
Discovery of a Novel Acetylcholinesterase Inhibitor by Fragment-Based Design and Virtual Screening

 ,
,  ,
,
Abstract
:1. Introduction
2. Results
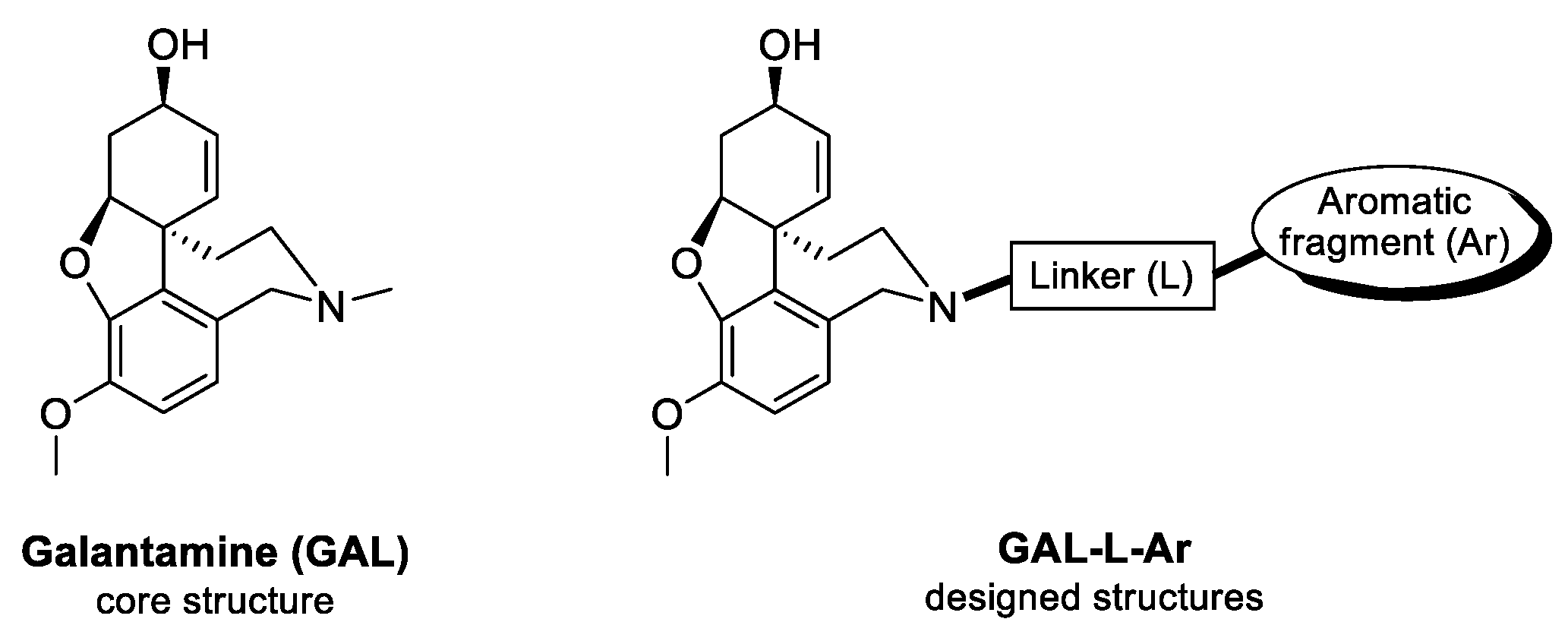
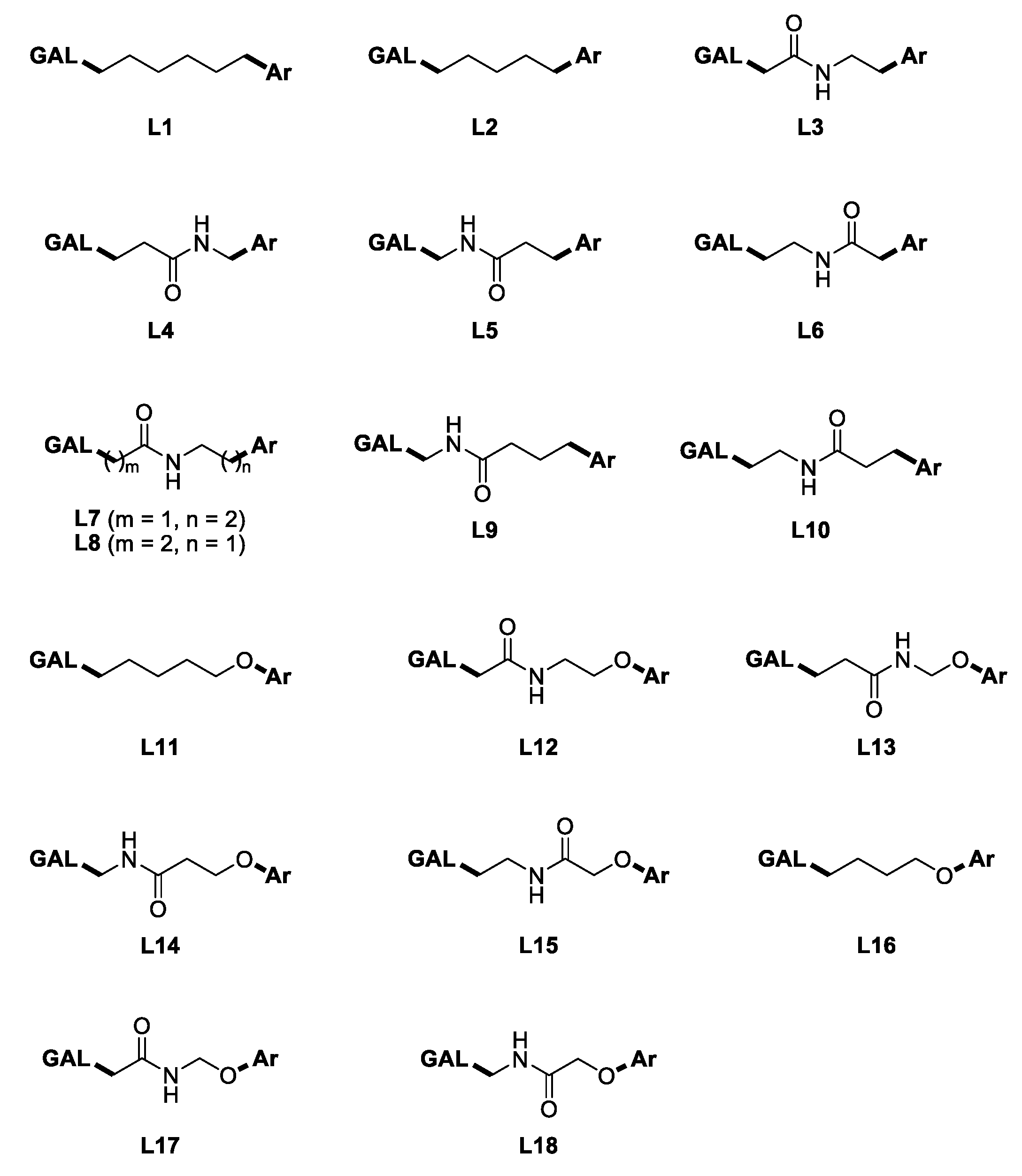
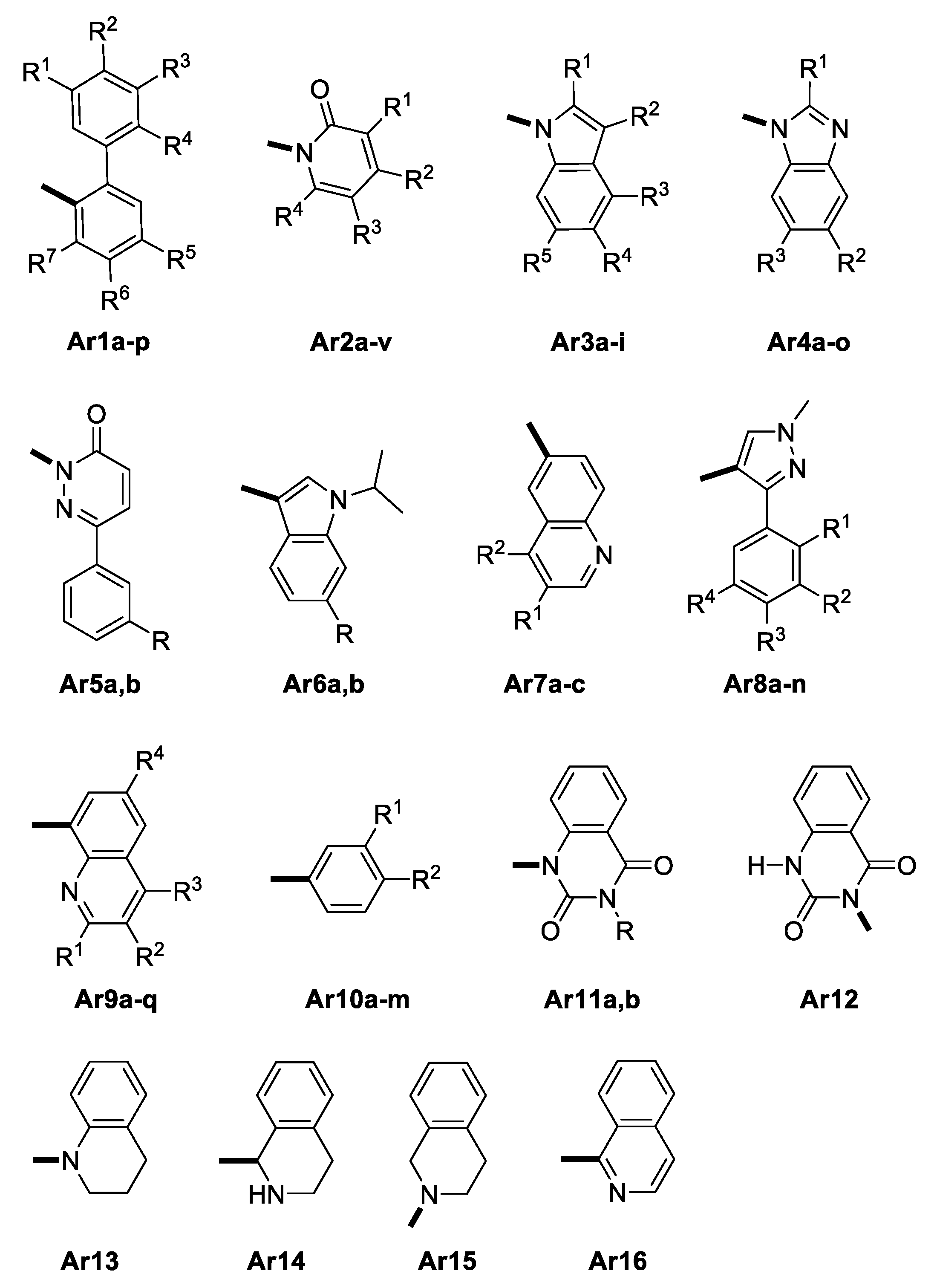
2.1. Design of GAL Derivatives Using Fragment-Based Libraries
2.2. Blood–Brain Barrier (BBB) Permeability
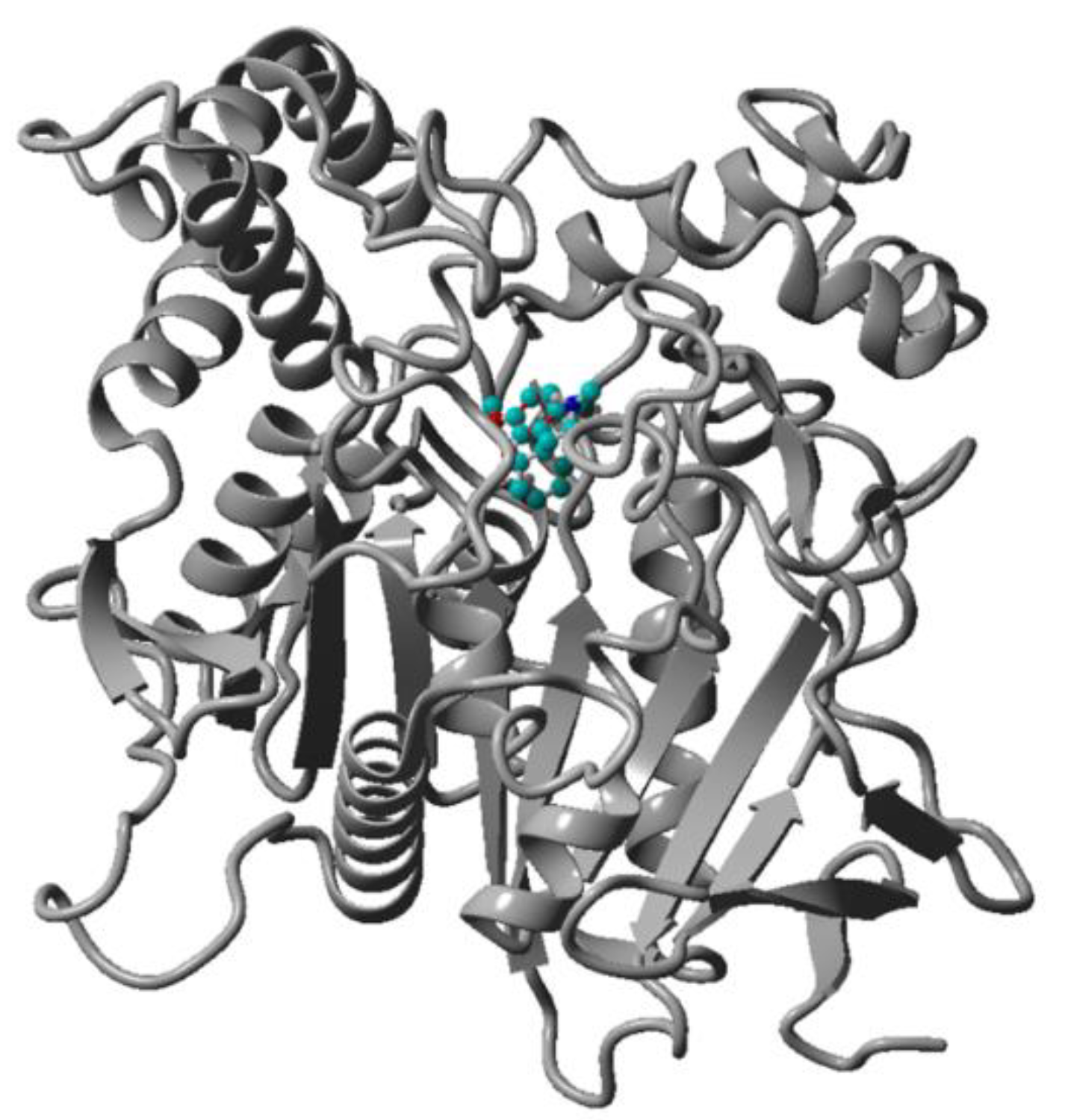
2.3. Virtual Screening by Molecular Docking
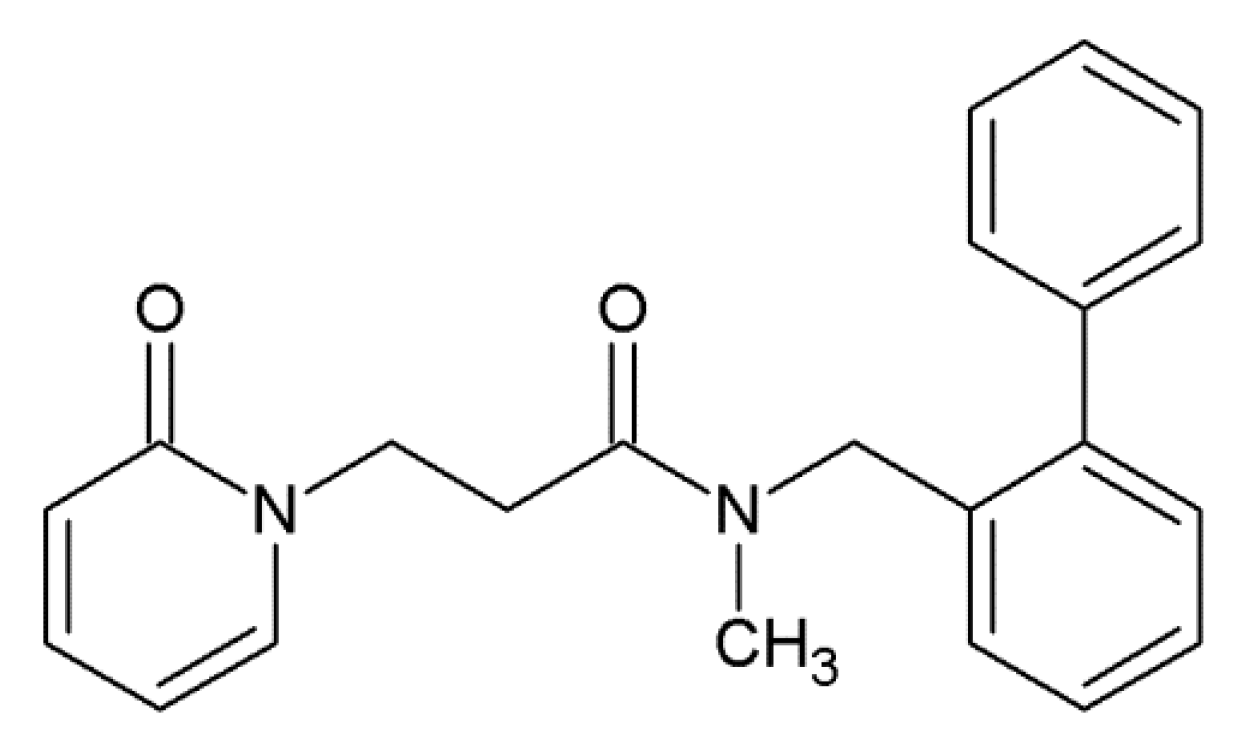
2.4. GAL Derivative Selected for Synthesis
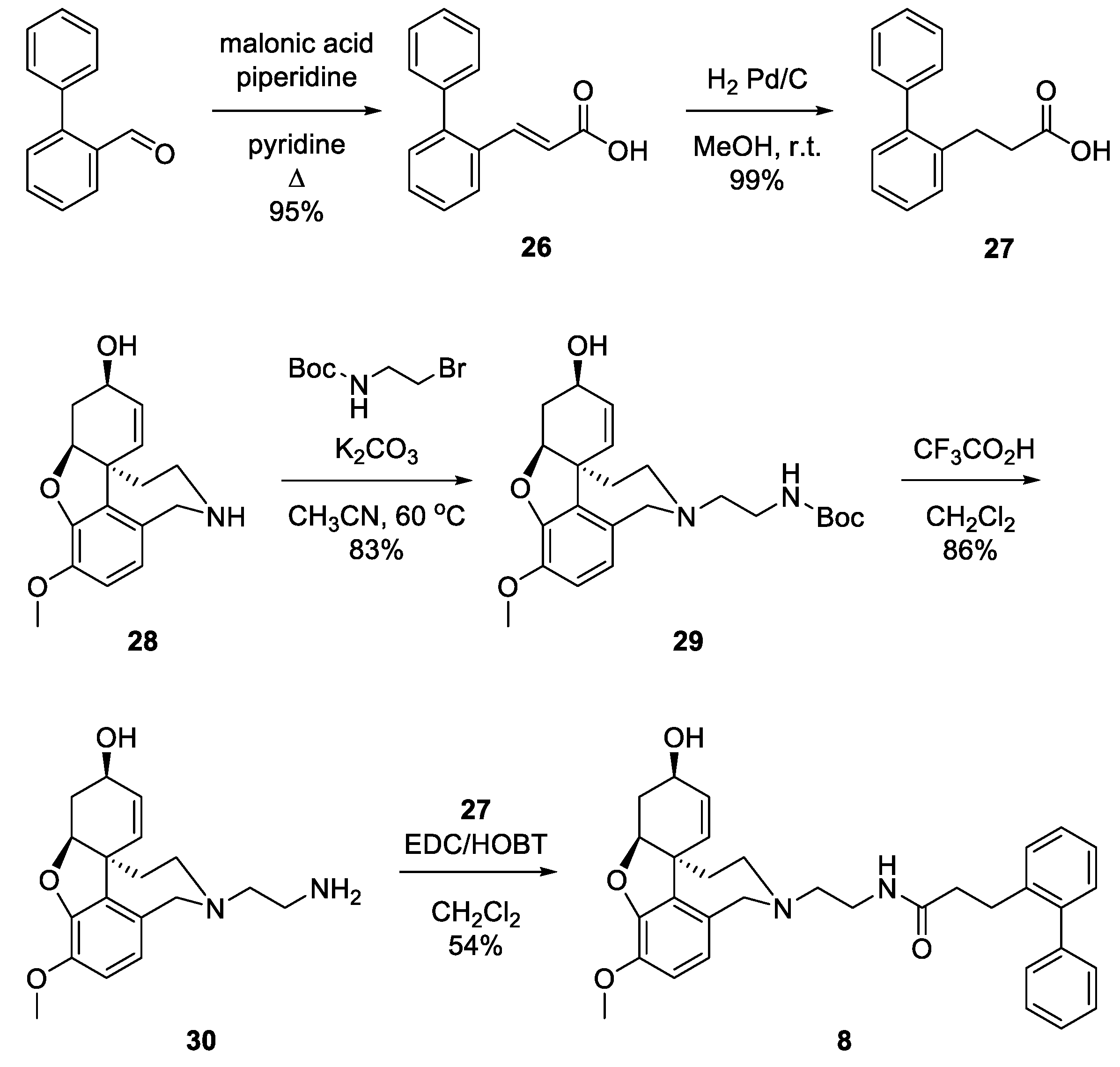
2.5. Synthesis of the Selected GAL Derivative
2.6. Neurotoxicity of the Selected GAL Derivative
2.7. Anti-AChE Activity of the Selected GAL Derivative
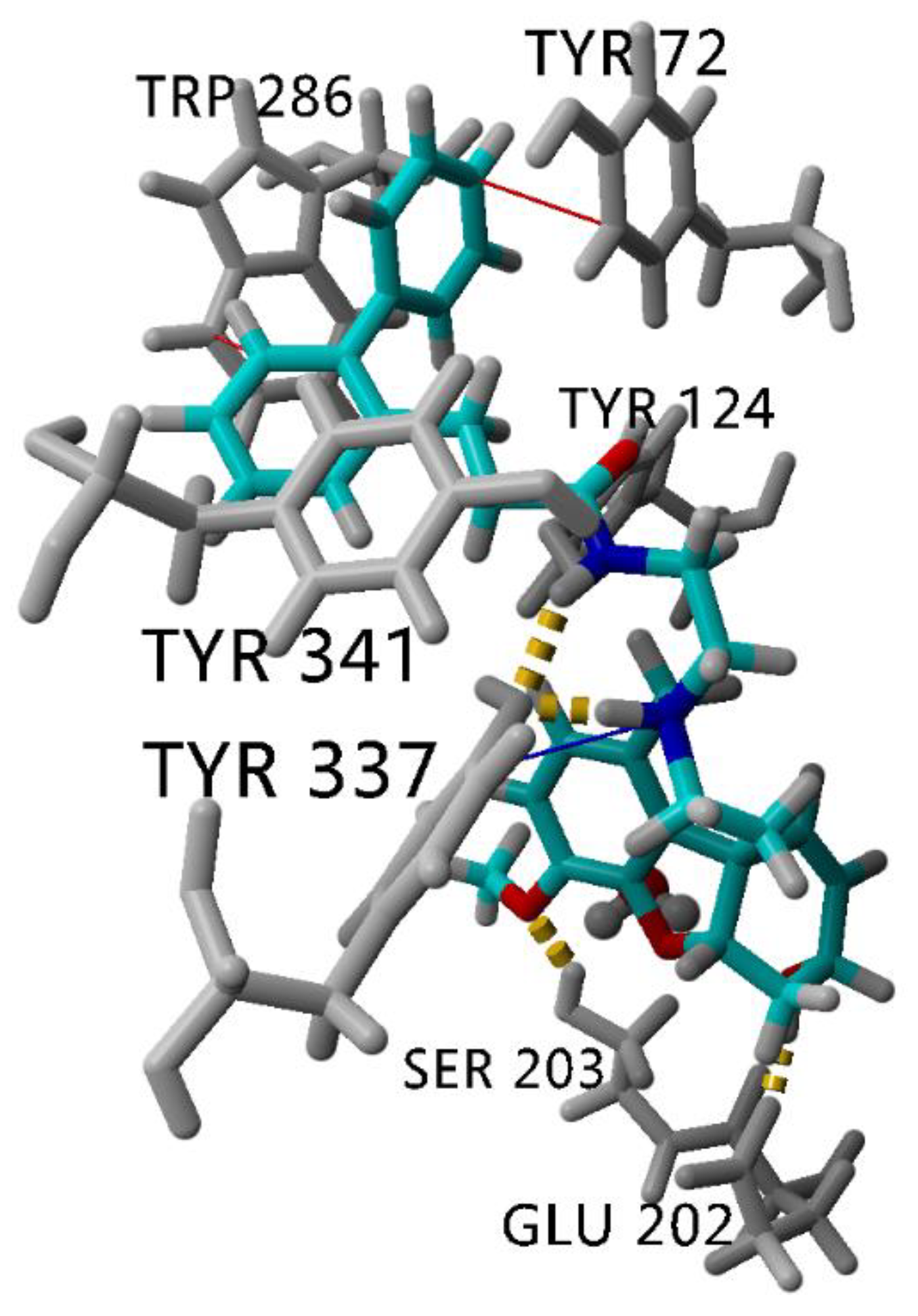
2.8. Stability of the Complex AChE-Compound 8 assessed by Molecular Dynamics Simulation
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Synthesis and Analytical Data of (E)-3-(biphenyl-2-yl)acrylic acid 26

4.3. Synthesis and Analytical Data of 3-(biphenyl-2-yl)propanoic acid 27

4.4. Synthesis and Analytical Data of tert-butyl {2-[(4aS,6R,8aS)-6-hydroxy-3-methoxy- 5,6,9,10-tetrahydro-4aH-[1]benzofuro[3a,3,2-ef][2]benzazepin-11(12H)-yl]ethyl}carbamate 29

4.5. Synthesis and Analytical Data of 3-(biphenyl-2-yl)-N-{2-[(4aS,6R,8aS)-6-hydroxy- 3-methoxy-5,6,9,10-tetrahydro-4aH-[1]benzofuro[3a,3,2-ef][2]benzazepin-11(12H)-yl]ethyl}propanamide 8

