
Engineering of Challenging G Protein-Coupled Receptors for Structure Determination and Biophysical Studies


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. The Need for G Protein-Coupled Receptors (GPCRs) with Favorable Biophysical Properties
2. Engineering GPCRs Harnessing Directed Evolution
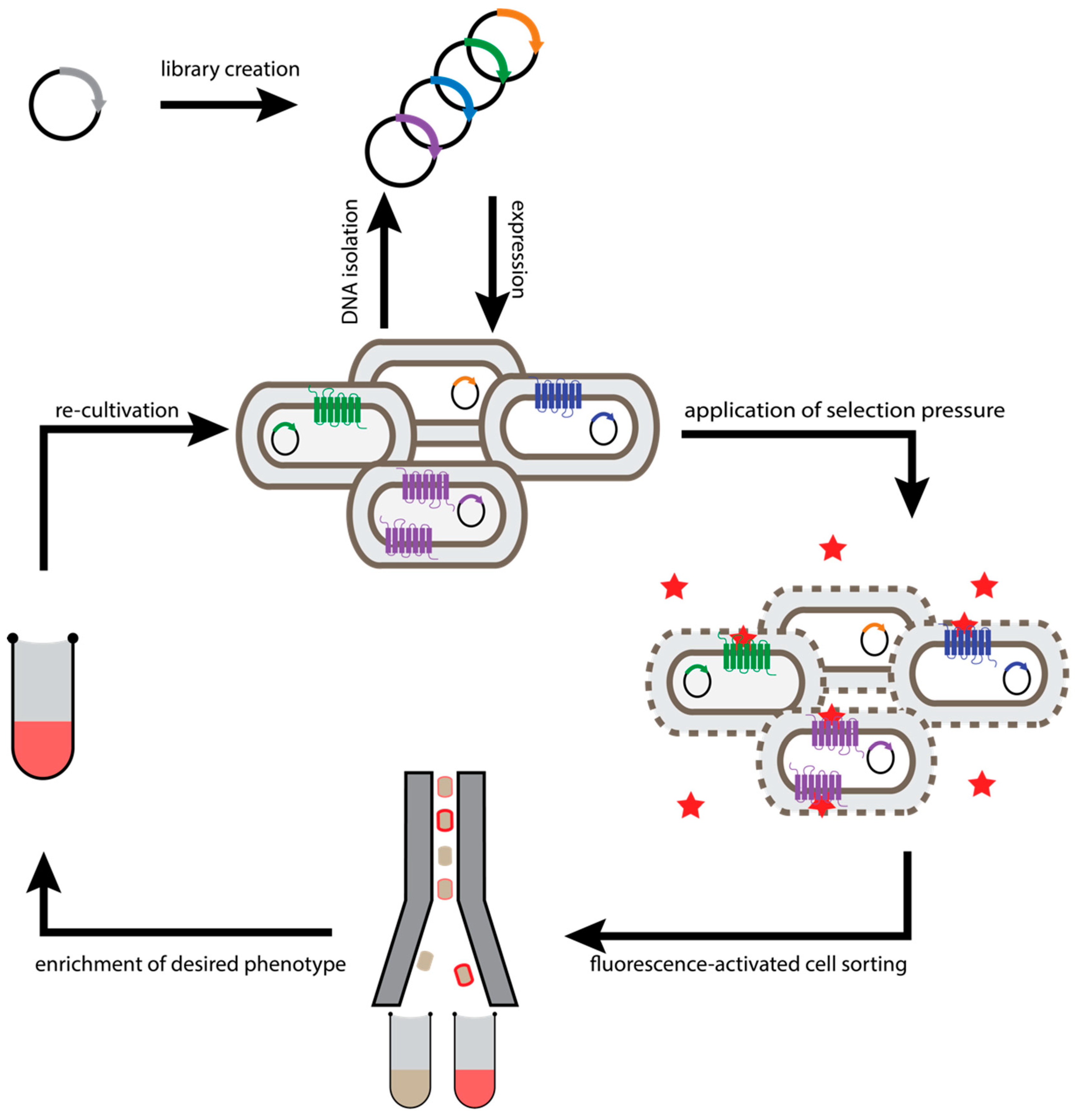
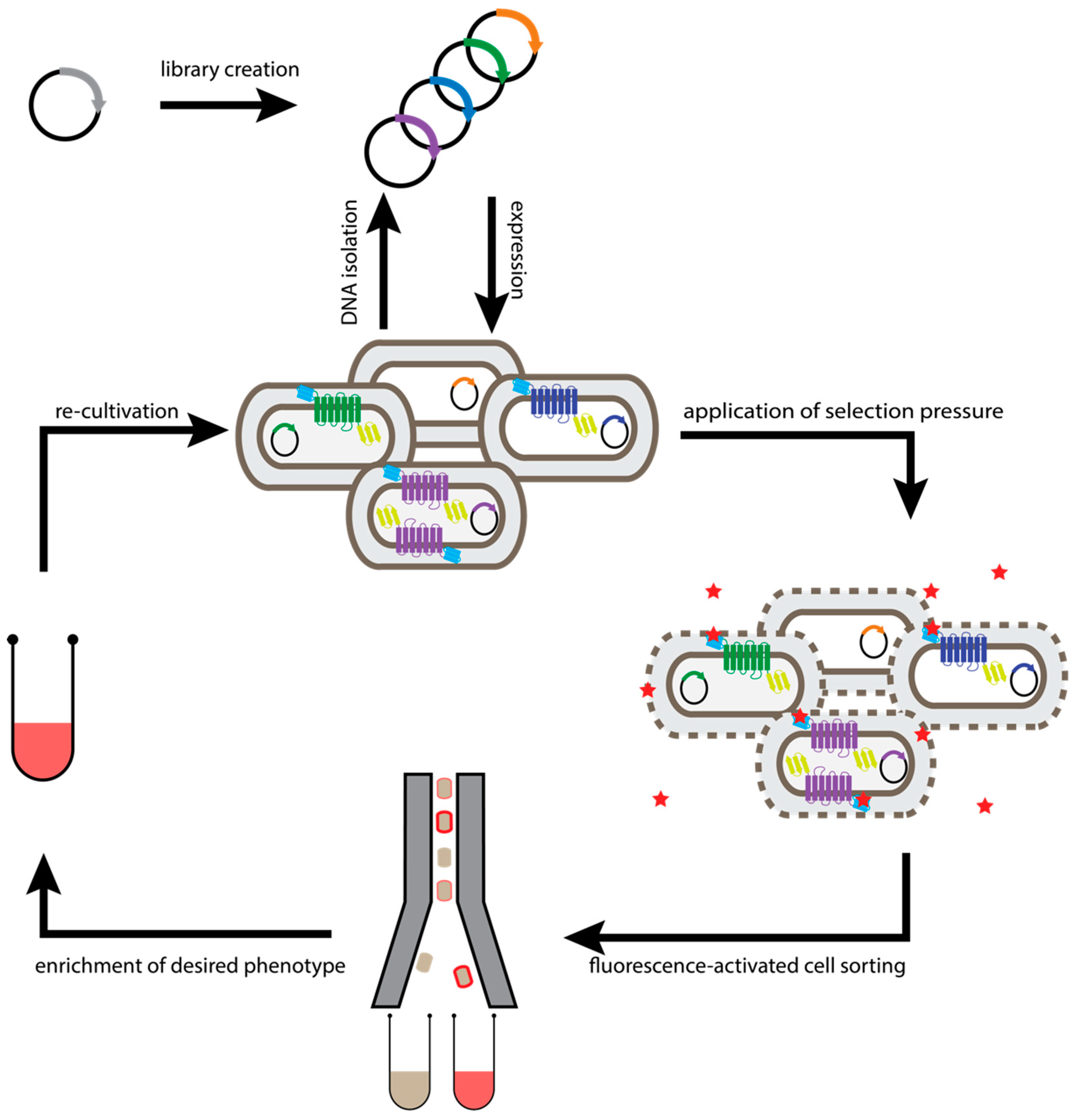
2.1. Escherichia Coli-Based Directed Evolution
2.2. Generic Selection of GPCRs
2.3. Cellular High-Throughput Encapsulation, Solubilization and Screening (CHESS)
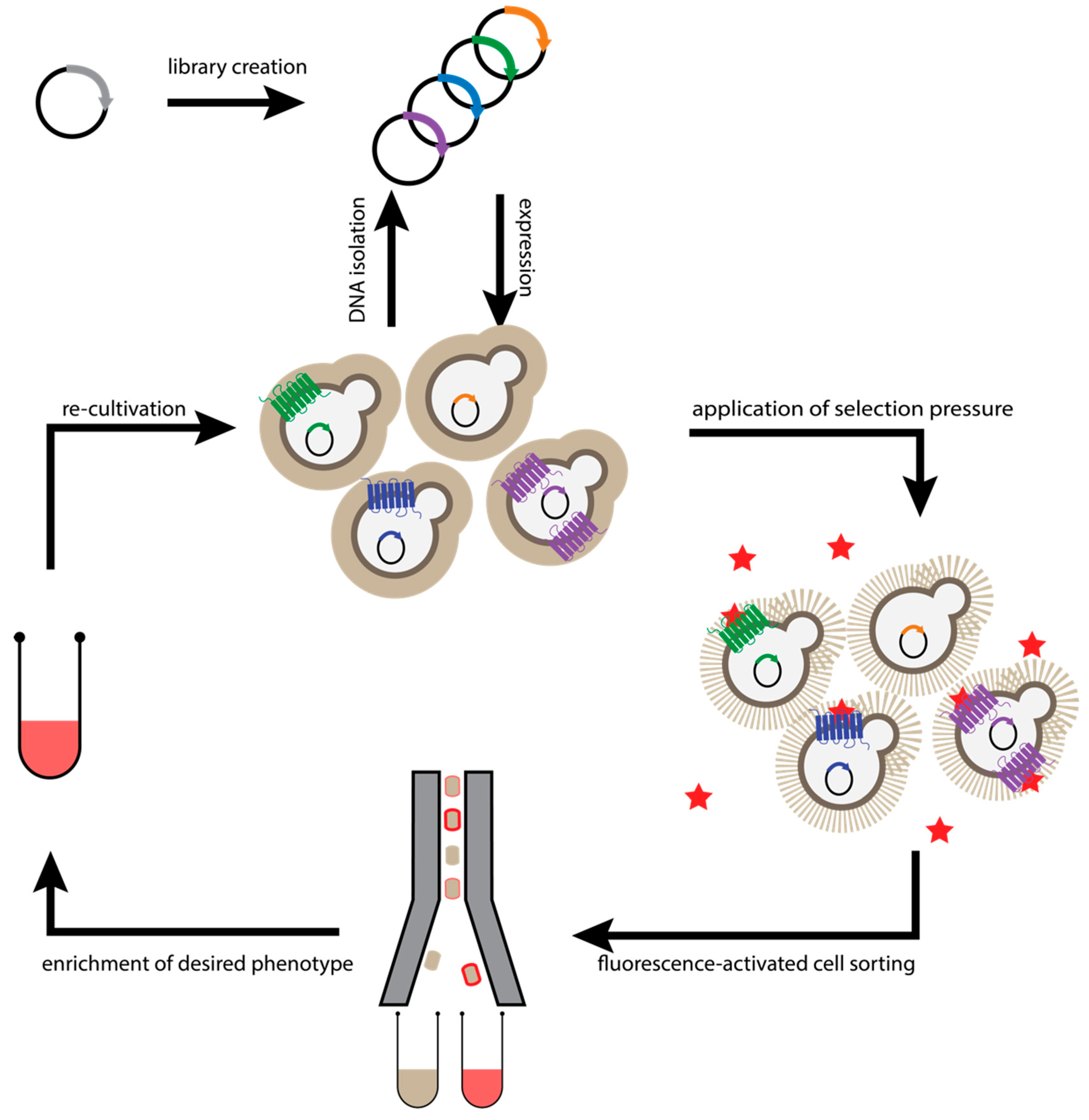
2.4. Saccharomyces Cerevisiae-Based Receptor Evolution (SaBRE)
3. Insight Obtained from High-Resolution GPCR Structures
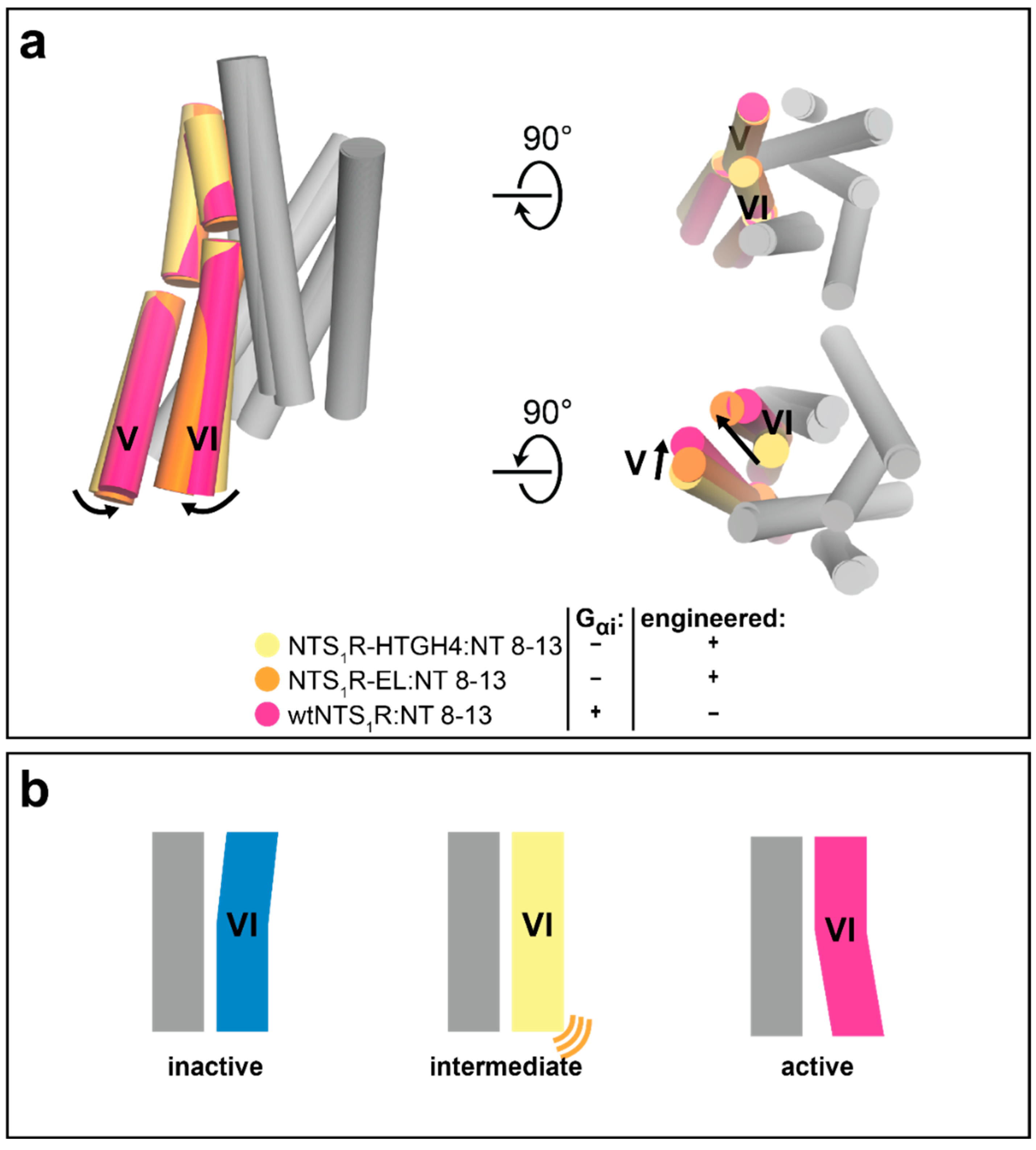
3.1. Advances in Understanding Class A GPCR Function
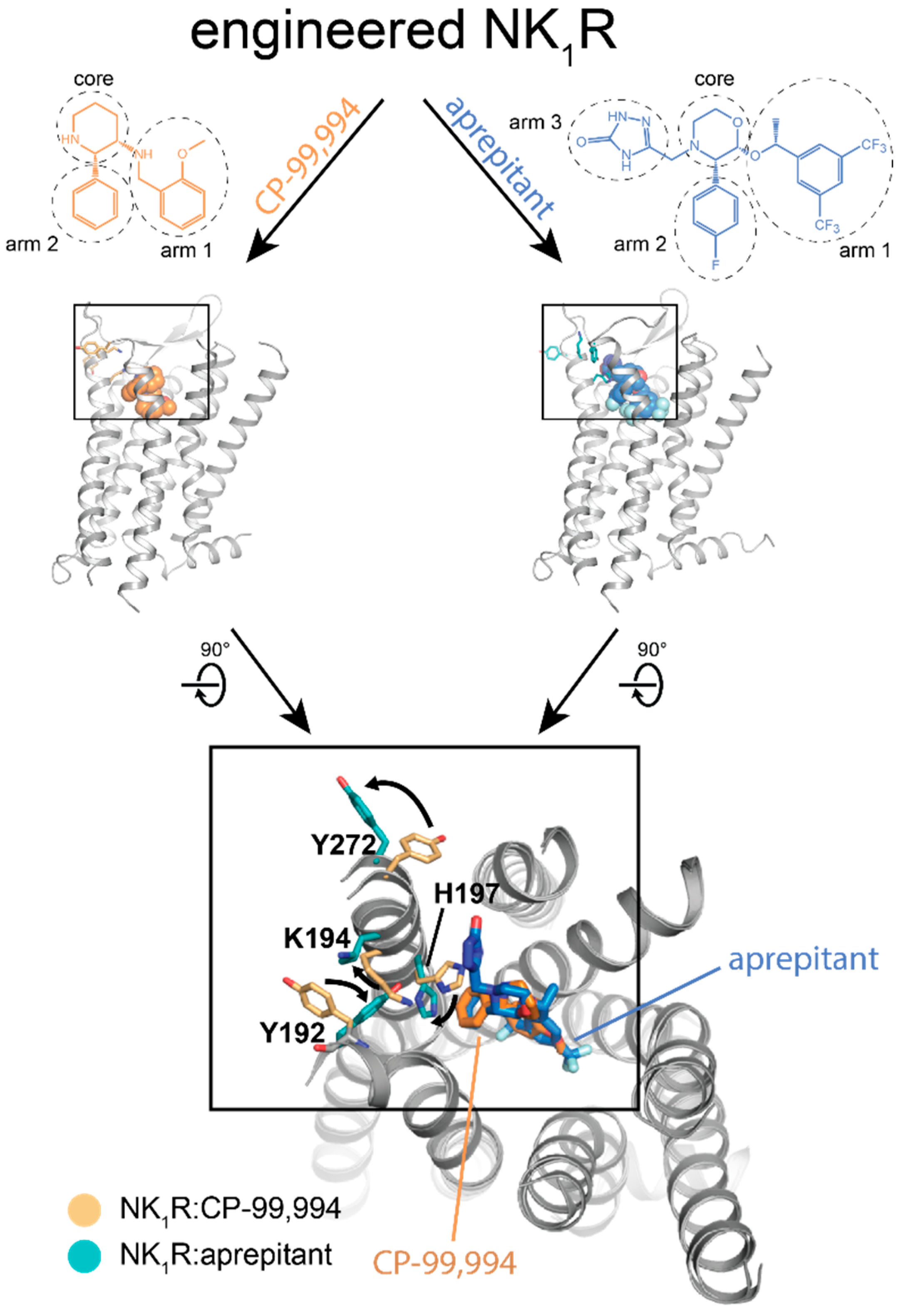
3.2. Insights into Receptor–Ligand Interaction
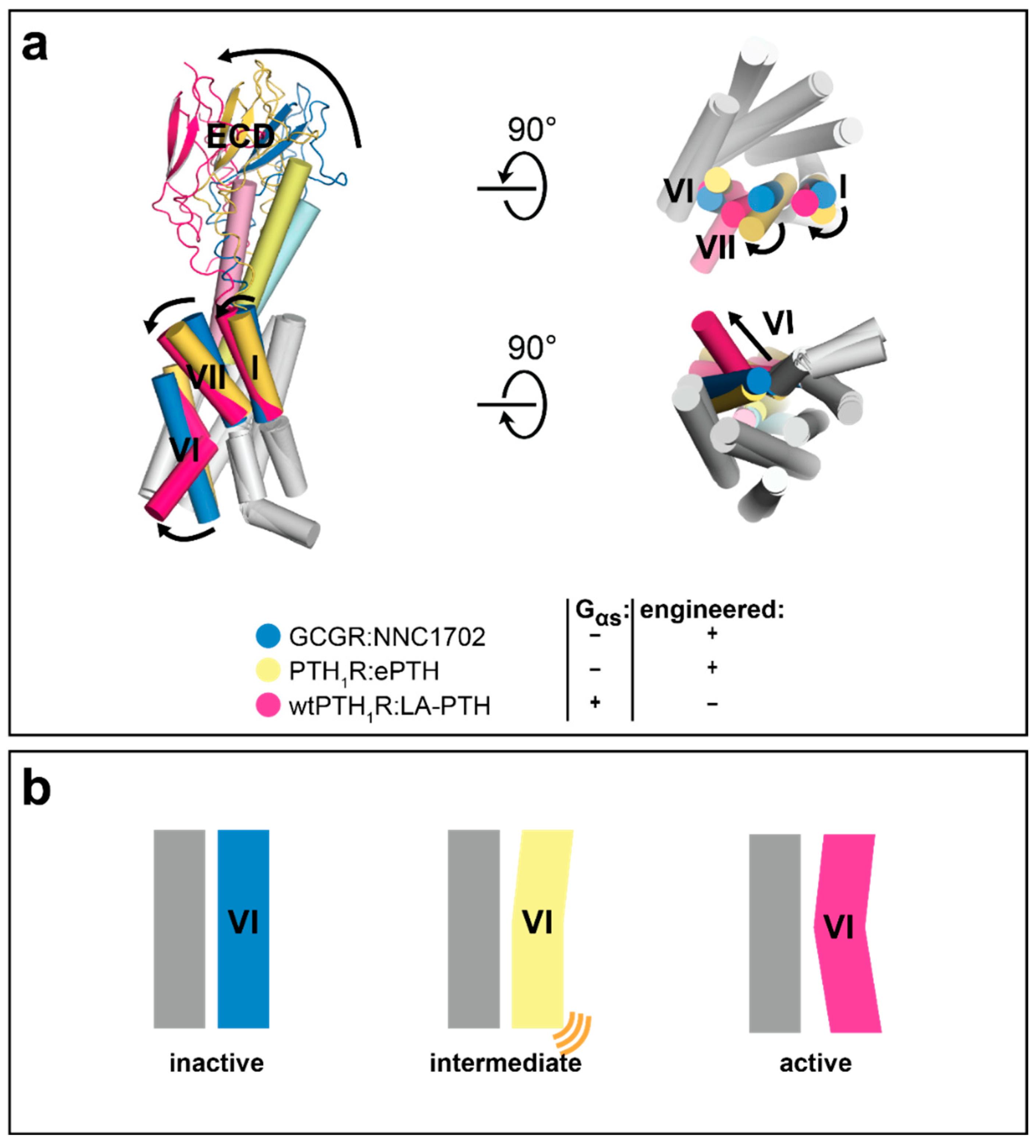
3.3. Advances in Understanding Class B GPCR Function
4. Engineered Receptors Outside Crystallography
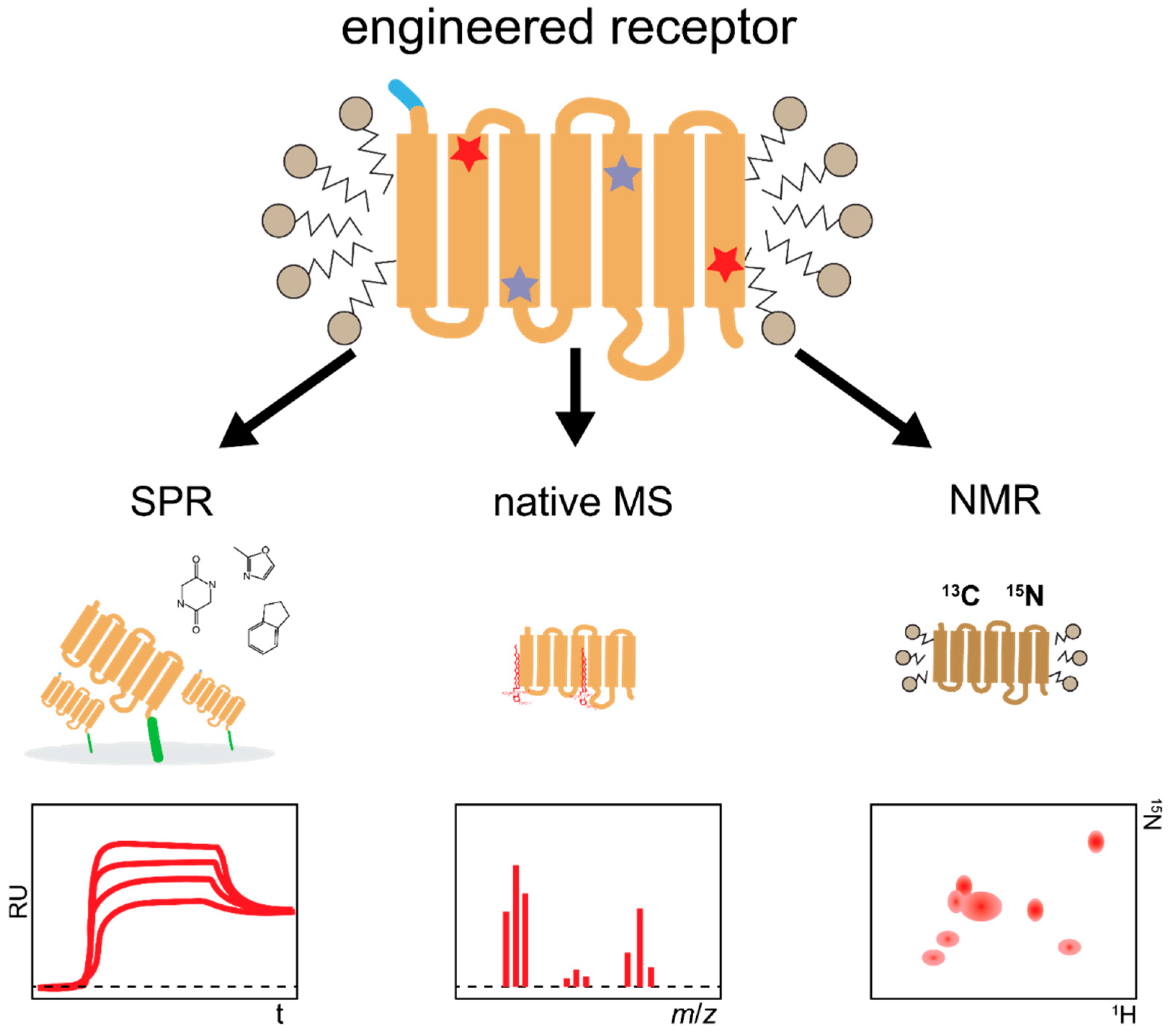
4.1. Biophysical Techniques for Structural and Functional Studies
4.2. Drug Screening
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Foord, S.M. Receptor classification: Post genome. Curr. Opin. Pharmacol. 2002, 2, 561–566. [Google Scholar] [CrossRef]
- Fredriksson, R.; Gloriam, D.E.I.; Höglund, P.J.; Lagerström, M.C.; Schiöth, H.B. There exist at least 30 human G-protein-coupled receptors with long Ser/Thr-rich N-termini. Biochem. Biophys. Res. Commun. 2003, 301, 725–734. [Google Scholar] [CrossRef]
- Sriram, K.; Insel, P.A. G protein-coupled receptors as targets for approved drugs: How many targets and how many drugs? Mol. Pharmacol. 2018, 93, 251–258. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, K.A. New paradigms in GPCR drug discovery. Biochem. Pharmacol. 2015, 98, 541–555. [Google Scholar] [CrossRef] [Green Version]
- Congreve, M.; De Graaf, C.; Swain, N.A.; Tate, C.G. Impact of GPCR structures on drug discovery. Cell 2020, 181, 81–91. [Google Scholar] [CrossRef]
- García-Nafría, J.; Tate, C.G. Cryo-electron microscopy: Moving beyond X-ray crystal structures for drug receptors and drug development. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 51–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renaud, J.-P.; Chari, A.; Ciferri, C.; Liu, W.-T.; Rémigy, H.-W.; Stark, H. Cryo-EM in drug discovery: Achievements, limitations and prospects. Nat. Rev. Drug Discov. 2018, 17, 471–492. [Google Scholar] [CrossRef] [PubMed]
- Casanas, A.; Warshamanage, R.; Finke, A.D.; Panepucci, E.; Olieric, V.; Nöll, A. EIGER detector: Application in macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2016, 72, 1036–1048. [Google Scholar] [CrossRef] [PubMed]
