
Coumarin-Chalcone Hybrids as Inhibitors of MAO-B: Biological Activity and In Silico Studies

 , ,
, ,  , and
, and 
Abstract
:1. Introduction
2. Results and Discussion
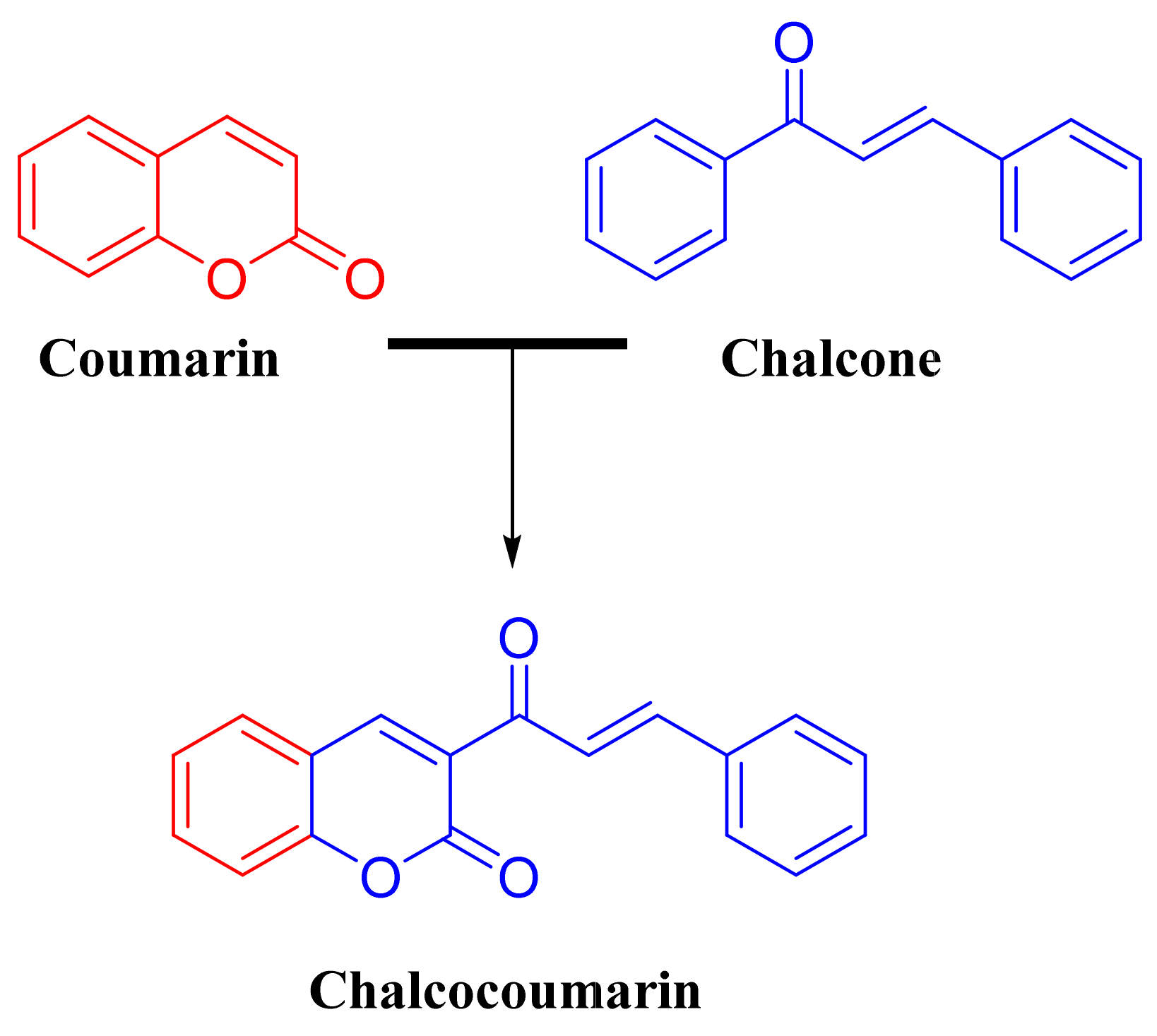
2.1. Chemistry
2.2. Biological Analysis in Rat MAO
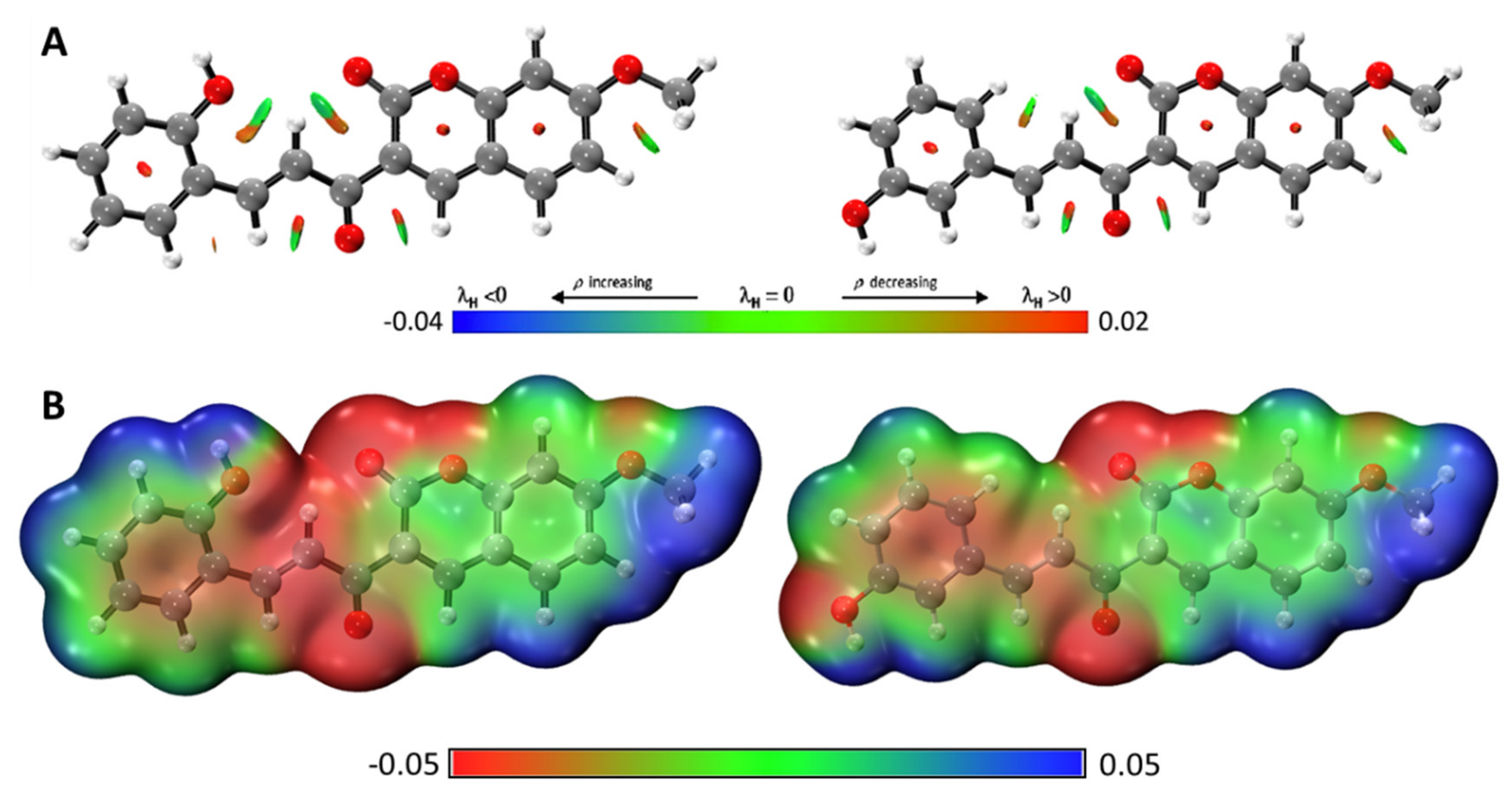
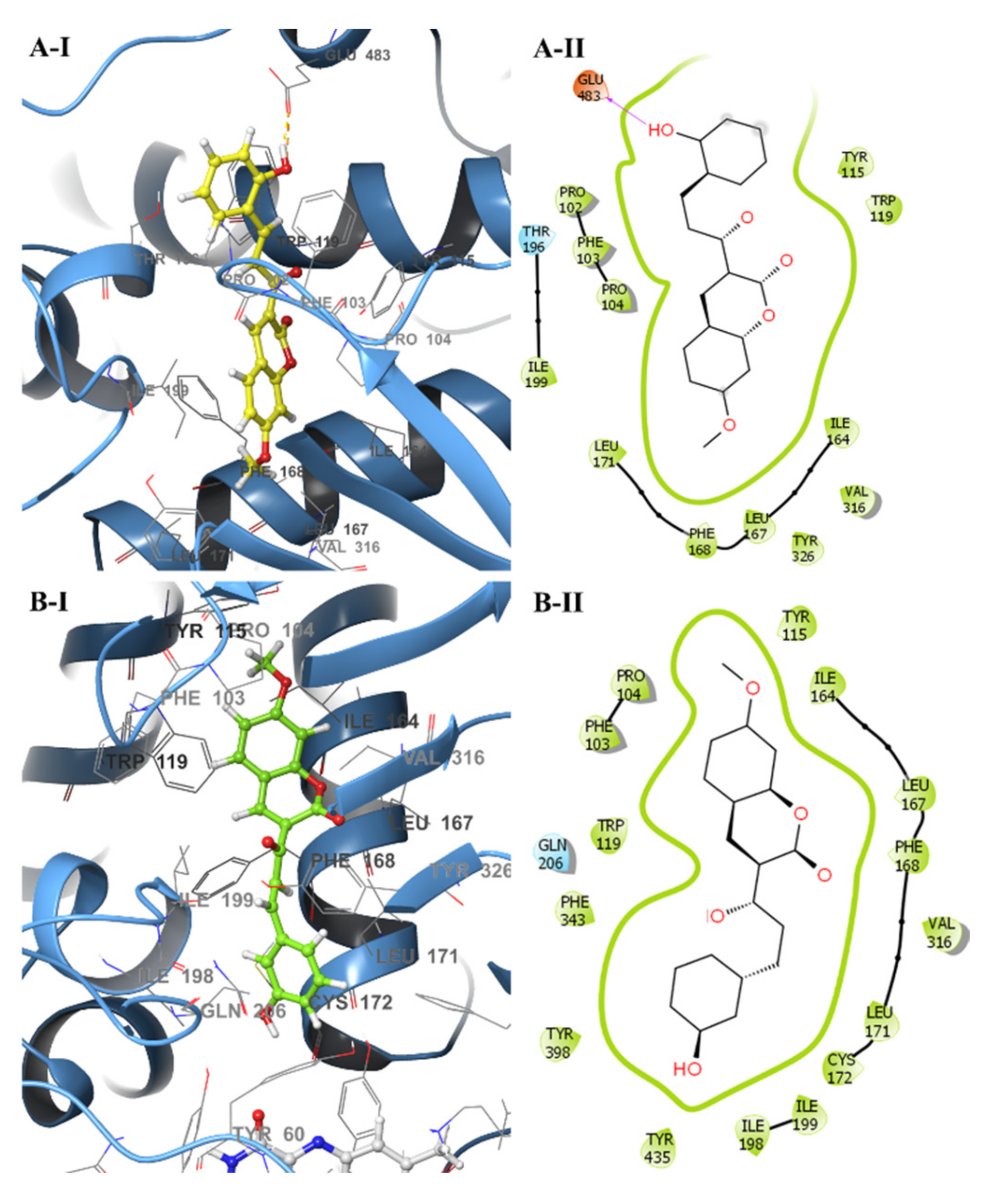
2.3. Molecular Docking and Ligand Efficiency Analysis
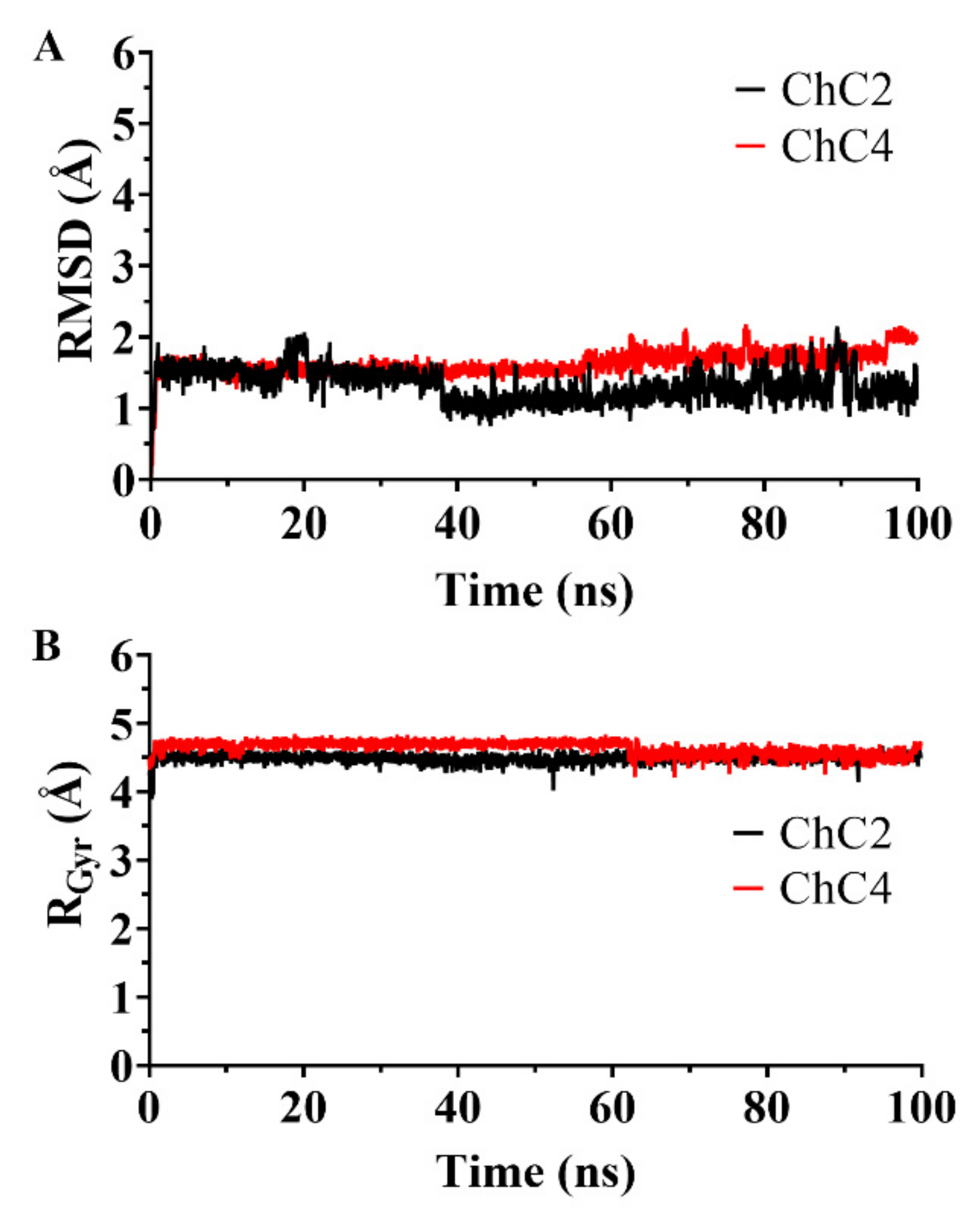
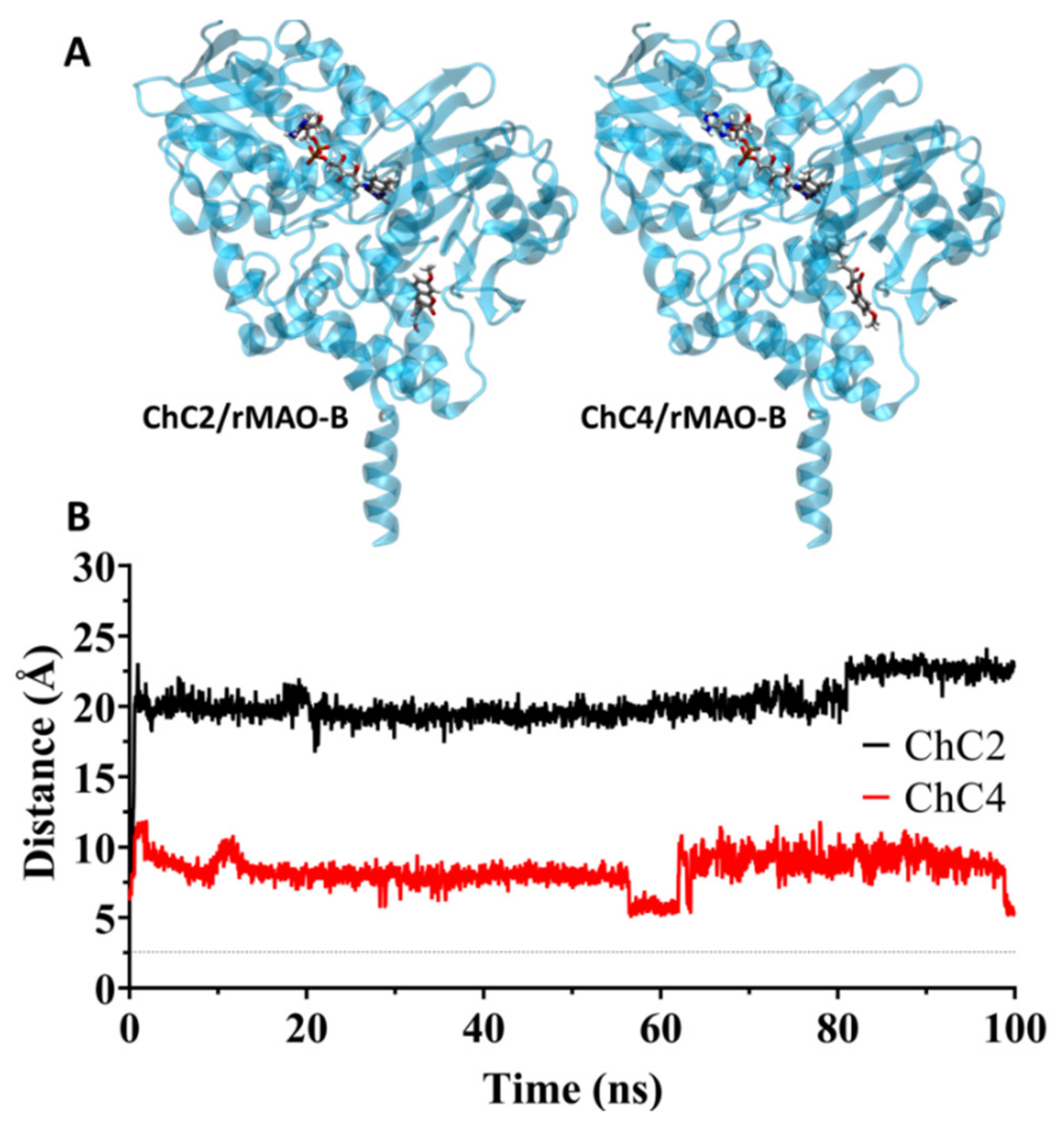
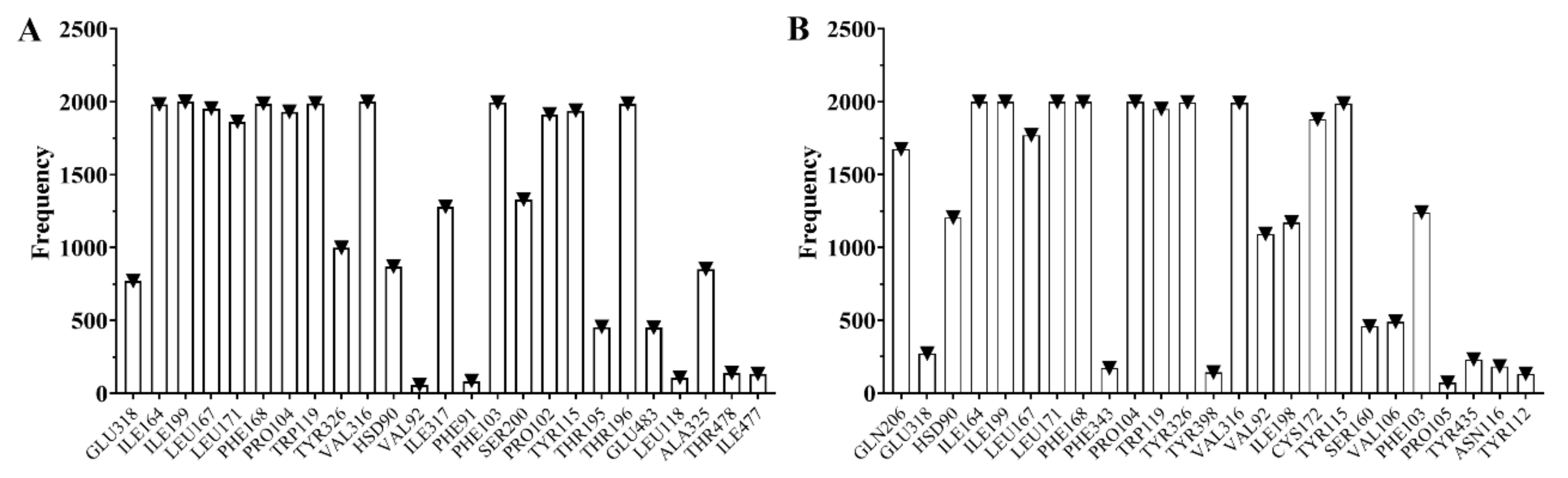
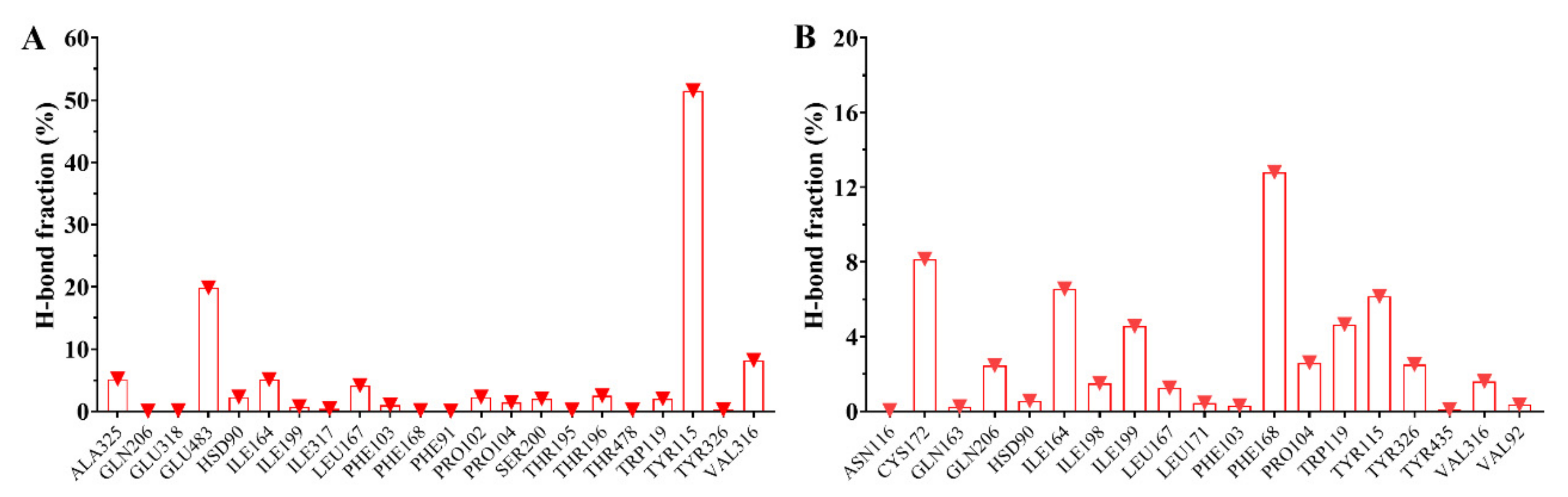
2.4. Analysis of Molecular Dynamics Simulations
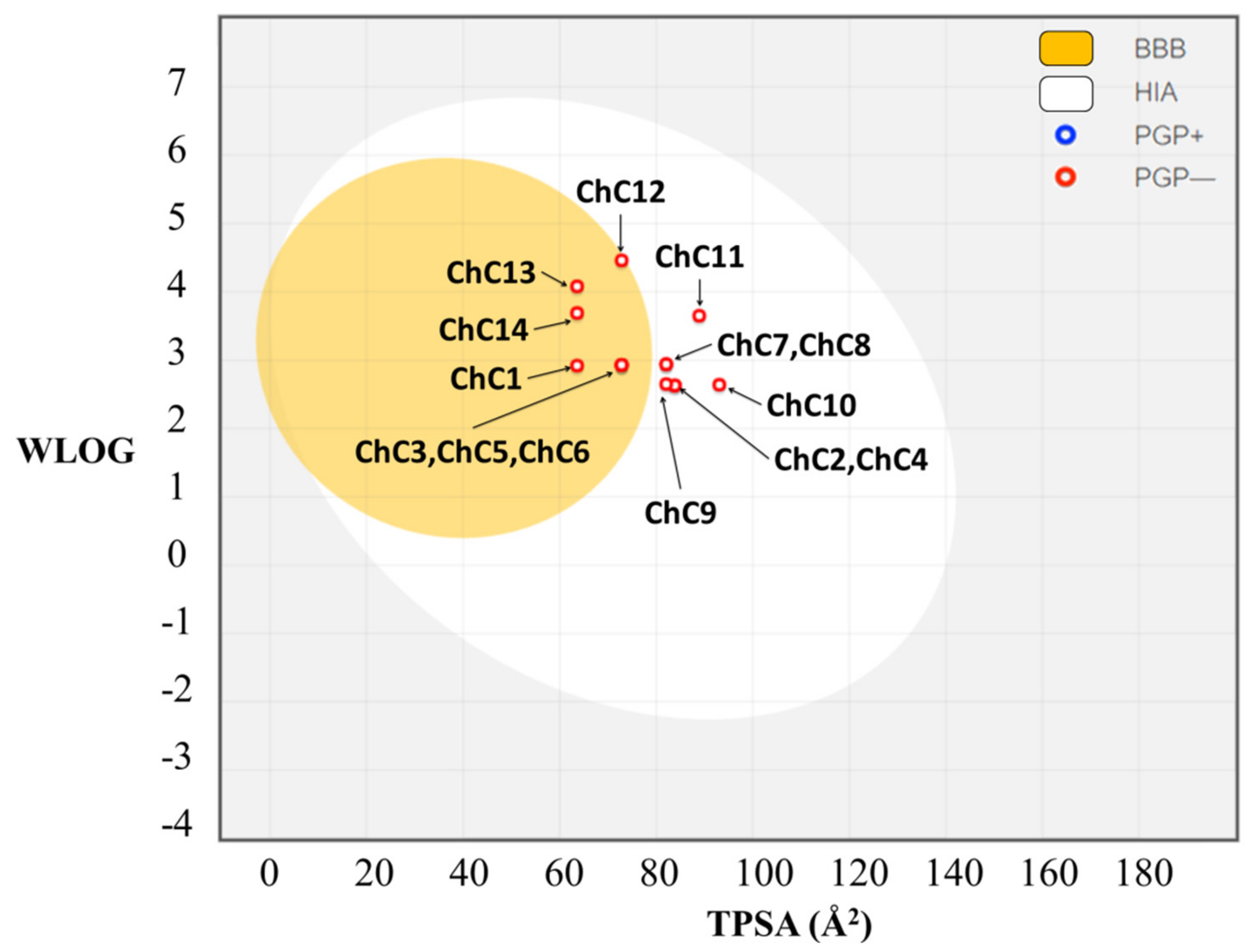
2.5. In Silico Pharmacokinetic Prediction
3. Materials and Methods
3.1. Solvents and Reagents
3.2. Synthesis
3.3. Biological Assessment
3.4. Computational Analysis
3.4.1. Homology Modeling
3.4.2. Molecular Docking
3.4.3. Ligand Efficiency Approach
3.4.4. Molecular Dynamic Simulations
3.4.5. Free Energy Calculation
3.4.6. ADMET Prediction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Sample Availability
References
- Rodríguez-Enríquez, F.; Costas-Lago, M.C.; Besada, P.; Alonso-Pena, M.; Torres-Terán, I.; Viña, D.; Fontenla, J.Á.; Sturlese, M.; Moro, S.; Quezada, E.; et al. Novel coumarin-pyridazine hybrids as selective MAO-B inhibitors for the Parkinson’s disease therapy. Bioorg. Chem. 2020, 104, 104203. [Google Scholar] [CrossRef] [PubMed]
- Garćia-Beltran, O.; Mena, N.P.; Aguirre, P.; Barriga-Gonzalez, G.; Galdamez, A.; Nagles, E.; Adasme, T.; Hidalgo, C.; Nuñez, M.T. Development of an iron-selective antioxidant probe with protective effects on neuronal function. PLoS ONE 2017, 12, e0189043. [Google Scholar] [CrossRef] [Green Version]
- Aguirre, P.; García-Beltrán, O.; Tapia, V.; Muñoz, Y.; Cassels, B.K.; Núñez, M.T. Neuroprotective Effect of a New 7,8-Dihydroxycoumarin-Based Fe2+/Cu2+ Chelator in Cell and Animal Models of Parkinson’s Disease. ACS Chem. Neurosci. 2017, 8, 178–185. [Google Scholar] [CrossRef]
