
Cyan-Emitting Cu(I) Complexes and Their Luminescent Metallopolymers
by
 , , , , and
, , , , and
Federico Ferrari
1 ,
,
Jonas Braun
2 ,
,
Christopher E. Anson
2,
Bodo D. Wilts
3,
Dafni Moatsou
1,* and
Claudia Bizzarri
1,* 1
Karlsruhe Institute of Technology, Institute of Organic Chemistry, Fritz-Haber-Weg 6, 76131 Karlsruhe, Germany
2
Karlsruhe Institute of Technology, Institute of Inorganic Chemistry, Engesserstrasse 15, 76131 Karlsruhe, Germany
3
Adolphe Merkle Institute, University of Fribourg, Chemin des Verdiers 4, 1700 Fribourg, Switzerland
*
Authors to whom correspondence should be addressed.
Molecules 2021, 26(9), 2567; https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26092567
Submission received: 19 March 2021
/
Revised: 19 April 2021
/
Accepted: 26 April 2021
/
Published: 28 April 2021
(This article belongs to the Special Issue Synthesis and Investigation of Novel Coordination Compounds)
Abstract
:Copper complexes have shown great versatility and a wide application range across the natural and life sciences, with a particular promise as organic light-emitting diodes. In this work, four novel heteroleptic Cu(I) complexes were designed in order to allow their integration in advanced materials such as metallopolymers. We herein present the synthesis and the electrochemical and photophysical characterisation of these Cu(I) complexes, in combination with ab initio calculations. The complexes present a bright cyan emission (λem ~ 505 nm) in their solid state, both as powder and as blends in a polymer matrix. The successful synthesis of metallopolymers embedding two of the novel complexes is shown. These copolymers were also found to be luminescent and their photophysical properties were compared to those of their polymer blends. The chemical nature of the polymer backbone contributes significantly to the photoluminescence quantum yield, paving a route for the strategic design of novel luminescent Cu(I)-based polymeric materials.
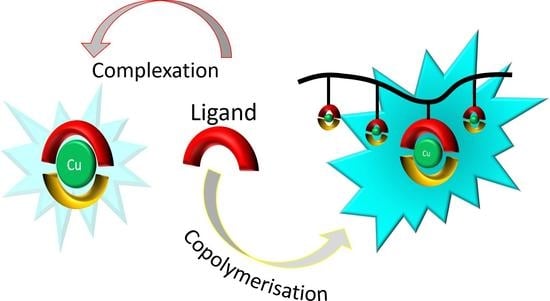
1. Introduction
Luminescent transition metal complexes represent a significant class of materials that is exploited in a vast range of applications, from optoelectronic technologies, such as organic light-emitting diodes (OLEDs) [1,2], to bioimaging and sensors [3,4] or in photo(redox) catalysis [5,6,7]. Copper (I) complexes are a desirable cost-effective choice for these applications, particularly in contrast to well established light-emitting materials based on noble metals, like iridium, platinum or ruthenium [8,9]. Their high availability paired with the significantly lower price could enable the implementation of Cu(I) complexes into everyday life applications, such as electronic devices. Cu(I) complexes present tuneable emission at room temperature in solid state and solution. However, the photoluminescence quantum yield in solution often suffers from the Jahn–Teller distortion that these complexes undergo in their excited state [10,11,12]. This distortion is a consequence of the population of a metal-to-ligand charge-transfer state (MLCT), where the metal centre formally loses one electron that goes to an empty π* orbital of the coordinated ligand. Thus, the valence state of the Cu centre in its excited state can be considered +2. The structural change might activate non-radiative pathways, thereby decreasing the photoluminescence quantum yield of these complexes.
A strategy to reduce the disadvantages of the population of the MLCT, minimising the Jahn–Teller distortion, is to functionalise the coordinated ligands with bulky substituents [13,14] or increase the rigidity of ancillary ligands, such as chelating diphosphines [15,16,17]. In any case, this inconvenience is moderate in solid state. Thus, Cu(I) complexes have been indicated as promising emitters in OLED technologies [9,18,19,20].
Polymers are often used as scaffolds onto which functional moieties are immobilised to improve the stability and solubility of these functional groups [21]. The so-formed functional polymers combine the components’ properties; for example, hybrid materials that combine the properties of a coordination metal complex with those of an organic polymer have been previously reported [22]. Indeed, the covalent attachment of metal–organic complexes to polymer chains has been shown to prevent phase separation and to enhance the properties of the functional groups (e.g., in catalysis). At the same time, unique properties such as unimolecular micellization have also been reported [23,24,25]. Furthermore, such hybrids improve processability and give the metal complex increased stability [26]. Two approaches are commonly pursued: the functional group is either attached to the polymer in a post-polymerisation reaction or through direct copolymerisation, which first requires introducing a polymerisable moiety onto the functional group [21]. Both approaches have been reported for a range of polymerisation techniques. However, one commonly used approach is radical (co)polymerisation, as it is a robust reaction that is generally tolerant towards a range of functionalities. Despite the increasing number of highly emissive Cu(I) complexes in the scientific literature, very few works report luminescent metallopolymers based on copper [27,28,29,30,31].
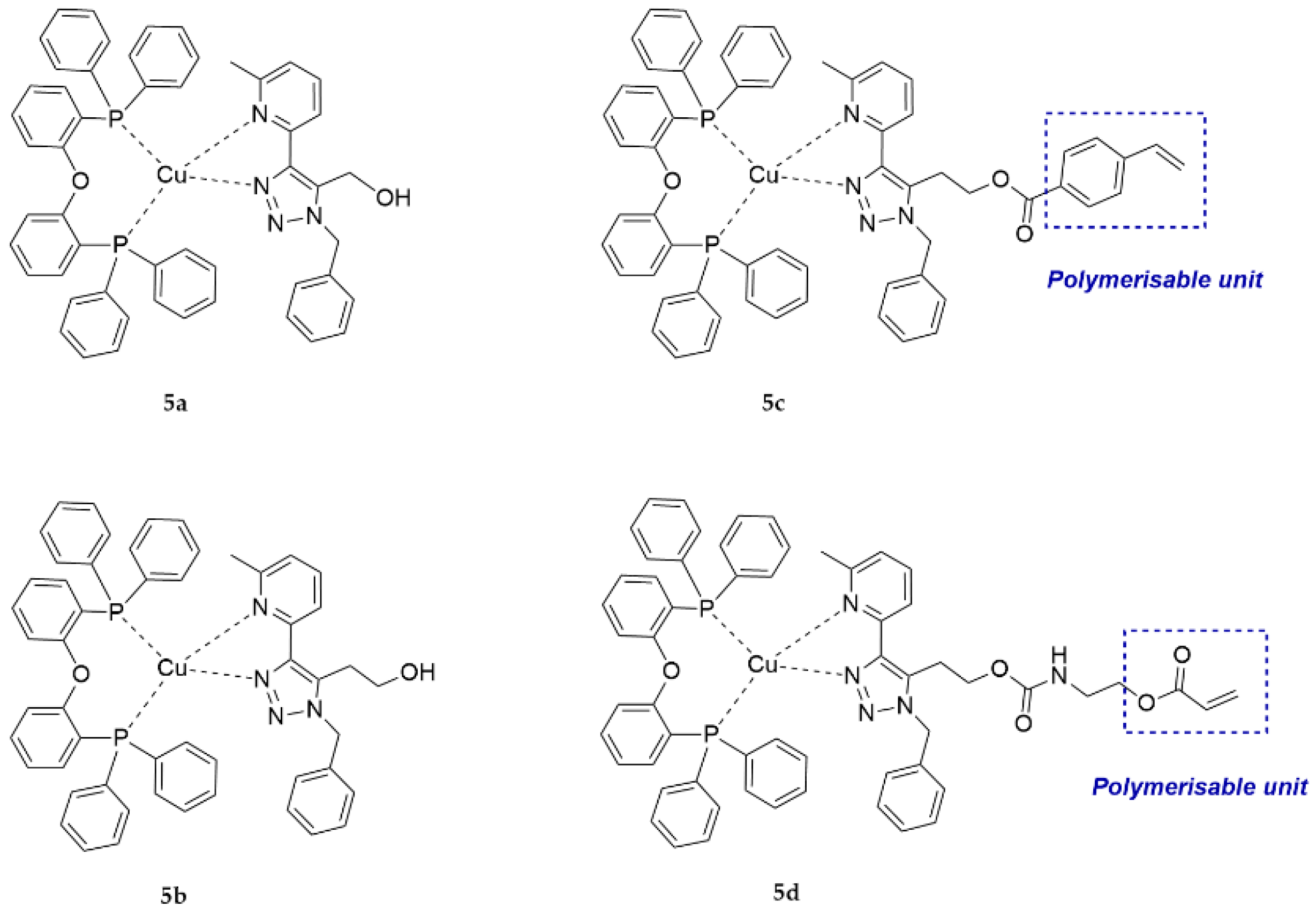
In this work, four novel heteroleptic copper (I) complexes giving a bright cyan emission in their solid state were synthesised (Figure 1). The coordination of Cu(I) to the diphosphine bis[(2-diphenylphosphino)phenyl]ether (DPEPhos) gave more stability to the final complex since the steric hindrance of this chelating diphosphine is elevated. The chelating diimines, whose electronic properties determine the emission of the complex, are based on pyridinyl-1,2,3-triazole. Two of those diimines carry a hydroxyl group (5a and 5b), which can be used in carbamate or esterification reactions. Further functionalisation of the diimine ligand with vinyl derivatives gave access to polymerisable complexes (5c and 5d). Herein, we present the electrochemical and the photophysical characterisation together with computational studies of the new Cu(I) complexes. Radical copolymerisation of the polymerisable ligands with suitable comonomers yielded functional polymers with controlled loadings. The properties of the copper complexes in bulk were directly compared to those of their respective copolymers. Moreover, polymer blends of the complexes 5c and 5d were also investigated to compare the metal complex-functional repeat unit of the corresponding copolymer.
2. Results and Discussion
2.1. Synthesis
2.1.1. Ligand Synthesis
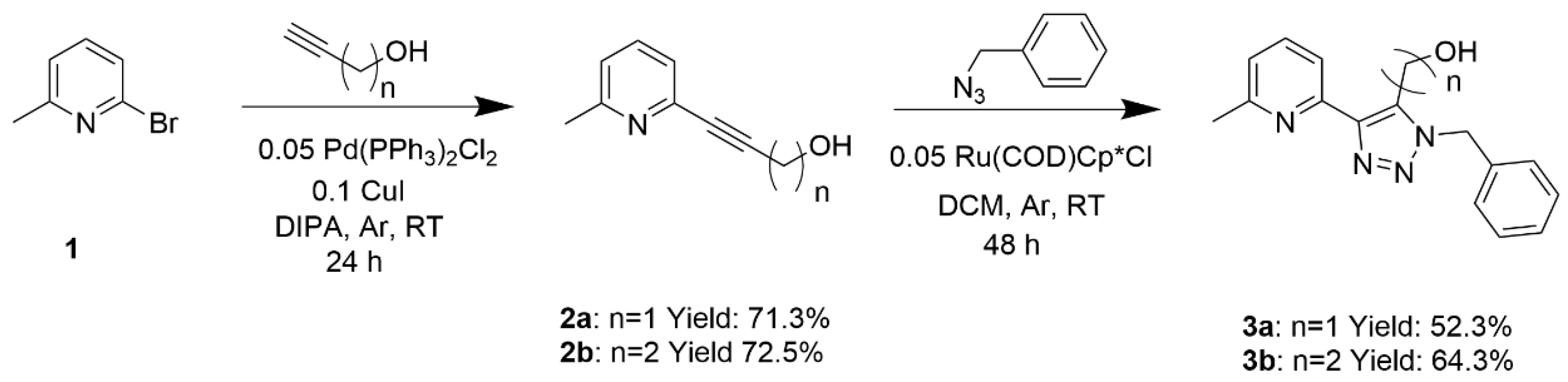
The chelating diimine ligands were synthesised in two steps, as shown in Scheme 1. First, a Sonogashira cross-coupling reaction [32,33] was carried out on the substrate 2-bromo-6-methylpyridine, 1, with one equivalent of alkynyl alcohols, propargyl alcohol or 3-butynol, to give the products 2a and 2b, respectively. The following step was forming the 1,2,3-triazoles 3a and 3b, obtained from a cyclisation reaction between 2a or 2b and benzyl azide via a ruthenium–alkyne–azide-cycloaddition (RuAAC) [34]. A Cu-catalysed cycloaddition [35] was not possible because the triple bonds of 2a and 2b are not terminal. As all organic azides, benzyl azide is a hazardous and potentially explosive material. Therefore, particular care is needed [36]. These ligands differ in the number of methylene units that separate the triazole ring from the hydroxyl group, which was subsequently found to drastically affect the reactivity of the latter, e.g., in esterification reactions.
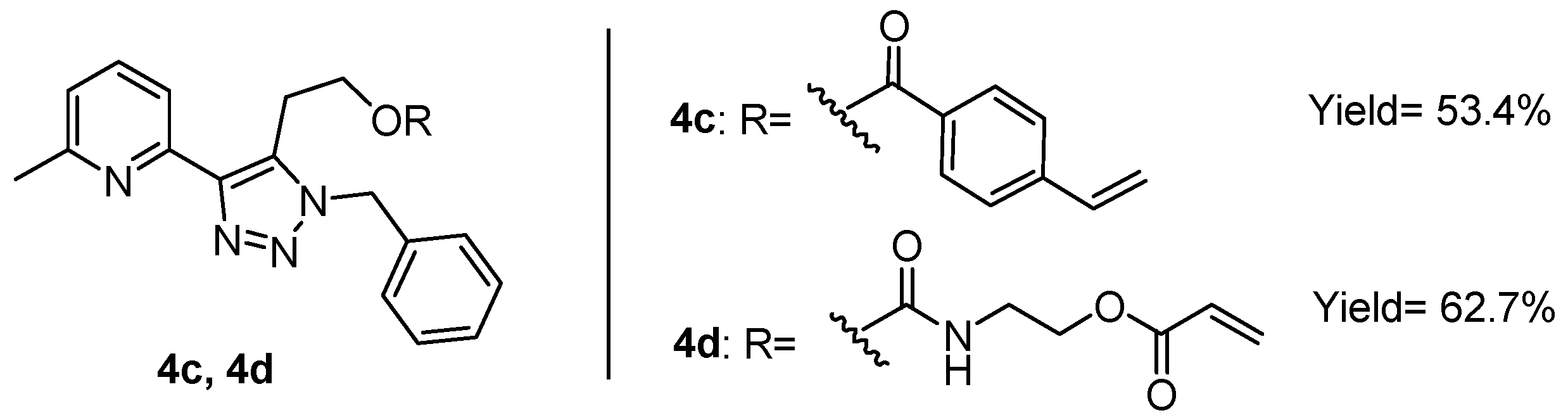
The alcohol function of 3a and 3b was used to introduce a polymerisable unit, an acrylate or styrene, via carbamate formation or a Steglich esterification [37]. These two functionalities were chosen as they can readily undergo radical polymerisation [38]. The acrylate was particularly interesting as its corresponding polymer does not absorb in the near UV. This UV transparency was anticipated to prevent the undesirable quenching of the complex fluorescence. In contrast, styrene and its polymers absorb at 295 nm, which was envisioned to interfere with the photophysical properties of the metal complex. The modification of 3a was found to suffer from poor yields and the subsequent polymerisation of the product was unsuccessful. For the styrenic monomer, vinyl benzoic acid was used to perform a Steglich esterification with triazole 3b to yield monomer 4c (Figure 2). To obtain the acrylate monomer (4d), triazole 3b was reacted with 2-isocyanatoethyl acrylate yielding the corresponding urethane in reasonable yield and purity (see Figure 2).
