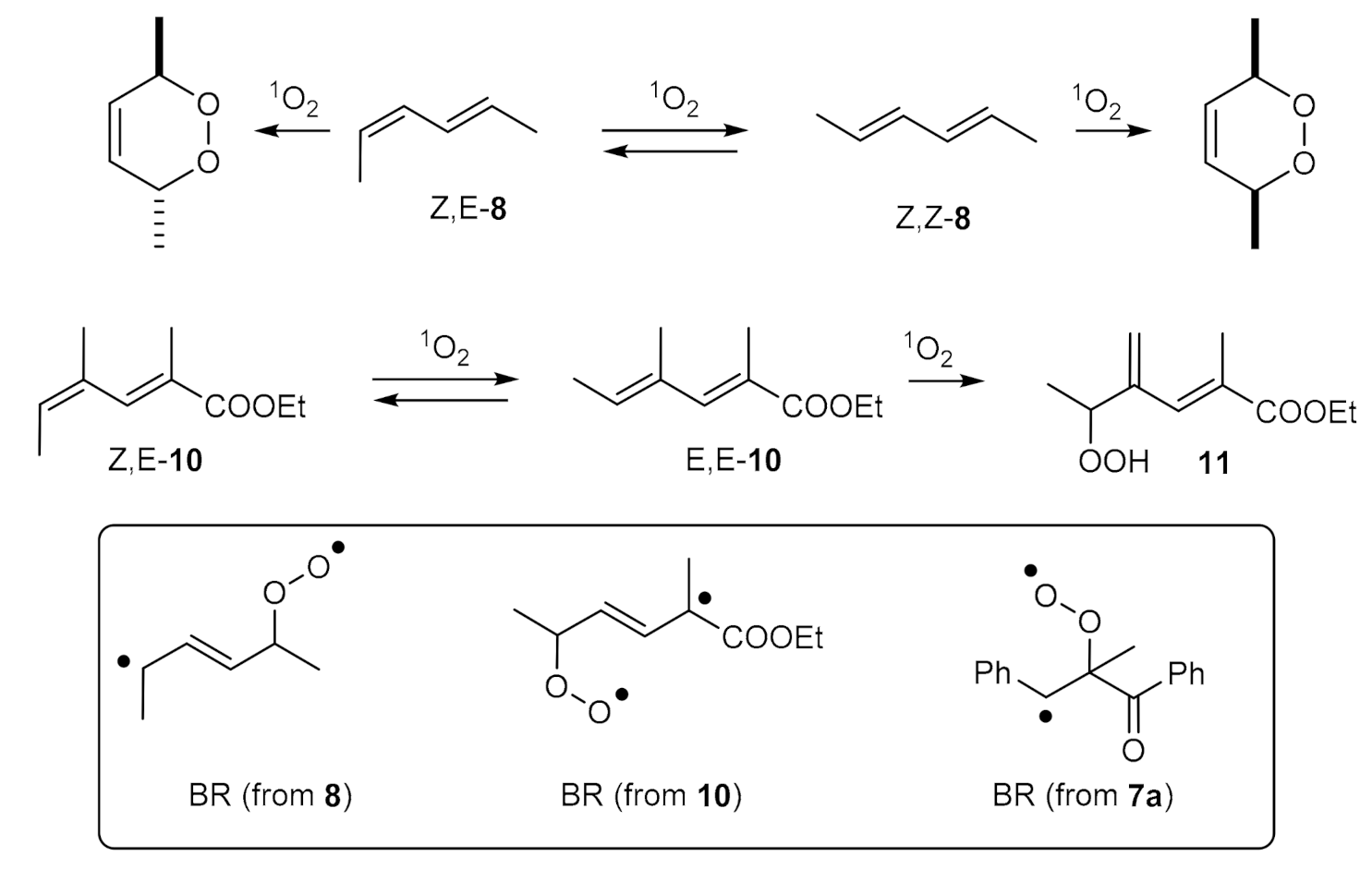
:1. Introduction
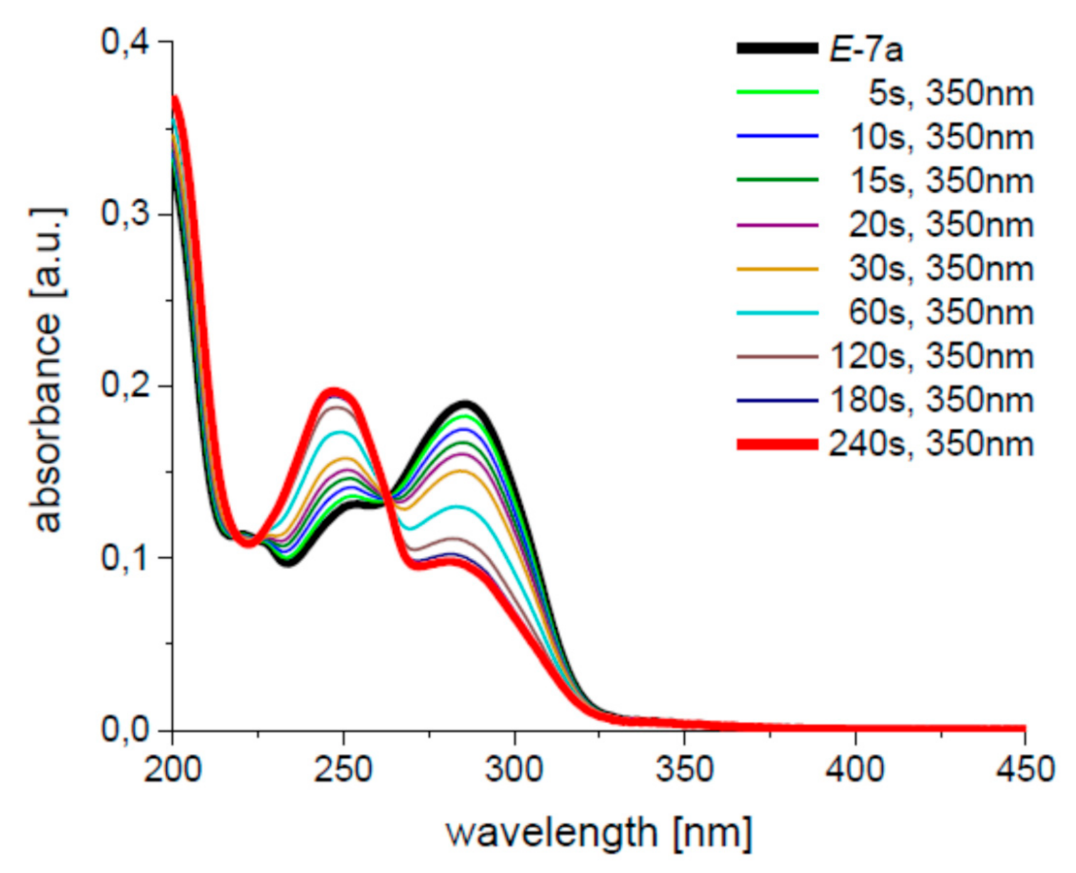
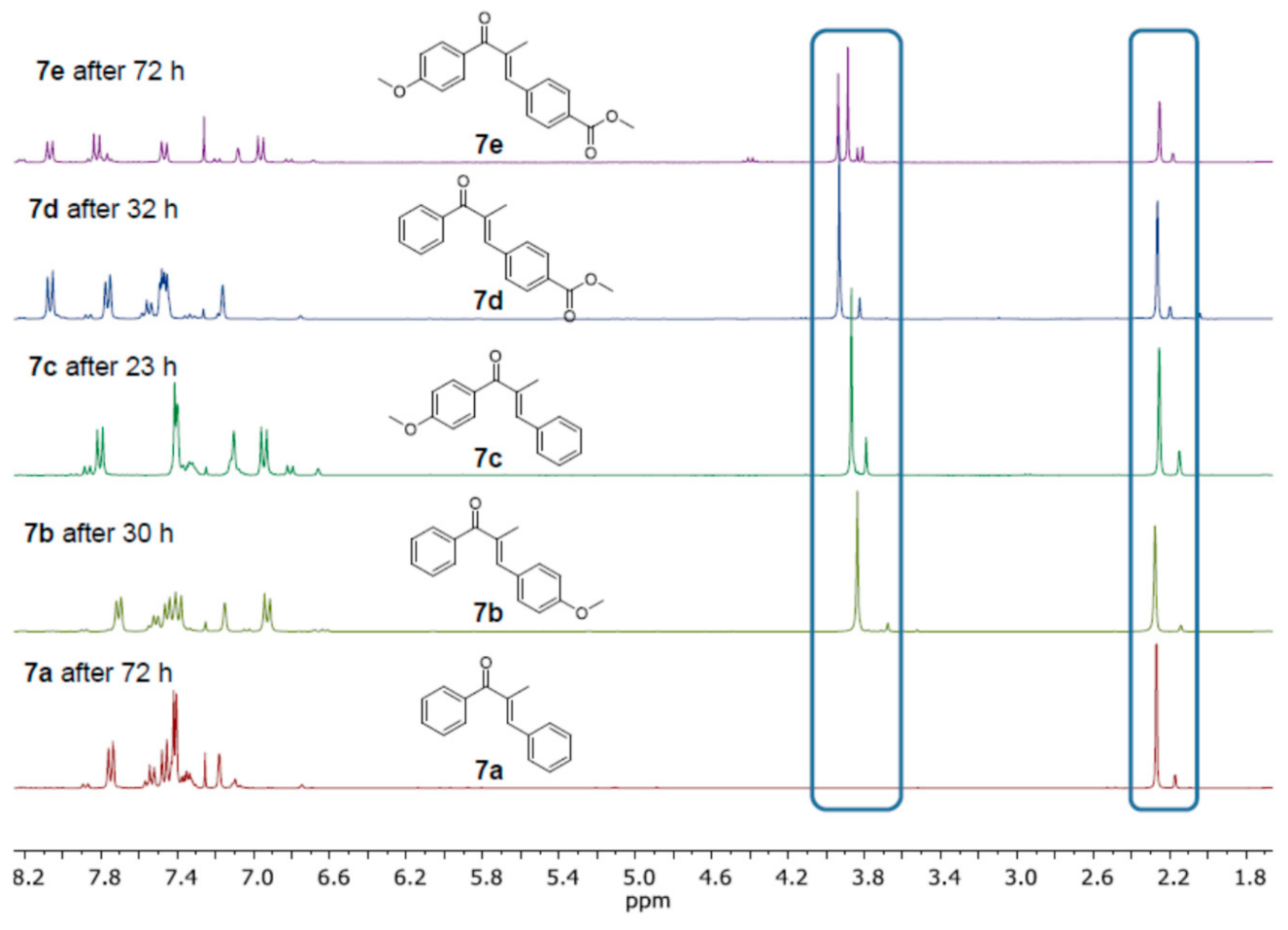
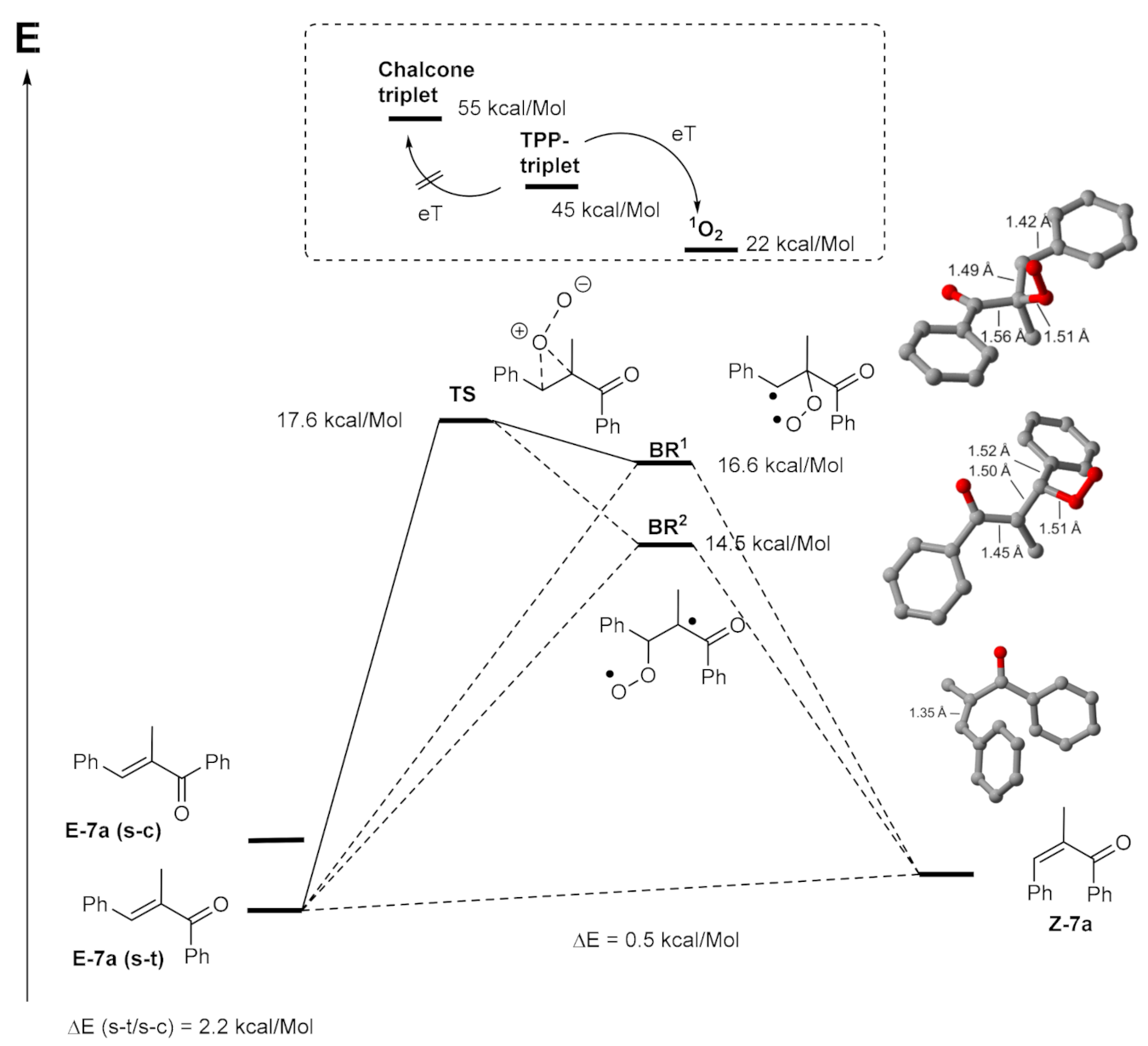
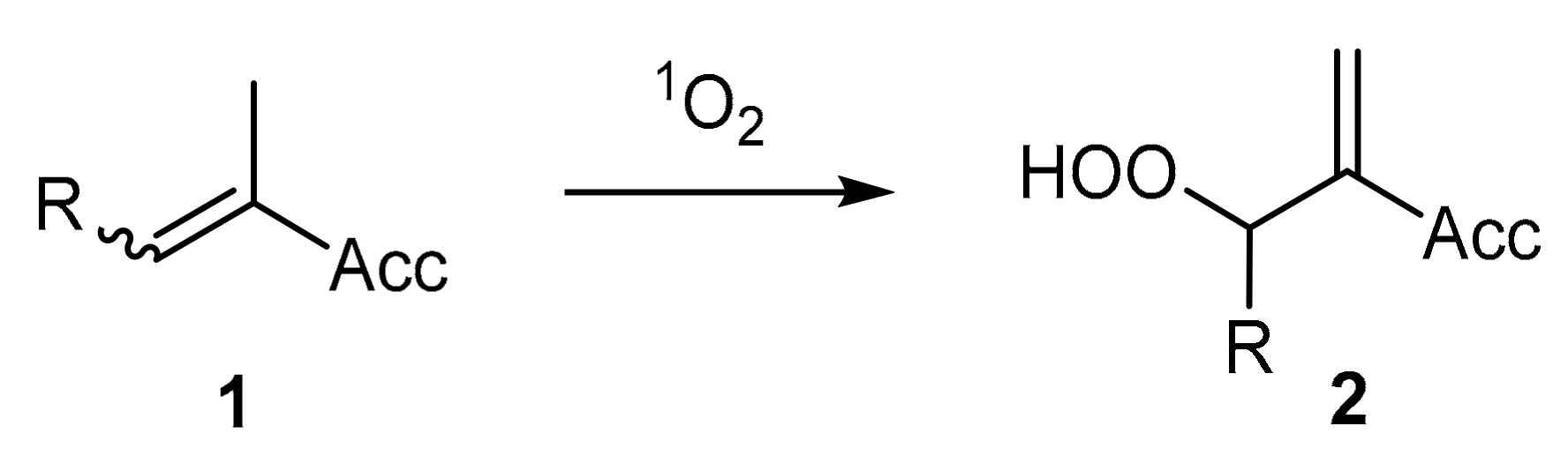
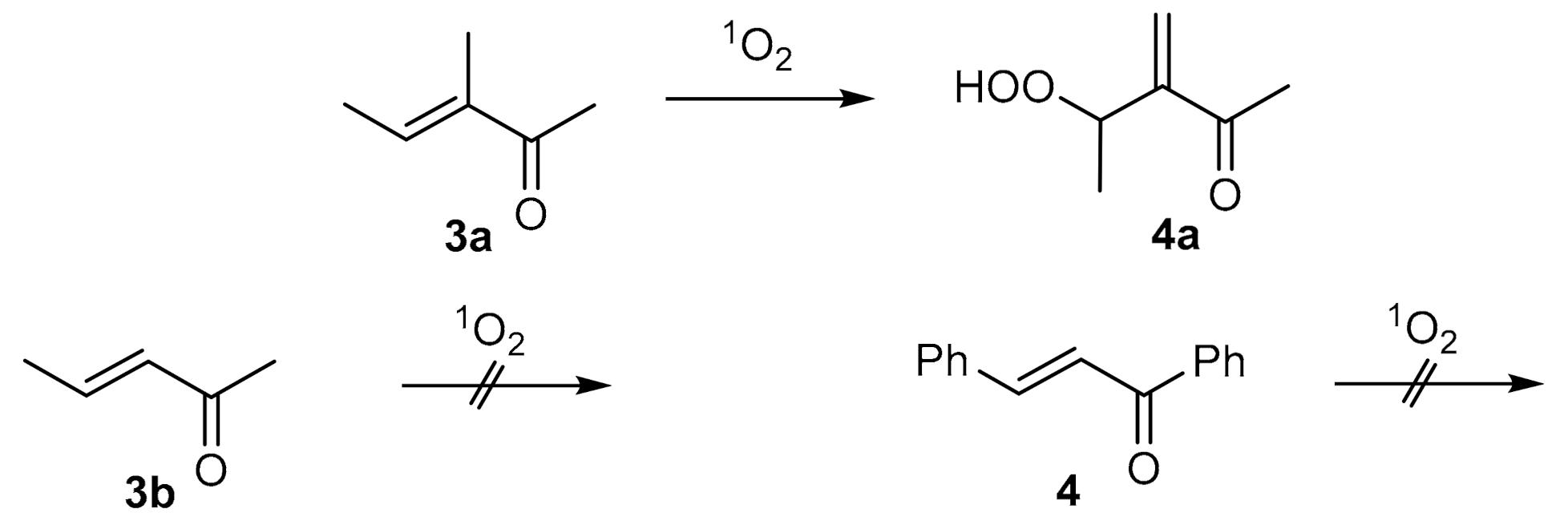
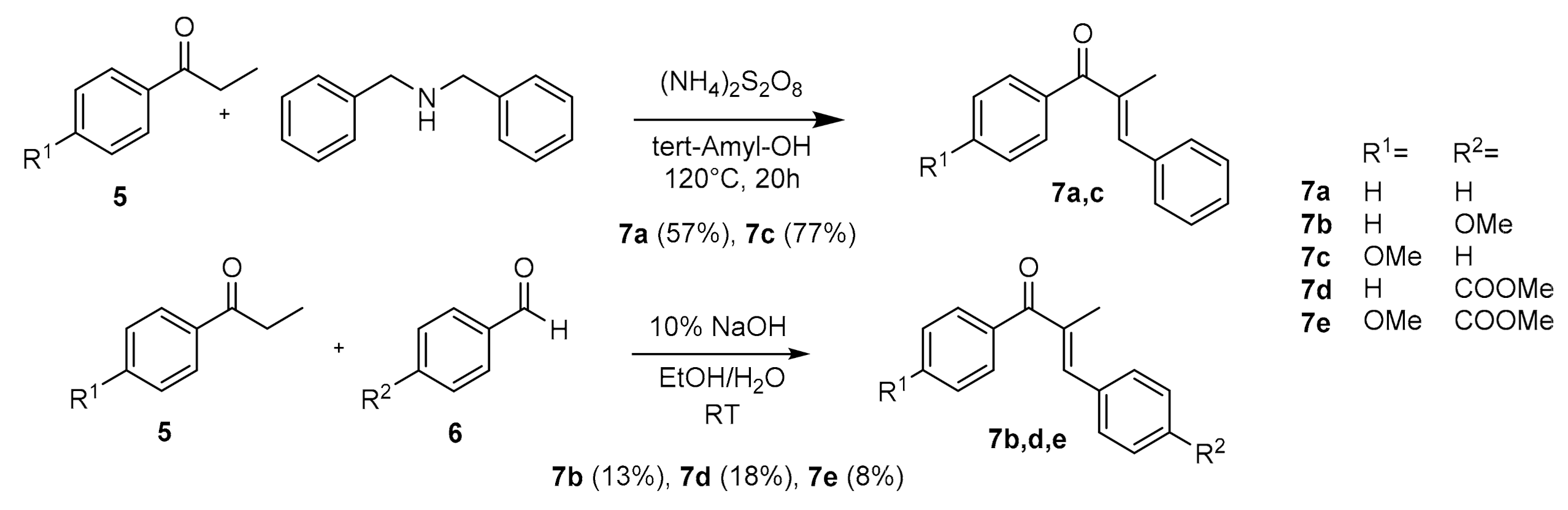
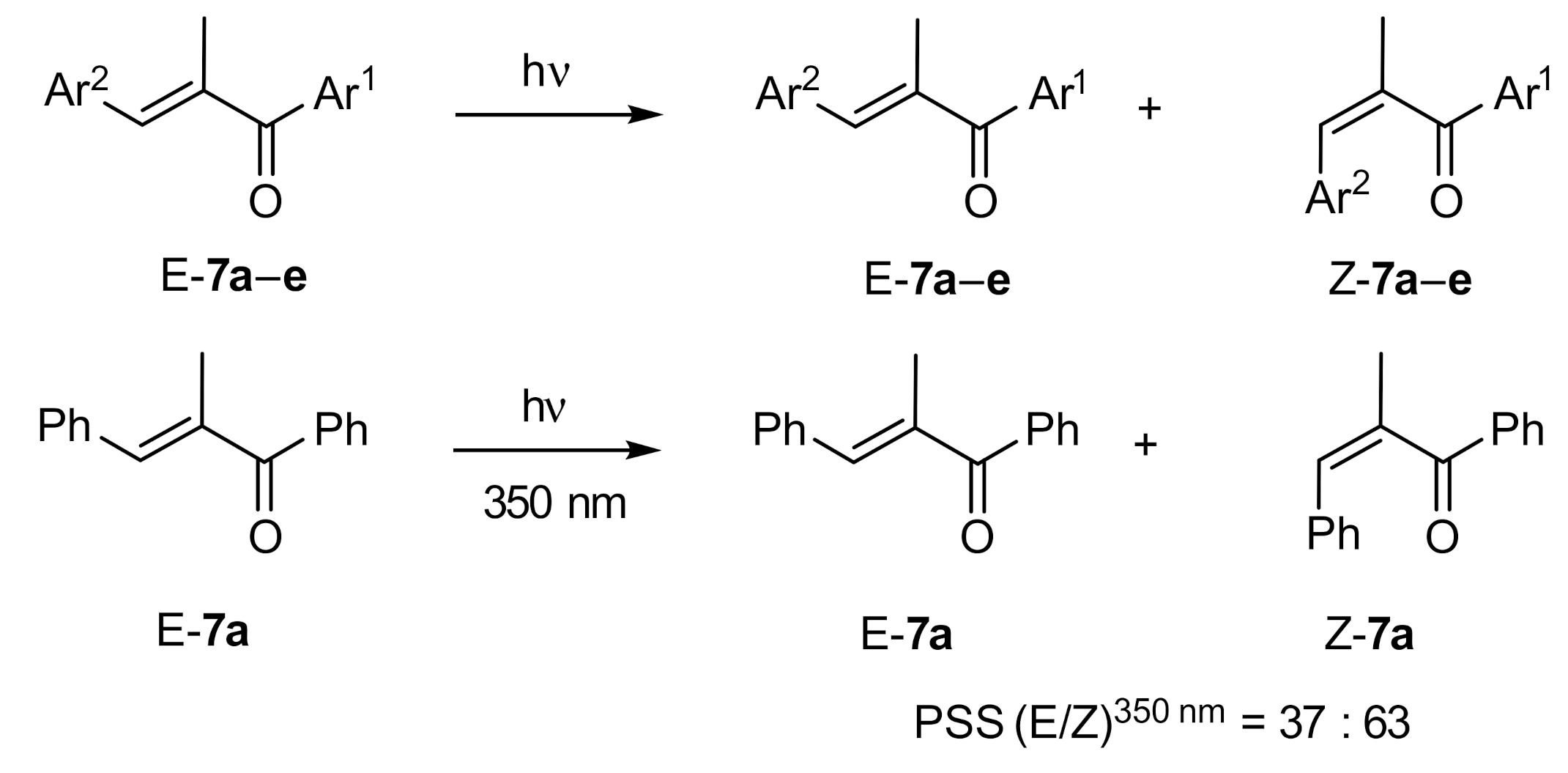
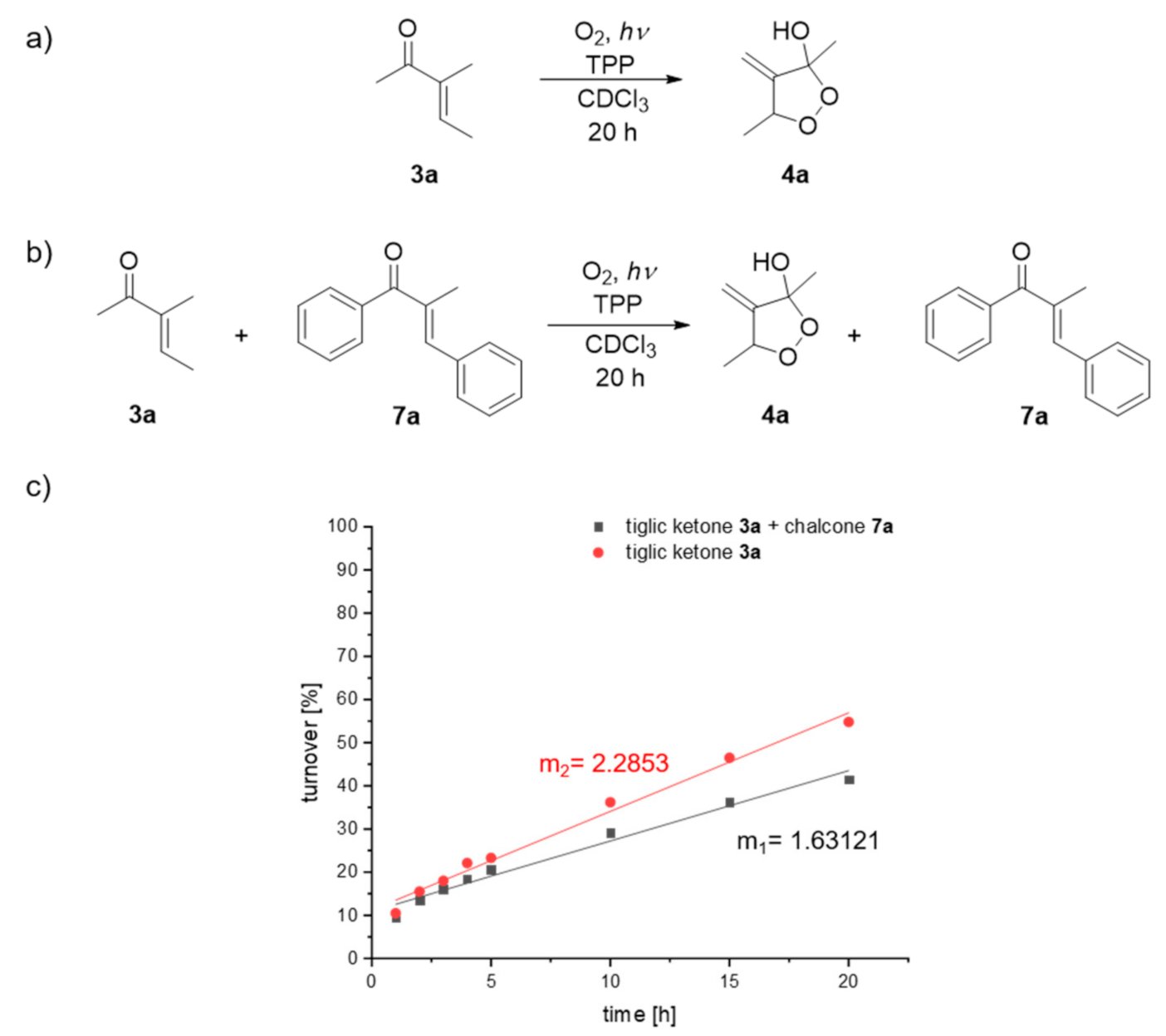
2. Results and Discussion
3. Conclusions
4. Experimental Section
5. Computational Part
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Greer, A. Christopher Foote´s discovery of the role of singlet oxygen [1O2 (1∆g)] in photosensitized oxidation reactions. Acc. Chem. Res. 2006, 39, 797–804. [Google Scholar]
- Schmidt, R. Photosensitized Generation of Singlet Oxygen. Photochem. Photobiol. 2007, 82, 1161–1177. [Google Scholar]
- Kanofyky, J.R. Photosensitization. In Singlet Oxygen: Application in Biosciences and Nanosciences; Flors, C., Nonell, S., Eds.; The Royal Society of Chemistry: Cambridge, UK, 2016; Volume 1, pp. 93–106. [Google Scholar]
- Schweitzer, C.; Schmidt, R. Physical Mechanisms of Generation and Deactivation of Singlet Oxygen. Chem. Rev. 2003, 103, 1685–1757. [Google Scholar]
- Ghogara, A.A.; Greer, A. Using Singlet Oxygen to Synthesize Natural Products and Drugs. Chem. Rev. 2016, 116, 9994–10034. [Google Scholar]