- Al-Warhi, T.; Sabt, A.; Elkaeed, E.B.; Eldehna, W.M. Recent advancements of coumarin-based anticancer agents: An up-to-date review. Bioorg. Chem. 2020, 103, 104163. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.L.; Liu, L.; Hu, Y.; Wang, G.X. Evaluation of the antiparasitic activity of coumarin derivatives against Dactylogyrus intermedius in goldfish (Carassius auratus). Aquaculture 2021, 533, 736069. [Google Scholar] [CrossRef]
- Sahoo, C.R.; Sahoo, J.; Mahapatra, M.; Lenka, D.; Kumar Sahu, P.; Dehury, B.; Nath Padhy, R.; Kumar Paidesetty, S. Coumarin derivatives as promising antibacterial agent(s). Arab. J. Chem. 2021, 14, 102922. [Google Scholar] [CrossRef]
- Cione, E.; La Torre, C.; Cannataro, R.; Caroleo, M.C.; Plastina, P.; Gallelli, L. Quercetin, Epigallocatechin Gallate, Curcumin, and Resveratrol: From Dietary Sources to Human MicroRNA Modulation. Molecules 2020, 25, 63. [Google Scholar] [CrossRef] [Green Version]
- Oliver, D.M.A.; Reddy, P.H. Small molecules as therapeutic drugs for Alzheimer’s disease. Mol. Cell. Neurosci. 2019, 96, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Lv, D.; Hu, Z.; Lu, L.; Lu, H.; Xu, X. Three-dimensional cell culture: A powerful tool in tumor research and drug discovery. Oncol. Lett. 2017, 14, 6999–7010. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Enríquez, F.; Viña, D.; Uriarte, E.; Fontenla, J.A.; Matos, M.J. Discovery and optimization of 3-thiophenylcoumarins as novel agents against Parkinson’s disease: Synthesis, in vitro and in vivo studies. Bioorg. Chem. 2020, 101, 103986. [Google Scholar] [CrossRef]
- Patil, P.O.; Bari, S.B.; Firke, S.D.; Deshmukh, P.K.; Donda, S.T.; Patil, D.A. A comprehensive review on synthesis and designing aspects of coumarin derivatives as monoamine oxidase inhibitors for depression and Alzheimer’s disease. Bioorg. Med. Chem. 2013, 21, 2434–2450. [Google Scholar] [CrossRef]
- Carotti, A.; Altomare, C.; Catto, M.; Gnerre, C.; Summo, L.; De Marco, A.; Rose, S.; Jenner, P.; Testa, B. Lipophilicity plays a major role in modulating the inhibition on monoamine oxidase B by 7-substituted coumarins. Chem. Biodivers. 2006, 3, 134–149. [Google Scholar] [CrossRef] [PubMed]