2.1.2. Copper(I) Complexes Synthesis
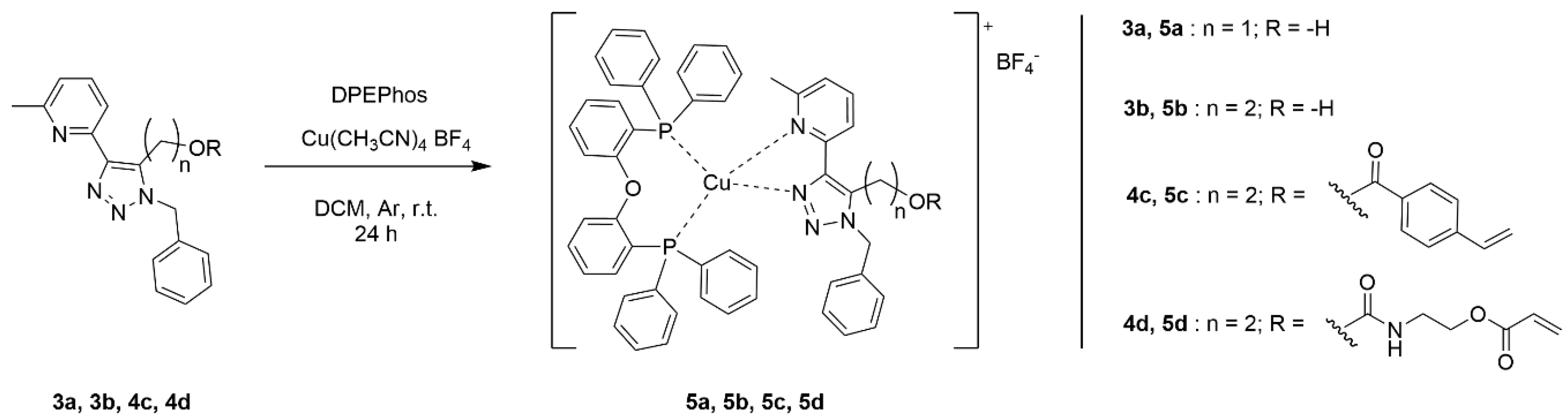
Ligands 3a and 3b, as well as monomer-ligands 4c and 4d, were used to obtain the respective heteroleptic copper complexes. The synthesis of all the complexes started from the formation of the diphosphine complex by mixing bis[(2-diphenylphosphino)phenyl]ether (DPEPhos) and a copper precursor tetrakis(acetonitrile) copper(I) tetrafluoroborate [39]. Subsequently, the chelating ligands (3a, 3b, 4c, and 4d) were added to the stirring mixture, thus allowing the ligand exchange with one of the DPEPhos and resulting in the formation of the respective heteroleptic complexes (5a, 5b, 5c and 5d) that are shown in Scheme 2. The evaporation of the solvent gave pale yellow powders, which were found to exhibit luminescence when placed under UV light (350 nm). Purification was done by recrystallisation.
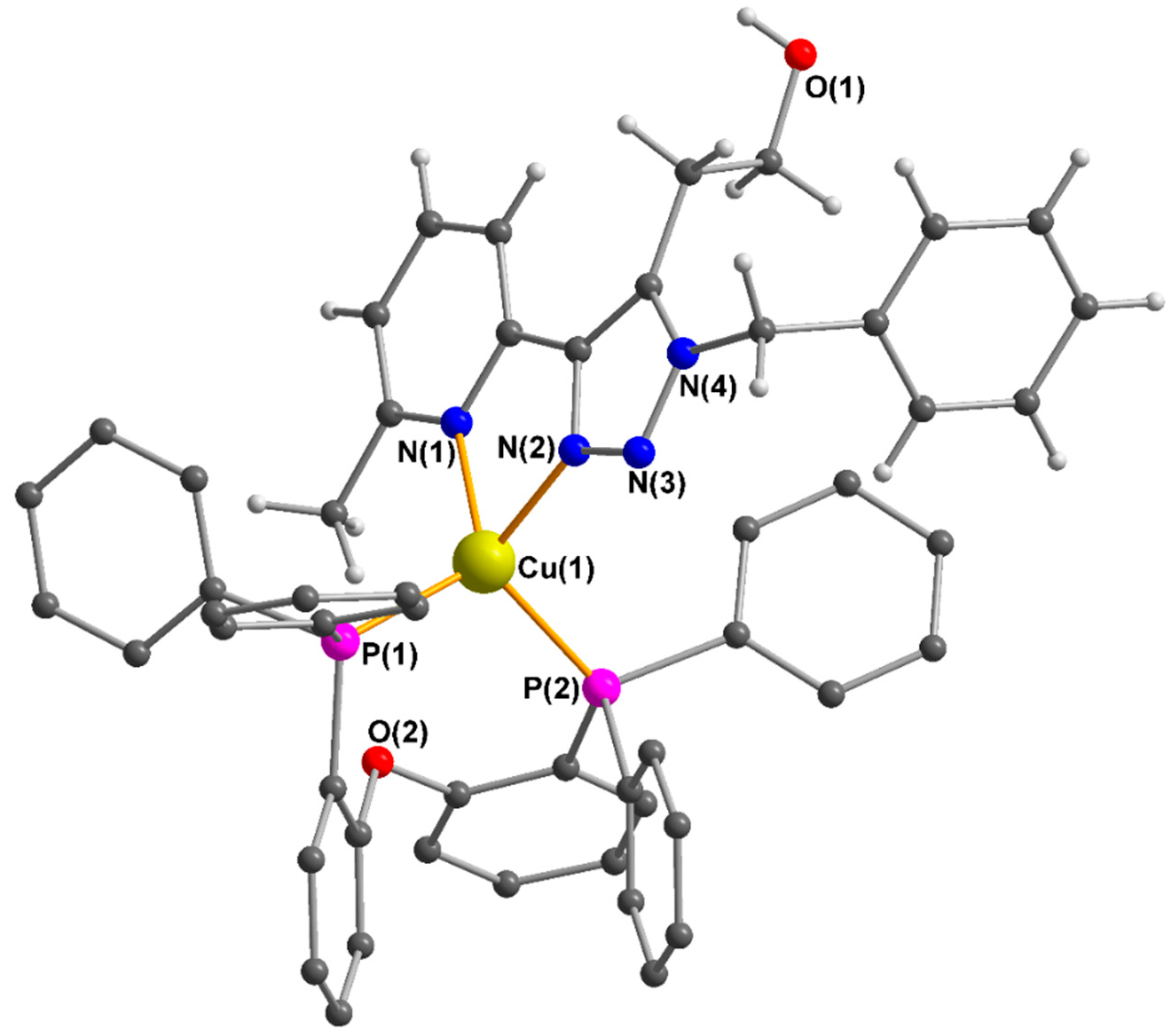
Needle-shaped crystals of 5b were obtained by the slow diffusion of cyclohexane into a concentrated solution in dichloromethane. The molecular structure of complex 5b was determined by X-ray diffraction, shown in Figure 3, and agrees with heteroleptic Cu(I) complexes with similar ligands [40,41]. Bond valence sum analysis for Cu(1) in 5b gave a calculated oxidation state of 1.007 [42]. Further parameters are described in the experimental section.
2.2. Synthesis and Structural Characterisation of Metallopolymers with Cu(I) Complexes
In order to access functional polymeric materials with different physical properties, but also to take advantage of the different photophysical properties of the complexes (vide infra), styrene (St) and methyl acrylate (MA) were copolymerised with the corresponding styrenic (4c) and acrylate-functional (4d) ligands, respectively, to yield two copolymers: poly(St-co-4c) (6c) and poly(MA-co-4d) (6d) (molecular weights are reported in Table 1). These were subsequently used to form the heteroleptic Cu(I) complexes, similarly to 5c and 5d, by the addition of DPEPhos and Cu(CH3CN)4BF4. Consequently, the polymer-supported complexes were obtained, namely 7c and 7d, with a targeted complex loading of 10 mol%. It is noted that the direct copolymerisation of the corresponding complexes (5c and 5d) was unsuccessful, presumably due to the copper interfering with the radical process [43].
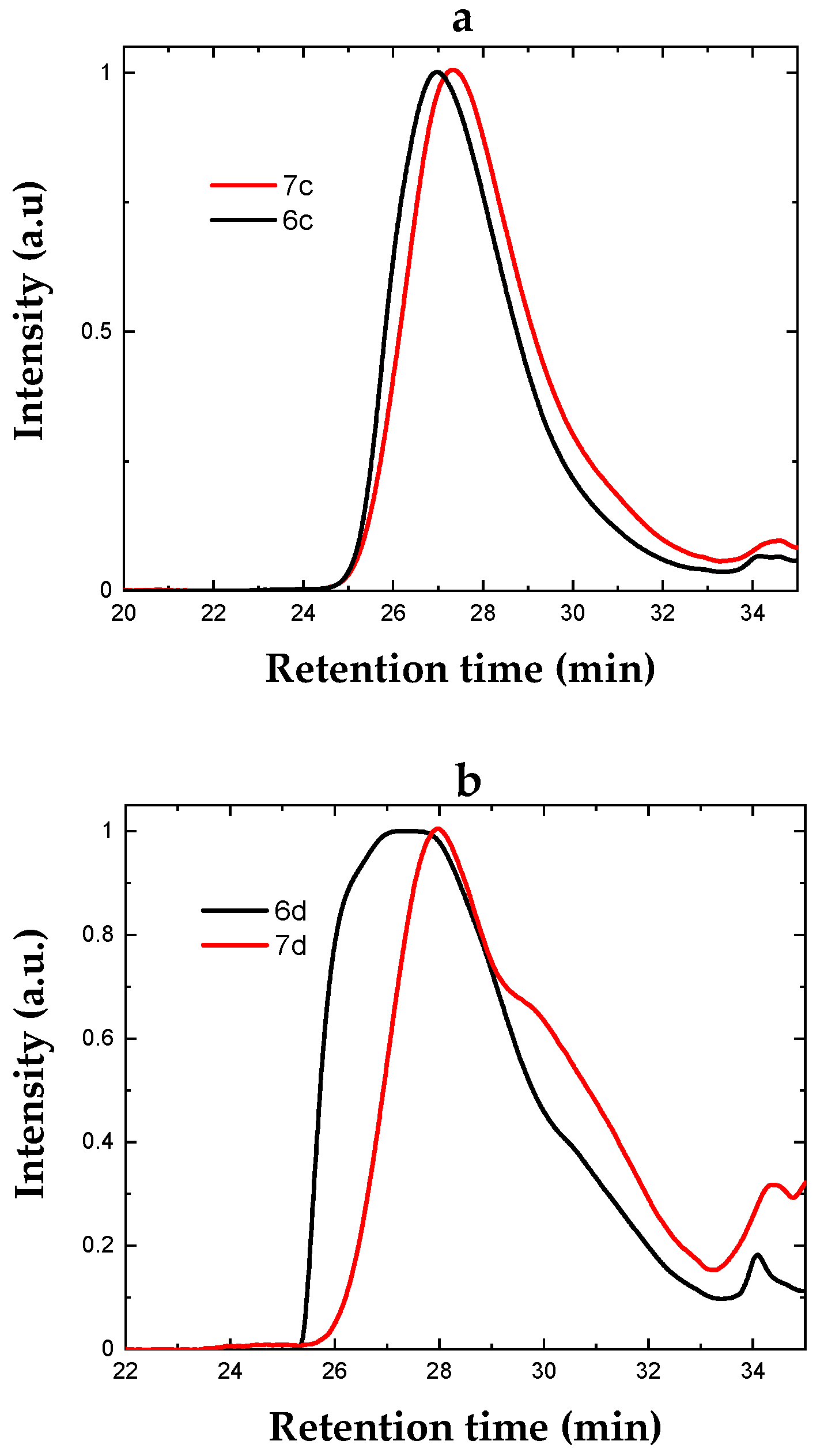
The copolymerisation of the styrenic ligand 5c with styrene yielded a copolymer with a single molecular weight distribution in the size exclusion chromatogram (SEC, Figure 4), indicating the reasonable control of the reaction, in contrast to the copolymerisation of the acrylate ligand 5d with methyl acrylate that resulted in a broad and multimodal distribution. It is noted that higher molecular weights are attainable by reducing the ratio of the initiator (AIBN) equivalents to the monomers. Nonetheless, the NMR analysis of both copolymers indicated ligand loading in reasonable agreement with the targeted value, namely 8 mol% for 6c and 11 mol% for 6d (Supporting Information, Figures S14 and S15). Subsequent complexation, however, was found highly efficient for the styrenic copolymer with an overall complex loading of 7 mol% for 7c, whereas, for the acrylate copolymer 7d, it was 5 mol% (Supporting Information, Figures S16 and S17). This was attributed to the dispersity of copolymer 6d as we hypothesised that the uncontrolled polymerisation reaction might have resulted in the poor distribution of the ligand along the polymer chain. The SEC data of both complexed copolymers indicated a decrease in the apparent molecular weight, attributed to the interaction of the newly introduced species with the column material.