4.6. Prediction of Blood–Brain Barrier (BBB) Permeability
4.7. Virtual Screening by Molecular Docking
4.8. Neurotoxicity Test
4.9. Assessment of AChE Inhibitory Activity
4.10. Molecular Dynamics Protocol
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Geula, C.; Mesulam, M.M. Cholinergic systems in Alzheimer’s disease. In Alzheimer Disease, 2nd ed.; Terry, R.D., Katzman, R., Bick, K.L., Sisodia, S.S., Eds.; Lippincott, Williams & Wilkins: Philadelphia, PA, USA, 1999; pp. 269–292. [Google Scholar] [CrossRef]
- Janeczek, M.; Gefen, T.; Samimi, M.; Kim, G.; Weintraub, S.; Bigio, E.; Rogalski, E.; Mesulam, M.-M.; Geula, C. Variations in Acetylcholinesterase Activity within Human Cortical Pyramidal Neurons Across Age and Cognitive Trajectories. Cereb. Cortex 2017, 28, 1329–1337. [Google Scholar] [CrossRef]
- Palop, J.J.; Mucke, L. Amyloid-β-induced neuronal dysfunction in Alzheimer’s disease: From synapses toward neural net-works. Nat. Neurosci. 2010, 13, 812–818. [Google Scholar] [CrossRef]
- Wozniak, M.A.; Itzhaki, R.F.; Shipley, S.J.; Dobson, C.B. Herpes simplex virus infection causes cellular β-amyloid accumu-lation and secretase upregulation. Neurosci. Lett. 2007, 429, 95–100. [Google Scholar] [CrossRef]
