
The Fluoride Anion-Catalyzed Sulfurization of Thioketones with Elemental Sulfur Leading to Sulfur-Rich Heterocycles: First Sulfurization of Thiochalcones †

 , and
, and
Abstract
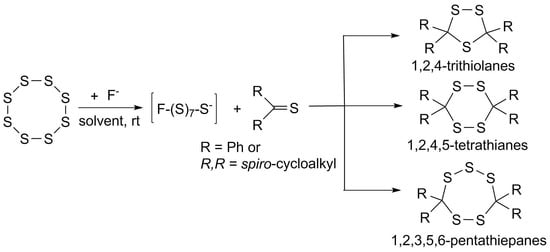
:
1. Introduction
2. Results and Discussion
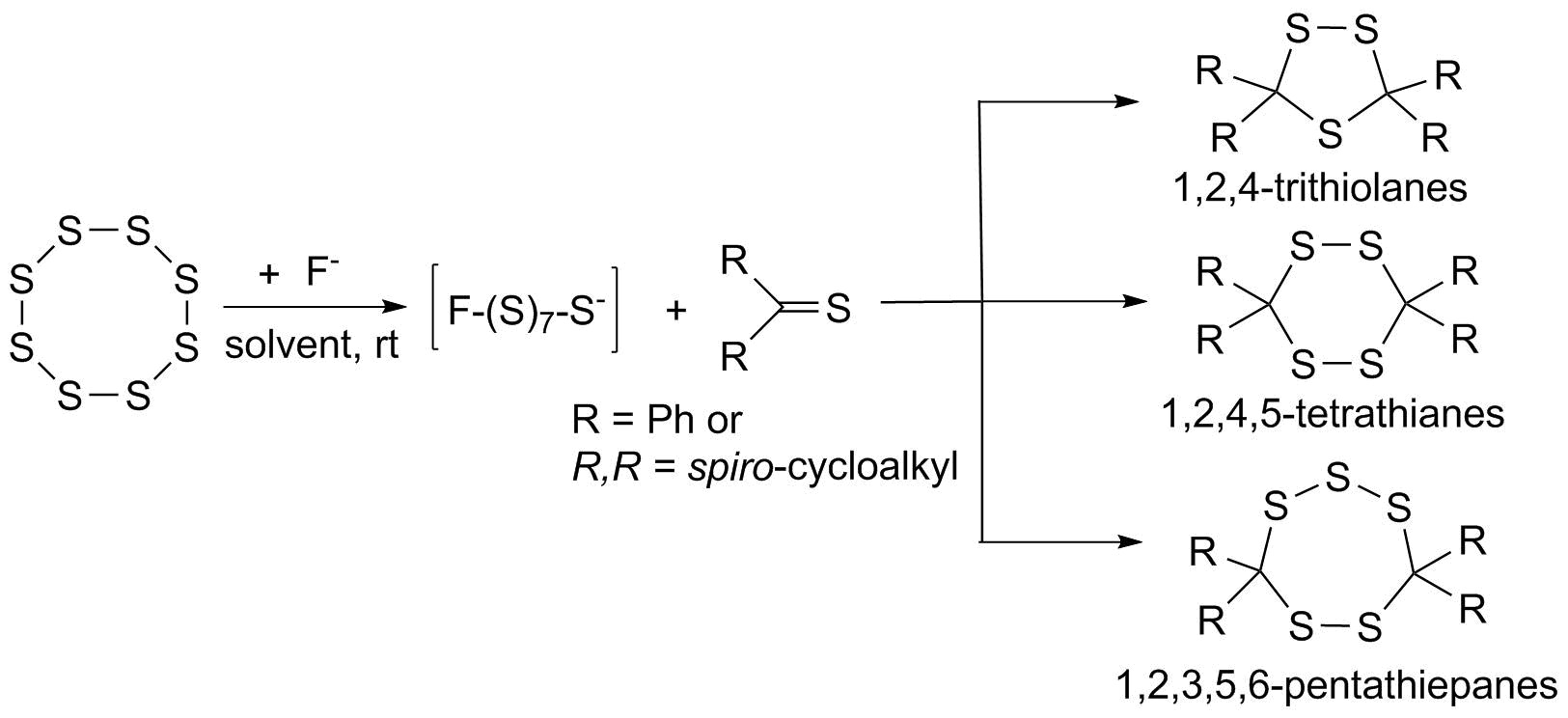
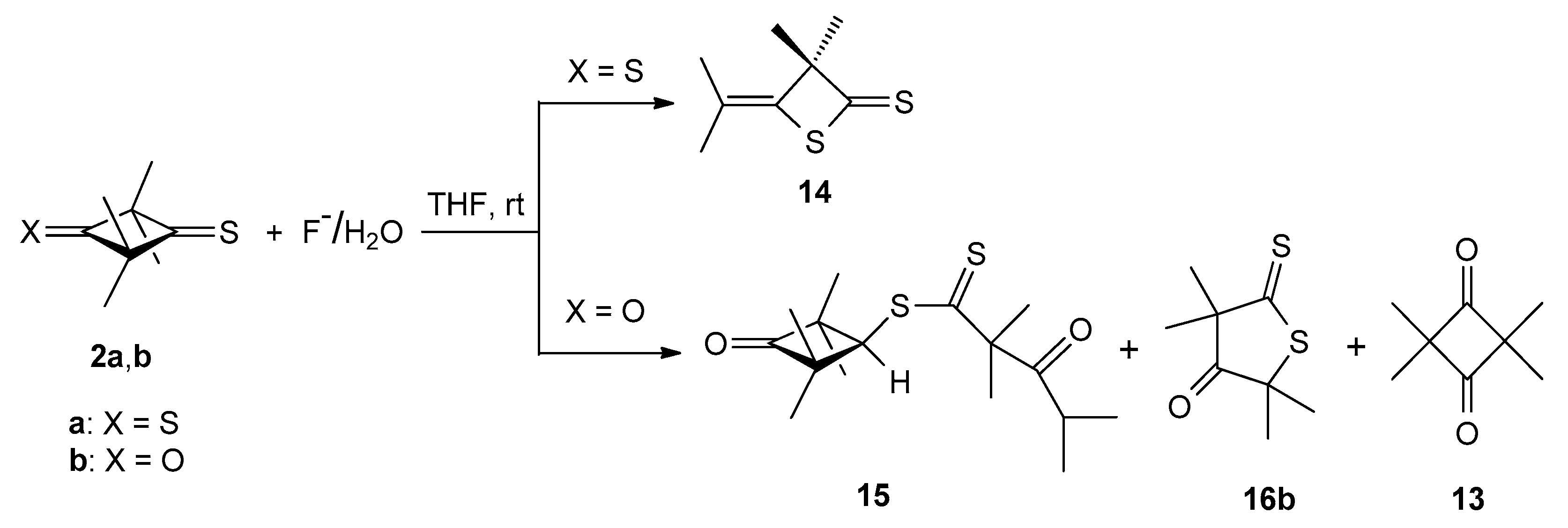
2.1. Conversions of Thioketones 2a and 2b upon Tretament with Catalytic Amounts of TBAF in the Absence of Elmental Sulfur
2.2. Activation of Elemental Sulfur with Fluoride Anion
2.3. Sulfurization of Thioketones with Elemental Sulfur
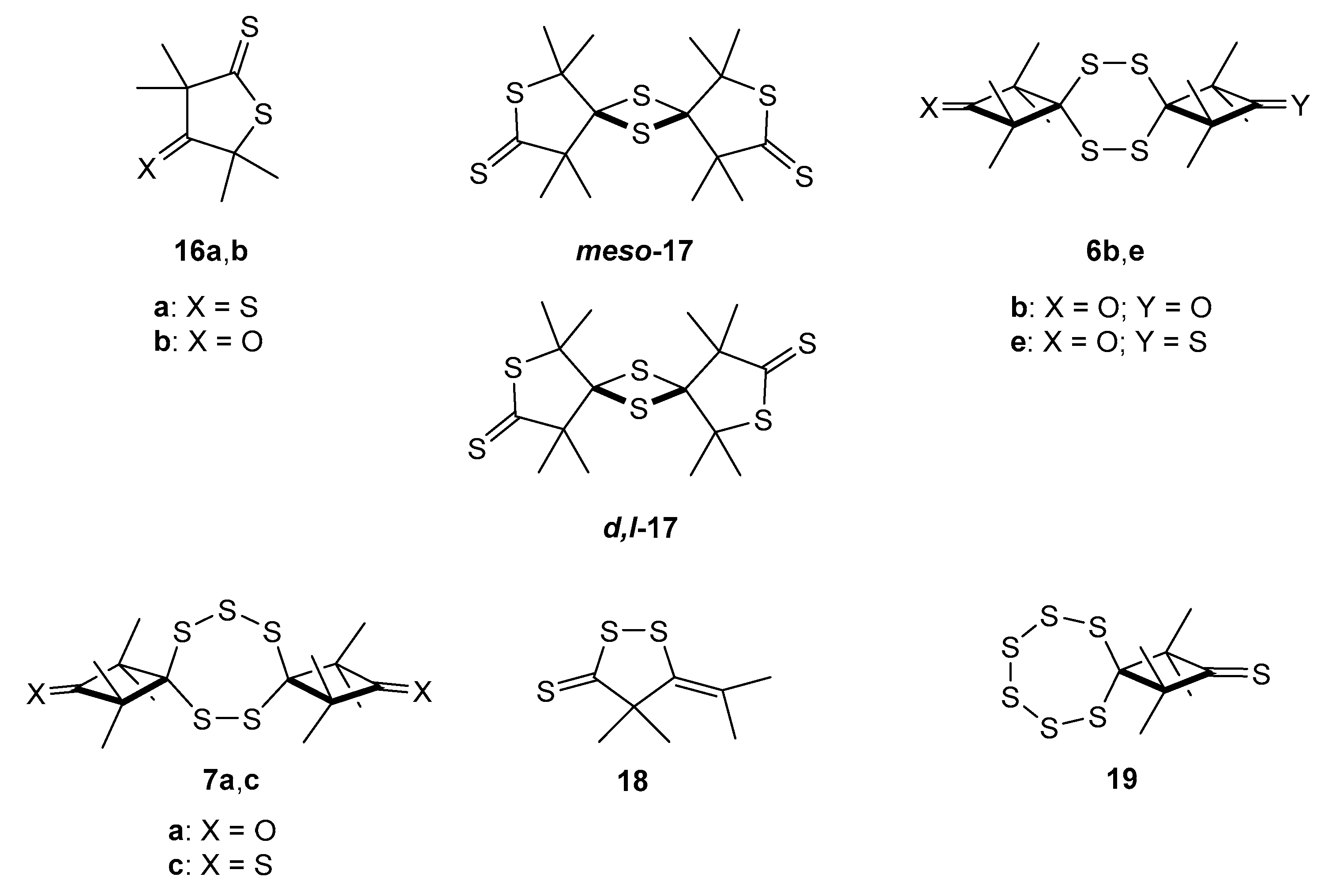
2.3.1. Sulfurization of ‘Dithione’ 2a and ‘Monothione’ 2b
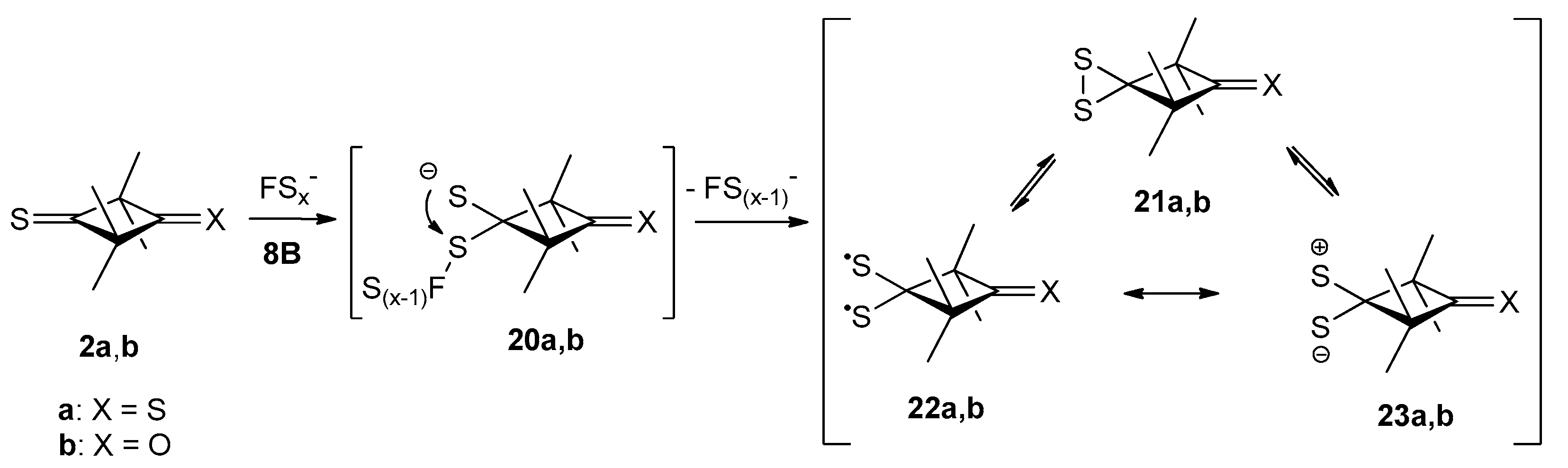
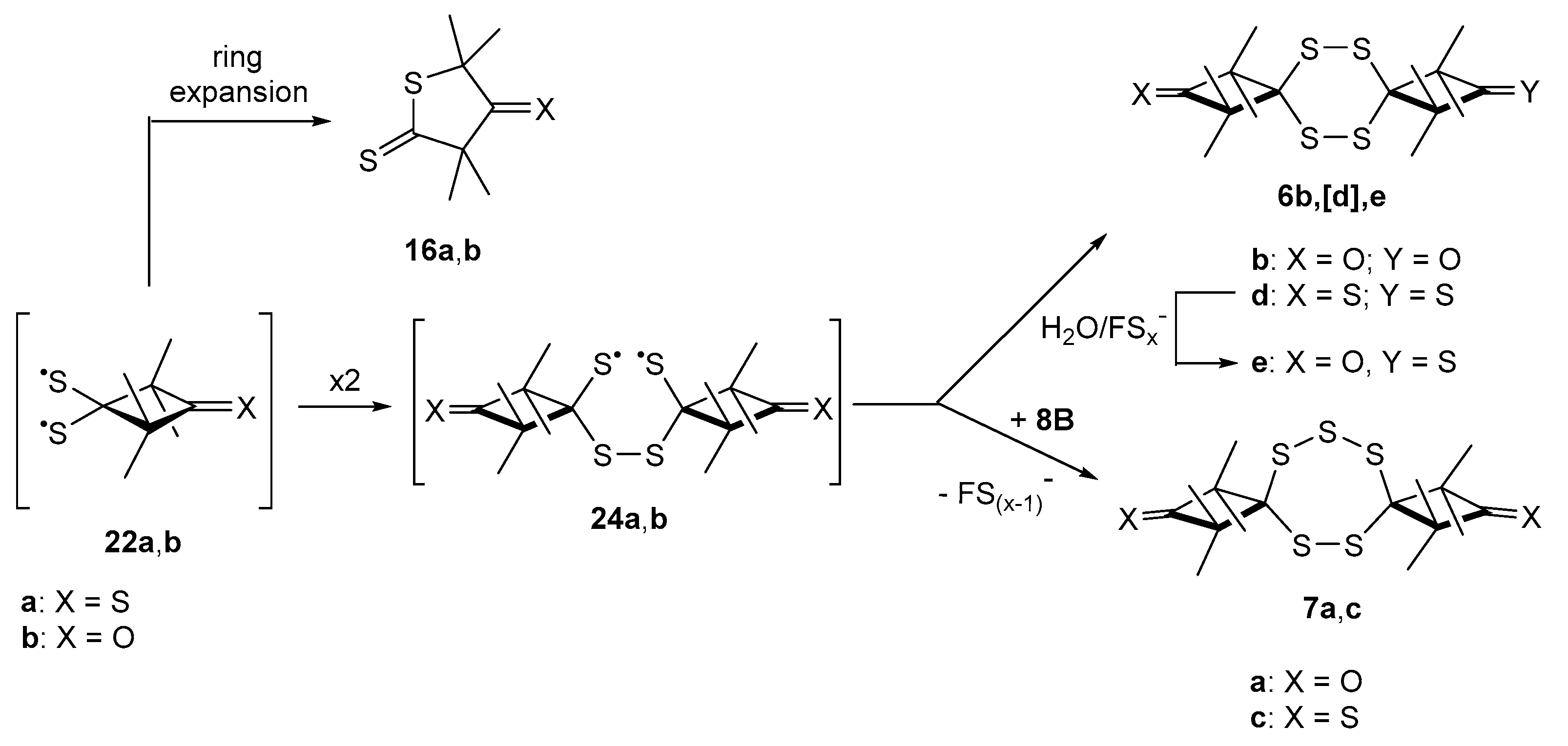
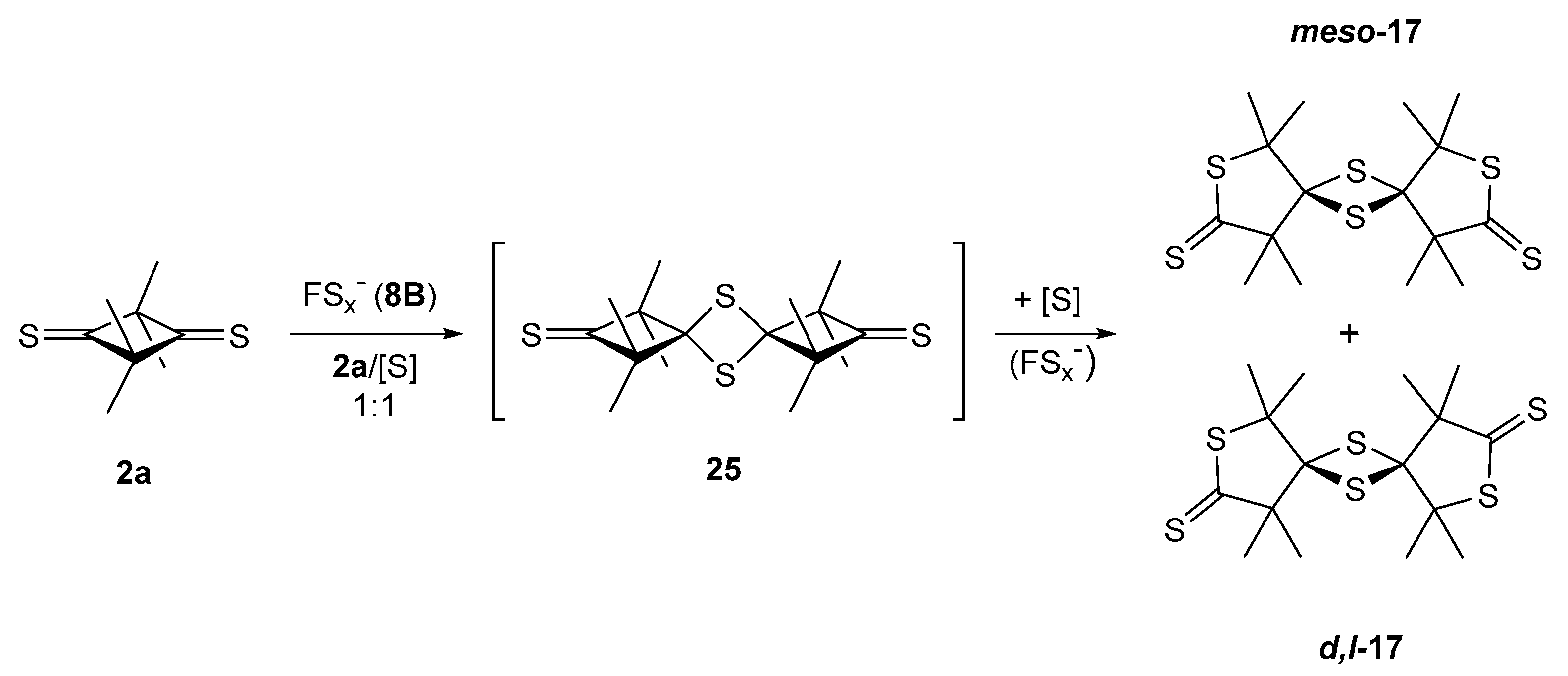
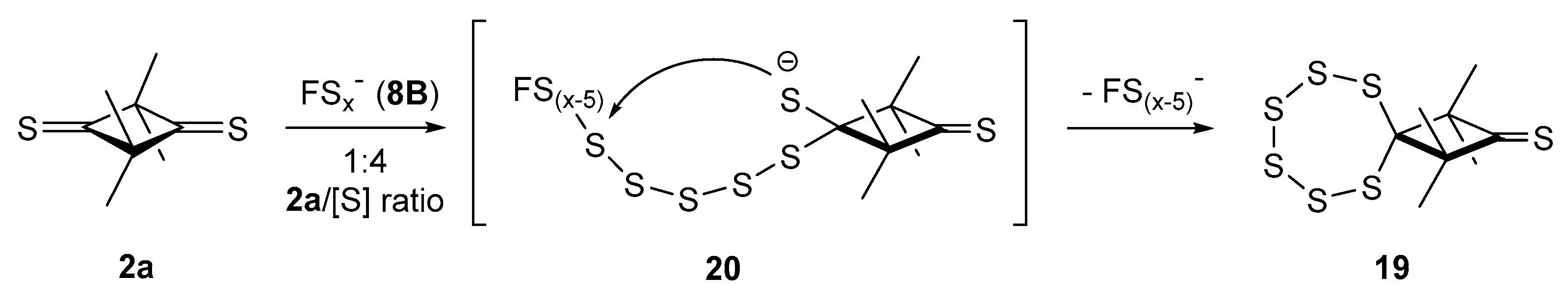
2.3.2. Formation of Sulfur-Rich Heterocycles from Thioketones 2a and 2b; Mechanistic Interpretations
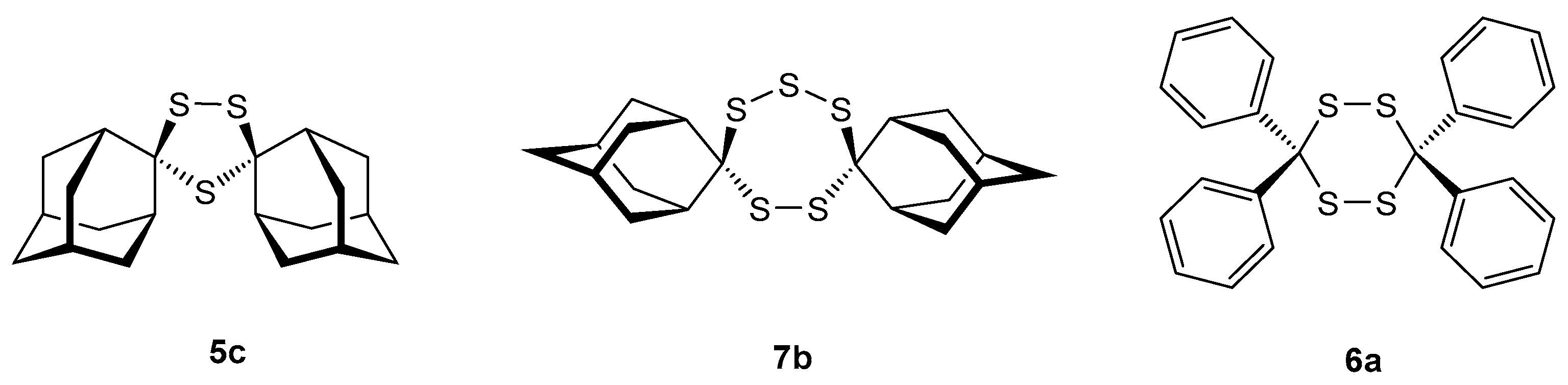
2.3.3. Sulfurization of ‘Adamantanethione (2c) and Thiobenzophenone (1a)
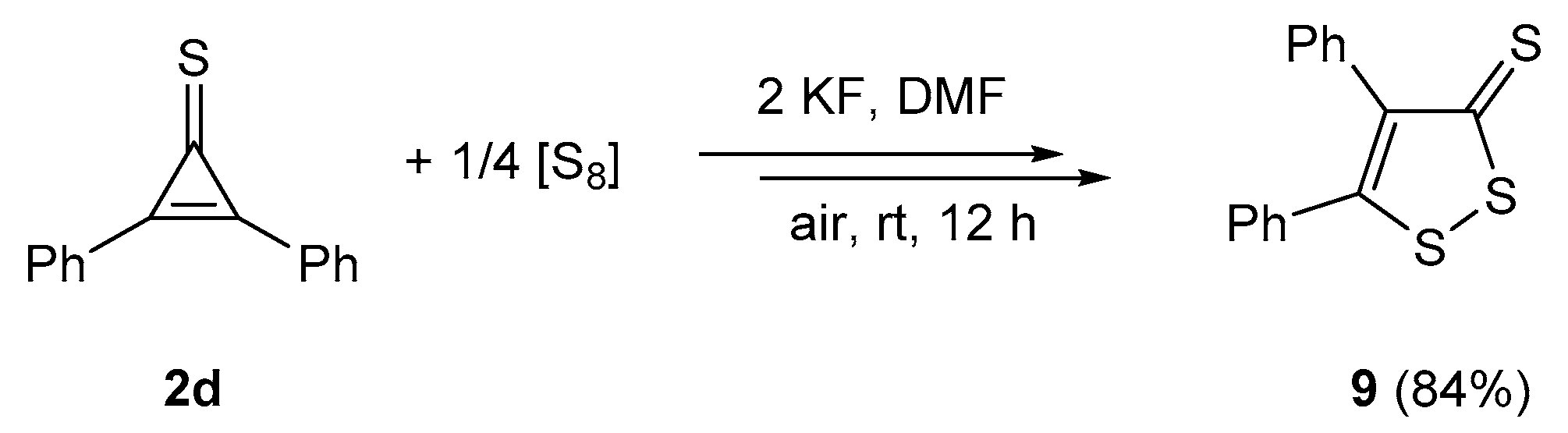
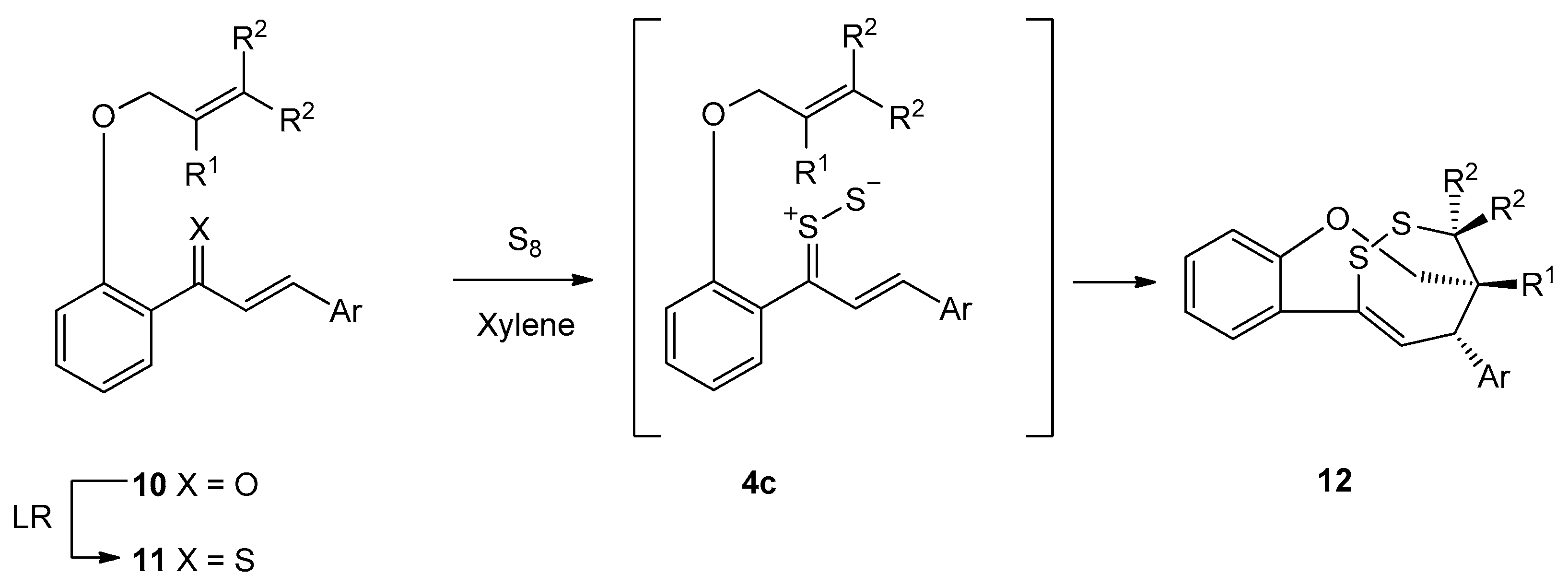
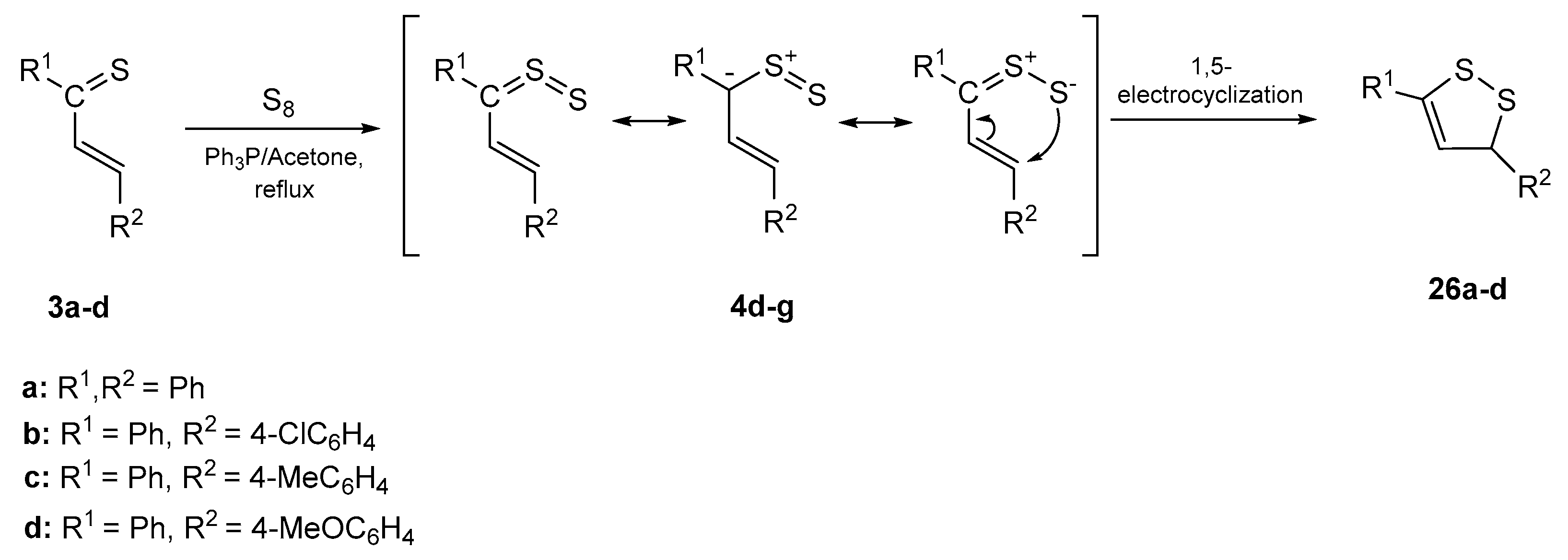
2.4. Sulfurization of Thiochalcones 3
3. Materials and Methods
3.1. Materials
3.2. Analytical Methods and Equipment
3.3. Conversions of Thioketones 2a,2b in the Presence of Fluoride Anion and Absence of S8
3.3.1. Procedure I: TBAF/THF
3.3.2. Procedure II: CsF/DMF
3.3.3. Product Characterization
3.4. General Procedures of the Fluoride Anion Catalyzed Sulfurization of Thioketones 1 and 2 with Elemental Sulfur (S8)
3.4.1. Procedure I: TBAF/THF
3.4.2. Procedure II: CsF/DMF
3.4.3. Product Characterization
