
UHPLC-UV/PDA Method Validation for Simultaneous Quantification of Luteolin and Apigenin Derivatives from Elaeis guineensis Leaf Extracts: An Application for Antioxidant Herbal Preparation




Abstract
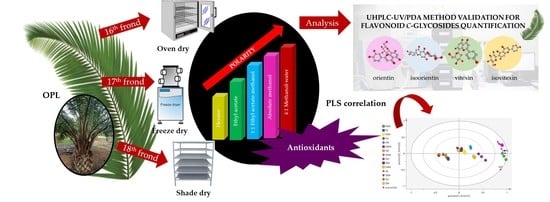
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Preparation of Various OPL Extracts
2.3. Development of UHPLC-UV/PDA Method
2.4. UHPLC-MS/MS for Identification of Luteolin and Apigenin Derivatives
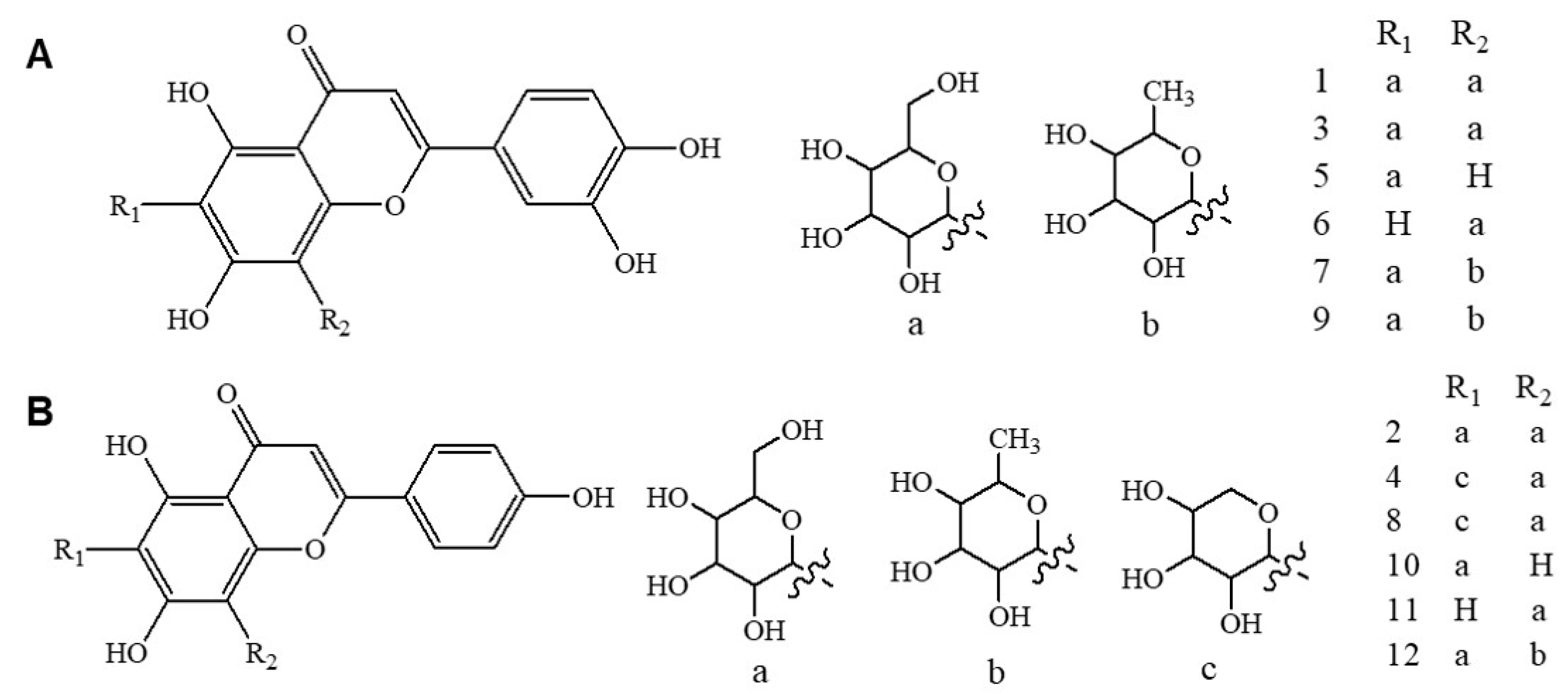
2.5. Identification of Luteolin and Apigenin Derivatives
2.6. Validation of UHPLC-UV/PDA Method
2.6.1. Specificity
2.6.2. Linearity
2.6.3. Limit of Detection (LOD) and Limit of Quantification (LOQ)
2.6.4. Accuracy
2.6.5. Repeatability and Intermediate Precision
2.6.6. Robustness
2.7. Quantification of Luteolin and Apigenin Derivatives
2.8. Determination of Total Phenolic Content
2.9. Determination of Total Flavonoid Content
2.10. Determination of 2,2-Diphenyl-1-picrylhydrazyl (DPPH) Free Radical-Scavenging Activity
2.11. Determination of Nitric Oxide (NO) Free Radical-Scavenging Activity
2.12. Statistical Analysis
3. Results and Discussion