- Martiel, I.; Buntschu, D.; Meier, N.; Gobbo, A.; Panepucci, E.; Schneider, R. The TELL automatic sample changer for macromolecular crystallography. J. Synchrotron. Radiat. 2020, 27, 860–863. [Google Scholar] [CrossRef]
- Rucktooa, P.; Cheng, R.K.Y.; Segala, E.; Geng, T.; Errey, J.C.; Brown, G.A. Towards high throughput GPCR crystallography: In meso soaking of Adenosine A2A receptor crystals. Sci. Rep. 2018, 8, 41. [Google Scholar] [CrossRef] [Green Version]
- Scott, D.J.; Kummer, L.; Tremmel, D.; Plückthun, A. Stabilizing membrane proteins through protein engineering. Curr. Opin. Chem. Biol. 2013, 17, 427–435. [Google Scholar] [CrossRef]
- Tate, C.G.; Schertler, G.F.X. Engineering G protein-coupled receptors to facilitate their structure determination. Curr. Opin. Struct. Biol. 2009, 19, 386–395. [Google Scholar] [CrossRef]
- Grisshammer, R. New approaches towards the understanding of integral membrane proteins: A structural perspective on G protein-coupled receptors. Protein Sci. 2017, 26, 1493–1504. [Google Scholar] [CrossRef] [Green Version]
- Tate, C.G. A crystal clear solution for determining G-protein-coupled receptor structures. Trends Biochem. Sci. 2012, 37, 343–352. [Google Scholar] [CrossRef]
- Thal, D.M.; Vuckovic, Z.; Draper-Joyce, C.J.; Liang, Y.-L.; Glukhova, A.; Christopoulos, A. Recent advances in the determination of G protein-coupled receptor structures. Curr. Opin. Chem. Biol. 2018, 51, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Ehrenmann, J.; Schöppe, J.; Klenk, C.; Rappas, M.; Kummer, L.; Doré, A.S. High-resolution crystal structure of parathyroid hormone 1 receptor in complex with a peptide agonist. Nat. Struct. Mol. Biol. 2018, 25, 1086–1092. [Google Scholar] [CrossRef] [PubMed]
- Egloff, P.; Hillenbrand, M.; Klenk, C.; Batyuk, A.; Heine, P.; Balada, S. Structure of signaling-competent neurotensin receptor 1 obtained by directed evolution in Escherichia coli. Proc. Natl. Acad. Sci. USA 2014, 111, E655–E662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waltenspühl, Y.; Schöppe, J.; Ehrenmann, J.; Kummer, L.; Plückthun, A. Crystal structure of the human oxytocin receptor. Sci. Adv. 2020, 6, eabb5419. [Google Scholar] [CrossRef] [PubMed]
- Schöppe, J.; Ehrenmann, J.; Klenk, C.; Rucktooa, P.; Schutz, M.; Doré, A.S. Crystal structures of the human neurokinin 1 receptor in complex with clinically used antagonists. Nat. Commun. 2019, 10, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, R.K.Y.; Fiez-Vandal, C.; Schlenker, O.; Edman, K.; Aggeler, B.; Brown, D.G. Structural insight into allosteric modulation of protease-activated receptor 2. Nature 2017, 545, 112–115. [Google Scholar] [CrossRef]
- Doré Andrew, S.; Robertson, N.; Errey James, C.; Ng, I.; Hollenstein, K.; Tehan, B. Structure of the adenosine A2A receptor in complex with ZM241385 and the xanthines XAC and caffeine. Structure 2011, 19, 1283–1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jazayeri, A.; Doré, A.S.; Lamb, D.; Krishnamurthy, H.; Southall, S.M.; Baig, A.H. Extra-helical binding site of a glucagon receptor antagonist. Nature 2016, 533, 274–277. [Google Scholar] [CrossRef]