- Eimer, W.A.; Kumar, D.K.V.; Shanmugam, N.K.N.; Rodriguez, A.S.; Mitchell, T.; Washicosky, K.J.; György, B.; Breakefield, X.O.; Tanzi, R.E.; Moir, R.D. Alzheimer’s Disease-Associated β-Amyloid Is Rapidly Seeded by Herpesviridae to Protect against Brain Infection. Neuron 2018, 99, 56–63. [Google Scholar] [CrossRef] [Green Version]
- Guo, T.; Noble, W.; Hanger, D.P. Roles of tau protein in health and disease. Acta Neuropathol. 2017, 133, 665–704. [Google Scholar] [CrossRef] [Green Version]
- Sulzer, D.; Edwards, R.H. The physiological role of α-synuclein and its relationship to Parkinson’s Disease. J. Neurochem. 2019, 150, 475–486. [Google Scholar] [CrossRef] [Green Version]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Irwin, D.J.; Lee, V.M.-Y.; Trojanowski, J.Q. Parkinson’s disease dementia: Convergence of α-synuclein, tau and amyloid-β pa-thologies. Nat. Rev. Neurosci. 2013, 14, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Colović, M.B.; Krstić, D.Z.; Lazarević-Pašti, T.D.; Bondžić, A.M.; Vasić, V.M. Acetylcholinesterase inhibitors: Pharmacology and toxicology. Curr. Neuropharmacol. 2013, 11, 315–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moss, D.E. Improving Anti-Neurodegenerative Benefits of Acetylcholinesterase Inhibitors in Alzheimer’s Disease: Are Irreversible Inhibitors the Future? Int. J. Mol. Sci. 2020, 21, 3438. [Google Scholar] [CrossRef] [PubMed]
- Galimberti, D.; Scarpini, E. Old and new acetylcholinesterase inhibitors for Alzheimer’s disease. Expert Opin. Investig. Drugs 2016, 25, 1181–1187. [Google Scholar] [CrossRef]