3.1. Validation of Developed UHPLC-UV/PDA Method
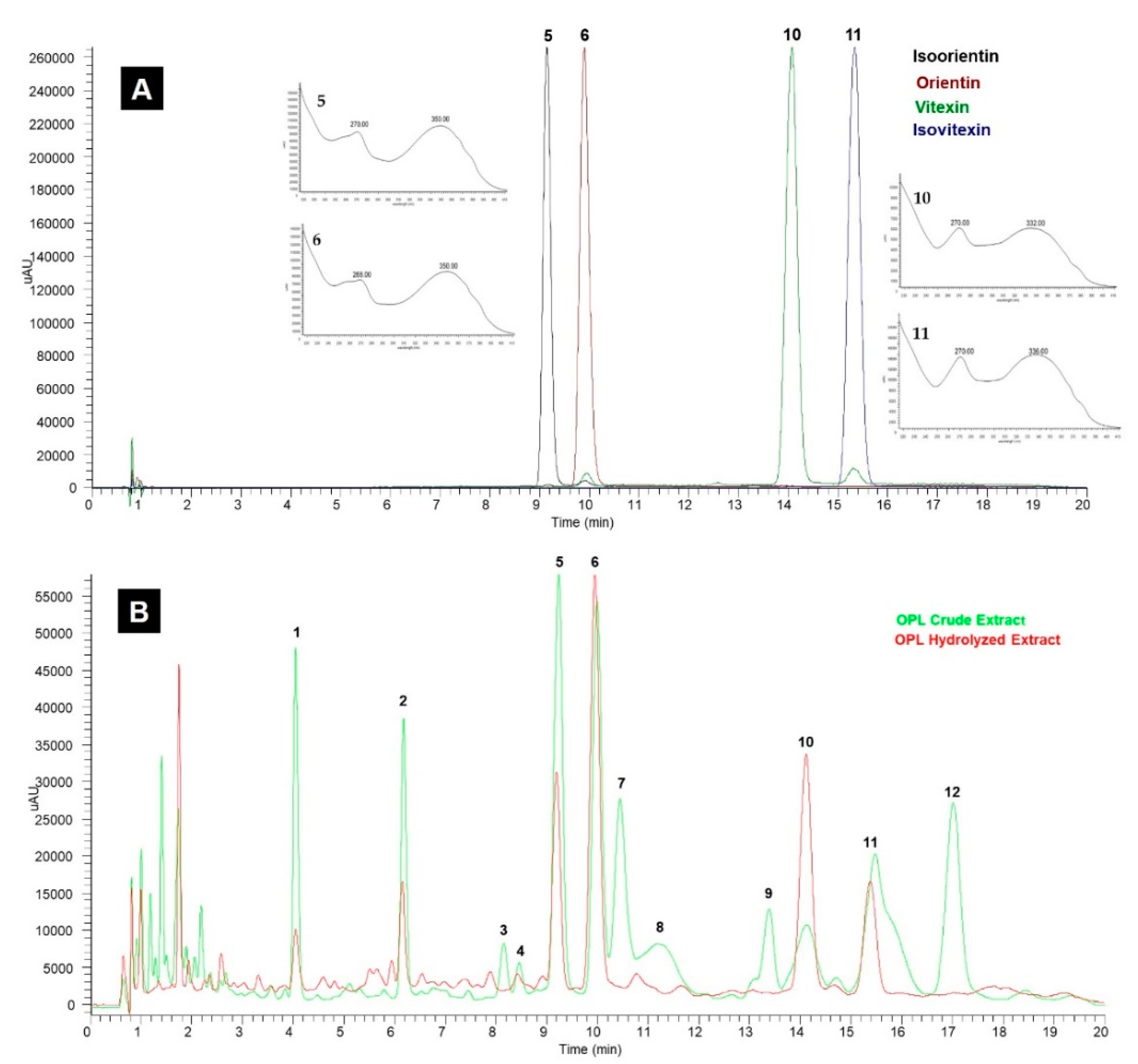
3.1.1. Specificity
3.1.2. Linearity, Limits of Detection (LOD), and Limits of Quantification (LOQ)
3.1.3. Accuracy, Precision, and Robustness
3.2. Application of UHPLC-UV/PDA Method for Quantification of Luteolin and Apigenin Derivatives in Various OPL Extracts
3.3. Polyphenolic Content and Antioxidant Activity in Various OPL Extracts
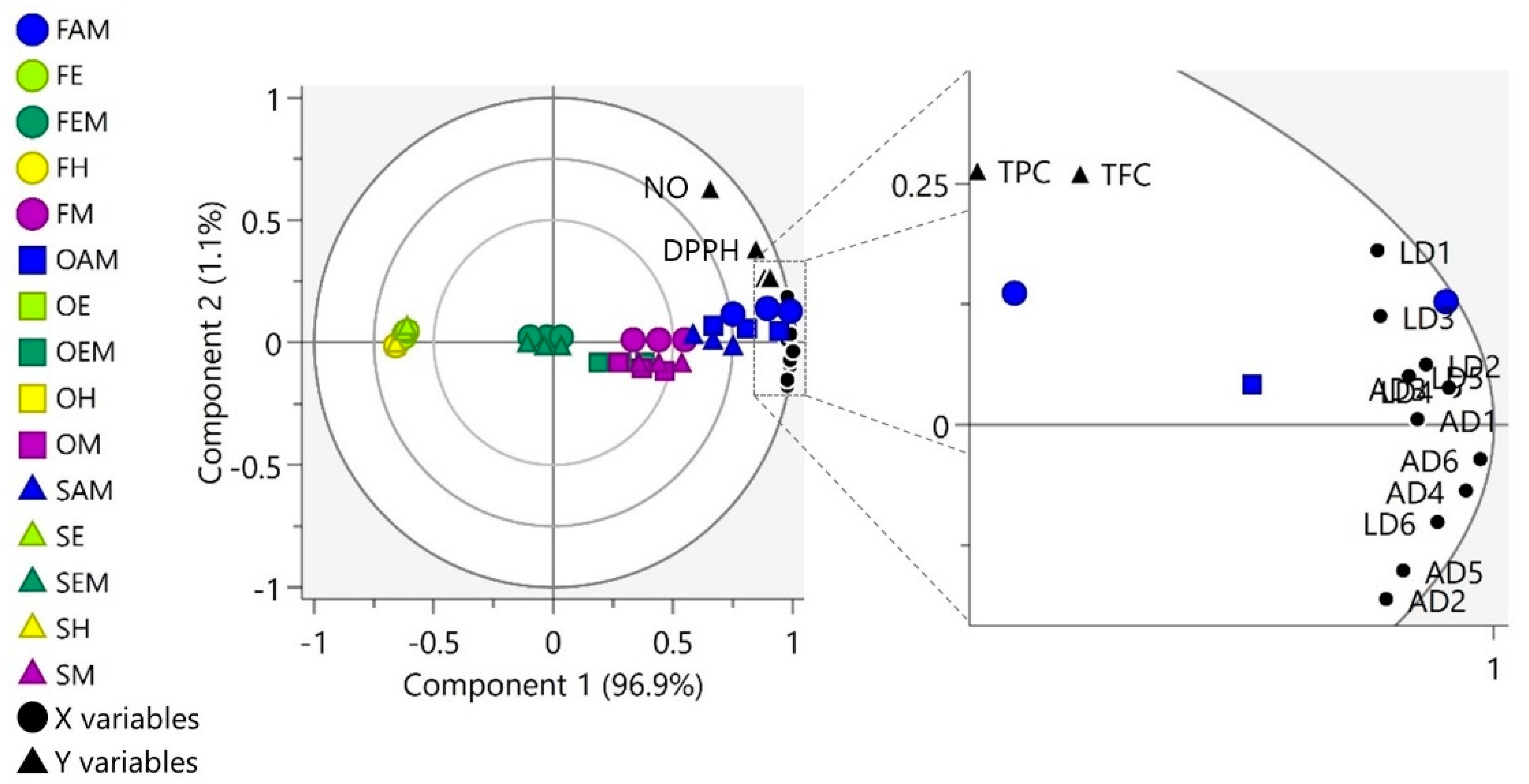
3.4. Partial Least Square Analysis (PLS) Correlation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yin, N.G.S.; Abdullah, S.; Phin, C.K. Phytochemical constituents from Leaves of Elaeis guineensis and their antioxidant and antimicrobial activities. Int. J. Pharm. Pharm. Sci. 2013, 5, 137–140. [Google Scholar]
- Mohamed, S. Oil Palm Leaf: A New Functional Food Ingredient for Health and Disease Prevention. J. Food Process. Technol. 2014, 5, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Suhaila, M.; Juliana, J.; Noordin, M.M.; Intan, N.A.; Myrsyida, A.R. Extract from Palm Leaves and a Method for Producing the Same. Malaysia Patent MY164863A, 11 July 2007. [Google Scholar]