- Jazayeri, A.; Rappas, M.; Brown, A.J.H.; Kean, J.; Errey, J.C.; Robertson, N.J. Crystal structure of the GLP-1 receptor bound to a peptide agonist. Nature 2017, 546, 254–258. [Google Scholar] [CrossRef]
- Robertson, N.; Rappas, M.; Doré, A.S.; Brown, J.; Bottegoni, G.; Koglin, M. Structure of the complement C5a receptor bound to the extra-helical antagonist NDT9513727. Nature 2018, 553, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Warne, T.; Serrano-Vega, M.J.; Baker, J.G.; Moukhametzianov, R.; Edwards, P.C.; Henderson, R. Structure of a β1-adrenergic G protein-coupled receptor. Nature 2008, 454, 486–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, J.F.; Noinaj, N.; Shibata, Y.; Love, J.; Kloss, B.; Xu, F. Structure of the agonist-bound neurotensin receptor. Nature 2012, 490, 508–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deupi, X.; Kobilka, B. Activation of G protein-coupled receptors. Adv. Protein Chem. 2007, 74, 137–166. [Google Scholar] [PubMed]
- Weis, W.I.; Kobilka, B.K. The molecular basis of G protein–coupled receptor activation. Annu. Rev. Biochem. 2018, 87, 897–919. [Google Scholar] [CrossRef] [PubMed]
- Magnani, F.; Serrano-Vega, M.J.; Shibata, Y.; Abdul-Hussein, S.; Lebon, G.; Miller-Gallacher, J. A mutagenesis and screening strategy to generate optimally thermostabilized membrane proteins for structural studies. Nat. Protoc. 2016, 11, 1554–1571. [Google Scholar] [CrossRef]
- Magnani, F.; Shibata, Y.; Serrano-Vega, M.J.; Tate, C.G. Co-evolving stability and conformational homogeneity of the human adenosine A2A receptor. Proc. Natl. Acad. Sci. USA 2008, 105, 10744–10749. [Google Scholar] [CrossRef] [Green Version]
- Serrano-Vega, M.J.; Magnani, F.; Shibata, Y.; Tate, C.G. Conformational thermostabilization of the β1-adrenergic receptor in a detergent-resistant form. Proc. Natl. Acad. Sci. USA 2008, 105, 877–882. [Google Scholar] [CrossRef] [Green Version]
- Shibata, Y.; Gvozdenovic-Jeremic, J.; Love, J.; Kloss, B.; White, J.F.; Grisshammer, R. Optimising the combination of thermostabilising mutations in the neurotensin receptor for structure determination. Biochim. Biophys. Acta Biomembr. 2013, 1828, 1293–1301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, C.A.; Dodevski, I.; Kenig, M.; Dudli, S.; Mohr, A.; Hermans, E. Directed evolution of a G protein-coupled receptor for expression, stability, and binding selectivity. Proc. Natl. Acad. Sci. USA 2008, 105, 14808–14813. [Google Scholar] [CrossRef] [Green Version]
- Schlinkmann, K.M.; Hillenbrand, M.; Rittner, A.; Künz, M.; Strohner, R.; Plückthun, A. Maximizing detergent stability and functional expression of a GPCR by exhaustive recombination and evolution. J. Mol. Biol. 2012, 422, 414–428. [Google Scholar] [CrossRef] [PubMed]