3.5. General Procedures for Sulfurization of Thiochalcones 3 with Elemental Sulfur (S8)
3.5.1. Procedure I: Ph3P/Acetone
3.5.2. Procedure II: PhSK/Acetone
3.5.3. Procedure III: Ph3P/Butanone
3.5.4. Procedure IV: CsF/DMF
3.5.5. Product Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Chung, W.J.; Griebel, J.J.; Kim, E.T.; Yoon, H.; Simmonds, A.G.; Ji, H.J.; Dirlam, P.T.; Glass, R.S.; Wie, J.J.; Nguyen, N.A.; et al. The use of elemental sulfur as an alternative feedstock for polymeric materials. Nat. Chem. 2013, 5, 518–524. [Google Scholar] [CrossRef]
- Nguyen, T.B. Recent advances in organic reactions involving elemental sulfur. Adv. Synth. Catal. 2017, 359, 1066–1130. [Google Scholar] [CrossRef]
- Huisgen, R.; Rapp, J. The conversion of thioketones to 1,2,4,5-tetrathianes and its mechanism. Heterocycles 1997, 45, 507–525. [Google Scholar] [CrossRef]
- Huisgen, R.; Rapp, J.; Huber, H. Sulfurization of thiones: 2,2,4,4-tetramethyl-3-thioxocyclobutanone. Liebigs Ann. Recl. 1997, 1997, 1517–1523. [Google Scholar] [CrossRef]
- Okuma, K.; Kojima, K.; Shibata, S. An efficient synthesis of 1,2-dithiolanes and 1,2,4-trithiolanes by the reaction of phosphorus ylides with elemental sulfur. Heterocycles 2000, 53, 2753–2758. [Google Scholar] [CrossRef]
- Ishii, A.; Oshida, H.; Nakayama, J. Structure elucidation of 6-t-butyl-6-phenylpentathiane monoxides by X-ray crystallography and DFT calculations. J. Bull. Chem. Soc. Jpn. 2002, 75, 319–328. [Google Scholar] [CrossRef]
- Oshida, H.; Ishii, A.; Nakayama, J. Oxidation of cis- and trans-3,5-di-tert-butyl-3,5-diphenyl-1,2,4-trithiolanes: Isolation and properties of the 1-oxides and the 1,2-dioxides. J. Org. Chem. 2004, 69, 1695–1703. [Google Scholar] [CrossRef]