- Griesbeck, A.G.; Kleczka, M.; de Kiff, A.; Vollmer, M.; Eske, A.; Sillner, S. Singlet oxygen and natural substrates: Functional polyunsaturated models for the photooxidative degradation of carotenoids. Pure Appl. Chem. 2015, 87, 639–647. [Google Scholar]
- Prein, M.; Adam, W. The Schenck ene reaction: Diastereoselective oxyfunctionalization with singlet oxygen in synthetic applications. Angew. Chem. Int. Ed. 1996, 35, 477–494. [Google Scholar]
- DeRosa, M.C.; Crutchley, R.J. Photosensitized singlet oxygen and its applications. Coord. Chem. Rev. 2002, 233–234, 351–371. [Google Scholar]
- Iesce, M.; Cermola, F.; Temussi, F. Photooxygenation of Heterocycles. Curr. Org. Chem. 2005, 9, 109–139. [Google Scholar]
- Clennan, E.L.; Pace, A. Advances in singlet oxygen chemistry. Tetrahedron 2005, 61, 6665–6691. [Google Scholar]
- Tzirakis, M.D.; Alberti, M.N. On the mechanism of the [4+2] cycloaddition of 1O2 with 1,3-dienes: Identifying the putative biradical/dipolar intermediate by employing the gem-diphenylcyclopropyl-carbinyl radical clock as a mechanistic probe. Arkivoc 2015, 2015, 83–100. [Google Scholar]
- Bartlett, P.D.; Schaap, A.P. Stereospecific Formation of 1,2-Dioxetanes from cis- and trans-Diethoxy-ethylenes by Singlet Oxygen. J. Am. Chem. Soc. 1970, 92, 3223–3225. [Google Scholar]
- Bayer, P.; Perez-Ruiz, R.; Jacobi von Wangelin, A. Stereoselective Photooxidations by the Schenck Ene Reaction. ChemPhotoChem 2018, 2, 559–570. [Google Scholar]