- Schlinkmann, K.M.; Plückthun, A. Directed evolution of G protein-coupled receptors for high functional expression and detergent stability. Methods Enzymol. 2013, 520, 67–97. [Google Scholar] [CrossRef] [PubMed]
- Klenk, C.; Ehrenmann, J.; Schütz, M.; Plückthun, A. A generic selection system for improved expression and thermostability of G protein-coupled receptors by directed evolution. Sci. Rep. 2016, 6, 21294. [Google Scholar] [CrossRef] [Green Version]
- Schütz, M.; Schöppe, J.; Sedlák, E.; Hillenbrand, M.; Nagy-Davidescu, G.; Ehrenmann, J. Directed evolution of G protein-coupled receptors in yeast for higher functional production in eukaryotic expression hosts. Sci. Rep. 2016, 6, 21508. [Google Scholar] [CrossRef] [Green Version]
- Scott, D.J.; Plückthun, A. Direct molecular evolution of detergent-stable G protein-coupled receptors using polymer encapsulated cells. J. Mol. Biol. 2013, 425, 662–677. [Google Scholar] [CrossRef]
- Dodevski, I.; Plückthun, A. Evolution of three human GPCRs for higher expression and stability. J. Mol. Biol. 2011, 408, 599–615. [Google Scholar] [CrossRef]
- Schlinkmann, K.M.; Honegger, A.; Türeci, E.; Robison, K.E.; Lipovšek, D.; Plückthun, A. Critical features for biosynthesis, stability, and functionality of a G protein-coupled receptor uncovered by all-versus-all mutations. Proc. Natl. Acad. Sci. USA 2012, 109, 9810–9815. [Google Scholar] [CrossRef] [Green Version]
- Klepsch, M.M.; Persson, J.O.; De Gier, J.-W.L. Consequences of the overexpression of a eukaryotic membrane protein, the human KDEL receptor, in Escherichia coli. J. Mol. Biol. 2011, 407, 532–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlegel, S.; Rujas, E.; Ytterberg, A.J.; Zubarev, R.A.; Luirink, J.; De Gier, J.-W. Optimizing heterologous protein production in the periplasm of E. coli by regulating gene expression levels. Microb. Cell Fact. 2013, 12, 24. [Google Scholar] [CrossRef] [Green Version]
- Wagner, S.; Baars, L.; Ytterberg, A.J.; Klussmeier, A.; Wagner, C.S.; Nord, O. Consequences of membrane protein overexpression in Escherichia coli. Mol. Cell. Proteom. 2007, 6, 1527–1550. [Google Scholar] [CrossRef] [Green Version]
- Schütz, M.; Batyuk, A.; Klenk, C.; Kummer, L.; De Picciotto, S.; Gülbakan, B. Generation of fluorogen-activating designed ankyrin repeat proteins (FADAs) as versatile sensor tools. J. Mol. Biol. 2016, 428, 1272–1289. [Google Scholar] [CrossRef]
- Scott, D.J.; Kummer, L.; Egloff, P.; Bathgate, R.A.D.; Plückthun, A. Improving the apo-state detergent stability of NTS1 with CHESS for pharmacological and structural studies. Biochim. Biophys. Acta Biomembr. 2014, 1838, 2817–2824. [Google Scholar] [CrossRef] [Green Version]