- Okuma, K.; Shibata, S.; Shioji, K.; Yokomori, Y. A new simple synthesis of cis- and trans-3,5-di-tert-butyl-3,5-diaryl-1,2,4-trithiolanes from ketones and tetraphosphorus decasulfide. Chem. Commun. 2000, 16, 1535–1536. [Google Scholar] [CrossRef]
- Mlostoń, G.; Heimgartner, H. Thioaldehyde and thioketone S-sulfides (Thiosulfines). In Science of Synthesis; Thieme Verlag: Stuttgart, Germany, 2014; Volume 27, pp. 403–411. [Google Scholar]
- Mlostoń, G.; Heimgartner, H. Bildung von 1,2,4-Trithiolanen in Dreikomponenten-Gemischen aus Phenyl-azid, aromatischen Thioketonen und 2,2,4,4-Tetramethylcyclobutanthionen: Eine Schwefel-Transfer-Reaktion unter Bildung von ‘Thiocarbonylthiolaten’ ((Alkylidensulfonio) thiolaten) als reaktive Zwischenstufen. Helv. Chim. Acta 1995, 78, 1298–1310. [Google Scholar]
- Takikawa, Y.; Makabe, T.; Hirose, N.; Hiratsuka, T.; Takoh, R.; Shimada, K. A novel method for the generation of thial S-sulfides from 2,4,6-trisubstituted 5,6-dihydro-1,3,5-dithiazines. Chem. Lett. 1988, 17, 1517–1520. [Google Scholar] [CrossRef]
- Liu, X.; Xu, C.; Wang, M.; Liu, Q. Trifluoromethyltrimethylsilane: Nucleophilic trifluoromethylation and beyond. Chem. Rev. 2015, 115, 683–730. [Google Scholar] [CrossRef] [PubMed]
- Uneyama, K. Fluoride ion-catalyzed desilylative-defluorination for synthetic organic chemistry. J. Fluor. Chem. 2007, 128, 1087–1090. [Google Scholar] [CrossRef]
- Clarck, J.H. Fluoride anion as a base in organic synthesis. Chem. Rev. 1980, 80, 429–452. [Google Scholar] [CrossRef]
- Petrov, V.A.; Marshall, W. Remarkable effect of metal fluoride catalyst on reaction of hexafluoropropene, sulfur and vinyl ethers. Convenient synthesis of 2,2-bis(trifluoromethyl)-4-R-thietanes, 3,3-bis(trifluoromethyl)-5-R-1,2-dithiolanes and 2,2-bis(trifluoromethyl)-4-R-1,3-dithiolanes. J. Fluor. Chem. 2010, 131, 1144–1155. [Google Scholar]