- Boix-Garriga, E.; Rodríguez-Amigo, B.; Planas, O.; Nonell, S. Properties of Singlet Oxygen. In Singlet Oxygen: Application in Biosciences and Nanosciences; Flors, C., Nonell, S., Eds.; The Royal Society of Chemistry: Cambridge, UK, 2016; Volume 1, pp. 23–46. [Google Scholar]
- Kearns, D.R. Physical and chemical properties of singlet molecular oxygen. Chem. Rev. 1971, 71, 395–427. [Google Scholar]
- Kearns, D.R.; Merkel, P.M. Radiationless decay of singlet molecular oxygen in solution. An experimental and theoretical study of electronic-to-vibrational energy transfer. J. Am. Chem. Soc. 1972, 94, 7244–7253. [Google Scholar]
- Gorman, A.A.; Rodgers, M.A.J. Singlet molecular oxygen. Chem. Soc. Rev. 1981, 10, 205–231. [Google Scholar]
- Griesbeck, A.G.; Schlundt, V.; Neudörfl, J.M. Functionalized polar 1,2,4-trioxanes as bulding blocks by singlet oxygenation of 4-hydroxy tiglic acid using the deuterium isotope trick. RSC Adv. 2013, 3, 7265–7270. [Google Scholar]
- Alberti, M.N.; Orfanopoulos, M. Recent Mechanistic Insights in the Singlet Oxygen Ene Reaction. Chem. Eur. J. 2010, 16, 9414–9421. [Google Scholar]
- Adam, W.; Bottke, N.; Krebs, O. The new skew and the established cis and gem regioselectivities in the ene reaction of trisubstituted olefins: Comparison of the singlet oxygen, triazolinedione, and nitrosoarene enophiles. J. Am. Chem. Soc. 2000, 122, 6791–6792. [Google Scholar]
- Orfanopoulos, M.; Stratakis, M.; Elemes, Y. Geminal selectivity of singlet oxygen ene reactions—The nonbonding large group effect. J. Am. Chem. Soc. 1990, 112, 6417–6419. [Google Scholar]