- Deluigi, M.; Klipp, A.; Klenk, C.; Merklinger, L.; Eberle, S.A.; Morstein, L. Complexes of the neurotensin receptor 1 with small-molecule ligands reveal structural determinants of full, partial and inverse agonism. Sci. Adv. 2021, 7, eabe5504. [Google Scholar] [CrossRef] [PubMed]
- Manglik, A.; Kim Tae, H.; Masureel, M.; Altenbach, C.; Yang, Z.; Hilger, D. Structural insights into the dynamic process of β2-adrenergic receptor signaling. Cell 2015, 161, 1101–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibata, Y.; White, J.F.; Serrano-Vega, M.J.; Magnani, F.; Aloia, A.L.; Grisshammer, R. Thermostabilization of the neurotensin receptor NTS1. J. Mol. Biol. 2009, 390, 262–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krumm, B.E.; Lee, S.; Bhattacharya, S.; Botos, I.; White, C.F.; Du, H. Structure and dynamics of a constitutively active neurotensin receptor. Sci. Rep. 2016, 6, 38564. [Google Scholar] [CrossRef] [Green Version]
- Kato, H.E.; Zhang, Y.; Hu, H.; Suomivuori, C.-M.; Kadji, F.M.N.; Aoki, J. Conformational transitions of a neurotensin receptor 1–Gi1 complex. Nature 2019, 572, 80–85. [Google Scholar] [CrossRef]
- Hilger, D.; Kumar, K.K.; Hu, H.; Pedersen, M.F.; O’Brien, E.S.; Giehm, L. Structural insights into differences in G protein activation by family A and family B GPCRs. Science 2020, 369, eaba3373. [Google Scholar] [CrossRef]
- Mattedi, G.; Acosta-Gutiérrez, S.; Clark, T.; Gervasio, F.L. A combined activation mechanism for the glucagon receptor. Proc. Natl. Acad. Sci. USA 2020, 117, 15414–15422. [Google Scholar] [CrossRef]
- Chang, R.; Zhang, X.; Qiao, A.; Dai, A.; Belousoff, M.J.; Tan, Q. Cryo-electron microscopy structure of the glucagon receptor with a dual-agonist peptide. J. Biol. Chem. 2020. [Google Scholar] [CrossRef]
- Liang, Y.-L.; Khoshouei, M.; Glukhova, A.; Furness, S.G.B.; Zhao, P.; Clydesdale, L. Phase-plate cryo-EM structure of a biased agonist-bound human GLP-1 receptor–Gs complex. Nature 2018, 555, 121–125. [Google Scholar] [CrossRef]
- Liang, Y.-L.; Khoshouei, M.; Radjainia, M.; Zhang, Y.; Glukhova, A.; Tarrasch, J. Phase-plate cryo-EM structure of a class B GPCR–G-protein complex. Nature 2017, 546, 118–123. [Google Scholar] [CrossRef]
- Liang, Y.-L.; Zhao, P.; Draper-Joyce, C.; Baltos, J.-A.; Glukhova, A.; Truong, T.T. Dominant negative G proteins enhance formation and purification of agonist-GPCR-G protein complexes for structure determination. ACS Pharmacol. Transl. Sci. 2018, 1, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Qiao, A.; Han, S.; Li, X.; Li, Z.; Zhao, P.; Dai, A. Structural basis of Gs and Gi recognition by the human glucagon receptor. Science 2020, 367, 1346–1352. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Song, X.; Zhang, D.; Chen, X.; Li, X.; Sun, Y. Cryo-EM structures of PAC1 receptor reveal ligand binding mechanism. Cell Res. 2020, 30, 436–445. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Sun, B.; Feng, D.; Hu, H.; Chu, M.; Qu, Q. Cryo-EM structure of the activated GLP-1 receptor in complex with a G protein. Nature 2017, 546, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.H.; Ma, S.; Sutkeviciute, I.; Shen, D.D.; Zhou, X.E.; De Waal, P.W. Structure and dynamics of the active human parathyroid hormone receptor-1. Science 2019, 364, 148–153. [Google Scholar] [CrossRef]