- Wu, J.; Gao, W.-X.; Huang, X.-B.; Zhou, Y.-B.; Liu, M.-C.; Wu, H.-Y. Selective [3+2] cycloaddition of cyclopropenone derivatives and elemental chalcogens. Org. Lett. 2020, 22, 5555–5560. [Google Scholar] [CrossRef]
- Saito, T.; Nagashima, M.; Karakasa, T.; Motoki, S. α,β-Unsaturated thiocarbonyl S-sulfides (thiosulfines). Their generation, intramolecular trapping by a non-activated C=C dipolarophile via [5+2] cycloaddition and trans-sulfurization ability. J. Chem. Soc. Chem. Commun. 1992, 5, 411–413. [Google Scholar] [CrossRef]
- Mloston, G.; Prakash, G.K.S.; Olah, G.; Heimgartner, H. Studies on reactions of thioketones with trimethyl (trifluoromethyl) silane catalyzed by fluoride ions. Helv. Chim. Acta 2002, 85, 1644–1658. [Google Scholar] [CrossRef]
- Karl, O.; Christe, K.-O.; William, W.; Wilson, W.W.; Richard, D.; Wilson, R.D.; Robert Ban, R.; Feng, J. Syntheses, properties, and structures of anhydrous tetramethylammonium fluoride and its 1:1 adduct with trans-3-amino-2-butenenitrile. J. Am. Chem. Soc. 1990, 112, 7619–7625. [Google Scholar]
- Pilcher, A.S.; Ammon, H.L.; DeShong, P. Utilization of tetrabutylammonium triphenylsilyl difluoride as a fluoride source for nucleophilic fluorination. J. Am. Chem. Soc. 1995, 117, 5166–5167. [Google Scholar] [CrossRef]
- Okuma, K.; Nojima, A.; Shigetomi, T.; Yokomori, Y. Novel formation of 1,2-dithiolane-3-thione from β-dithiolactone. Isolation of dithiolato-palladium and -platinum complexes. Tetrahedron 2007, 63, 11748–11753. [Google Scholar] [CrossRef]