- Akyüz, M.A.; Erdem, S.S.; Edmondson, D.E. The aromatic cage in the active site of monoamine oxidase B: Effect on the structural and electronic properties of bound benzylamine and p-nitrobenzylamine. J. Neural Transm. 2007, 114, 693. [Google Scholar] [CrossRef] [PubMed]
- Can, N.O.; Osmaniye, D.; Levent, S.; Sa Saǧlik, B.N.; Inci, B.; Ilgin, S.; Özkay, Y.; Kaplancikli, Z.A. Synthesis of new hydrazone derivatives for MAO enzymes inhibitory activity. Molecules 2017, 22, 1381. [Google Scholar] [CrossRef] [PubMed]
- Billett, E.E. Monoamine Oxidase (MAO) in Human Peripheral Tissues. Neurotoxicology 2004, 25, 139–148. [Google Scholar] [CrossRef]
- Binda, C.; Wang, J.; Pisani, L.; Caccia, C.; Carotti, A.; Salvati, P.; Edmondson, D.E.; Mattevi, A. Structures of human monoamine oxidase B complexes with selective noncovalent inhibitors: Safinamide and coumarin analogs. J. Med. Chem. 2007, 50, 5848–5852. [Google Scholar] [CrossRef]
- Novaroli, L.; Daina, A.; Favre, E.; Bravo, J.; Carotti, A.; Leonetti, F.; Catto, M.; Carrupt, P.A.; Reist, M. Impact of species-dependent differences on screening, design, and development of MAO B inhibitors. J. Med. Chem. 2006, 49, 6264–6272. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Parada, M.; Fierro, A.; Iturriaga-Vasquez, P.; Cassels, B. Monoamine Oxidase Inhibition in the Light of New Structural Data. Curr. Enzym. Inhib. 2006, 1, 85–95. [Google Scholar] [CrossRef]
- Shih, J.C.; Chen, K.; Ridd, M.J. Monoamine oxidase: From genes to behavior. Annu. Rev. Neurosci. 1999, 22, 197–217. [Google Scholar] [CrossRef] [Green Version]
- Matos, M.J.; Vazquez-Rodriguez, S.; Uriarte, E.; Santana, L.; Viña, D. MAO inhibitory activity modulation: 3-Phenylcoumarins versus 3-benzoylcoumarins. Bioorg. Med. Chem. Lett. 2011, 21, 4224–4227. [Google Scholar] [CrossRef]