- Zhao, P.; Liang, Y.L.; Belousoff, M.J.; Deganutti, G.; Fletcher, M.M.; Willard, F.S. Activation of the GLP-1 receptor by a non-peptidic agonist. Nature 2020, 577, 432–436. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Qiao, A.; Yang, L.; Van Eps, N.; Frederiksen, K.S.; Yang, D. Structure of the glucagon receptor in complex with a glucagon analogue. Nature 2018, 553, 106–110. [Google Scholar] [CrossRef]
- Latorraca, N.R.; Venkatakrishnan, A.J.; Dror, R.O. GPCR dynamics: Structures in motion. Chem. Rev. 2017, 117, 139–155. [Google Scholar] [CrossRef] [PubMed]
- Bumbak, F.; Keen, A.C.; Gunn, N.J.; Gooley, P.R.; Bathgate, R.A.D.; Scott, D.J. Optimization and 13CH3 methionine labeling of a signaling competent neurotensin receptor 1 variant for NMR studies. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1372–1383. [Google Scholar] [CrossRef]
- Goba, I.; Goricanec, D.; Schum, D.; Hillenbrand, M.; Plückthun, A.; Hagn, F. Probing the conformational states of neurotensin receptor 1 variants by NMR site-directed methyl labeling. ChemBioChem 2020, 22, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Vaid, T.M.; Chalmers, D.K.; Scott, D.J.; Gooley, P.R. INPHARMA-based determination of ligand binding modes at α1-adrenergic receptors explains the molecular basis of subtype selectivity. Chemistry 2020, 26, 11796–11805. [Google Scholar] [CrossRef]
- Yong, K.J.; Vaid, T.M.; Shilling, P.J.; Wu, F.-J.; Williams, L.M.; Deluigi, M. Determinants of ligand subtype-selectivity at α1A-adrenoceptor revealed using saturation transfer difference (STD) NMR. ACS Chem. Biol. 2018, 13, 1090–1102. [Google Scholar] [CrossRef] [PubMed]
- Horst, R.; Liu, J.J.; Stevens, R.C.; Wüthrich, K. β2-adrenergic receptor activation by agonists studied with 19F NMR spectroscopy. Angew. Chem. Int. Ed. 2013, 52, 10762–10765. [Google Scholar] [CrossRef] [Green Version]
- Isogai, S.; Deupi, X.; Opitz, C.; Heydenreich, F.M.; Tsai, C.-J.; Brueckner, F. Backbone NMR reveals allosteric signal transduction networks in the β1-adrenergic receptor. Nature 2016, 530, 237–241. [Google Scholar] [CrossRef]
- Kofuku, Y.; Ueda, T.; Okude, J.; Shiraishi, Y.; Kondo, K.; Maeda, M. Efficacy of the β2-adrenergic receptor is determined by conformational equilibrium in the transmembrane region. Nat. Commun. 2012, 3, 1045. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.J.; Horst, R.; Katritch, V.; Stevens, R.C.; Wüthrich, K. Biased signaling pathways in β2-adrenergic receptor characterized by 19F-NMR. Science 2012, 335, 1106–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solt, A.S.; Bostock, M.J.; Shrestha, B.; Kumar, P.; Warne, T.; Tate, C.G. Insight into partial agonism by observing multiple equilibria for ligand-bound and Gs-mimetic nanobody-bound β1-adrenergic receptor. Nat. Commun. 2017, 8, 1795. [Google Scholar] [CrossRef] [PubMed]