- Zain, M.S.C.; Lee, S.Y.; Teo, C.Y.; Shaari, K. Adsorption and Desorption Properties of Total Flavonoids from Oil Palm (Elaeis guineensis Jacq.) Mature Leaf on Macroporous Adsorption Resins. Molecules 2020, 25, 1–17. [Google Scholar]
- Zain, M.S.C.; Lee, S.Y.; Sarian, M.N.; Fakurazi, S.; Shaari, K. In vitro wound healing potential of flavonoid c-glycosides from oil palm (Elaeis guineensis jacq.) leaves on 3t3 fibroblast cells. Antioxidants 2020, 9, 1–18. [Google Scholar]
- Tahir, N.I.; Shaari, K.; Abas, F.; Parveez, G.K.A.; Ishak, Z.; Ramli, U.S. Characterization of apigenin and luteolin derivatives from oil palm (Elaeis guineensis Jacq.) Leaf using LC-ESI-MS/MS. J. Agric. Food Chem. 2012, 60, 11201–11210. [Google Scholar] [CrossRef]
- Jaffri, J.M.; Mohamed, S.; Ahmad, I.N.; Mustapha, N.M.; Manap, Y.A.; Rohimi, N. Effects of catechin-rich oil palm leaf extract on normal and hypertensive rats’ kidney and liver. Food Chem. 2011, 128, 433–441. [Google Scholar] [CrossRef]
- Mohamed, S.; Lee Ming, T.; Jaffri, J.M. Cognitive enhancement and neuroprotection by catechin-rich oil palm leaf extract supplement. J. Sci. Food Agric. 2013, 93, 819–827. [Google Scholar] [CrossRef]
- Anyanji, V.U.; Mohamed, S.; Zokti, J.A.; Ado, M.A. Anti-inflammatory properties of oil palm leaf (Elaeis guineensis Jacq.) extract in aged rats. Int. J. Pharm. Pharm. Sci. 2013, 5, 134–136. [Google Scholar]
- Nováková, L.; Spáčil, Z.; Seifrtová, M.; Opletal, L.; Solich, P. Rapid qualitative and quantitative ultra high performance liquid chromatography method for simultaneous analysis of twenty nine common phenolic compounds of various structures. Talanta 2010, 80, 1970–1979. [Google Scholar] [CrossRef]
- Shams Eldin, S.M.; Radwan, M.M.; Wanas, A.S.; Habib, A.A.M.; Kassem, F.F.; Hammoda, H.M.; ElSohly, M.A. A Validated UPLC-PDA Method for Simultaneous Determination of 3 Biologically Active Isoflavans in Trigonella stellata Extract. Nat. Prod. Commun. 2020, 15, 1–6. [Google Scholar] [CrossRef]
- International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use. In Validation of Analytical Procedures: Text and Methodology Q2(R1); European Medicines Agency: Amsterdam, The Netherlands, 2005; Volume 7, pp. 1–13.