- Griesbeck, A.G.; Goldfuss, B.; Jäger, C.; Brüllingen, E.; Lippold, T.; Kleczka, M. Strong Asymmetry in the Perepoxide Bifurcation Mechanism: The Large-Group Effect in the Singlet Oxygen Ene Reaction with Allylic Alcohols. ChemPhotoChem 2017, 1, 213–221. [Google Scholar]
- Garavelli, M.; Bernardi, F.; Olivucci, M.; Robb, M.A. DFT Study of the Reactions between Singlet-Oxygen and a Carotenoid Model. J. Am. Chem. Soc. 1998, 120, 10210–10222. [Google Scholar]
- Iesce, M.R.; Cremola, F. Photooxygenation, [2+2] and [4+2]. In CRC Handbook of Organic Photochemistry and Photobiology; Griesbeck, A.A., Oelgemöller, M., Ghetti, F., Eds.; CRC Press Taylor and Francis Group: Boca Raton, FL, USA, 2012; pp. 727–764. [Google Scholar]
- Petrou, A.L.; Petrou, P.L.; Ntanos, T.; Liapis, A. A possible role for singlet oxygen in the degradation of various antioxidants. A meta-analysis and review of literature data. Antioxidants 2018, 7, 35. [Google Scholar] [CrossRef] [Green Version]
- Islamian, J.P.; Mehrali, H. Lycopene as a carotenoid provides radioprotectant and antioxidant effects by quenching radiation-induced free radical singlet oxygen: An overview. Cell J. 2015, 16, 386–391. [Google Scholar]
- Gaonkar, S.L.; Vignesh, U.N. Synthesis and pharmacological properties of chalcones: A review. Res. Chem. Intermed. 2017, 43, 6043–6077. [Google Scholar]