- Sounier, R.; Mas, C.; Steyaert, J.; Laeremans, T.; Manglik, A.; Huang, W. Propagation of conformational changes during μ-opioid receptor activation. Nature 2015, 524, 375–378. [Google Scholar] [CrossRef] [Green Version]
- Ye, L.; Van Eps, N.; Zimmer, M.; Ernst, O.P.; Scott Prosser, R. Activation of the A2A adenosine G-protein-coupled receptor by conformational selection. Nature 2016, 533, 265–268. [Google Scholar] [CrossRef] [PubMed]
- Schuster, M.; Deluigi, M.; Pantić, M.; Vacca, S.; Baumann, C.; Scott, D.J. Optimizing the α1B-adrenergic receptor for solution NMR studies. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183354. [Google Scholar] [CrossRef]
- Goricanec, D.; Stehle, R.; Egloff, P.; Grigoriu, S.; Plückthun, A.; Wagner, G. Conformational dynamics of a G-protein α subunit is tightly regulated by nucleotide binding. Proc. Natl. Acad. Sci. USA 2016, 113, E3629. [Google Scholar] [CrossRef] [Green Version]
- Yen, H.-Y.; Hoi, K.K.; Liko, I.; Hedger, G.; Horrell, M.R.; Song, W. PtdIns(4,5)P2 stabilizes active states of GPCRs and enhances selectivity of G-protein coupling. Nature 2018, 559, 423–427. [Google Scholar] [CrossRef]
- Gavriilidou, A.F.M.; Hunziker, H.; Mayer, D.; Vuckovic, Z.; Veprintsev, D.B.; Zenobi, R. Insights into the basal activity and activation mechanism of the β1 adrenergic receptor using native mass spectrometry. J. Am. Soc. Mass Spectrom. 2019, 30, 529–537. [Google Scholar] [CrossRef] [Green Version]
- Lagerström, M.C.; Schiöth, H.B. Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat. Rev. Drug Discov. 2008, 7, 339–357. [Google Scholar] [CrossRef]
- Russ, A.P.; Lampel, S. The druggable genome: An update. Drug Discov. Today 2005, 10, 1607–1610. [Google Scholar] [CrossRef]
- Zheng, C.J.; Han, L.Y.; Yap, C.W.; Xie, B.; Chen, Y.Z. Trends in exploration of therapeutic targets. Drug News Perspect. 2005, 18, 109–127. [Google Scholar] [CrossRef]
- Navratilova, I.; Besnard, J.; Hopkins, A.L. Screening for GPCR ligands using surface plasmon resonance. ACS Med. Chem. Lett. 2011, 2, 549–554. [Google Scholar] [CrossRef]
- Navratilova, I.; Sodroski, J.; Myszka, D.G. Solubilization, stabilization, and purification of chemokine receptors using biosensor technology. Anal. Biochem. 2005, 339, 271–281. [Google Scholar] [CrossRef]
- Shepherd, C.A.; Hopkins, A.L.; Navratilova, I. Fragment screening by SPR and advanced application to GPCRs. Prog. Biophys. Mol. Biol. 2014, 116, 113–123. [Google Scholar] [CrossRef] [Green Version]