- Tong, J.; Meyer, J.H.; Furukawa, Y.; Boileau, I.; Chang, L.J.; Wilson, A.A.; Houle, S.; Kish, S.J. Distribution of monoamine oxidase proteins in human brain: Implications for brain imaging studies. J. Cereb. Blood Flow Metab. 2013, 33, 863–871. [Google Scholar] [CrossRef] [Green Version]
- Viña, D.; Matos, M.J.; Ferino, G.; Cadoni, E.; Laguna, R.; Borges, F.; Uriarte, E.; Santana, L. 8-Substituted 3-Arylcoumarins as Potent and Selective MAO-B Inhibitors: Synthesis, Pharmacological Evaluation, and Docking Studies. ChemMedChem 2012, 7, 464–470. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, A.; Yabanoglu, S.; Sinha, B.N.; Ucar, G.; Basu, A.; Jayaprakash, V. Towards development of selective and reversible pyrazoline based MAO-inhibitors: Synthesis, biological evaluation and docking studies. Bioorg. Med. Chem. Lett. 2010, 20, 132–136. [Google Scholar] [CrossRef]
- Chimenti, F.; Secci, D.; Bolasco, A.; Chimenti, P.; Granese, A.; Carradori, S.; Befani, O.; Turini, P.; Alcaro, S.; Ortuso, F. Synthesis, molecular modeling studies, and selective inhibitory activity against monoamine oxidase of N,N′-bis[2-oxo-2H-benzopyran]-3-carboxamides. Bioorg. Med. Chem. Lett. 2006, 16, 4135–4140. [Google Scholar] [CrossRef] [PubMed]
- García-Beltrán, O.; Cassels, B.K.; Mena, N.; Nuñez, M.T.; Yañez, O.; Caballero, J. A coumarinylaldoxime as a specific sensor for Cu2+ and its biological application. Tetrahedron Lett. 2014, 55, 873–976. [Google Scholar] [CrossRef]
- García-Beltrán, O.; Mena, N.; Pérez, E.G.; Cassels, B.K.; Nuñez, M.T.; Werlinger, F.; Zavala, D.; Aliaga, M.E.; Pavez, P. The development of a fluorescence turn-on sensor for cysteine, glutathione and other biothiols. A kinetic study. Tetrahedron Lett. 2011, 52, 6606–6609. [Google Scholar] [CrossRef]
- Konidala, S.K.; Kotra, V.; Danduga, R.C.S.R.; Kola, P.K. Coumarin-chalcone hybrids targeting insulin receptor: Design, synthesis, anti-diabetic activity, and molecular docking. Bioorg. Chem. 2020, 104, 104207. [Google Scholar] [CrossRef]