- Mloston, G.; Majchrzak, A.; Senning, A.; Søtofte, I. Highly sulfurated heterocycles via dithiiranes and trithietanes as key intermediates. J. Org. Chem. 2002, 67, 5690–5695. [Google Scholar] [CrossRef]
- Mlostoń, G.; Romański, J.; Reisenauer, H.-P.; Maier, G. Thioformaldehyd S-sulfid (Thiosulfin). Angew. Chem. Int. Ed. Engl. 2001, 40, 393–395. [Google Scholar] [CrossRef]
- Mlostoń, G.; Heimgartner, H.; Romański, J. Sulfur centered 1,3-dipoles. An efficient trapping of adamantanethione S-sulfide generated in the reaction of adamantanethione with organic azides. Pol. J. Chem. 1996, 70, 437–445. [Google Scholar]
- Grzelak, P.; Utecht, G.; Jasiński, M.; Mlostoń, G. First (3+2)-cycloadditions of thiochalcones as C=S dipolarophiles: Efficient synthesis of 1,3,4-thiadiazoles via reactions with fluorinated nitrile imines. Synthesis 2017, 49, 2129–2137. [Google Scholar]
- Mlostoń, G.; Grzelak, P.; Heimgartner, H. Hetero-Diels–Alder reactions of hetaryl thiochalcones with acetylenic dienophiles. J. Sulfur. Chem. 2017, 38, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Mlostoń, G.; Urbaniak, K.; Jasiński, M.; Würthwein, E.-U.; Heimgartner, H.; Zimmer, R.; Reissig, H.-U. The [4+2]-cycloadditionof α-nitrosoalkenes with thiochalcones as a prototype of periselective hetero-Diels-Alder reactions—Experimental and computational studies. Chem. Eur. J. 2020, 26, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Mlostoń, G.; Romański, J.; Weigand, W.; Heimgartner, H. Organic and coordination chemistry of 1,2,4-trithiolanes. Eur. J. Org. Chem. 2019, 2019, 1867–1875. [Google Scholar] [CrossRef] [Green Version]
- Mlostoń, G.; Heimgartner, H. Syntheses of five-membered sulfur-heterocycles via 1,5-dipolar electrocyclization of thioca bonyl ylides and related processes. Curr. Org. Chem. 2011, 15, 675–693. [Google Scholar] [CrossRef]