In order to assess the effect of the covalent immobilisation of the metal complexes onto the polymers, the respective blends were also prepared by simply mixing in solution and subsequently allowing the drying of poly(styrene) (PS) (Mn = 12.9 kg/mol, Ð = 1.50) with 5c, yielding PS/5c, and poly(methyl acrylate) (PMA) (Mn = 17.0 kg/mol, Ð = 1.68) with 5d, yielding PMA/5d, targeting in both cases an average loading comparable to that of the ligand-copolymers (10 mol%).
2.3. Photophysical Characterisation of the Cu(I) Complexes
2.3.1. In Solution
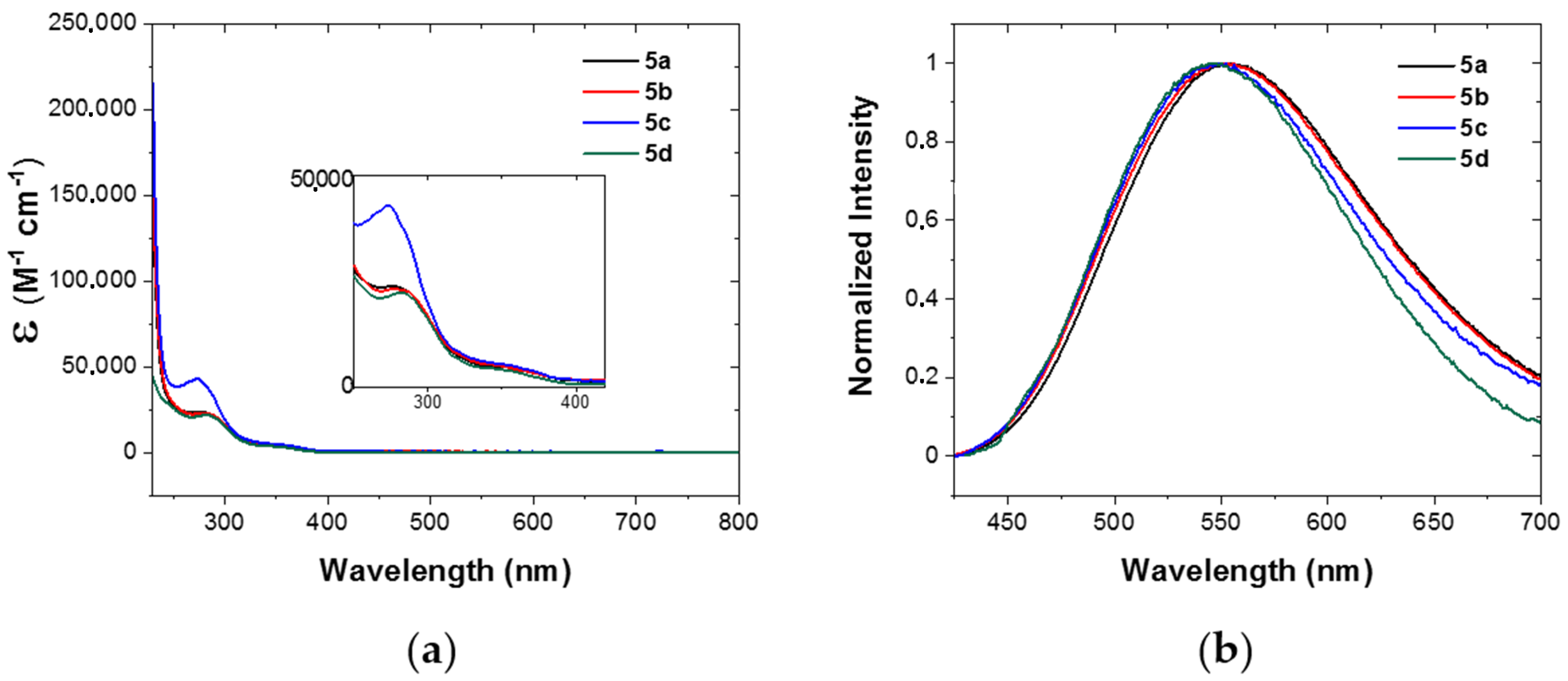
The electronic absorption spectroscopy of the four new Cu(I) complexes was investigated in dichloromethane (DCM) solutions at room temperature. The molar extinction coefficients are reported in Table 2 and shown in Figure 5a. As expected from the strong similarity of their chemical structures, we can identify many correspondences in their UV–vis absorption spectra. All the complexes present very intense extinction coefficients below 300 nm. These are attributed to spin-allowed π–π* transitions, i.e., ligand-centred transitions (1LC), mainly located on the chelating phosphine DPEPhos and the diimine ligand [15]. The additional contribution of the vinyl-phenyl moiety on the ligand of complex 5c is highlighted by the absorption band with a maximum at 273 nm. A low-intensity shoulder is present in all the complexes, ranging from 320 nm to 400 nm. This shoulder was assigned to the electronic transitions from the copper centre to the π* orbital of the diimine ligand, populating a singlet metal-to-ligand charge-transfer state (1MLCT), typical for Cu(I) complexes [12,14].
All the new complexes are luminescent at room temperature in Ar-saturated solutions (Figure 5b). In particular, when excited at their MLCT band, they show an approximately 180 nm-broad structureless emission band centred at about 550 nm, with only slight differences among the complexes (Table 2). This emission profile is typical for Cu(I) complexes of type Cu(PP)(NN), and it is assigned to the excited MLCT state. The Stokes shifts are substantial (1.33 eV for 5a and 5b, 1.24 eV for 5c and 1.30 eV for 5d), which, together with the observation that the luminescence is quenched in air-equilibrated solutions, are firm evidence of a triplet nature of the excited state. However, as several Cu(I) complexes have been demonstrated to show thermally activated fluorescence (TADF), [19,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58] this cannot be excluded at this stage. The excited state lifetimes in solution are in the order of one microsecond, in addition to complex 5c which shows a lifetime of 83 ns. Further analyses are ongoing to investigate the processes involved in the excited state in more detail and whether these complexes show TADF.
The photoluminescence quantum yields (Φ) of compound 5b and its acrylate derivative 5d are very similar (3.7% and 3.5% for 5b and 5d, respectively). The low emission of Cu(I) complexes even in non-coordinating solvents, such as DCM, is attributed to the Jahn–Teller distortion that the complex undergoes upon excitation. The presence of the methyl substituent α to the pyridine might have impeded the distortion to some extent, as this was also demonstrated in other studies [40,59,60,61]. Complex 5c presents a Φ of only 0.9%. The presence of the styrene moiety on the diimine is likely responsible for the lower quantum yield with respect to the one of the precursor complex 5b. This effect was also observed with other Cu(I) complexes containing a styrene moiety [27]. Moreover, ab initio computational studies confirm that styrene is involved in the lowest unoccupied molecular orbital (LUMO) of complex 5c (vide infra; see Section 2.4 and the Supporting Information, Figures S44–S46).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 2.
Photophysical properties of Cu(I) complexes 5a, 5b, 5c and 5d in solution at room temperature.
Table 2.
Photophysical properties of Cu(I) complexes 5a, 5b, 5c and 5d in solution at room temperature.
| Sample | λabs (nm) | ε ×10−3 (M−1 cm−1) | λem a (nm) | Φ b | τ (ns) c |
|---|---|---|---|---|---|
| 5a | 347 276 250 | 4.3 23.5 27.3 | 552 | n.a. | 850 |
| 5b | 347 276 250 | 4.4 22.7 28.5 | 552 | 0.037 | 1050 |
| 5c | 354 273 250 | 4.6 43.0 38.6 | 548 | 0.009 | 83 |
| 5d | 347 281 240 | 3.6 22.0 30.7 | 545 | 0.035 | 1500 |
a in Ar-saturated DCM; b photoluminescence quantum yields were measured with the relative method using Ru(bpy)3Cl2 in aerated water solution as standard (Φ = 0.040) [62]; c lifetimes were measured with TCSPC and NanoLED excitation at 366 nm.
2.3.2. In Solid State
We carried out the photoluminescence quantum yield measurements from the samples in bulk in order to assess the photophysical properties of the complexes and their metallopolymers in the solid state (Table 3). For the sake of accuracy, the homopolymers of PMA and PS were also measured and exhibited no luminescence, as expected. Initially, the photoluminescence quantum yields of the crystalline powders of the four Cu(I) complexes were estimated. The highest quantum yield of 17% was obtained for the complex bearing the acrylate functionality (5d) (Figures S52–S55). Overall, the trend of data was in agreement with the measurements carried out in solution.
Furthermore, drop-casted films of the metallopolymers 7c and 7d were evaluated and compared with those films of polymeric blends with complexes 5c and 5d (a photograph of drop-casted PMA/5d is shown in Figure S75). The emission colour of these films is cyano-green, as the chromaticity diagram CIE 1931 reports (Figure S66). The polymeric blends were obtained dispersing 5c or 5d in both homopolymers, PS and PMA, at comparable loading (10%). Neither drop-casted film of blends PS/5c nor PMA/5c exhibited luminescence when excited at 350 nm, while the metallopolymer 7c (loading 8%) presents an emission with Φ of 10% (Figures S56–S61). This points to the significant effect of the conjugation in the styrene moiety of the free 5c, which appears to be responsible for the low or absent photoluminescence. This hypothesis was validated by computational analysis (vide infra). On the contrary, the emission of the metallopolymer 7c is encouraging and further supports the development of metallopolymers as materials with advanced properties.
In contrast, the double bond of the acrylate unit of complex 5d does not have an unfavourable effect on its emission. In fact, both the PS/5d and the PMA/5d blends were emissive. In particular, PMA/5d exhibited a Φ of 21%, which was almost twice the Φ of PS/5d (Φ = 13%). This difference was attributed to the different chemical environment provided by the respective polymers. Thus, PS induces nonradiative pathways of 5d and this quenching may be due to π-stacking between the aryl groups of the complex interacting with the pendant phenyl rings of the PS backbone. Alike the metallopolymer 7c, the film of 7d was luminescent and presented a Φ of 10%. It must be remarked that the complex loading of 7d was only 5%, due to the poor yield of the DPEPhosCu+ coordination onto the copolymer 6d (see Section 2.2). Thus, the metallopolymer 7d also shows promising emissive properties in line with the respective blend in PMA.
2.4. Computational Studies of Cu(I) Complexes
To gain further insight into the photophysical and electronic properties of the new copper complexes, theoretical ab initio quantum chemical calculations were performed. We omitted the investigation of 5a since this complex was not further functionalised with a monomeric unit (e.g., an acrylate or a styrene moiety) and therefore was not part of the final metallopolymers studied in this work. Moreover, 5a differs from complex 5b only by a -CH2- unit, having in position 5 of the triazole ring a hydroxymethyl substituent, while 5b has a hydroxyethyl at the same position. Therefore, we expect 5a to have comparable properties to 5b. The geometry of complexes 5b, 5c and 5d was then optimised at the density functional theory (DFT) level (PBE0/6-31G**/LANL2DZ, in vacuum) using the QChem package [64].
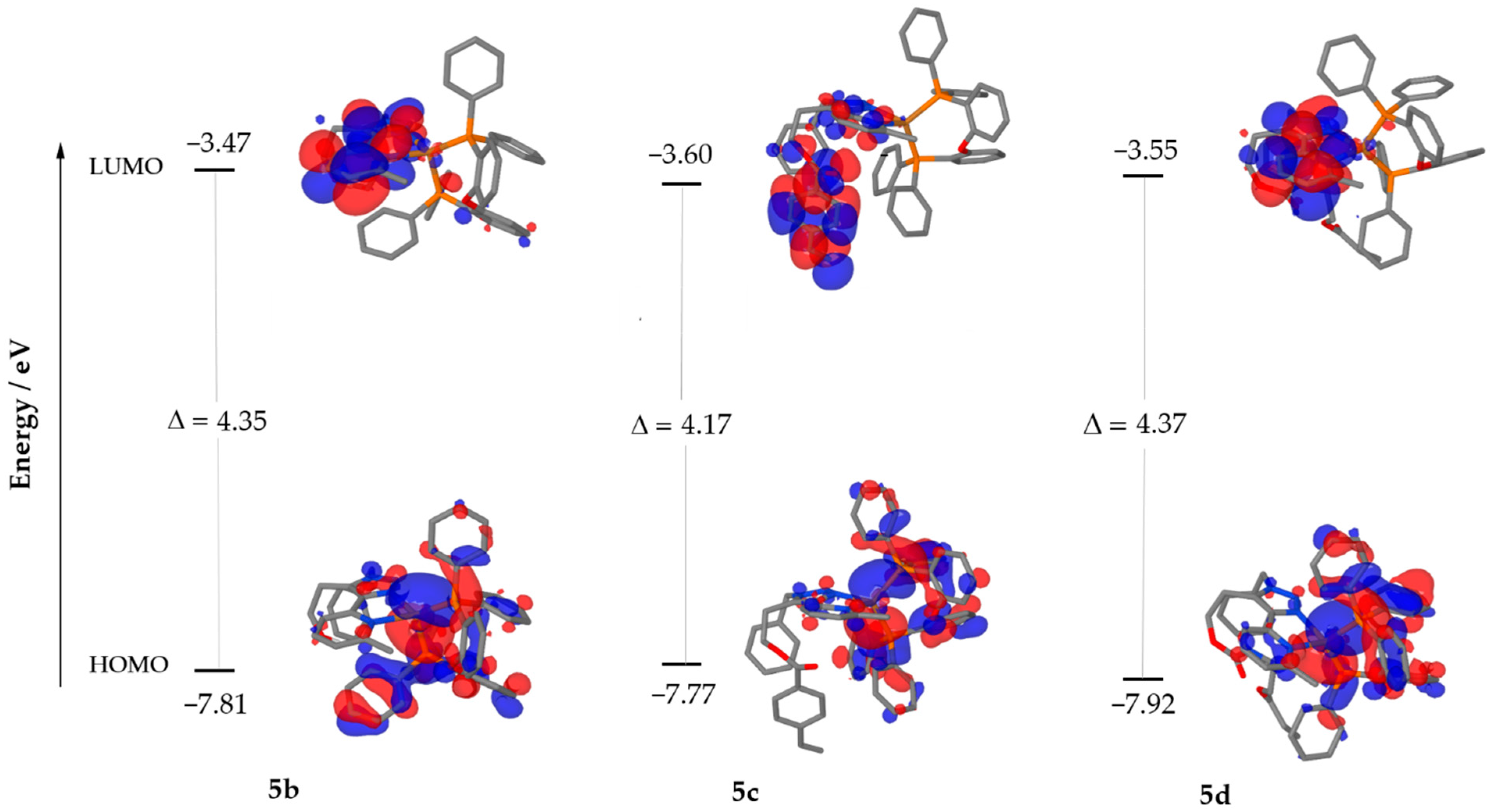
The optimised geometries were used to predict absorption spectra via time-dependent DFT (TDDFT), from where the energies and molecular orbital compositions of triplet and singlet states were obtained. The calculations indicate that the transition to the first excited singlet is described by the electronic transition from the highest occupied molecular orbital (HOMO) to the LUMO. As shown in Figure 6, the HOMO of the three complexes is mainly located on the Cu centre with some contribution from the chelating phosphine, whilst in contrast, the LUMO is completely delocalised over the diimine ligand. Thus, upon HOMO → LUMO excitation, the charge is transferred from Cu(PP) to the pyridinyl-triazole (NN) ligand, indicating an MLCT character of the excited state. The convoluted data, obtained from the predicted vertical transitions, are in accordance with the experimental electronic absorption spectra. This agreement is presented in Figures S44–S46 for 5b, 5c and 5d.