- Numonov, S.R.; Qureshi, M.N.; Aisa, H.A. Development of HPLC protocol and simultaneous quantification of four free flavonoids from Dracocephalum heterophyllum benth. Int. J. Anal. Chem. 2015, 2015, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batista, A.N.D.L.; Colombo, R.; Pascoli, I.C.D.; Teles, H.L.; Silva, G.H.; Bomfim, G.C.; Reimberg, M.C.H. Development and validation of a HPLC method for standardization of herbal and commercial extracts of Myrcia uniflora. Braz. J. Pharm. 2011, 21, 402–406. [Google Scholar] [CrossRef] [Green Version]
- Landim, L.P.; Feitoza, G.S.; da Costa, J.G.M. Development and validation of a HPLC method for the quantification of three flavonoids in a crude extract of Dimorphandra gardneriana. Rev. Bras. Farmacogn. 2013, 23, 58–64. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Zhao, Y.; Ma, C.; Wang, W.; Xing, D.; Du, L. Acid hydrolytic method for determination of ginkgo biloba total flavonoids in Rat plasma by HPLC for pharmacokinetic studies. Tsinghua Sci. Technol. 2010, 15, 452–459. [Google Scholar] [CrossRef]
- Zhao, Z.; Dong, L.; Lin, F.; Wu, Y. Simultaneous characterization and quantification of flavonoids in Euonymus alatus (Thunb.) Siebold from different origins by HPLC-PAD-MS. Arab. J. Chem. 2013, 6, 205–209. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.H.; Wang, Y.X.; Liu, H.J. Validation and application by HPLC for simultaneous determination of vitexin-2″-O-glucoside, vitexin-2″-O-rhamnoside, rutin, vitexin, and hyperoside. J. Pharm. Anal. 2011, 1, 291–296. [Google Scholar] [CrossRef] [Green Version]
- Tuszyńska, M. Validation of the analytical method for the determination of flavonoids in broccoli. J. Hortic. Res. 2014, 22, 131–140. [Google Scholar] [CrossRef] [Green Version]
- Verdu, C.F.; Gatto, J.; Freuze, I.; Richomme, P.; Laurens, F.; Guilet, D. Comparison of two methods, UHPLC-UV and UHPLC-MS/MS, for the quantification of polyphenols in cider apple juices. Molecules 2013, 18, 10213–10227. [Google Scholar] [CrossRef] [Green Version]
- Villedieu-Percheron, E.; Ferreira, V.; Campos, J.F.; Destandau, E.; Pichon, C.; Berteina-Raboin, S. Quantitative Determination of Andrographolide and Related Compounds in Andrographis paniculata Extracts and Biological Evaluation of Their Anti-Inflammatory Activity. Foods 2019, 8, 683. [Google Scholar] [CrossRef] [Green Version]
- Mediani, A.; Abas, F.; Khatib, A.; Tan, C.P.; Ismail, I.S.; Shaari, K.; Lajis, N.H. Relationship Between Metabolites Composition and Biological Activities of Phyllanthus niruri Extracts Prepared by Different Drying Methods and Solvents Extraction. Plant Foods Hum. Nutr. 2015, 70, 184–192. [Google Scholar] [CrossRef]
- Anwar, F.; Kalsoom, U.; Sultana, B.; Mushtaq, M.; Mehmood, T.; Arshad, H.A. Effect of drying method and extraction solvent on the total phenolics and antioxidant activity of cauliflower (Brassica oleracea L.) Extracts. Int. Food Res. J. 2013, 20, 653–659. [Google Scholar]