- Han, S.; Sweeney, J.; Bachman, E.; Schweiger, E.; Forloni, G.; Coyle, J.; Davis, B.; Joullie, M. Chemical and pharmacological characterization of galanthamine, an acetylcholinesterase inhibitor, and its derivatives. A potential application in Alzheimer’s disease? Eur. J. Med. Chem. 1992, 27, 673–687. [Google Scholar] [CrossRef]
- Dajas-Bailador, F.A.; Heimala, K.; Wonnacott, S. The Allosteric Potentiation of Nicotinic Acetylcholine Receptors by Galantamine Is Transduced into Cellular Responses in Neurons: Ca2+ Signals and Neurotransmitter Release. Mol. Pharmacol. 2003, 64, 1217–1226. [Google Scholar] [CrossRef] [Green Version]
- Simeonova, R.; Zheleva, D.; Valkova, I.; Stavrakov, G.; Philipova, I.; Atanasova, M.; Doytchinova, I. A Novel Galantamine-Curcumin Hybrid as a Potential Multi-Target Agent against Neurodegenerative Disorders. Molecules 2021, 26, 1865. [Google Scholar] [CrossRef]
- Takata, K.; Kitamura, Y.; Saeki, M.; Terada, M.; Kagitani, S.; Kitamura, R.; Fujikawa, Y.; Maelicke, A.; Tomimoto, H.; Taniguchi, T.; et al. Galantamine-induced Amyloid-β Clearance Mediated via Stimulation of Microglial Nicotinic Acetylcholine Receptors*. J. Biol. Chem. 2010, 285, 40180–40191. [Google Scholar] [CrossRef] [Green Version]
- Bores, G.M.; Kosley, R.W. Galanthamine derivatives for the treatment of Alzheimer’s disease. Drugs Future 1996, 21, 621–635. [Google Scholar] [CrossRef]