Interestingly, in complex 5c, the LUMO is strongly localised on the styrene unit appended to the diimine ligand through the ester functionality, showing less contribution of the pyridinyl-triazole moiety directly coordinating the metal centre. This result justifies the experimental observation of the lower luminescence of 5c when compared to 5b and 5d. In other words, the styrene unit is responsible for a significant electronic involvement with the consequent deactivation of the excited state. Furthermore, the acrylate unit of 5d does not participate in the electronic transitions, explaining why the photoluminescence quantum yield of 5d is virtually identical to 5b. The HOMO and LUMO values are reported in Table 4. Furthermore, the trend of electronic band gaps obtained from theoretical calculations is in agreement with the electrochemical data presented subsequently.
2.5. Electrochemical Properties of Cu(I) Complexes
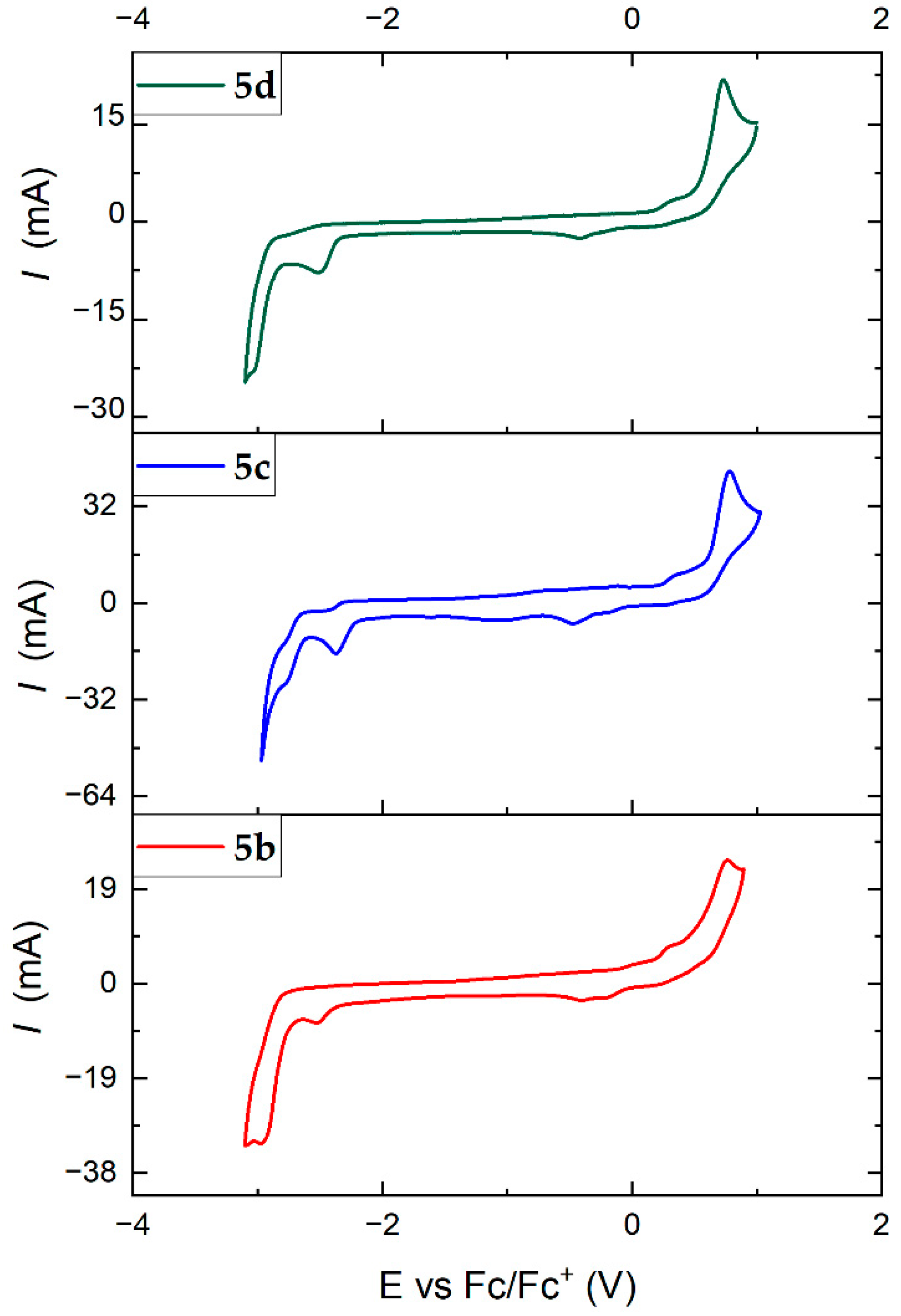
The electrochemical behaviour of the new Cu(I) complexes was investigated by cyclic voltammetry (CV), which was performed in DCM (Figure S47) and in N,N-dimethylformamide (DMF) solutions with 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) as the supporting electrolyte. The three-electrode cell was equipped with glassy-carbon as the working electrode, and a Pt wire was used as the auxiliary electrode. Since Ag wire was used as a quasi-reference electrode, we reported all the values versus the ferrocene/ferricinium (Fc/Fc+) redox couple used as the internal standard. The obtained results are displayed in Figure 7, and the derived values are reported in Table 4. We observe an irreversible oxidation process around 0.6 V for the three investigated complexes, attributed to the oxidation of Cu(I) to Cu(II). Note that this oxidation process becomes reversible when the measurement is performed in dichloromethane. Therefore, although the electrochemical window of DCM is too small in reduction, a more detailed study of the oxidation process was carried out in this solvent for all the complexes. As is displayed in the Supporting Information (Figures S47–S51), we performed the electrochemical measurement at different scan rates. The peak intensity of the anodic process was plotted versus the square root of the scan-rate. An excellent linear fit confirms that the oxidation processes of all four complexes are diffusion-controlled (Figures S48–S51), as the anodic peak currents of their oxidation process follow the Randles–Sevcik equation [65]. In the reduction, two irreversible processes are observed for the complexes 5b, 5c and 5d, which are attributed to the reduction in the respective diimine chelating ligand. In particular, the first reduction can be associated with the LUMO of the complex. Thus, the electrochemical band gaps were estimated to be 3.15 V, 3.01 V and 3.12 V for complexes 5b, 5c and 5d, respectively. Although these band gaps deviate considerably from those calculated from the computational studies, there is a correlation, revealing that the electrochemical and theoretical HOMO–LUMO gaps are consistent.
3. Materials and Methods
3.1. Materials
Solvents of p.a. quality (per analysis) were commercially acquired from Sigma Aldrich, Carl Roth, or Acros Fisher Scientific and unless otherwise stated, used without further purification. Anhydrous solvents were purchased from Carl Roth, Acros, or Sigma Aldrich (less than 50 ppm of H2O, kept over molecular sieves). 2-Bromo-6-methylpyridine (98%) and benzyl azide (94%) were bought from Alfa Aesar. Propargyl alcohol (98%), butynyl alcohol (97%), bis(triphenylphosphine)palladium(II) dichloride (98%), and chloro(pentamethylcyclopentadienyl)(cyclooctadiene)ruthenium(II) (99.5%) were purchased from Sigma Aldrich.
Reaction mixtures were purified by flash chromatography. For the stationary phase of the column, silica gel, produced by Merck (silica gel 60, 0.040 × 0.063 mm, 260–400 mesh ASTM) and sea sand by Riedel de Haën (baked out and washed with hydrochloric acid) were used.
Air- and moisture-sensitive reactions were carried out under argon atmosphere in previously baked out apparatuses with standard Schlenk techniques. Liquid reagents and solvents were injected with syringes and stainless-steel cannulas of different sizes.
3.2. Synthetic Procedures
3.2.1. 2-(2-Propyn-1-ol)-6-methylpyridine (2a) and 2-(3-Butynyl-1-ol)-6-methylpyridine (2b)
In a typical procedure, into a flame-dried 50 mL two-neck round bottom flask, 25 mL of dry diisopropylamine (DIPA) was added under argon, followed by the addition of 1-bromo-6-methylpyridine 1 (1.99 mL, 17.44 mmol, 3.00 g, 1 equiv.), the alcohol (propargyl alcohol 8a or 3-butynol 8b) (17.44 mmol, 1 equiv.), copper(I) iodide (1.74 mmol, 0.33 g, 0.1 equiv.) and bis(triphenylphosphine)palladium(II) dichloride (0.872 mmol, 0.61 g, 0.05 equiv.). The reaction mixture was allowed to stir at room temperature for 24 h. After the removal of the solvent under reduced pressure, the residue was dissolved in 5 mL of DCM and cyclohexane was added dropwise. The biphasic solution obtained was then left in the fridge for 48 h until a precipitated was formed. The pure product was isolated as a brown solid. Yields are shown in Table 5.
2a:
1H-NMR (CDCl3, 400 MHz) δ (ppm): 7.50 (1H, t, 3J = 7.7 Hz, -N=C-CH=CH-), 7.22 (1H, d, 3J = 7.7 Hz, CH3-C=CH-), 7.07 (1H, d, 3J = 7.7 Hz, -N=C-CH=CH-), 4.53 (2H, s, CH2), 2.51 (3H, s, CH3).
13C-NMR (CDCl3, 101 MHz) δ (ppm): 158.78, 142.04, 136.78, 124.39, 88.58, 84.25, 50.99, 24.24.
ESI-MS m/z [M+H]+ calculated for C9H9NO: 148.08, found: 148.08.
IR ν (cm−1) I3186.1 (m), 3056.6 (w), 2955.8 (w), 2916.7 (w), 2852.9 (w), 1590.0 (m), 1569.4 (m), 1460.4 (s), 1439.8 (s), 1376.1 (m), 1355.5 (s), 1289.7 (w), 1236.2 (m), 1168.3 (s), 1094.3 (w), 1042.9 (s), 1018.2 (s), 997.6 (vs), 962.6 (m), 892.7 (w), 851.6 (w), 796.0 (vs), 746.7 (w), 695.2 (vs), 602.7 (w), 575.9 (vs), 541.0 (m), 532.7 (m), 440.2 (w).
1H-NMR, 13C-NMR, IR spectra are shown in the Supporting Information: Figures S2, S18, S28, respectively.
2b:
1H-NMR (CDCl3, 400 MHz) δ (ppm): 7.50 (1H, t, 3J = 7.7 Hz,-N=C-CH=CH-), 7.20 (1H, d, 3J = 7.7 Hz, CH3-C=CH-), 7.06 (1H, d, 3J = 7.7 Hz, -N-C-CH=CH-), 3.86 (2H, t, 3J = 6.6 Hz, -CH2-CH2-OH), 3.41 (1H, br, -OH), 2.71 (2H, t, 3J = 6.6 Hz, -CH2-CH2-OH), 2.52 (3H, s, CH3).
13C-NMR (CDCl3, 101 MHz) δ (ppm): 158.74, 142.70, 136.74, 122.57, 88.58, 84.25, 50.99, 24.24.
ESI-MS m/z [M+H]+ calculated for C10H11NO: 162.08, found: 162.08.
IR ν (cm−1) 3186.1 (w), 3182.0 (w), 3073.0 (w), 3058.6 (w), 2935.2 (w), 2910.5 (w), 2867.3 (w), 2840.6 (w), 2231.7 (w), 1631.1 (vw), 1585.9 (s), 1571.5 (s), 1503.6 (w), 1456.3 (vs), 1427.5 (s), 1376.1 (m), 1300.0 (w), 1289.7 (w), 1256.8 (w), 1236.2 (m), 1180.7 (w), 1166.3 (m), 1119.0 (w), 1100.4 (w), 1059.3 (vs), 1044.9 (vs), 1012.0 (s), 999.7 (s), 915.3 (w), 847.4 (w), 796.0 (vs), 775.5 (m), 752.8 (m), 719.9 (s), 695.2 (vs), 600.6 (m), 573.9 (w), 553.3 (m), 541.0 (s), 487.5 (w), 471.0 (w), 442.2 (s), 419.6 (s).
1H-NMR, 13C-NMR, IR spectra are shown in the Supporting Information: Figures S3, S19 and S29, respectively.
3.2.2. (1-Benzyl-4-(6-methylpyridin-2-yl)-1H-1,2,3-triazol-5yl)methanol (3a) and 2-(1-Benzyl-5-(6-methylpyridin-2-yl)-1H-1,2,3-triazol-4-yl)ethan-1-ol (3b)
In a typical procedure, in a flame-dried 50 mL two-neck round bottom flask, 25 mL of dichloromethane (DCM) was added under argon, followed by the addition of the corresponding alkyne (2a or 2b) (0.67 mmol, 1 equiv.), benzyl azide (9) (0.30 mL, 1.34 mmol, 0.31 g, 2.0 equiv.), and chloro(pentamethylcyclopentadienyl)(cyclooctadiene)ruthenium(II) (Ru(COD)Cp*Cl) (0.03 mmol, 0.013 g, 0.05 equiv.). The reaction mixture was allowed to stir at room temperature for 48 h. After the removal of the solvent under reduced pressure, the residue was purified by column chromatography on silica gel using a mixture of dichloromethane and methanol and the desired isomer was isolated (Rf,3a = 0.20, Rf,3b = 0.22, Rf,3e = 0.46, Rf, 3f = 0.33 in 5% v/v of methanol), yields are shown in Table 6.