- Emam, S.H.; Sonousi, A.; Osman, E.O.; Hwang, D.; Do Kim, G.; Hassan, R.A. Design and synthesis of methoxyphenyl- and coumarin-based chalcone derivatives as anti-inflammatory agents by inhibition of NO production and down-regulation of NF-κB in LPS-induced RAW264.7 macrophage cells. Bioorg. Chem. 2021, 107, 104630. [Google Scholar] [CrossRef]
- Kurt, B.Z.; Ozten Kandas, N.; Dag, A.; Sonmez, F.; Kucukislamoglu, M. Synthesis and biological evaluation of novel coumarin-chalcone derivatives containing urea moiety as potential anticancer agents. Arab. J. Chem. 2020, 13, 1120–1129. [Google Scholar] [CrossRef]
- Vilches-Herrera, M.; Miranda-Sepúlveda, J.; Rebolledo-Fuentes, M.; Fierro, A.; Lühr, S.; Iturriaga-Vasquez, P.; Cassels, B.K.; Reyes-Parada, M. Naphthylisopropylamine and N-benzylamphetamine derivatives as monoamine oxidase inhibitors. Bioorg. Med. Chem. 2009, 17, 2452–2460. [Google Scholar] [CrossRef] [PubMed]
- Lühr, S.; Vilches-Herrera, M.; Fierro, A.; Ramsay, R.R.; Edmondson, D.E.; Reyes-Parada, M.; Cassels, B.K.; Iturriaga-Vásquez, P. 2-Arylthiomorpholine derivatives as potent and selective monoamine oxidase B inhibitors. Bioorg. Med. Chem. 2010, 18, 1388–1395. [Google Scholar] [CrossRef] [PubMed]
- Binda, C.; Aldeco, M.; Mattevi, A.; Edmondson, D.E. Interactions of Monoamine Oxidases with the Antiepileptic Drug Zonisamide: Specificity of Inhibition and Structure of the Human Monoamine Oxidase B Complex. J. Med. Chem. 2011, 54, 909–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binda, C.; Newton-Vinson, P.; Hubálek, F.; Edmondson, D.E.; Mattevi, A. Structure of human monoamine oxidase B, a drug target for the treatment of neurological disorders. Nat. Struct. Biol. 2002, 9, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Abad-Zapatero, C. Ligand efficiency indices for effective drug discovery. Expert Opin. Drug Discov. 2007, 2, 469–488. [Google Scholar] [CrossRef]
- Abad-Zapatero, C.; Perišić, O.; Wass, J.; Bento, A.P.; Overington, J.; Al-Lazikani, B.; Johnson, M.E. Ligand efficiency indices for an effective mapping of chemico-biological space: The concept of an atlas-like representation. Drug Discov. Today 2010, 15, 804–811. [Google Scholar] [CrossRef]