- Von Kostanecki, S.; Tambor, J. Über die sechs isomeren Monooxybenzalacetophenone (Monooxychalkone). Ber. Dtsch. Chem. Ges. 1899, 32, 1921–1926. [Google Scholar]
- Mahapatra, D.K.; Asati, V.; Bharti, K.S. Chalcones and their therapeutic targets for the management of diabetes: Structural and pharmacological perspectives. Eur. J. Med. Chem. 2015, 92, 839–865. [Google Scholar]
- Zhuang, C.; Zhang, W.; Sheng, C.; Zhang, W.; Xing, C.; Miao, Z. Chalcone: A privileged structure in medicinal chemistry. Chem. Rev. 2017, 117, 7762–7810. [Google Scholar]
- Vever-Bizet, C.; Dellinger, M.; Brault, D.; Rougee, M.; Bensasson, R.V. Singlet molecular oxygen quenching by saturated and unsaturated fatty-acids and by cholesterol. Photochem. Photobiol. 1989, 50, 321–325. [Google Scholar]
- Chawla, H.M.; Chakrabarty, K. Dye-sensitized Photo-oxygenation of Chalcones. J. Chem. Soc. Perkin Trans. I 1984, 132, 1511–1513. [Google Scholar]
- Ren, B.-Z.; Ablise, M.; Yang, X.-C.; Liao, B.; Yang, Z. Synthesis and biological evaluation of α-methyl-chalcone for anti-cervical cancer activity. Med. Chem. Res. 2017, 26, 1871–1883. [Google Scholar]
- Wei, Y.; Tang, J.; Cong, X.; Zeng, X. Practical metal-free synthesis of chalcone derivatives via a tandem cross-dehydrogenative-coupling/elimination reaction. Green Chem. 2013, 15, 3165–3169. [Google Scholar]