3a:
1H-NMR (CDCl3, 400 MHz) δ (ppm): 7.70 (1H, t, 3J = 7.8 Hz, para-pyridine), 7.47 (1H, d, 3J = 7.8 Hz, ortho-pyridine), 7.30–7.10 (6H, m, Ar), 5.91 (2H, s, -N-CH2-), 5.01 (1H, br, -OH), 4.81 (2H, app s, -CH2-OH), 2.63 (3H, s, CH3).
13C-NMR (CDCl3, 101 MHz) δ 158.83, 146.54, 145.80, 137.64, 135.59, 134.59, 128.71, 128.01, 127.29, 123.32, 121.33, 56.02, 52.95, 24.37.
ESI-MS m/z [M+H]+ calculated for C16H16N4O: 281.14, found: 281.14.
IR ν (cm−1) 2844.7 (w), 1604.4 (m), 1577.6 (m), 1550.9 (w), 1495.4 (w), 1474.8 (m), 1454.2 (s), 1446.0 (s), 1376.1 (w), 1339.0 (w), 1316.4 (w), 1295.9 (w), 1281.5 (w), 1252.7 (m), 1242.4 (m), 1203.3 (w), 1158.0 (m), 1088.1 (w), 1071.6 (w), 1055.2 (m), 1038.7 (vs), 1009.9 (m), 985.3 (w), 970.9 (w), 935.9 (w), 909.2 (w), 855.7 (w), 808.4 (vs), 756.9 (s), 740.5 (s), 715.8 (vs), 707.6 (vs), 695.2 (s), 680.8 (m), 647.9 (m), 617.1 (w), 588.3 (w), 578.0 (m), 559.5 (w), 538.9 (vw), 512.2 (w), 469.0 (w).
1H-NMR, 13C-NMR, IR spectra are shown in the Supporting Information: Figures S4, S20 and S30, respectively.
3b:
1H-NMR (CDCl3, 400 MHz) δ (ppm): 7.99 (1H, d, 3J = 7.8 Hz, ortho-pyridine), 7.71 (1H, t, 3J = 7.8 Hz, para-pyridine) 7.39–7.29 (m, 3H, m- and p-benzyl), 7.22–7.16 (m, 2H, o-benzyl), 7.11 (1H, d, 3J = 7.8 Hz, ortho-pyridine), 6.68 (1H, br, -CH2-OH), 5.59 (2H, s, -CH2), 3.68 (2H, t, 3J = 8.8 Hz, -CH2-OH), 3.16 (2H, t, 3J = 8.8 Hz, -CH2-CH2-OH), 2.54 (3H, s, -CH3).
13C-NMR (CDCl3, 101 MHz) δ 157.25, 149.89, 145.48, 138.01, 135.01, 133.78, 129.20, 128.58, 127.17, 122.57, 119.13, 61.33, 52.15, 26.51, 23.70.
ESI-MS [M+H]+ m/z calculated for C17H18N4O: 295.15, found: 295.15.
IR ν (cm−1) 3200.5 (w), 3089.5 (w), 3058.6 (w), 3033.9 (w), 3005.1 (w), 2953.7 (w), 2924.9 (w), 2855.0 (w), 1602.3 (m), 1575.6 (s), 1497.4 (w), 1478.9 (m), 1450.1 (vs), 1376.1 (m), 1355.5 (m), 1334.9 (w), 1289.7 (w), 1238.3 (m), 1205.3 (w), 1182.7 (w), 1158.0 (m), 1108.7 (w), 1092.2 (w), 1059.3 (vs), 1047.0 (vs), 1016.1 (s), 997.6 (s), 962.6 (w), 905.0 (w), 892.7 (w), 866.0 (m), 851.6 (w), 796.0 (vs), 748.7 (s), 724.0 (vs), 695.2 (vs), 619.1 (m), 602.7 (m), 575.9 (s), 541.0 (m), 530.7 (m), 491.6 (m), 458.7 (m), 440.2 (m), 417.6 (w), 407.3 (w).
1H-NMR, 13C-NMR, IR spectra are shown in the Supporting Information: Figures S5, S21 and S31, respectively.
3.2.3. 2-(1-Benzyl-4-(6-methylpyridin-2-yl)-1H-1,2,3-triazol-5-yl)ethyl 4-vinylbenzoate (4c)
In a flame-dried 50 mL two-neck round bottom flask, 25 mL of DCM was added under argon, followed by the addition of 3b (0.67 mmol, 0.2 g, 1 equiv.), 4-vinylbenzoic acid (1.02 mmol, 151.0 mg, 1.5 equiv.), N-(3-dimethylaminopropyl)-N′-ethyl carbodiimide (EDCI, 0.240 mL, 1.36 mmol, 210.96 mg, 2 equiv.) and 4-(dimethylamino)pyridine (DMAP, 0.136 mmol, 16.6 mg, 0.2 equiv.). The suspension was allowed to stir at room temperature for 48 h. After the removal of the solvent under reduced pressure, the residue was dissolved in 20 mL of DCM and washed twice with 20 mL of NaOH solution (1.0 M) and twice with brine. The solution was dried over anhydrous magnesium sulphate and after filtration, the solvent was removed under reduced pressure. The product was isolated as a yellow viscous liquid with a yield of 53.4%
1H-NMR (CDCl3, 400 MHz) δ (ppm): 7.99 (1H, d, 3J = 7.8 Hz, ortho-pyridine ), 7.81 (2H, d, 3J = 8.2 Hz , meta-styrene), 7.57 (1H, t, 3J = 7.8 Hz, para-pyridine ), 7.38 (2H, d, 3J = 8.2 Hz, ortho-styrene ), 7.31-7.16 (m, 5H, benzyl), 6.96 (1H, d, 3J = 7.8 Hz, ortho-pyridine ), 6.70 (1H, dd, 3J = 17.6 Hz, 10.9 Hz, CH2=CH-), 5.82 (1H, app d, 3J = 17.6 Hz CH2=CH-), 5.61 (2H, s, -N-CH2-C-), 5.34 (1H, app d, 3J = 10.9 Hz, CH2=CH-), 4.49 (2H, t, 3J = 6.6 Hz, -CH2-CH2-O-), 3.53 (2H, t, 3J = 6.6 Hz, -CH2-CH2-O-), 2.46 (3H, s, -N=C-CH3).
13C-NMR (CDCl3, 101 MHz) δ 165.97, 157.55, 150.62, 144.49, 141.92, 136.64, 135.75, 134.81, 132.30, 129.85, 129.70, 129.60, 128.90, 128.85, 128.81, 128.31, 128.28, 128.22, 127.02, 126.99, 125.95, 121.41, 121.35, 117.60, 116.50, 63.12, 62.68, 51.76, 24.24, 23.60.
ESI-MS [M+H]+ m/z calculated for C26H24N4O2: 425.20, found: 425.20.
IR ν (cm−1) 2957.8 (vw), 2920.8 (vw), 1711.3 (vs), 1651.7 (w), 1629.1 (w), 1604.4 (m), 1575.6 (m), 1497.4 (w), 1481.0 (w), 1448.1 (m), 1404.9 (w), 1374.0 (w), 1357.6 (w), 1334.9 (w), 1310.2 (w), 1267.1 (vs), 1203.3 (w), 1178.6 (s), 1162.2 (w), 1102.5 (vs), 1073.7 (m), 1036.7 (m), 1016.1 (m), 995.5 (m), 917.4 (w), 859.8 (s), 802.2 (m), 781.6 (s), 746.7 (m), 724.0 (vs), 713.7 (vs), 695.2 (s), 635.6 (w), 619.1 (w), 580.0 (w), 555.4 (w), 541.0 (m), 508.1 (w), 493.7 (w), 483.4 (w), 456.6 (m), 401.1 (w).
1H-NMR, 13C-NMR, IR spectra are shown in the Supporting Information: Figures S6, S22, S32, respectively.
3.2.4. 2-(((2-(1-benzyl-4-(6-methylpyridin-2-yl)-1H-1,2,3-triazol-5-yl)ethoxy)carbonyl)amino)ethyl acrylate (4d)
In a flame-dried 50 mL two-neck round bottom flask, 25 mL of DCM was added under argon, followed by the addition of 3b (1.02 mmol, 0.3 g, 1 equiv.), slow addition of 2-isocyanatoethyl acrylate (0.20 mL, 1.53 mmol, 215.8 mg, 1.5 equiv.) and one drop of dibutyltin dilaurate (DBTDL). The reaction mixture was allowed to stir at room temperature for 48 h. After the removal of the solvent under reduced pressure, the residue was purified by column chromatography on silica gel using a mixture of DCM and methanol (Rf = 0.35 in 5% v/v of methanol). The product was isolated as a colourless viscous liquid with a yield of 62.7%.
1H-NMR (CDCl3, 400 MHz) δ (ppm): 8.03 (1H, d, 3J = 7.8 Hz, ortho-pyridine), 7.64 (1H, t, 3J = 7.8 Hz, para-pyridine), 7.38–7.30 (3H, m, -meta- and para-benzyl), 7.27–7.23 (2H, m, ortho-benzyl), 7.04 (1H, d, 3J = 7.8 Hz, CH3-C=CH-), 6.43 (1H, dd, 3J = 17.3 Hz, 2J = 1.4 Hz O=C-CH=CH2), 6.13 (1H, dd, 3J = 17.3 Hz, 2J =10.4 Hz, O=C-CH=CH2), 5.88 (1H, dd, 3J = 10.4 Hz, 2J = 1.4 Hz O=C-CH=CH2), 5.62 (2H, s, -N-CH2-), 4.55 (1H, br, -NH-), 4.33 (2H, 3J = 6.5 Hz, -O-CH2-CH2-C-), 4.19 (2H, t, 3J = 5.3 Hz, -O-CH2-CH2-NH-), 3.42 (2H, m, -O-CH2-CH2-NH- and -O-CH2-CH2-C-), 2.51 (2H, s, -N=C-CH3).
13C-NMR (CDCl3, 101 MHz) δ 166.07, 157.75, 156.25, 150.80, 144.29, 136.89, 135.40, 133.01, 131.46, 128.95, 128.21, 128.00, 127.26, 121.56, 117.77, 63.42, 63.20, 51.66, 40.12, 24.46, 24.20.
ESI-MS [M+H]+ m/z calculated for C23H25N5O4: 436.20, found: 436.20.
IR ν (cm−1) 3346.6 (vw), 2959.9 (w), 2927.0 (w), 1715.5 (vs), 1602.3 (m), 1575.6 (m), 1530.3 (m), 1497.4 (w), 1483.0 (m), 1450.1 (s), 1406.9 (s), 1374.0 (w), 1359.6 (w), 1332.9 (w), 1267.1 (vs), 1180.7 (vs), 1156.0 (s), 1102.5 (s), 1071.6 (s), 1044.9 (s), 1016.1 (m), 985.3 (s), 919.4 (w), 859.8 (m), 804.3 (s), 779.6 (m), 746.7 (m), 724.0 (vs), 695.2 (s), 674.7 (m), 617.1 (w), 578.0 (m), 555.4 (m), 541.0 (m), 483.4 (m), 456.6 (m), 417.6 (w), 409.3 (w).
1H-NMR, 13C-NMR, IR spectra are shown in the Supporting Information: Figures S7, S23, S33, respectively.
3.2.5. Heteroleptic Cu(I) Complexes 5a, 5b, 5c, 5d
In a typical procedure, into a flame-dried 50 mL two-neck round bottom flask, 25 mL of DCM was added under argon, followed by the addition of tetrakis(acetonitrile)copper(I) tetrafluoroborate (0.04 mmol, 12.6 mg, 1 equiv.) (synthesised according to the literature [66]) and diphenyl phosphine ether (0.04 mmol, 21.5 mg, 1 equiv.). After 30 min under stirring, the ligands 3a, 3b, 4c, or 4d (0.04 mmol, 1 equiv.) were added. The reaction mixture was allowed to stir at room temperature for 48 h. After the removal of the solvent under reduced pressure, the residue was dissolved in 1 mL of DCM and cyclohexane was added dropwise. The biphasic solution obtained was then left in the fridge for 48 h until a precipitated was formed. The precipitate was then collected by filtration and washed with cold cyclohexane. The product was isolated as a white (5d) yellowish (5b and 5c) or brown solid (5a), yields are reported in Table 7.
5a:
1H-NMR (CD2Cl2, 400 MHz) δ 7.89 (2H, m, para-,ortho-pyridine ), 7.49–6.63 (34H, m, Ar), 5.74 (2H, s, N-CH2-C-), 4.79 (2H, s, -C-CH2-OH), 3.76 (1H, br, -N-CH2-OH), 2.04 (3H, s, -N=C-CH3).
13C-NMR (CDCl3, 101 MHz) δ 158.97, 158.55, 158.49, 158.43, 146.85, 143.78, 143.76, 139.08, 138.80, 135.46, 134.53, 134.44, 131.85, 130.92, 130.35, 129.73, 129.04, 128.53, 128.00, 127.79, 125.00, 124.98, 124.95, 124.52, 124.37, 124.23, 120.37, 120.06, 77.48, 77.16, 76.84, 52.93, 51.90, 25.29.
ESI [M]+ m/z calculated for C52H44CuN4O2P2: 881.22, found: 881.22 .
IR ν (cm−1) 3346.6 (vw), 2959.9 (w), 2927.0 (w), 1715.5 (vs), 1602.3 (m), 1575.6 (m), 1530.3 (m), 1497.4 (w), 1483.0 (m), 1450.1 (s), 1406.9 (s), 1374.0 (w), 1359.6 (w), 1332.9 (w), 1267.1 (vs), 1180.7 (vs), 1156.0 (s), 1102.5 (s), 1071.6 (s), 1044.9 (s), 1016.1 (m), 985.3 (s), 919.4 (w), 859.8 (m), 804.3 (s), 779.6 (m), 746.7 (m), 724.0 (vs), 695.2 (s), 674.7 (m), 617.1 (w), 578.0 (m), 555.4 (m), 541.0 (m), 483.4 (m), 456.6 (m), 417.6 (w), 409.3 (w)
19F NMR (CDCl3, 377 MHz) δ −153.15
31P NMR (CDCl3, 162 MHz) δ −13.90
11B NMR (CDCl3, 128 MHz) δ −0.81
1H-NMR, 13C-NMR, IR spectra are shown in the Supporting Information: Figures S8, S24, S34, respectively.
Emission and excitation spectra in Ar-saturated DCM are shown in Figure S40.