- Abad-Zapatero, C. Ligand Efficiency Indices for Drug Discovery: Towards an Atlas-Guided Paradigm; Elsevier: Amsterdam, The Netherlands, 2013; ISBN 978-0-12-404635-1. [Google Scholar]
- Carugo, O. How root-mean-square distance (r.m.s.d.) values depend on the resolution of protein structures that are compared. J. Appl. Crystallogr. 2003, 36, 125–128. [Google Scholar] [CrossRef]
- Shalaby, R.; Petzer, J.P.; Petzer, A.; Ashraf, U.M.; Atari, E.; Alasmari, F.; Kumarasamy, S.; Sari, Y.; Khalil, A. SAR and molecular mechanism studies of monoamine oxidase inhibition by selected chalcone analogs. J. Enzyme Inhib. Med. Chem. 2019, 34, 863–876. [Google Scholar] [CrossRef] [Green Version]
- Binda, C.; Hubálek, F.; Li, M.; Edmondson, D.E.; Mattevi, A. Crystal structure of human monoamine oxidase B, a drug target enzyme monotopically inserted into the mitochondrial outer membrane. FEBS Lett. 2004, 564, 225–228. [Google Scholar] [CrossRef] [Green Version]
- Milczek, E.M.; Binda, C.; Rovida, S.; Mattevi, A.; Edmondson, D.E. The ‘gating’ residues Ile199 and Tyr326 in human monoamine oxidase B function in substrate and inhibitor recognition. FEBS J. 2011, 278, 4860–4869. [Google Scholar] [CrossRef] [Green Version]
- Fierro, A.; Osorio-Olivares, M.; Cassels, B.K.; Edmondson, D.E.; Sepúlveda-Boza, S.; Reyes-Parada, M. Human and rat monoamine oxidase-A are differentially inhibited by (S)-4-alkylthioamphetamine derivatives: Insights from molecular modeling studies. Bioorg. Med. Chem. 2007, 15, 5198–5206. [Google Scholar] [CrossRef] [Green Version]
- Morales-Camilo, N.; Salas, C.O.; Sanhueza, C.; Espinosa-Bustos, C.; Sepúlveda-Boza, S.; Reyes-Parada, M.; Gonzalez-Nilo, F.; Caroli-Rezende, M.; Fierro, A. Synthesis, biological evaluation, and molecular simulation of chalcones and aurones as selective MAO-B inhibitors. Chem. Biol. Drug Des. 2015, 85, 685–695. [Google Scholar] [CrossRef]
- GraphPad One-Way ANOVA with Dunnett’s Post Test was Performed Using GraphPad Prism, version 9.00 for Windows 2021; GraphPad: San Diego, CA, USA.