- Mary, A.; Renko, D.Z.; Guillou, C.; Thal, C. Potent acetylcholinesterase inhibitors: Design, synthesis, and structure–Activity relationships of bis-interacting ligands in the galanthamine series. Bioorganic Med. Chem. 1998, 6, 1835–1850. [Google Scholar] [CrossRef]
- Guillou, C.; Mary, A.; Renko, D.Z.; Gras, E.; Thal, C. Potent acetylcholinesterase inhibitors: Design, synthesis and structure-activity relationships of alkylene linked bis-galanthamine and galanthamine-galanthaminium salts. Bioorganic Med. Chem. Lett. 2000, 10, 637–639. [Google Scholar] [CrossRef]
- Herlem, D.; Martin, M.-T.; Thal, C.; Guillou, C. Synthesis and structure–activity relationships of open D-Ring galanthamine analogues. Bioorganic Med. Chem. Lett. 2003, 13, 2389–2391. [Google Scholar] [CrossRef]
- Jia, P.; Sheng, R.; Zhang, J.; Fang, L.; He, Q.; Yang, B.; Hu, Y. Design, synthesis and evaluation of galanthamine derivatives as acetylcholinesterase inhibitors. Eur. J. Med. Chem. 2009, 44, 772–784. [Google Scholar] [CrossRef] [PubMed]
- Atanasova, M.; Yordanov, N.; Dimitrov, I.; Berkov, S.; Doytchinova, I. Molecular Docking Study on Galantamine Derivatives as Cholinesterase Inhibitors. Mol. Inform. 2015, 34, 394–403. [Google Scholar] [CrossRef]
- Atanasova, M.; Stavrakov, G.; Philipova, I.; Zheleva, D.; Yordanov, N.; Doytchinova, I. Galantamine derivatives with indole moiety: Docking, design, synthesis and acetylcholinesterase inhibitory activity. Bioorganic Med. Chem. 2015, 23, 5382–5389. [Google Scholar] [CrossRef]