5b:
1H-NMR (CDCl3, 400 MHz) δ 7.94 (1H, t, 3J = 7.8 Hz, -CH=CH-CH-, para pyridine), 7.76 (1H, d, 3J = 7.8 Hz, -CH=C-CH3, meta-pyridine), 7.49 (4H, br, para-phenyl), 7.41–7.01 (21H, br, meta-phenyl, ortho-phenyl, para-benzyl), 7.01–6.91 (4H, br, meta- and ortho-benzyl), 6.81–6.66 (5H, br, -N=C-CH, -O-C-CH=CH-), 5.66 (2H, s, -C-CH2-N), 3.66 (2H, m, -CH2-CH2-OH), 3.50 (1H, br, -CH2-OH) 3.15 (2H, t, 3J = 6.9 Hz, -CH2-CH2-OH), 2.02 (3H, s, N=C-CH3).
13C-NMR (CDCl3, 101 MHz) δ 159.05, 158.64, 158.58, 158.52, 147.11, 142.84, 139.43, 134.67, 134.60, 134.32, 131.83, 129.13, 128.59, 127.67, 124.99, 124.65, 124.51, 124.38, 120.40, 118.76, 77.48, 77.16, 76.84, 59.91, 52.65, 27.05, 25.42.
IR ν (cm−1) 3533.8 (vw), 3060.7 (vw), 2922.9 (w), 2859.1 (w), 1631.1 (vw), 1608.5 (w), 1573.5 (w), 1483.0 (w), 1458.3 (m), 1433.7 (vs), 1258.8 (w), 1215.6 (s), 1094.3 (s), 1053.1 (vs), 997.6 (s), 878.3 (w), 804.3 (m), 744.6 (vs), 724.0 (s), 693.2 (vs), 520.4 (s), 508.1 (vs), 483.4 (s), 475.1 (vs), 466.9 (s), 419.6 (s), 411.4 (s)
ESI [M]+ m/z calculated for C53H46CuN4O2P2: 895.24 , found: 895.24 .
19F NMR (CDCl3, 377 MHz) δ −153.16
31P NMR (CDCl3, 162 MHz) δ −13.91
11B NMR (CDCl3, 128 MHz) δ −0.81
Crystal data for 5b: C55H50BCl4CuF4N4O2P2, 1153.08 g mol−1, orthorhombic, Pbca, a = 26.5017(15), b = 12.1623(4), c = 33.0708(11) Å, Z = 8, V = 10659.4(8) Å3, T = 100(2) K, dcalc = 1.437 g cm-3, F(000) = 4736, μ(Mo-Kα) = 0.730 mm−1. Colourless rod, 0.42×0.17×0.05 mm3, 24885 data, 2θmax = 52.74°, 10557 unique (Rint = 0.0524), 660 parameters, final wR2 = 0.2082, S = 1.008 (all data), R1 (6502 data with I > 2σ(I)) = 0.0686, max. diff. peak/hole +0.93/-0.64 eÅ-3. Full crystallographic data for the structure have been deposited with the Cambridge Crystallographic Data Centre as CCDC 2067971 (copies available free of charge from https://www.ccdc.cam.ac.uk/structures/, accessed on 1 March 2021)
1H-NMR, 13C-NMR, IR spectra are shown in the Supporting Information: Figures S9, S25 and S35, respectively.
Emission and excitation spectra in Ar-saturated DCM are shown in Figure S41.
5c:
1H-NMR (CDCl3, 400 MHz) δ (ppm): 7.96 (1H, d, 3J = 7.8 Hz, ortho-pyridine), 7.89 (1H, d, 3J = 7.8 Hz, para-pyridine), 7.74 (2H, d, 3J = 8.1 Hz, meta-styrene), 7.58-6.89 (29 H, br, phenyl, ortho- and meta-benzyl, ortho-styrene, ortho-O-Ar), 6.80-6.63 (8H, br, ortho-pyridine, meta-O-Ar, para-O-Ar, -CH=CH2) 5.80 (1H, app d, 3J = 17.6 Hz CH2=CH-), 5.73 (2H, s, -N-CH2-C-), 5.36 (1H, app d, 3J = 10.9 Hz CH2=CH-), 4.25 (2H, t, 3J = 6.9 Hz, -O-CH2-CH2-), 3.47 (2H, t, 3J = 6.9 Hz, CH2-CH2-C), 1.99 (3H, s, N-C-CH3).
13C-NMR (CDCl3, 101 MHz) δ 166.15, 159.45, 146.66, 142.32, 139.37, 135.95, 134.53, 131.90, 130.04, 129.28, 128.82, 127.53, 126.24, 125.04, 124.77, 118.65, 61.37, 52.86, 25.41, 22.77.
IR ν (cm−1) 3398.0 (vw), 3048.3 (vw), 2961.9 (vw), 2927.0 (vw), 2855.0 (vw), 1717.5 (w), 1604.4 (w), 1565.3 (w), 1478.9 (w), 1460.4 (m), 1433.7 (vs), 1380.2 (w), 1267.1 (m), 1215.6 (m), 1180.7 (w), 1160.1 (w), 1094.3 (s), 1053.1 (vs), 1034.6 (vs), 997.6 (s), 870.1 (w), 802.2 (m), 744.6 (vs), 724.0 (s), 693.2 (vs), 619.1 (w), 580.0 (w), 543.0 (m), 510.1 (vs), 485.4 (s), 475.1 (s), 466.9 (s), 450.5 (m), 438.1 (m), 419.6 (s) cm-1
ESI [M]+ m/z calculated for C62H52CuN4O3P2 : 1025.28, found: 1025.28.
19F NMR (CDCl3, 377 MHz) δ −153.04
31P NMR (CDCl3, 162 MHz) δ −13.93
11B NMR (CDCl3, 128 MHz) δ −1.11
1H-NMR, 13C-NMR, IR spectra are shown in the Supporting Information: Figures S10, S26 and S36, respectively.
Emission and excitation spectra in Ar-saturated DCM are shown in Figure S42.
5d:
1H-NMR (CDCl3, 400 MHz) δ (ppm): 7.92 (1H, t 3J = 7.8 Hz, para-pyridine), 7.70 (1H, d, 3J = 7.8 Hz, ortho-pyridine), 7.58–6.93 (29H, br, phenyl, benzyl, meta-O-Ar, para-O-Ar), 6.85–6.74 (3H, br, ortho-O-Ar, ortho-pyridine) 6.74–6.68 (2H, br, meta-O-Ar) 6.37 (1H, dd, 3J = 17.2 Hz, 2J = 1.4 Hz CH2=CH-), 6.11 (1H, app d, 3J = 17.2 Hz, 2J = 10.4 Hz CH2=CH-), 5.82 (1H, dd, 3J = 10.4 Hz, 2J = 1.4 Hz CH2=CH-) 5.69 (2H, s, -N-CH2-C-), 5.25 (1H, br, C-NH-CH2-), 4.15 (2H, t, 3J = 6.9 Hz, -O-CH2-CH2-NH-), 3.97 (2H, t, 3J = 6.9 Hz, -C-CH2-CH2-O-), 3.37 (2H, m, -O-CH2-CH2-NH-), 3.21 (2H, t, 3J = 6.9 Hz, -C-CH2-CH2-O-), 2.06 (3H, s, N-C-CH3).
13C-NMR (CD2Cl2, 101 MHz) δ 166.49, 160.31, 159.03, 158.97, 158.91, 156.40, 147.07, 139.46, 134.98, 134.93, 134.65, 134.16, 133.24, 132.44, 131.38, 129.68, 128.53, 127.89, 125.44, 125.42, 125.29, 124.71, 124.57, 124.42, 120.91, 118.61, 63.73, 61.61, 54.54, 54.27, 54.00, 53.73, 53.46, 53.29, 40.63, 27.47, 25.78.
IR ν (cm−1) 3398.0 (vw), 3048.3 (vw), 2961.9 (vw), 2927.0 (vw), 2855.0 (vw), 1717.5 (w), 1604.4 (w), 1565.3 (w), 1478.9 (w), 1460.4 (m), 1433.7 (vs), 1380.2 (w), 1267.1 (m), 1215.6 (m), 1180.7 (w), 1160.1 (w), 1094.3 (s), 1053.1 (vs), 1034.6 (vs), 997.6 (s), 870.1 (w), 802.2 (m), 744.6 (vs), 724.0 (s), 693.2 (vs), 619.1 (w), 580.0 (w), 543.0 (m), 510.1 (vs), 485.4 (s), 475.1 (s), 466.9 (s), 450.5 (m), 438.1 (m), 419.6 (s)
ESI [M]+ m/z calculated for C59H53CuN5O5P2: 1036.28, found: 1036.28 .
19F NMR (CDCl3, 377 MHz) δ –153.20
31P NMR (CDCl3, 162 MHz) δ –13.84
11B NMR (CDCl3, 128 MHz) δ –0.79
1H-NMR, 13C-NMR, IR spectra are shown in the Supporting Information: Figures S11, S27, S37, respectively.
Emission and excitation spectra in Ar-saturated DCM are shown in Figure S43.
3.2.6. Synthesis of Poly(methyl acrylate) Homopolymer PMA
In a flame-dried 25 mL two-neck round bottom flask, 5 mL of toluene was added followed by the addition of methyl acrylate (MA, 0.50 mL, 5.51 mmol, 0.475 g, 100 equiv.), and 2,2′-azobis(2-methylpropionitrile) (AIBN, 0.055 mmol, 9.05 mg, 1 equiv.). Argon was bubbled through the solution for 5 min and then the reaction mixture was allowed to stir at 70 °C for 4 h under an argon blanket. The reaction was then cooled and quenched by being opened to air and the polymer was then precipitated by dropwise addition into 15 mL of stirring cold hexane. The product was dried and a transparent solid was obtained with a yield of 83.4% (Mn = 11.71 kg/mol, Ð = 2.46).
1H-NMR spectrum and SEC chromatogram are shown in the Supporting Information: Figures S12, S38, respectively.
3.2.7. Synthesis of Poly(styrene) Homopolymer PS
In a flame-dried 25 mL two-neck round bottom flask, 5 mL of toluene was added followed by the addition of styrene (Sty, 0.57 g, 0.63 mL, 5.51 mmol, 100 equiv.), and 2,2′-azobis(2-methylpropionitrile) (AIBN, 0.055 mmol, 9.05 mg, 1 equiv.). Argon was bubbled through the solution for 5 min and then the reaction mixture was allowed to stir at 70 °C for 4 h under an argon blanket. The reaction was then cooled and quenched by opening it to air and the polymer was then precipitated by dropwise addition into 15 mL of stirring cold hexane. The product was dried and a transparent solid was obtained with a yield of 57.8% (Mn = 14.5 kg/mol, Ð = 1.44).
1H-NMR spectrum and SEC chromatogram are shown in the Supporting Information: Figures S13, S39, respectively.
3.2.8. Synthesis of Poly(Sty-co-4c): Copolymer 6c
In a flame-dried 25 mL two-neck round bottom flask, 430 µL of toluene was added, followed by the addition of styrene (St, 52.63 µL, 0.46 mmol, 47.8 mg, 10 equiv.), M2 (20.0 mg, 0.046 mmol, 1 equiv.) and AIBN (0.003 mmol, 0.42 mg, 0.07 equiv.). Argon was bubbled through the solution for 5 min and then the reaction mixture was allowed to stir at 70 °C for 24 h under an argon blanket. The polymer was then precipitated in 1.5 mL of cold hexane. The product was isolated as a yellowish solid at a yield of 64.4% (Mn = 13.5 g/mol, Ð = 1.65). The monomer ratio in the copolymer was determined by 1H-NMR spectroscopy and found to correspond to 8% 4c.
1H-NMR spectrum shown in the Supporting Information: Figure S14.
3.2.9. Synthesis of Poly(MA-co-4d): Copolymer 6d
In a flame-dried 25 mL two-neck round bottom flask, 403 µL of toluene was added, followed by the addition of MA (39.83 µL, 0.44 mmol, 37.8 mg, 10 equiv.), 4d (20.0 mg, 0.044 mmol, 1 equiv.) and AIBN (0.002 mmol, 0.40 mg, 0.06 equiv.). Argon was bubbled through the solution for 5 min and then the reaction mixture was allowed to stir at 70 °C for 24 h. The polymer was then precipitated in 1.5 mL of cold hexane. The product was isolated as a transparent solid at a yield of 83.4% (Mn = 9.54 kg/mol, Ð = 1.92). The monomer ratio in the copolymer was determined by 1H-NMR spectroscopy and found to correspond to 11% 4c.
1H-NMR spectrum is shown in the Supporting Information: Figure S15.
3.2.10. Post-Polymerisation Complexation to Obtain Functional Copolymers 7c and 7d
In a typical complexation, in a flame-dried 25 mL two-neck round bottom flask, 10 mL of DCM was added under argon, followed by the addition of tetrakis(acetonitrile)copper(I) tetrafluoroborate (0.09 mmol, 0.048 g, 1 equiv. with respect to the functional groups in the polymer) and DPEPhos (0.09 mmol, 0.073 g, 1 equiv. with respect to the functional groups in the polymer). After 30 min under stirring, 6 (0.14 mmol, 1 equiv. with respect to the functional groups in the polymer) was added. The reaction mixture was allowed to stir at room temperature for 72 h. After removal of the solvent under reduced pressure, the residue was dissolved in 2 mL of THF and precipitated in 10 mL of cold n-hexane. After filtration, the product was obtained as a white (7c) or colourless (7d) solid.
1H-NMR spectra are shown in the Supporting Information: Figures S16 and S17, respectively.
4. Conclusions
In this work, four novel heteroleptic copper (I) complexes were successfully synthesised, two of which bear polymerisable groups: a styrenic or an acrylate moiety. The electrochemical and the photophysical investigation of the complexes were performed and showed a good correlation with the TD-DFT calculations. Overall, the complexes present reasonable quantum yields in solution and in solid state, emitting a bright cyan colour. The copolymerisation of the diimine ligands bearing the polymerisable groups and the corresponding monomers, followed by the coordination of the Cu(I)-diphosphine moiety, gave two luminescent metallopolymers. The emission quantum yields of these copolymers were compared to the corresponding polymer blends, showing the significance of the chemical environment and the final structure of the emitter. Especially the copolymer made with the acrylate unit presents the same emission behaviour as the corresponding blend. On the contrary, the copolymer made with the less emissive Cu(I) complex, the one bearing the styrenic unit, presented a notable improvement in respect to the corresponding blend. This effect was attributed to the loss of the significant electronic involvement of the double bond in the styrene, thanks to the polymerisation.