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geer, L.Y.; Marchler-Bauer, A.; Geer, R.C.; Han, L.; He, J.; He, S.; Liu, C.; Shi, W.; Bryant, S.H. The NCBI BioSystems database. Nucleic Acids Res. 2009, 38, D492–D496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corpet, F. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 1988, 16, 10881–10890. [Google Scholar] [CrossRef]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [Green Version]
- Melo, F.; Devos, D.; Depiereux, E.; Feytmans, E. ANOLEA: A www server to assess protein structures. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1997, 5, 187–190. [Google Scholar]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Ma, J.; Yoshimura, M.; Yamashita, E.; Nakagawa, A.; Ito, A.; Tsukihara, T. Structure of Rat Monoamine Oxidase A and Its Specific Recognitions for Substrates and Inhibitors. J. Mol. Biol. 2004, 338, 103–114. [Google Scholar] [CrossRef]
- Gordon, J.C.; Myers, J.B.; Folta, T.; Shoja, V.; Heath, L.S.; Onufriev, A. H++: A server for estimating p Ka s and adding missing hydrogens to macromolecules. Nucleic Acids Res. 2005, 33, W368–W371. [Google Scholar] [CrossRef] [PubMed]
- Anandakrishnan, R.; Onufriev, A. Analysis of Basic Clustering Algorithms for Numerical Estimation of Statistical Averages in Biomolecules. J. Comput. Biol. 2008, 15, 165–184. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods V: Modification of NDDO approximations and application to 70 elements. J. Mol. Model. 2007, 13, 1173–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Řezáč, J.; Hobza, P. Advanced corrections of hydrogen bonding and dispersion for semiempirical quantum mechanical methods. J. Chem. Theory Comput. 2012, 8, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.J.P. MOPAC 2016, Stewart Computational Chemistry: Colorado Springs, CO, USA.
- Reynolds, C.H.; Tounge, B.A.; Bembenek, S.D. Ligand Binding Efficiency: Trends, Physical Basis, and Implications. J. Med. Chem. 2008, 51, 2432–2438. [Google Scholar] [CrossRef]
- Cavalluzzi, M.M.; Mangiatordi, G.F.; Nicolotti, O.; Lentini, G. Ligand efficiency metrics in drug discovery: The pros and cons from a practical perspective. Expert Opin. Drug Discov. 2017, 12, 1087–1104. [Google Scholar] [CrossRef]
- Neria, E.; Fisher, S.; Karplus, M. Simulation of activation free energies in molecular systems. J. Chem. Phys. 1996, 105, 1902–1921. [Google Scholar] [CrossRef]
- Zhao, Y.; Schultz, N.E.; Truhlar, D.G. Design of Density Functionals by Combining the Method of Constraint Satisfaction with Parametrization for Thermochemistry, Thermochemical Kinetics, and Noncovalent Interactions. J. Chem. Theory Comput. 2006, 2, 364–382. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Dodda, L.S.; Vilseck, J.Z.; Tirado-Rives, J.; Jorgensen, W.L. 1.14*CM1A-LBCC: Localized Bond-Charge Corrected CM1A Charges for Condensed-Phase Simulations. J. Phys. Chem. B 2017, 121, 3864–3870. [Google Scholar] [CrossRef] [Green Version]
- Dodda, L.S.; Cabeza de Vaca, I.; Tirado-Rives, J.; Jorgensen, W.L. LigParGen web server: An automatic OPLS-AA parameter generator for organic ligands. Nucleic Acids Res. 2017, 45, W331–W336. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Tirado-Rives, J. Potential energy functions for atomic-level simulations of water and organic and biomolecular systems. Proc. Natl. Acad. Sci. USA 2005, 102, 6665–6670. [Google Scholar] [CrossRef] [Green Version]
- Bayly, C.I.; Cieplak, P.; Cornell, W.D.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- MacKerell, A.D., Jr.; Bashford, D.; Bellott, M.L.D.R.; Dunbrack, R.L., Jr.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef]
- Huang, J.; MacKerell, A.D., Jr. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalé, L.; Skeel, R.; Bhandarkar, M.; Brunner, R.; Gursoy, A.; Krawetz, N.; Phillips, J.; Shinozaki, A.; Varadarajan, K.; Schulten, K. NAMD2: Greater scalability for parallel molecular dynamics. J. Comput. Phys. 1999, 151, 283–312. [Google Scholar] [CrossRef]
- Hayes, J.M.; Archontis, G. MM-GB(PB)SA Calculations of Protein-Ligand Binding Free Energies. In Molecular Dynamics; Wang, L., Ed.; IntechOpen: Rijeka, Croatia, 2012. [Google Scholar]
- Song, L.; Lee, T.-S.; Zhu, C.; York, D.M.; Merz, K.M., Jr. Using AMBER18 for Relative Free Energy Calculations. J. Chem. Inf. Model. 2019, 59, 3128–3135. [Google Scholar] [CrossRef] [PubMed]
- Götz, A.W.; Williamson, M.J.; Xu, D.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput. 2012, 8, 1542–1555. [Google Scholar] [CrossRef] [PubMed]