- Huisgen, R.G.; Mlostoń, G.K.; Polborn, K.F.; Palacios-Gambra, F. Some cycloadditions of aromatic thione S-oxides. Liebigs Ann. Recl. 1997, 1997, 187–192. [Google Scholar] [CrossRef]
- Huisgen, R.; Rapp, J. The chemistry of thiocarbonyl S-sulfides. Tetrahedron 1997, 53, 939–960. [Google Scholar] [CrossRef]
- Fabian, J.; Mayer, R.; Bleisch, S.; Zahradnik, R. MO-LCAO Calculations on sulfur-containing pi-electron systems. 46. Electronic excitation of organosulfur radicals. Phosphorus Sulfur Siliconand Relat. Elem. 1982, 13, 107–117. [Google Scholar] [CrossRef]
- Amzil, J.; Catel, J.M.; Lecoustumer, G.; Mollier, Y.; Sauvé, J.P.; Flandrois, S. Organic conductors—Physical synthesis, structure and studies of tetracyanoquinodimethane radical-anion salts with 1,2-dithioylium and 1,2-diselenolylium. Mol. Cryst. Liq. Cryst. 1986, 133, 333–354. [Google Scholar] [CrossRef]
- Guemas, J.-P.; Quinou, H. Recherches sur les composes sulfurés organique. XV. Action de l’hydrogène sulfuré et de l’iode sur les β-dicétones. Bull. Chem. Soc. Fr. 1973, 592–597. [Google Scholar]
- Franek, W. 1,2,4,5-Tetrathiane—I. Synthesen und Reaktionen. Sulfur Rep. 1991, 10, 193–232. [Google Scholar] [CrossRef]
- Franek, W. 1,2,4,5-Tetrathiane—II. Strukturelle und spektroskopische Eigenschaften. Elektrochemie, Aromaforschung, Anwendungen. Sulfur Rep. 1991, 10, 233–278. [Google Scholar] [CrossRef]
- Penczek, S.; Duda, A.; Ślązak, R. Anionic copolymerisation of elemental sulphur. Nature 1978, 273, 738–739. [Google Scholar] [CrossRef]
- Duda, A.; Penczek, S. Anionic copolymerization of elemental sulfur with propylene sulfide. Macromolecules 1982, 15, 36–40. [Google Scholar] [CrossRef]