- Lei, T.; Zhou, C.; Huang, M.-Y.; Zhao, L.-M.; Yang, B.; Ye, C.; Xiao, H.; Meng, Q.-Y.; Ramamurthy, V.; Tung, C.-H.; et al. General and efficient intermolecular [2+2] photodimerization of chalcones and cinnamic acid derivatives in solution through visible-light catalysis. Angew. Chem. Int. Ed. 2017, 56, 15407–15410. [Google Scholar]
- Seely, R. The energetics of electron-transfer reactions of chlorophyll and other compounds. Photochem. Photobiol. 1978, 27, 639–654. [Google Scholar]
- Kleczka, M. Application of the Large-Group-Non-Bonding Effect in the Synthesis of Hydroperoxides via Ene Reaction. Ph.D. Thesis, University of Cologne, Cologne, Germany, 2016. [Google Scholar]
- Gollnick, K.; Griesbeck, A.G. [4+2]-Cycloadditionen of Singlet Oxygen to Conjugated Acyclic Hexadienes: Evidence of Singlet Oxygen Induced cis to trans Isomerization. Tetrahedron Lett. 1983, 24, 3303–3306. [Google Scholar]
- Eske, A.; Goldfuss, B.; Griesbeck, A.G.; de Kiff, A.; Kleczka, M.; Leven, M.; Neudörfl, J.M.; Vollmer, M. Ene-Diene Transmissive Cycloaddition Reactions with Singlet Oxygen: The Vinylogous Gem Effect and its Use for Polyoxyfunctionalization of Dienes. J. Org. Chem. 2014, 79, 1818–1829. [Google Scholar]
- Ducki, S.; Forrest, R.; Hadfield, J.A.; Kendall, A.; Lawrence, N.J.; McGown, A.T.; Rennison, D. Potent Antimitotic and Cell Growth Inibitory Properties of Substituted Chalcones. Bioorg. Med. Chem. Lett. 1998, 8, 1051–1056. [Google Scholar]
- Singleton, D.A.; Hang, C.; Szymanski, M.J.; Meyer, M.P.; Leach, A.G.; Kuwata, K.T.; Chen, J.S.; Greer, A.; Foote, C.S.; Houk, K.N. Mechanism of Ene Reactions of Singlet Oxygen. A Two-Step No-Intermediate Mechanism. J. Am. Chem. Soc. 2003, 125, 1319–1328. [Google Scholar]