- Abroshan, H.; Akbarzadeh, H.; Parsafar, G.A. Molecular dynamics simulation and MM-PBSA calculations of sickle cell hemoglobin in dimer form with Val, Trp, or Phe at the lateral contact. J. Phys. Org. Chem. 2010, 23, 866–877. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Zoete, V. A BOILED-Egg To Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
| Compounds | R1 | R2 | R3 | R4 |
| ChC1 | H | H | H | H |
| ChC2 | OH | H | H | H |
| ChC3 | OCH3 | H | H | H |
| ChC4 | H | OH | H | H |
| ChC5 | H | OCH3 | H | H |
| ChC6 | H | H | OCH3 | H |
| ChC7 | OCH3 | OCH3 | H | H |
| ChC8 | OCH3 | H | H | OCH3 |
| ChC10 | H | H | OH | OCH3 |
| ChC11 | H | H | SCH3 | H |
| ChC12 | H | Br | OCH3 | Br |
| ChC13 | H | H | N(CH3)2 | H |
| ChC14 | H | H | Br | H |
 | ||||
| Compounds | IC50 (µM) rMAO A | IC50 (µM) rMAO B |
|---|---|---|
| ChC1 | >10 | >10 |
| ChC2 | >10 | >10 |
| ChC3 | >10 | >10 |
| ChC4 | >10 | 0.76 ± 0.08 |
| ChC5 | >10 | 9.63 ± 0.90 |
| ChC6 | >10 | 6.96 ± 0.07 |
| ChC7 | >10 | >10 |
| ChC8 | >10 | >10 |
| ChC9 | >10 | 3.71 ± 0.68 |
| ChC10 | >10 | >10 |
| ChC11 | >10 | 5.88 ± 0.57 |
| ChC12 | >10 | >10 |
| ChC13 | >10 | >10 |
| ChC14 | >10 | >10 |
| Compound | Docking Results a | Ligand Efficiency | ||||
|---|---|---|---|---|---|---|
| rMAO-A | rMAO-B | rMAO-A | rMAO-B | |||
| ∆Ebinding (kcal·mol−1) | ∆Ebinding (kcal·mol−1) | Kd | LE (kcal·mol−1) | Kd | LE (kcal·mol−1) | |
| ChC1 | −6.4 | −9.3 | 2.0310−5 | 0.278 | 1.5210−7 | 0.404 |
| ChC2 | −6.7 | −9.8 | 1.2210−5 | 0.279 | 6.5710−8 | 0.408 |
| ChC3 | −6.7 | −9.1 | 1.2210−5 | 0.268 | 2.1410−7 | 0.364 |
| ChC4 | −6.3 | −9.8 | 2.4110−5 | 0.262 | 6.5710−8 | 0.408 |
| ChC5 | −6.5 | −9.6 | 1.7210−5 | 0.260 | 9.2110−8 | 0.384 |
| ChC6 | −6.6 | −9.3 | 1.4510−5 | 0.264 | 1.5210−7 | 0.372 |
| ChC7 | −6.2 | −8.7 | 2.8510−5 | 0.229 | 4.2010−7 | 0.322 |
| ChC8 | −6.7 | −9.6 | 1.2210−5 | 0.248 | 9.2110−8 | 0.355 |
| ChC9 | −6.8 | −9.9 | 1.0310−5 | 0.261 | 5.5510−8 | 0.380 |
| ChC10 | −6.4 | −9.2 | 2.0310−5 | 0.246 | 1.8010−7 | 0.353 |
| ChC11 | −6.4 | −8.9 | 2.0310−5 | 0.256 | 3.0010−7 | 0.356 |
| ChC12 | −6.7 | −7.2 | 1.2210−5 | 0.248 | 5.2810−6 | 0.266 |
| ChC13 | −7.0 | −9.9 | 7.4110−6 | 0.269 | 5.5510−8 | 0.380 |
| ChC14 | −6.6 | −9.4 | 1.4510−5 | 0.275 | 1.2910−7 | 0.391 |
| Compounds | Calculated Free Energy Descomposition (kcal·mol−1) | |||
|---|---|---|---|---|
| ΔGbinding | ΔEvdW | ΔEelect | ΔEpot | |
| ChC2 | −29.06 ± 0.10 | −39.69 ± 0.13 | 15.03 ± 0.17 | −24.65 ± 0.10 |
| ChC4 | −25.87 ± 0.09 | −44.94 ± 0.09 | 24.90 ± 0.04 | −20.03 ± 0.09 |
| Compounds | Log P | MW (g/mol) | TPSA (Å2) | HBA | HBD | RB | Log S | Log Kp (cm/s) | Nº Violations |
|---|---|---|---|---|---|---|---|---|---|
| ChC1 | 3.38 | 307.32 | 63.6 | 4 | 1 | 4 | −4.89 | −5.43 | 0 |
| ChC2 | 2.9 | 323.32 | 83.83 | 5 | 2 | 4 | −4.94 | −5.79 | 0 |
| ChC3 | 3.36 | 337.35 | 72.83 | 5 | 1 | 5 | −5.06 | −5.64 | 0 |
| ChC4 | 2.97 | 323.32 | 83.83 | 5 | 2 | 4 | −4.94 | −5.79 | 0 |
| ChC5 | 3.27 | 337.35 | 72.83 | 5 | 1 | 5 | −5.06 | −5.64 | 0 |
| ChC6 | 3.27 | 337.35 | 72.83 | 5 | 1 | 5 | −5.06 | −5.64 | 0 |
| ChC7 | 3.28 | 367.37 | 82.06 | 6 | 1 | 6 | −5.22 | −5.84 | 0 |
| ChC8 | 3.27 | 367.37 | 82.06 | 6 | 1 | 6 | −5.22 | −5.84 | 0 |
| ChC9 | 3.23 | 351.33 | 82.06 | 6 | 1 | 4 | −5.08 | −5.84 | 0 |
| ChC10 | 2.92 | 353.35 | 93.06 | 6 | 2 | 5 | −5.11 | −5.99 | 0 |
| ChC11 | 3.82 | 353.41 | 88.9 | 4 | 1 | 5 | −5.95 | −5.35 | 0 |
| ChC12 | 4.48 | 495.14 | 72.83 | 5 | 1 | 5 | −6.49 | −5.62 | 0,1 * |
| ChC13 | 4.24 | 351.42 | 63.6 | 4 | 1 | 6 | −5.86 | −5.04 | 0 |
| ChC14 | 3.91 | 386.22 | 63.6 | 4 | 1 | 4 | −5.61 | −5.43 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moya-Alvarado, G.; Yañez, O.; Morales, N.; González-González, A.; Areche, C.; Núñez, M.T.; Fierro, A.; García-Beltrán, O. Coumarin-Chalcone Hybrids as Inhibitors of MAO-B: Biological Activity and In Silico Studies. Molecules 2021, 26, 2430. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26092430
Moya-Alvarado G, Yañez O, Morales N, González-González A, Areche C, Núñez MT, Fierro A, García-Beltrán O. Coumarin-Chalcone Hybrids as Inhibitors of MAO-B: Biological Activity and In Silico Studies. Molecules. 2021; 26(9):2430. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26092430
Chicago/Turabian StyleMoya-Alvarado, Guillermo, Osvaldo Yañez, Nicole Morales, Angélica González-González, Carlos Areche, Marco Tulio Núñez, Angélica Fierro, and Olimpo García-Beltrán. 2021. "Coumarin-Chalcone Hybrids as Inhibitors of MAO-B: Biological Activity and In Silico Studies" Molecules 26, no. 9: 2430. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26092430