- Christopher, J.A.; Brown, J.; Doré, A.S.; Errey, J.C.; Koglin, M.; Marshall, F.H. Biophysical fragment screening of the β1-adrenergic receptor: Identification of high affinity arylpiperazine leads using structure-based drug design. J. Med. Chem. 2013, 56, 3446–3455. [Google Scholar] [CrossRef]
- Congreve, M.; Rich, R.L.; Myszka, D.G.; Figaroa, F.; Siegal, G.; Marshall, F.H. Fragment screening of stabilized G protein-coupled receptors using biophysical methods. Methods Enzymol. 2011, 493, 115–136. [Google Scholar] [CrossRef] [PubMed]
- Rich, R.L.; Errey, J.; Marshall, F.; Myszka, D.G. Biacore analysis with stabilized G-protein-coupled receptors. Anal. Biochem. 2011, 409, 267–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, S.; Casagrande, F.; Hug, M.N.; Wang, L.; Heine, P.; Kummer, L. SPR-based fragment screening with neurotensin receptor 1 generates novel small molecule ligands. PLoS ONE 2017, 12, e0175842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranganathan, A.; Heine, P.; Rudling, A.; Plückthun, A.; Kummer, L.; Carlsson, J. Ligand discovery for a peptide-binding GPCR by structure-based screening of fragment- and lead-like chemical libraries. ACS Chem. Biol. 2017, 12, 735–745. [Google Scholar] [CrossRef]
- Heine, P.; Witt, G.; Gilardi, A.; Gribbon, P.; Kummer, L.; Plückthun, A. High-throughput fluorescence polarization assay to identify ligands using purified G protein-coupled receptor. SLAS Discov. 2019, 24, 915–927. [Google Scholar] [CrossRef] [PubMed]
- Vanwetswinkel, S.; Heetebrij, R.J.; Van Duynhoven, J.; Hollander, J.G.; Filippov, D.V.; Hajduk, P.J. TINS, target immobilized NMR screening: An efficient and sensitive method for ligand discovery. Chem. Biol. 2005, 12, 207–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Ranganathan, A.; Ijzerman, A.P.; Siegal, G.; Carlsson, J. Complementarity between in silico and biophysical screening approaches in fragment-based lead discovery against the A2A adenosine receptor. J. Chem. Inf. Model. 2013, 53, 2701–2714. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Waltenspühl, Y.; Ehrenmann, J.; Klenk, C.; Plückthun, A. Engineering of Challenging G Protein-Coupled Receptors for Structure Determination and Biophysical Studies. Molecules 2021, 26, 1465. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26051465
Waltenspühl Y, Ehrenmann J, Klenk C, Plückthun A. Engineering of Challenging G Protein-Coupled Receptors for Structure Determination and Biophysical Studies. Molecules. 2021; 26(5):1465. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26051465
Chicago/Turabian StyleWaltenspühl, Yann, Janosch Ehrenmann, Christoph Klenk, and Andreas Plückthun. 2021. "Engineering of Challenging G Protein-Coupled Receptors for Structure Determination and Biophysical Studies" Molecules 26, no. 5: 1465. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26051465