- Stavrakov, G.; Philipova, I.; Zheleva, D.; Atanasova, M.; Konstantinov, S.; Doytchinova, I. Docking-based design of galantamine derivatives with dual-site binding to acetylcholinesterase. Mol. Inf. 2016, 35, 278–285. [Google Scholar] [CrossRef]
- Stavrakov, G.; Philipova, I.; Zheleva-Dimitrova, D.; Valkova, I.; Salamanova, E.; Konstantinov, S.; Doytchinova, I. Docking-based design and synthesis of galantamine-camphane hybrids as inhibitors of acetylcholinesterase. Chem. Biol. Drug Des. 2017, 90, 709–718. [Google Scholar] [CrossRef]
- Stavrakov, G.; Philipova, I.; Lukarski, A.; Valkova, I.; Atanasova, M.; Dimitrov, I.; Konstantinov, S.; Doytchinova, I. Acetylcholinesterase inhibitors selected by docking-based screening–proof-of-concept study. Bulg. Chem. Commun. 2018, 50, 40. [Google Scholar]
- Stavrakov, G.; Philipova, I.; Lukarski, A.; Atanasova, M.; Zheleva, D.; Zhivkova, Z.D.; Ivanov, S.; Atanasova, T.; Konstantinov, S.; Doytchinova, I. Galantamine-Curcumin Hybrids as Dual-Site Binding Acetylcholinesterase Inhibitors. Molecules 2020, 25, 3341. [Google Scholar] [CrossRef]
- Cheung, J.; Rudolph, M.J.; Burshteyn, F.; Cassidy, M.S.; Gary, E.N.; Love, J.; Franklin, M.C.; Height, J.J. Structures of Human Acetylcholinesterase in Complex with Pharmacologically Important Ligands. J. Med. Chem. 2012, 55, 10282–10286. [Google Scholar] [CrossRef] [PubMed]