- Abdul-Hamid, N.A.; Abas, F.; Ismail, I.S.; Shaari, K.; Lajis, N.H. Influence of Different Drying Treatments and Extraction Solvents on the Metabolite Profile and Nitric Oxide Inhibitory Activity of Ajwa Dates. J. Food Sci. 2015, 80, H2603–H2611. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Abas, F.; Khatib, A.; Ismail, I.S.; Shaari, K.; Zawawi, N. Metabolite profiling of Neptunia oleracea and correlation with antioxidant and α-glucosidase inhibitory activities using 1H NMR-based metabolomics. Phytochem. Lett. 2016, 16, 23–33. [Google Scholar] [CrossRef]
- Zeraik, M.L.; Yariwake, J.H. Quantification of isoorientin and total flavonoids in Passiflora edulis fruit pulp by HPLC-UV/DAD. Microchem. J. 2010, 96, 86–91. [Google Scholar] [CrossRef]
- Pereira, C.A.M.; Yariwake, J.H.; McCullagh, M. Distinction of the C-glycosylfavone isomer pairs orientin/isoorientin and vitexin/isovitexin using HPLC-MS exact mass measurement and in-source CID. Phytochem. Anal. 2005, 16, 295–301. [Google Scholar] [CrossRef]
- Ji, H.F.; Du, A.L.; Zhang, L.W.; Xu, C.Y.; Yang, M.D.; Li, F.F. Effects of drying methods on antioxidant properties in Robinia pseudoacacia L. flowers. J. Med. Plants Res. 2012, 6, 3233–3239. [Google Scholar]
- Mediani, A.; Abas, F.; Khatib, A.; Maulidiani, H.; Shaari, K.; Choi, Y.H.; Lajis, N.H. 1H-NMR-based metabolomics approach to understanding the drying effects on the phytochemicals in Cosmos caudatus. Food Res. Int. 2012, 49, 763–770. [Google Scholar] [CrossRef]
- Macias, M.A. Comparatives studies of different drying process of strawberry hot air drying freeze-drying and swell-drying: Application on the biological compounds preservation. Procedia Eng. 2012, 42, 297–312. [Google Scholar]
- Hung, P.V.; Duy, T.L. Effects of drying methods on bioactive compounds of vegetables and correlation between bioactive compounds and their antioxidants. Int. Food Res. J. 2012, 19, 327–332. [Google Scholar]
- Andersen, O.M.; Markham, K.R. Flavonoids. Chemistry, Biochemistry and Applications; Angewandte Chemie International Edition: Boca Raton, FL, USA, 2006; Volume 45, p. 1237. [Google Scholar]
- Do, Q.D.; Angkawijaya, A.E.; Tran-Nguyen, P.L.; Huynh, L.H.; Soetaredjo, F.E.; Ismadji, S.; Ju, Y.H. Effect of extraction solvent on total phenol content, total flavonoid content, and antioxidant activity of Limnophila aromatica. J. Food Drug. Anal. 2014, 22, 296–302. [Google Scholar] [CrossRef] [Green Version]
- Maulidiani Abas, F.; Khatib, A.; Shaari, K.; Lajis, N.H. Chemical characterization and antioxidant activity of three medicinal Apiaceae species. Ind. Crops Prod. 2014, 55, 238–247. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Flavonoid | Concentration Range (µg/mL) | Regression Equation | Correlation Coefficient (R2) | LOD (µg/mL) | LOQ (µg/mL) |
|---|---|---|---|---|---|
| Isoorientin | 16–500 | y = 1931.7x + 3571.4 | 0.9999 | 17.99 | 54.52 |
| Orientin | 31–800 | y = 2706.2x − 19677 | 0.9999 | 30.22 | 91.58 |
| Vitexin | 47–1500 | y = 534.05x − 6500.5 | 0.9997 | 80.63 | 244.35 |
| Isovitexin | 16–500 | y = 11136x + 13498 | 0.9999 | 17.69 | 53.61 |
| Flavonoids | Concentration (µg/mL) | Recovery (%) | Precision (RSD %) | Robustness p-Value (t-Test) | |||
|---|---|---|---|---|---|---|---|