Supplementary Materials
The following are available online. Details regarding the instrumentation used in this work and supporting data, and cif. and checkcif. Files.
Author Contributions
Conceptualisation, C.B. and D.M.; investigation, F.F., J.B., C.E.A., B.D.W. and C.B.; project administration, and supervision, C.B. and D.M.; writing—original draft preparation, F.F., D.M. and C.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG), Collaborative Centre SFB/TRR 88 “3MET” (Bizzarri, Project B9).
Data Availability Statement
Datasets related to this article can be provided upon request.
Acknowledgments
The authors acknowledge the help of Heidi Garbus with experimental procedures, as well as Maximiliane Frölich and Philipp Bohn for their help with the MS–ESI measurements. We thank Michael Gamer for measuring the X-ray diffraction dataset. Michael A.R. Meier and Stefan Bräse kindly provided laboratory space and some of the equipment used in this work. Patrick Théato is thanked for providing access to the SEC equipment. Letizia Sambri from the University of Bologna is acknowledged for her constant motivation and support. We acknowledge support by the KIT-Publication Fund of the Karlsruhe Institute of Technology.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Yam, V.W.; Au, V.K.; Leung, S.Y. Light-emitting self-assembled materials based on d(8) and d(10) transition metal complexes. Chem. Rev. 2015, 115, 7589–7728. [Google Scholar] [CrossRef] [PubMed]
- Yam, V.W.-W.; Wong, K.M.-C. Luminescent metal complexes of d6, d8 and d10 transition metal centres. Chem. Commun. 2011, 47, 11579–11592. [Google Scholar] [CrossRef] [PubMed]
- De Silva, A.P.; Gunaratne, H.Q.N.; Gunnlaugsson, T.; Huxley, A.J.M.; McCoy, C.P.; Rademacher, J.T.; Rice, T.E. Signaling recognition events with fluorescent sensors and switches. Chem. Rev. 1997, 97, 1515–1566. [Google Scholar] [CrossRef] [PubMed]
- Demas, J.N.; DeGraff, B.A. Applications of luminescent transition platinum group metal complexes to sensor technology and molecular probes. Coord. Chem. Rev. 2001, 211, 317–351. [Google Scholar] [CrossRef]
- Prier, C.K.; Rankic, D.A.; MacMillan, D.W.C. Visible light photoredox catalysis with transition metal complexes: Applications in organic synthesis. Chem. Rev. 2013, 113, 5322–5363. [Google Scholar] [CrossRef] [Green Version]
- Larsen, C.B.; Wenger, O.S. Photoredox catalysis with metal complexes made from earth-abundant elements. Chem. Eur. J. 2018, 24, 2039–2058. [Google Scholar] [CrossRef] [Green Version]
- Busch, J.; Knoll, D.M.; Zippel, C.; Bräse, S.; Bizzarri, C. Metal-supported and -assisted stereoselective cooperative photoredox catalysis. Dalton Trans. 2019, 48, 15338–15357. [Google Scholar] [CrossRef] [Green Version]
- Volz, D.; Wallesch, M.; Fléchon, C.; Danz, M.; Verma, A.; Navarro, J.M.; Zink, D.M.; Bräse, S.; Baumann, T. From iridium and platinum to copper and carbon: New avenues for more sustainability in organic light-emitting diodes. Green Chem. 2015, 17, 1988–2011. [Google Scholar] [CrossRef]
- Dumur, F. Recent advances in organic light-emitting devices comprising copper complexes: A realistic approach for low-cost and highly emissive devices? Org. Electron. 2015, 21, 27–39. [Google Scholar] [CrossRef]
- Iwamura, M.; Takeuchi, S.; Tahara, T. Real-Time Observation of the Photoinduced Structural Change of Bis(2,9-dimethyl-1,10-phenanthroline)copper(I) by Femtosecond Fluorescence Spectroscopy: A Realistic Potential Curve of the Jahn−Teller Distortion. J. Am. Chem. Soc. 2007, 129, 5248–5256. [Google Scholar] [CrossRef]
- Barbieri, A.; Accorsi, G.; Armaroli, N. Luminescent complexes beyond the platinum group: The d10 avenue. Chem. Commun. 2008, 19, 2185–2193. [Google Scholar] [CrossRef]
- Armaroli, N.; Accorsi, G.; Cardinali, F.; Listorti, A. Photochemistry and photophysics of coordination compounds: Copper. In Photochemistry and Photophysics of Coordination Compounds I; Balzani, V., Campagna, S., Eds.; Springer: Berlin/Heidelberg, Germany, 2007; pp. 69–115. [Google Scholar]
- Garakyaraghi, S.; McCusker, C.E.; Khan, S.; Koutnik, P.; Bui, A.T.; Castellano, F.N. Enhancing the visible-light absorption and excited-state properties of Cu(I) MLCT excited states. Inorg. Chem. 2018, 57, 2296–2307. [Google Scholar] [CrossRef]
- Garakyaraghi, S.; Danilov, E.O.; McCusker, C.E.; Castellano, F.N. Transient absorption dynamics of sterically congested Cu(I) MLCT excited states. J. Phys. Chem. A 2015, 119, 3181–3193. [Google Scholar] [CrossRef]
- Kuang, S.-M.; Cuttell, D.G.; McMillin, D.R.; Fanwick, P.E.; Walton, R.A. Synthesis and structural characterization of Cu(I) and Ni(II) complexes that contain the Bis[2-(diphenylphosphino)phenyl]ether ligand. Novel emission properties for the Cu(I) species. Inorg. Chem. 2002, 41, 3313–3322. [Google Scholar] [CrossRef]
- Kaeser, A.; Mohankumar, M.; Mohanraj, J.; Monti, F.; Holler, M.; Cid, J.-J.; Moudam, O.; Nierengarten, I.; Karmazin-Brelot, L.; Duhayon, C.; et al. Heteroleptic copper(I) complexes prepared from phenanthroline and bis-phosphine ligands. Inorg. Chem. 2013, 52, 12140–12151. [Google Scholar] [CrossRef] [PubMed]
- Leoni, E.; Mohanraj, J.; Holler, M.; Mohankumar, M.; Nierengarten, I.; Monti, F.; Sournia-Saquet, A.; Delavaux-Nicot, B.; Nierengarten, J.-F.i.; Armaroli, N. Heteroleptic copper(I) complexes prepared from phenanthroline and bis-phosphine ligands: Rationalization of the photophysical and electrochemical properties. Inorg. Chem. 2018, 57, 15537–15549. [Google Scholar] [CrossRef] [PubMed]
- Bizzarri, C.; Spuling, E.; Knoll, D.M.; Volz, D.; Bräse, S. Sustainable metal complexes for organic light-emitting diodes (OLEDs). Coord. Chem. Rev. 2018, 373, 49–82. [Google Scholar] [CrossRef]
- Bizzarri, C.; Hundemer, F.; Busch, J.; Bräse, S. Triplet emitters versus TADF emitters in OLEDs: A comparative study. Polyhedron 2018, 140, 51–66. [Google Scholar] [CrossRef]
- Ravaro, L.P.; Zanoni, K.P.S.; de Camargo, A.S.S. Luminescent copper(I) complexes as promising materials for the next generation of energy-saving OLED devices. Energy Rep. 2020, 6, 37–45. [Google Scholar] [CrossRef]
- Blasco, E.; Sims, M.B.; Goldmann, A.S.; Sumerlin, B.S.; Barner-Kowollik, C. 50th Anniversary perspective: Polymer functionalization. Macromolecules 2017, 50, 5215–5252. [Google Scholar] [CrossRef]
- Whittell, G.R.; Hager, M.D.; Schubert, U.S.; Manners, I. Functional soft materials from metallopolymers and metallosupramolecular polymers. Nat. Mater. 2011, 10, 176–188. [Google Scholar] [CrossRef]
- Neumann, L.N.; Urban, D.A.; Lemal, P.; Ramani, S.; Petri-Fink, A.; Balog, S.; Weder, C.; Schrettl, S. Preparation of metallosupramolecular single-chain polymeric nanoparticles and their characterization by Taylor dispersion. Polym. Chem. 2020, 11, 586–592. [Google Scholar] [CrossRef] [Green Version]
- Willenbacher, J.; Altintas, O.; Roesky, P.W.; Barner-Kowollik, C. Single-chain self-folding of synthetic polymers induced by metal–ligand complexation. Macromol. Rapid Commun. 2014, 35, 45–51. [Google Scholar] [CrossRef]
- Sanchez-Sanchez, A.; Arbe, A.; Colmenero, J.; Pomposo, J.A. Metallo-folded single-chain nanoparticles with catalytic selectivity. ACS Macro Lett. 2014, 3, 439–443. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Astruc, D.; Abd-El-Aziz, A.S. Metallopolymers for advanced sustainable applications. Chem. Soc. Rev. 2019, 48, 558–636. [Google Scholar] [CrossRef] [PubMed]
- Volz, D.; Hirschbiel, A.F.; Zink, D.M.; Friedrichs, J.; Nieger, M.; Baumann, T.; Bräse, S.; Barner-Kowollik, C. Highly efficient photoluminescent Cu(i)–PyrPHOS-metallopolymers. J. Mater. Chem. C 2014, 2, 1457–1462. [Google Scholar] [CrossRef]
- Elmas, S.; Macdonald, T.J.; Skinner, W.; Andersson, M.; Nann, T. Copper metallopolymer catalyst for the electrocatalytic hydrogen evolution reaction (HER). Polymers 2019, 11, 110. [Google Scholar] [CrossRef] [Green Version]
- De Hatten, X.; Bell, N.; Yufa, N.; Christmann, G.; Nitschke, J.R. A Dynamic covalent, luminescent metallopolymer that undergoes sol-to-gel transition on temperature rise. J. Am. Chem. Soc. 2011, 133, 3158–3164. [Google Scholar] [CrossRef]
- Greenfield, J.L.; Rizzuto, F.J.; Goldberga, I.; Nitschke, J.R. Self-assembly of conjugated metallopolymers with tunable length and controlled regiochemistry. Angew. Chem. Int. Ed. 2017, 56, 7541–7545. [Google Scholar] [CrossRef]
- Asil, D.; Foster, J.A.; Patra, A.; de Hatten, X.; del Barrio, J.; Scherman, O.A.; Nitschke, J.R.; Friend, R.H. Temperature- and Voltage-Induced Ligand Rearrangement of a Dynamic Electroluminescent Metallopolymer. Angew. Chem. Int. Ed. 2014, 53, 8388–8391. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.M.; Sujatha, A.; Anilkumar, G. Recent advances and perspectives in copper-catalyzed Sonogashira coupling reactions. RSC Adv. 2014, 4, 21688–21698. [Google Scholar] [CrossRef]
- Chinchilla, R.; Nájera, C. The sonogashira reaction: A booming methodology in synthetic organic chemistry. Chem. Rev. 2007, 107, 874–922. [Google Scholar] [CrossRef]
- Johansson, J.R.; Beke-Somfai, T.; Said Stalsmeden, A.; Kann, N. Ruthenium-catalyzed azide alkyne cycloaddition reaction: Scope, mechanism, and applications. Chem. Rev. 2016, 116, 14726–14768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise huisgen cycloaddition process: Copper(I)-catalyzed regioselective “Ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Bräse, S.; Gil, C.; Knepper, K.; Zimmermann, V. Organic Azides: An exploding diversity of a unique class of compounds. Angew. Chem. Int. Ed. 2005, 44, 5188–5240. [Google Scholar] [CrossRef] [PubMed]
- Neises, B.; Steglich, W. Simple Method for the esterification of carboxylic acids. Angew. Chem. Int. Ed. Engl. 1978, 17, 522–524. [Google Scholar] [CrossRef]
- Ballard, N.; Asua, J.M. Radical polymerization of acrylic monomers: An overview. Prog. Polym. Sci. 2018, 79, 40–60. [Google Scholar] [CrossRef]
- Jones, P.G.; Crespo, O. Tetrakis(acetonitrile-N)copper(I) tetrafluoroborate. Acta Crystallogr. Sect. C 1998, 54, 18–20. [Google Scholar] [CrossRef]
- Bizzarri, C.; Arndt, A.P.; Kohaut, S.; Fink, K.; Nieger, M. Mononuclear and dinuclear heteroleptic Cu(I) complexes based on pyridyl-triazole and DPEPhos with long-lived excited-state lifetimes. J. Organomet. Chem. 2018, 871, 140–149. [Google Scholar] [CrossRef]
- Sun, Y.; Lemaur, V.; Beltran, J.I.; Cornil, J.; Huang, J.; Zhu, J.; Wang, Y.; Frohlich, R.; Wang, H.; Jiang, L.; et al. Neutral mononuclear copper(I) complexes: Synthesis, crystal structures, and photophysical properties. Inorg. Chem. 2016, 55, 5845–5852. [Google Scholar] [CrossRef]
- Shields, G.P.; Raithby, P.R.; Allen, F.H.; Motherwell, W.D.S. The assignment and validation of metal oxidation states in the Cambridge Structural Database. Acta Crystallogr. Sect. B 2000, 56, 455–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matyjaszewski, K.; Woodworth, B.E. Interaction of propagating radicals with copper(I) and copper(II) species. Macromolecules 1998, 31, 4718–4723. [Google Scholar] [CrossRef]
- Gernert, M.; Balles-Wolf, L.; Kerner, F.; Müller, U.; Schmiedel, A.; Holzapfel, M.; Marian, C.M.; Pflaum, J.; Lambert, C.; Steffen, A. Cyclic (Amino)(aryl)carbenes enter the field of chromophore ligands: Expanded π system leads to unusually deep red emitting CuI compounds. J. Am. Chem. Soc. 2020, 142, 8897–8909. [Google Scholar] [CrossRef] [PubMed]
- Gernert, M.; Muller, U.; Haehnel, M.; Pflaum, J.; Steffen, A. A Cyclic Alkyl(amino)carbene as two-atom pi-chromophore leading to the first phosphorescent linear Cu(I) complexes. Chemistry 2017, 23, 2206–2216. [Google Scholar] [CrossRef] [PubMed]