- Inestrosa, N.C.; Dinamarca, M.C.; Alvarez, A. Amyloid-cholinesterase interactions. Implications for Alzheimer’s disease. FEBS J. 2008, 275, 625–632. [Google Scholar] [CrossRef]
- Doytchinova, I.; Atanasova, M.; Valkova, I.; Stavrakov, G.; Philipova, I.; Zhivkova, Z.; Zheleva-Dimitrova, D.; Konstantinov, S.; Dimitrov, I. Novel hits for acetylcholinesterase inhibition derived by docking-based screening on ZINC database. J. Enzym. Inhib. Med. Chem. 2018, 33, 768–776. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Wang, L.; Lv, M.; Pei, R.; Li, P.; Pei, Z.; Wang, Y.; Su, W.; Xie, X.-Q. AlzPlatform: An Alzheimer’s disease do-main-specific chemogenomics knowledgebase for polypharmacology and target identification research. J. Chem. Inf. Model. 2014, 54, 1050–1060. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.; Andres, V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- López, S.; Bastida, J.; Viladomat, F.; Codina, C. Acetylcholinesterase inhibitory activity of some Amaryllidaceae alkaloids and Narcissus extracts. Life Sci. 2002, 71, 2521–2529. [Google Scholar] [CrossRef]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. Amber; University of California: San Francisco, CA, USA, 2018; Available online: https://ambermd.org/doc12/Amber18.pdf (accessed on 8 March 2021).