| Intra-Day | Inter-Day | CT (°C) | λ (nm) | Day | |||
| Isoorientin | High | 102.34 | 1.25 | 0.01 | 0.06 | 0.07 | 0.37 |
| Medium | 100.21 | 0.81 | 0.04 | ||||
| Low | 98.32 | 1.49 | 0.66 | ||||
| Orientin | High | 95.61 | 0.45 | 0.32 | 0.15 | 0.07 | 0.24 |
| Medium | 97.71 | 0.87 | 0.50 | ||||
| Low | 99.33 | 0.34 | 0.28 | ||||
| Vitexin | High | 99.55 | 0.48 | 0.72 | 0.24 | 0.24 | 0.10 |
| Medium | 99.22 | 0.05 | 0.56 | ||||
| Low | 100.21 | 1.29 | 1.74 | ||||
| Isovitexin | High | 102.79 | 1.15 | 1.31 | 0.15 | 0.18 | 0.14 |
| Medium | 101.18 | 0.09 | 1.49 | ||||
| Low | 100.68 | 0.17 | 0.80 | ||||
| Drying | Solvent | Peak 1 | Peak 3 | * Peak 5 | * Peak 6 | Peak 7 | Peak 9 | TLC (µg/mg) |
|---|---|---|---|---|---|---|---|---|
| O | E | 0.61 ± 0.11 Aa | ND | 0.74 ± 0.09 Aa | 0.80 ± 0.23 Aa | ND | ND | 2.15 ± 0.43 Aa |
| EM | 2.22 ± 0.18 Ab | 0.97 ± 0.19 Aa | 10.14 ± 0.85 Ab | 7.99 ± 0.23 Ab | 5.35 ± 0.79 Aa | 2.38 ± 0.31 Aa | 29.05 ± 2.55 Ab | |
| M | 2.36 ± 0.14 Ab | 0.98 ± 0.10 Aa | 10.05 ± 1.17 Ab | 8.16 ± 0.41 Ab | 5.42 ± 0.42 Aa | 3.47 ± 0.50 Ab | 30.44 ± 2.74 Ab | |
| AM | 3.90 ± 0.15 Ac | 1.66 ± 0.12 Ab | 21.85 ± 2.19 Ac | 13.26 ± 1.18 Ac | 7.27 ± 1.04 Ab | 3.93 ± 0.39 Ab | 51.87 ± 5.07 Ac | |
| F | E | 0.60 ± 0.13 Aa | ND | 0.72 ± 0.10 Aa | 0.72 ± 0.20 Aa | ND | ND | 2.04 ± 0.43 Aa |
| EM | 1.47 ± 0.16 Bb | 0.89 ± 0.13 Aa | 9.66 ± 1.13 Ab | 6.51 ± 0.10 Bb | 3.20 ± 0.22 Ba | 1.63 ± 0.09 Ba | 23.36 ± 1.83 Bb | |
| M | 2.49 ± 0.21 Ac | 1.20 ± 0.08 Ab | 18.58 ± 1.19 Bc | 11.48 ± 0.89 Bc | 5.64 ± 0.69 Ab | 2.75 ± 0.48 Ab | 42.14 ± 3.54 Bc | |
| AM | 4.40 ± 0.18 Bd | 1.70 ± 0.05 Ac | 23.54 ± 3.39 Ad | 13.90 ± 1.30 Ad | 9.06 ± 2.11 Bc | 3.70 ± 0.29 Ac | 56.30 ± 7.32 Ad | |
| S | E | 0.65 ± 0.15 Aa | ND | 0.70 ± 0.05 Aa | 0.75 ± 0.15 Aa | ND | ND | 2.10 ± 0.35 Aa |
| EM | 1.32 ± 0.11 Bb | 0.75 ± 0.09 Ba | 8.36 ± 1.15 Ab | 6.38 ± 0.21 Bb | 3.14 ± 0.39 Ba | 1.79 ± 0.24 Ba | 21.74 ± 2.19 Bb | |
| M | 2.32 ± 0.14 Ac | 1.09 ± 0.12 Ab | 15.54 ± 2.19 Bc | 10.63 ± 1.08 Bc | 4.92 ± 0.81 Ab | 2.96 ± 0.19 Ab | 37.46 ± 4.53 Bc | |
| AM | 3.25 ± 0.13 Cd | 1.24 ± 0.07 Bb | 16.39 ± 1.59 Bc | 11.20 ± 1.12 Ac | 7.52 ± 1.19 Ac | 3.66 ± 0.26 Ac | 43.26 ± 4.36 Bc |
| Drying | Solvent | Peak 2 | Peak 4 | Peak 8 | * Peak 10 | * Peak 11 | Peak 12 | TAC (µg /mg) |
|---|---|---|---|---|---|---|---|---|
| O | E | ND | ND | ND | ND | 0.01 ± 0.02 Aa | 1.83 ± 0.13 Aa | 1.84 ± 0.15 Aa |
| EM | 17.50 ± 1.31 Aa | 3.80 ± 0.45 Aa | 17.65 ± 3.01 Aa | 22.04 ± 1.11 Aa | 1.54 ± 0.11 Ab | 36.61 ± 2.34 Ab | 99.14 ± 8.33 Ab | |
| M | 22.46 ± 0.85 Ab | 3.61 ± 0.31 Aa | 17.95 ± 3.20 Aa | 25.50 ± 2.32 Ab | 1.62 ± 0.19 Ab | 40.70 ± 3.18 Ab | 111.84 ± 10.1 Ab | |
| AM | 30.93 ± 2.11 Ac | 4.77 ± 0.41 Ab | 29.11 ± 5.12 Ab | 29.33 ± 2.18 Ac | 1.66 ± 0.25 Ab | 57.99 ± 4.12 Ac | 153.79 ± 14.2 Ac | |
| F | E | ND | ND | ND | ND | 0.02 ± 0.01 Aa | 1.83 ± 0.11 Aa | 1.85 ± 0.12 Aa |
| EM | 10.78 ± 1.21 Ba | 1.95 ± 0.21 Ba | 12.76 ± 2.21 Ba | 14.07 ± 1.13 Ba | 0.65 ± 0.14 Bb | 21.87 ± 2.19 Bb | 62.08 ± 7.09 Bb | |
| M | 18.95 ± 2.10 Ab | 3.39 ± 0.12 Ab | 23.66 ± 4.76 Bb | 21.62 ± 2.10 Ab | 1.49 ± 0.15 Ac | 40.27 ± 3.91 Ac | 109.38 ± 13.1 Ac | |