- Harvey, J.N. Spin-forbidden Reactions: Computational Insight into Mechanism and Kinetics. WIREs Comput. Mol. Sci. 2014, 4, 1–14. [Google Scholar]
- Harvey, J.N.; Aschi, M.; Schwarz, H.; Koch, W. The Singlet and Triplet States of Phenyl Cation. A Hybrid Approach for Locating Minimum Energy Crossing Points Between Non-interacting Potential Energy Surfaces. Theor. Chem. Acc. 1998, 99, 95–99. [Google Scholar]
- Clennan, E.L.; Noe, L.J.; Szneier, E.; Wen, T. Hydrazines: New Charge-Transfer Physical Quenchers of Singlet Oxygen. J. Am. Chem. Soc. 1990, 112, 5080–5085. [Google Scholar]
- Öngel, B.; Brüllingen, E.; Griesbeck, A.G. Photochromic α-Methyl Chalcones: Photoswitching and Halochromic Behaviour. ChemPhotoChem. manuscript in preparation.
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision B.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Tao, J.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the density functional ladder: Nonempirical meta-generalized gradient approximation designed for molecules and solids. Phys. Rev. Lett. 2003, 91, 146401. [Google Scholar] [CrossRef] [Green Version]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of damping function in dispersion corrected density functional theory. J. Comp. Chem. 2011, 32, 1456–1465. [Google Scholar]