- So, G.K.; Cheng, G.; Wang, J.; Chang, X.; Kwok, C.C.; Zhang, H.; Che, C.M. Efficient color-tunable copper(I) complexes and their applications in solution-processed organic light-emitting diodes. Chem. Asian J. 2017, 12, 1490–1498. [Google Scholar] [CrossRef] [Green Version]
- Wallesch, M.; Verma, A.; Flechon, C.; Flugge, H.; Zink, D.M.; Seifermann, S.M.; Navarro, J.M.; Vitova, T.; Gottlicher, J.; Steininger, R.; et al. Towards printed organic light-emitting devices: A solution-stable, highly soluble Cu(I) -NHetPHOS. Chemistry 2016, 22, 16400–16405. [Google Scholar] [CrossRef]
- Czerwieniec, R.; Leitl, M.J.; Homeier, H.H.H.; Yersin, H. Cu(I) complexes—Thermally activated delayed fluorescence. Photophysical approach and material design. Coord. Chem. Rev. 2016, 325, 2–28. [Google Scholar] [CrossRef]
- Verma, A.; Zink, D.M.; Fléchon, C.; Leganés Carballo, J.; Flügge, H.; Navarro, J.M.; Baumann, T.; Volz, D. Efficient, inkjet-printed TADF-OLEDs with an ultra-soluble NHetPHOS complex. Appl. Phys. A 2016, 122, 1–5. [Google Scholar] [CrossRef]
- Leitl, M.J.; Zink, D.M.; Schinabeck, A.; Baumann, T.; Volz, D.; Yersin, H. Copper(I) complexes for thermally activated delayed fluorescence: From photophysical to device properties. Top. Curr. Chem. 2016, 374, 25. [Google Scholar] [CrossRef]
- Osawa, M.; Hoshino, M.; Hashimoto, M.; Kawata, I.; Igawa, S.; Yashima, M. Application of three-coordinate copper(I) complexes with halide ligands in organic light-emitting diodes that exhibit delayed fluorescence. Dalton Trans. 2015, 44, 8369–8378. [Google Scholar] [CrossRef] [PubMed]
- Leitl, M.J.; Kuchle, F.R.; Mayer, H.A.; Wesemann, L.; Yersin, H. Brightly blue and green emitting Cu(I) dimers for singlet harvesting in OLEDs. J. Phys. Chem. A 2013, 117, 11823–11836. [Google Scholar] [CrossRef]
- Busch, J.M.; Zink, D.M.; Di Martino-Fumo, P.; Rehak, F.R.; Boden, P.; Steiger, S.; Fuhr, O.; Nieger, M.; Klopper, W.; Gerhards, M.; et al. Highly soluble fluorine containing Cu(i) AlkylPyrPhos TADF complexes. Dalton Trans. 2019, 48, 15687–15698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huitorel, B.; El Moll, H.; Utrera-Melero, R.; Cordier, M.; Fargues, A.; Garcia, A.; Massuyeau, F.; Martineau-Corcos, C.; Fayon, F.; Rakhmatullin, A.; et al. Evaluation of ligands effect on the photophysical properties of copper iodide clusters. Inorg. Chem. 2018, 57, 4328–4339. [Google Scholar] [CrossRef]
- El Sayed Moussa, M.; Khalil, A.M.; Evariste, S.; Wong, H.-L.; Delmas, V.; Le Guennic, B.; Calvez, G.; Costuas, K.; Yam, V.W.-W.; Lescop, C. Intramolecular rearrangements guided by adaptive coordination-driven reactions toward highly luminescent polynuclear Cu(i) assemblies. Inorg. Chem. Front. 2020, 7, 1334–1344. [Google Scholar] [CrossRef]
- Evariste, S.; Khalil, A.M.; Moussa, M.E.; Chan, A.K.-W.; Hong, E.Y.-H.; Wong, H.-L.; Le Guennic, B.; Calvez, G.; Costuas, K.; Yam, V.W.-W.; et al. Adaptive coordination-driven supramolecular syntheses toward new polymetallic Cu(I) luminescent assemblies. J. Am. Chem. Soc. 2018, 140, 12521–12526. [Google Scholar] [CrossRef] [PubMed]
- Chakkaradhari, G.; Eskelinen, T.; Degbe, C.; Belyaev, A.; Melnikov, A.S.; Grachova, E.V.; Tunik, S.P.; Hirva, P.; Koshevoy, I.O. Oligophosphine-thiocyanate Copper(I) and Silver(I) complexes and their borane derivatives showing delayed fluorescence. Inorg. Chem. 2019, 58, 3646–3660. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Nobuyasu, R.S.; Zhang, B.; Geng, Y.; Yao, B.; Xie, Z.; Zhu, D.; Shan, G.; Che, W.; Yan, L.; et al. Thermally activated delayed fluorescence in CuI complexes originating from restricted molecular vibrations. Chem. Eur. J. 2017, 23, 11761–11766. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Chen, J.; Wu, X.Y.; Chen, X.L.; Yu, R.; Lu, C.Z. Outstanding blue delayed fluorescence and significant processing stability of cuprous complexes with functional pyridine-pyrazolate diimine ligands. Dalton Trans. 2015, 44, 6706–6710. [Google Scholar] [CrossRef]
- Keller, S.; Prescimone, A.; La Placa, M.-G.; Junquera-Hernández, J.M.; Bolink, H.J.; Constable, E.C.; Sessolo, M.; Ortí, E.; Housecroft, C.E. The shiny side of copper: Bringing copper(i) light-emitting electrochemical cells closer to application. RSC Adv. 2020, 10, 22631–22644. [Google Scholar] [CrossRef]
- Brunner, F.; Prescimone, A.; Constable, E.C.; Housecroft, C.E. Positional isomerism in the N^N ligand: How much difference does a methyl group make in [Cu(P^P)(N^N)](+) complexes? Molecules 2020, 25, 2760. [Google Scholar] [CrossRef]
- Ishida, H.; Bünzli, J.-C.; Beeby, A. Guidelines for measurement of luminescence spectra and quantum yields of inorganic and organometallic compounds in solution and solid state (IUPAC Technical Report). Pure Appl. Chem. 2016, 88, 701–711. [Google Scholar] [CrossRef] [Green Version]
- Fries, F.; Reineke, S. Statistical treatment of photoluminescence quantum yield measurements. Sci. Rep. 2019, 9, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Gan, Z.; Epifanovsky, E.; Gilbert, A.T.B.; Wormit, M.; Kussmann, J.; Lange, A.W.; Behn, A.; Deng, J.; Feng, X.; et al. Advances in molecular quantum chemistry contained in the Q-Chem 4 program package. Mol. Phys. 2015, 113, 184–215. [Google Scholar] [CrossRef] [Green Version]
- Bard, A.J.; Faulkner, L.A. Electrochemical Methods: Fundamentals and Applications, 2nd ed.; Wiley Inc.: New York, NY, USA, 2000. [Google Scholar]
- Kubas, G.J.; Monzyk, B.; Crumbliss, A.L. Tetrakis(Acetonitrile)Copper(I) Hexafluorophosphate. In Inorganic Syntheses, 2nd ed.; Shriver, D.F., Ed.; Inorganic Syntheses Organization, John Wiley & Sons: Toronto, ON, Canada, 1979; p. 90. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |
Figure 1.
Structures of the copper(I) complexes that were investigated in this work. The structures can be classified as those bearing a hydroxyl group (5a, 5b) or a polymerisable unit (5c, 5d).
Figure 1.
Structures of the copper(I) complexes that were investigated in this work. The structures can be classified as those bearing a hydroxyl group (5a, 5b) or a polymerisable unit (5c, 5d).

Scheme 1.
Synthetic procedure for the preparation of the ligands 3a and 3b.

Figure 2.
Structure of the ligands bearing a polymerisable unit. Ligand 4c has a styrenic functionality, while 4d has an acrylate unit.
Figure 2.
Structure of the ligands bearing a polymerisable unit. Ligand 4c has a styrenic functionality, while 4d has an acrylate unit.

Scheme 2.
Synthetic procedure for the synthesis of the Cu(I) complexes.

Figure 3.
Molecular structure of the Cu(I) complex in 5b (counteranion, lattice solvent and hydrogen atoms of the DPEPhos ligand omitted for clarity). Selected bond lengths (Å) and angle (°): Cu(1)-N(1) 2.112(3), Cu(1)-N(2) 2.048(4), Cu(1)-P(1) 2.2371(12), Cu(1)-P(2) 2.2459(12), N(1)-Cu(1)-N(2) 79.56(14).
Figure 3.
Molecular structure of the Cu(I) complex in 5b (counteranion, lattice solvent and hydrogen atoms of the DPEPhos ligand omitted for clarity). Selected bond lengths (Å) and angle (°): Cu(1)-N(1) 2.112(3), Cu(1)-N(2) 2.048(4), Cu(1)-P(1) 2.2371(12), Cu(1)-P(2) 2.2459(12), N(1)-Cu(1)-N(2) 79.56(14).

Figure 4.
(a,b) SEC of the styrenic and acrylate copolymers before (6c, 6d) and after complexation (7c, 7d), respectively.
Figure 4.
(a,b) SEC of the styrenic and acrylate copolymers before (6c, 6d) and after complexation (7c, 7d), respectively.

Figure 5.
UV–vis spectroscopy of complexes 5a (black curves); 5b (red curves); 5c (blue curves); and 5d (green curves) in DCM solutions at r.t. (a) Molar extinction coefficients (ε) in the UV–visible range of the electromagnetic spectrum—a zoom-in is shown in the inset; (b) emission of Ar-saturated solutions (concentration ~μM; λexc = 370 nm).
Figure 5.
UV–vis spectroscopy of complexes 5a (black curves); 5b (red curves); 5c (blue curves); and 5d (green curves) in DCM solutions at r.t. (a) Molar extinction coefficients (ε) in the UV–visible range of the electromagnetic spectrum—a zoom-in is shown in the inset; (b) emission of Ar-saturated solutions (concentration ~μM; λexc = 370 nm).

Figure 6.
Isovalue contours calculated for the HOMO and LUMO of 5b (left), 5c (centre) and 5d (right). Hydrogen atoms are omitted for clarity. Red indicates the negative and blue the positive sign of the wave-function.
Figure 6.
Isovalue contours calculated for the HOMO and LUMO of 5b (left), 5c (centre) and 5d (right). Hydrogen atoms are omitted for clarity. Red indicates the negative and blue the positive sign of the wave-function.

Figure 7.
Cyclic voltammetry of Cu(I) complexes 5b (bottom), 5c (centre), 5d (top) in N,N-dimethylformamide (0.1 M TBAPF6) at a scan rate of 100 mV/s.
Figure 7.
Cyclic voltammetry of Cu(I) complexes 5b (bottom), 5c (centre), 5d (top) in N,N-dimethylformamide (0.1 M TBAPF6) at a scan rate of 100 mV/s.

Table 1.
Number average molecular weight (Mn) and dispersity (Ð) of the copolymers obtained, determined by SEC in tetrahydrofuran (THF).
Table 1.
Number average molecular weight (Mn) and dispersity (Ð) of the copolymers obtained, determined by SEC in tetrahydrofuran (THF).
| Polymer | Mn (kg/mol) | Đ |
|---|---|---|
| 6c | 13.5 | 1.65 |
| 7c | 11.8 | 1.63 |
| 6d | 9.54 | 1.92 |
| 7d | 7.26 | 1.62 |
Table 3.
Quantum yield of Cu(I) complexes, metallopolymers and blends in solid state at room temperature.
Table 3.
Quantum yield of Cu(I) complexes, metallopolymers and blends in solid state at room temperature.
| Sample | λem (nm) | Φ a |
|---|---|---|
| 5a | 511 | 0.06 (±0.02) |
| 5b | 504 | 0.12 (±0.02) |
| 5c | 501 | 0.09 b (±0.12) |
| 5d | 503 | 0.17 (±0.01) |
| 7c | 508 | 0.09 (±0.01) |
| 7d | 503 | 0.10 (±0.01) |
| PS/5c | n.d. | n.d. |
| PMA/5c | n.d. | n.d. |
| PS/5d | 502 | 0.13 (±0.02) |
| PMA/5d | 507 | 0.21 (±0.01) |
a determined with an integrating sphere for λex = 350 nm; b a statistical error associated with these measurements was attributed to the granular form of the sample [63].
Table 4.
Electrochemical properties of complexes reported versus Fc/Fc+ a.
| Sample | Eox/V | Ered1/V | Ered2/V | ΔHOMO-LUMO/eV | ΔHOMO-LUMO c/eV |
|---|---|---|---|---|---|
| 5a | n.a.; 0.84 b | n.a. | n.a. | n.a. | n.a. |
| 5b | 0.68; 0.82 b | −2.47 | −2.84 | 3.15 | 4.35 |
| 5c | 0.68; 0.84 b | −2.33 | −2.70 | 3.01 | 4.17 |
| 5d | 0.64; 0.88 b | −2.48 | −2.94 | 3.12 | 4.37 |
a in DMF (0.1 M TBAPF6) unless otherwise noted; b in DCM (0.1 M TBAPF6); c HOMO and LUMO energies of complexes optimised at PBE0/6-31G**/LANL2DZ/Grimme dispersion level (vacuum).
Table 5.
Quantities of reagents used for the synthesis of 2a and 2b and yields.
| Compound | Alcohol | Quantity (g) | Yield (%) |
|---|---|---|---|
| 2a | 8a | 0.98 | 71.3 |
| 2b | 8b | 1.22 | 72.5 |
Table 6.
Synthetic yields of 3a and 3b.
| Compound | Yield (%) |
|---|---|
| 3a | 52.3 |
| 3b | 64.3 |
Table 7.
Synthetic yields of 5a, 5b, 5c and 5d.
| Product | Yield (%) |
|---|---|
| 5a | 63.6 |
| 5b | 68.3 |
| 5c | 59.2 |
| 5d | 65.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ferrari, F.; Braun, J.; Anson, C.E.; Wilts, B.D.; Moatsou, D.; Bizzarri, C. Cyan-Emitting Cu(I) Complexes and Their Luminescent Metallopolymers. Molecules 2021, 26, 2567. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26092567
AMA Style
Ferrari F, Braun J, Anson CE, Wilts BD, Moatsou D, Bizzarri C. Cyan-Emitting Cu(I) Complexes and Their Luminescent Metallopolymers. Molecules. 2021; 26(9):2567. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26092567
Chicago/Turabian StyleFerrari, Federico, Jonas Braun, Christopher E. Anson, Bodo D. Wilts, Dafni Moatsou, and Claudia Bizzarri. 2021. "Cyan-Emitting Cu(I) Complexes and Their Luminescent Metallopolymers" Molecules 26, no. 9: 2567. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26092567