- Wręczycki, J.; Bieliński, D.M.; Kozanecki, M.; Maczugowska, P.; Mlostoń, G. Anionic copolymerization of styrene sulfide with elemental sulfur (S8). Materials 2020, 13, e2597. [Google Scholar] [CrossRef] [PubMed]
- Mlostoń, G.; Celeda, M.; Jasiński, M.; Urbaniak, K.; Boratyński, P.J.; Schreiner, P.R.; Heimgartner, H. Synthesis of 2-unsubstituted imidazole N-oxides as novel precursors of chiral 3-alkoxyimidazol-2-ylidenes derived from trans-1,2-diaminocyclohexane and other chiral amino compounds. Molecules 2019, 24, e4398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mlostoń, G.; Celeda, M.; Poper, W.; Kowalczyk, M.; Gach-Janczak, K.; Janecka, A.; Jasiński, M. Synthesis, selected transformations, and biological activity of alkoxy analogues of lepidilines A and C. Materials 2020, 13, e4190. [Google Scholar] [CrossRef] [PubMed]















{kind=link}
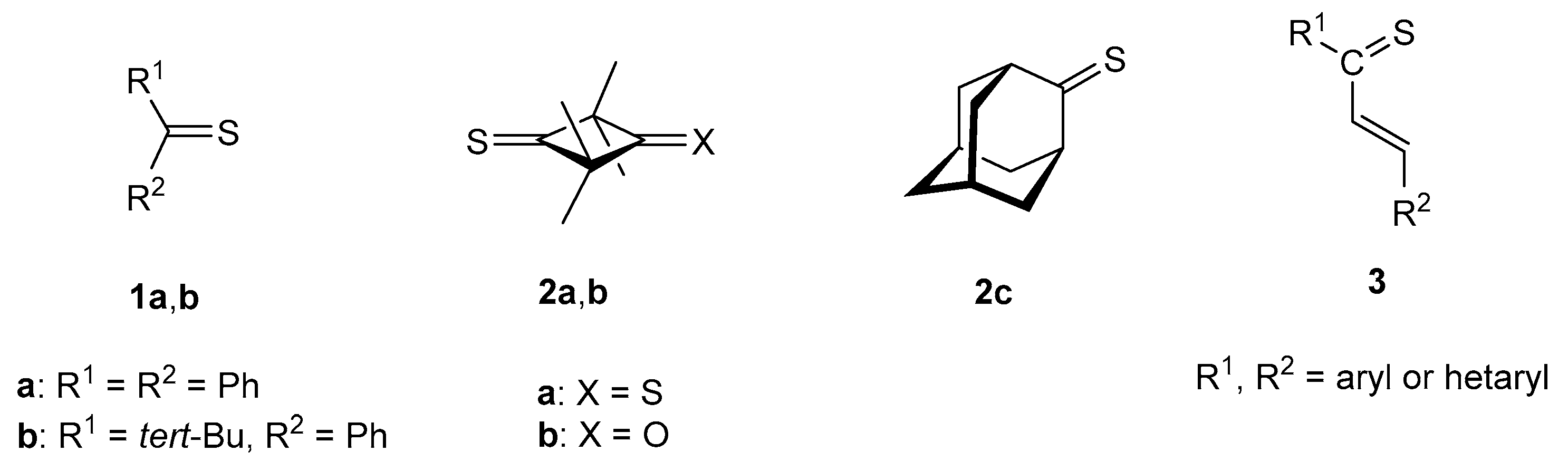{kind=link}
{kind=link}
{kind=link}
{kind=link}
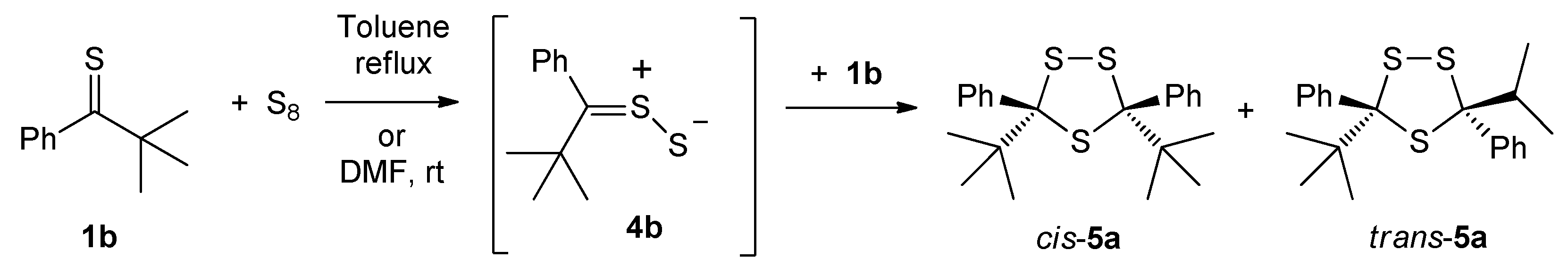{kind=link}
{kind=link}
{kind=link}
{kind=link}
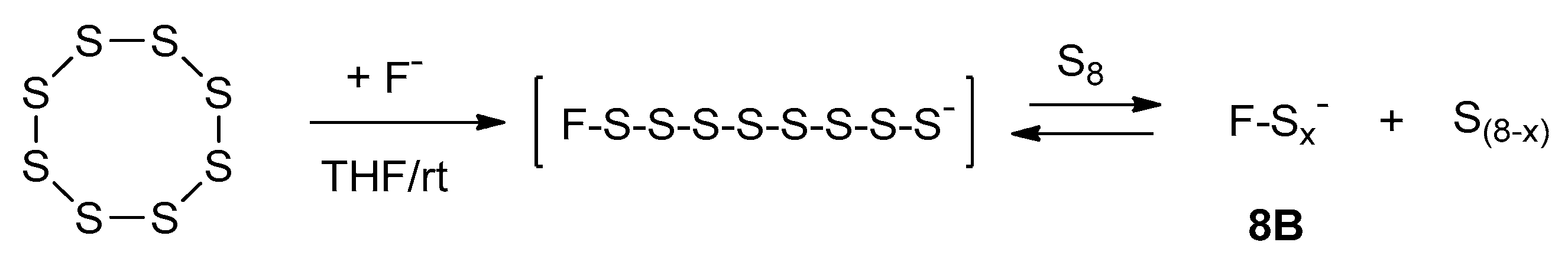{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Thioketone | Entry | RT [h], rt | Product | Yield (%) |
|---|---|---|---|---|
| 2a | A | 1 | 14 | 63 a |
| 16a | 11 a | |||
| B | 1.5 | 16a | 8 b | |
| 6e | 15 b | |||
| meso-17,D,L-17 | 43 b | |||
| C | 2 | 14 | 9 a | |
| 16a | 8 b | |||
| 18 | 5 a | |||
| 7c | 23 b | |||
| C′ | 24 | 7c | 54 b | |
| D | 2 | 14 | 3 a | |
| 16a | 5 a | |||
| 18 | 2 a | |||
| 7c | 46 b | |||
| 19 | 4 b | |||
| 2b | A | 1.5 | 15 | 30 a |
| 16b | 10 a | |||
| 6b | 12 a | |||
| B | 3 | 15 | 20 a | |
| 16b | 23 a | |||
| 6b | 40 a | |||
| C | 48 | 7a | 34 b | |
| C′ | 24 | 7a | 43 b | |
| D | 22 | 7a | 52 b | |
| E | 6 | 7a | 60 b |