- Legault, C.Y. CYLview, 1.0b; Université de Sherbrooke: Sherbrooke, QC, Canada, 2009; Available online: http://www.cylview.org (accessed on 21 November 2020).










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound: | E/Z (350 nm) | E/Z (1O2-induced) |
|---|---|---|
| 7a | 37:63 | 91:9 |
| 7b | 44:56 | 89:11 |
| 7c | 27:73 | 90:10 |
| 7d | 29:71 | 90:10 |
| 7e | 26:74 | 89:11 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Griesbeck, A.G.; Öngel, B.; Brüllingen, E.; Renner, M. New Photochromic α-Methylchalcones Are Highly Photostable, Even under Singlet Oxygen Conditions: Breaking the α-Methyl Michael-System Reactivity by Reversible Peroxybiradical Formation. Molecules 2021, 26, 642. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26030642
Griesbeck AG, Öngel B, Brüllingen E, Renner M. New Photochromic α-Methylchalcones Are Highly Photostable, Even under Singlet Oxygen Conditions: Breaking the α-Methyl Michael-System Reactivity by Reversible Peroxybiradical Formation. Molecules. 2021; 26(3):642. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26030642
Chicago/Turabian StyleGriesbeck, Axel G., Banu Öngel, Eric Brüllingen, and Melissa Renner. 2021. "New Photochromic α-Methylchalcones Are Highly Photostable, Even under Singlet Oxygen Conditions: Breaking the α-Methyl Michael-System Reactivity by Reversible Peroxybiradical Formation" Molecules 26, no. 3: 642. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26030642