- Krieger, E.; Vriend, G. YASARA View—molecular graphics for all devices—from smartphones to workstations. Bioinformatics 2014, 30, 2981–2982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daina, A.; Zoete, V. A BOILED-Egg To Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.; Wang, L.; Xie, X.-Q. Ligand Classifier of Adaptively Boosting Ensemble Decision Stumps (LiCABEDS) and Its Application on Modeling Ligand Functionality for 5HT-Subtype GPCR Families. J. Chem. Inf. Model. 2011, 51, 521–531. [Google Scholar] [CrossRef] [Green Version]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Momekov, G.; Ferdinandov, D.; Bakalova, A.; Zaharieva, M.; Konstantinov, S.; Karaivanova, M. In vitro toxicological evalu-ation of a dinuclear platinum(II) complex with acetate ligands. Arch. Toxicol. 2006, 80, 555–560. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound No. | GoldScore | Linker | Aromatic Fragment |
|---|---|---|---|
| 1 | 115.38 | L10 | Ar1d |
| 2 | 114.14 | L10 | Ar1h |
| 3 | 111.49 | L10 | Ar1n |
| 4 | 111.28 | L10 | Ar1m |
| 5 | 110.42 | L5 | Ar1p |
| 6 | 109.51 | L10 | Ar1i |
| 7 | 108.14 | L10 | Ar1o |
| 8 | 105.56 | L10 | Ar1a |
| 9 | 105.53 | L6 | Ar1a |
| 10 | 103.90 | L6 | Ar10e |
| 11 | 103.73 | L1 | Ar8l |
| 12 | 103.24 | L2 | Ar4e |
| 13 | 102.87 | L10 | Ar1e |
| 14 | 102.87 | L7 | Ar13 |
| 15 | 102.84 | L6 | Ar1j |
| 16 | 102.49 | L2 | Ar8n |
| 17 | 102.31 | L2 | Ar8e |
| 18 | 102.08 | L6 | Ar1k |
| 19 | 101.87 | L10 | Ar6a |
| 20 | 101.87 | L3 | Ar10e |
| 21 | 101.67 | L3 | Ar10l |
| 22 | 101.03 | L6 | Ar1b |
| 23 | 100.60 | L1 | Ar9o |
| 24 | 100.31 | L1 | Ar3d |
| 25 | 100.04 | L1 | Ar15 |
| GAL | 77.73 |
| Parameter | Compound 8 | GAL |
|---|---|---|
| MW1 | 524.65 | 287.35 |
| pKa | 7.10 | 7.92 |
| logP | 4.49 | 1.75 |
| logD7.4 | 4.34 | 1.12 |
| Polar surface area (Å) | 71.03 | 41.93 |
| Hydrogen-bond donors (HBD) | 2 | 1 |
| Hydrogen-bond acceptors (HBA) | 6 | 4 |
| Neurotoxicity on neuro-2A cells (IC50) | >100 µM | >50 µM 2 |
| Inhibitory activity on eeAChE (IC50) | 27.79 ± 5.49 nM | 1.92 ± 0.1 µM |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stavrakov, G.; Philipova, I.; Lukarski, A.; Atanasova, M.; Georgiev, B.; Atanasova, T.; Konstantinov, S.; Doytchinova, I. Discovery of a Novel Acetylcholinesterase Inhibitor by Fragment-Based Design and Virtual Screening. Molecules 2021, 26, 2058. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26072058
Stavrakov G, Philipova I, Lukarski A, Atanasova M, Georgiev B, Atanasova T, Konstantinov S, Doytchinova I. Discovery of a Novel Acetylcholinesterase Inhibitor by Fragment-Based Design and Virtual Screening. Molecules. 2021; 26(7):2058. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26072058
Chicago/Turabian StyleStavrakov, Georgi, Irena Philipova, Atanas Lukarski, Mariyana Atanasova, Borislav Georgiev, Teodora Atanasova, Spiro Konstantinov, and Irini Doytchinova. 2021. "Discovery of a Novel Acetylcholinesterase Inhibitor by Fragment-Based Design and Virtual Screening" Molecules 26, no. 7: 2058. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26072058