| Thioketone | Entry | RT (h) | Product | Yield (%) | Product | Yield (%) | ||
|---|---|---|---|---|---|---|---|---|
| 2c | A | 72 | 5c | 59 | 7b | 37 | ||
| B | 46 | - | - | 7b | 47 | |||
| C | 24 | - | - | 7b | 51 | |||
| C′ | 24 | 5c | 59 | - | - | |||
| 1a | B | 72 | 6a | 28 | - | - | ||
| C | 72 | 6a | 52 | - | - | |||
| C′ | 48 | 6a | 56 | - | - | |||
| Thiochalcone | Procedure | RT [h] | Product | Yield (%) |
|---|---|---|---|---|
| 3a | I | 3.5 | 26a | 40 a (63 b) |
| 3a | II | 4.0 | 26a | 15 a |
| 3a | III | 0.5 | 26a | 41 a |
| 3a | IV | 72 | 26a | 15 a |
| 3b | I | 4.0 | 26b | 15 a |
| 3c | I | 3.5 | 26c | 20 a |
| 3c | IV | 72 | 26c | 17 a |
| 3d | I | 3.0 | 26d | 12 a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mlostoń, G.; Wręczycki, J.; Urbaniak, K.; Bieliński, D.M.; Heimgartner, H. The Fluoride Anion-Catalyzed Sulfurization of Thioketones with Elemental Sulfur Leading to Sulfur-Rich Heterocycles: First Sulfurization of Thiochalcones. Molecules 2021, 26, 822. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26040822
Mlostoń G, Wręczycki J, Urbaniak K, Bieliński DM, Heimgartner H. The Fluoride Anion-Catalyzed Sulfurization of Thioketones with Elemental Sulfur Leading to Sulfur-Rich Heterocycles: First Sulfurization of Thiochalcones. Molecules. 2021; 26(4):822. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26040822
Chicago/Turabian StyleMlostoń, Grzegorz, Jakub Wręczycki, Katarzyna Urbaniak, Dariusz M. Bieliński, and Heinz Heimgartner. 2021. "The Fluoride Anion-Catalyzed Sulfurization of Thioketones with Elemental Sulfur Leading to Sulfur-Rich Heterocycles: First Sulfurization of Thiochalcones" Molecules 26, no. 4: 822. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26040822