| AM | 33.34 ± 2.31 Ac | 4.04 ± 0.25 Ac | 30.90 ± 3.14 Ac | 32.62 ± 1.98 Ac | 1.95 ± 0.11 Bd | 57.53 ± 4.32 Ad | 160.38 ± 12.1 Ad | |
| S | E | ND | ND | ND | ND | 0.001 ± 0.02 Ba | 2.14 ± 0.20 Aa | 2.14 ± 0.22 Aa |
| EM | 12.55 ± 0.75 Ba | 2.24 ± 0.18 Ba | 10.89 ± 2.25 Ba | 13.41 ± 1.41 Ba | 0.67 ± 0.13 Bb | 25.68 ± 2.11 Bb | 65.44 ± 6.83 Bb | |
| M | 23.09 ± 1.11 Ab | 4.22 ± 0.20 Bb | 20.24 ± 5.10 Bb | 23.33 ± 2.15 Ab | 1.69 ± 0.10 Ac | 46.42 ± 3.91 Ac | 118.99 ± 12.5 Ac | |
| AM | 35.16 ± 1.95 Ac | 3.72 ± 0.31 Ac | 24.11 ± 4.41 Bb | 28.97 ± 2.41 Ac | 1.92 ± 0.12 Bd | 54.69 ± 4.81 Ac | 148.57 ± 14.0 Ad |
| Drying | Solvent | TPC (mg GAE/g) | TFC (mg QCE/g) | DPPH (IC50 µg/mL) | NO (IC50 µg/mL) |
|---|---|---|---|---|---|
| O | H | 119.35 ± 2.86 Aa | 10.46 ± 2.13 Aa | 299.42 ± 5.57 Aa | 72.30 ± 2.49 Aa |
| E | 226.13 ± 5.77 Ab | 13.32 ± 2.05 Aa | 264.08 ± 8.56 Ab | 43.95 ± 5.40 Ab | |
| EM | 337.98 ± 2.47 Ac | 57.26 ± 2.19 Ab | 45.97 ± 0.36 Ac | 48.88 ± 4.44 Ab | |
| M | 381.73 ± 4.75 Ad | 57.58 ± 3.28 Ab | 42.63 ± 0.22 Ac | 49.63 ± 4.87 Ab | |
| AM | 393.27 ± 1.65 Ad | 107.66 ± 2.36 Ac | 19.32 ± 2.69 Ad | 19.34 ± 3.33 Ac | |
| F | H | 124.88 ± 4.95 Aa | 9.93 ± 0.10 Aa | 762.85 ± 9.63 Aa | 67.45 ± 3.67 Aa |
| E | 237.44 ± 3.50 Ab | 11.28 ± 0.01 Bb | 259.76 ± 4.67 Ab | 31.37 ± 2.03 Bb | |
| EM | 310.77 ± 3.40 Ac | 57.74 ± 0.63 Ac | 21.86 ± 0.28 Bc | 43.33 ± 6.23 Ac | |
| M | 460.83 ± 7.55 Bd | 91.23 ± 1.44 Bd | 35.24 ± 1.32 Bd | 41.20 ± 3.71 Ac | |
| AM | 552.80 ± 9.53 Be | 171.07 ± 0.63 Be | 14.29 ± 0.29 Bc | 14.02 ± 1.65 Bd | |
| S | H | 139.23 ± 6.54 Aa | 9.07 ± 0.16 Ba | 917.34 ± 7.33 Ba | 70.84 ± 2.78 Aa |
| E | 198.33 ± 8.88 Ab | 13.31 ± 0.06 Ab | 411.24 ± 7.43 Bb | 58.16 ± 4.90 Cb | |
| EM | 260.17 ± 1.88 Bc | 87.98 ± 1.04 Bc | 55.86 ± 0.79 Cc | 65.32 ± 7.96 Bb | |
| M | 251.55 ± 4.74 Cc | 82.50 ± 0.63 Cd | 44.56 ± 0.65 Ac | 49.63 ± 2.21 Ac | |
| AM | 404.11 ± 1.10 Ad | 83.93 ± 0.41 Cd | 30.44 ± 0.61 Cd | 20.15 ± 0.92 Ad |
Sample Availability: Not available. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Che Zain, M.S.; Osman, M.F.; Lee, S.Y.; Shaari, K. UHPLC-UV/PDA Method Validation for Simultaneous Quantification of Luteolin and Apigenin Derivatives from Elaeis guineensis Leaf Extracts: An Application for Antioxidant Herbal Preparation. Molecules 2021, 26, 1084. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26041084
Che Zain MS, Osman MF, Lee SY, Shaari K. UHPLC-UV/PDA Method Validation for Simultaneous Quantification of Luteolin and Apigenin Derivatives from Elaeis guineensis Leaf Extracts: An Application for Antioxidant Herbal Preparation. Molecules. 2021; 26(4):1084. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26041084
Chicago/Turabian StyleChe Zain, Mohamad Shazeli, Muhamad Faris Osman, Soo Yee Lee, and Khozirah Shaari. 2021. "UHPLC-UV/PDA Method Validation for Simultaneous Quantification of Luteolin and Apigenin Derivatives from Elaeis guineensis Leaf Extracts: An Application for Antioxidant Herbal Preparation" Molecules 26, no. 4: 1084. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26041084