
Applications of Hybrid Polymers Generated from Living Anionic Ring Opening Polymerization


Gelest Inc., 11 Steel Road East, Morrisville, PA 19067, USA
*
Author to whom correspondence should be addressed.
Molecules 2021, 26(9), 2755; https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26092755
Submission received: 23 March 2021
/
Revised: 20 April 2021
/
Accepted: 22 April 2021
/
Published: 7 May 2021
(This article belongs to the Special Issue Various Aspects of Silicon Polymer Chemistry: A Themed Issue in Honor of Professor Julian Chojnowski on the Occasion of His 85th Birthday)
Abstract
:Increasingly precise control of polymer architectures generated by “Living” Anionic Ring-Opening Polymerization (Living AROP) is leading to a broad range of commercial advanced material applications, particularly in the area of siloxane macromers. While academic reports on such materials remain sparse, a significant portion of the global population interacts with them on a daily basis—in applications including medical devices, microelectronics, food packaging, synthetic leather, release coatings, and pigment dispersions. The primary driver of this increased utilization of siloxane macromers is their ability to incorporate the properties of silicones into organic structures in a balanced manner. Compared to organic polymers, the differentiating properties of silicones—low Tg, hydrophobicity, low surface energy, and high free molal space—logically lend themselves to applications in which low modulus, release, permeability to oxygen and moisture, and tactile interaction are desired. However, their mechanical, structural and processing properties have until recently precluded practical applications. This review presents applications of “Living” AROP derived polymers from the perspective of historical technology development. Applications in which products are produced on a commercial scale—defined as not only offered for sale, but sold on a recurrent basis—are emphasized. Hybrid polymers with intriguing nanoscale morphology and potential applications in photoresist, microcontact printing, biomimetic soft materials, and liquid crystals are also discussed. Previously unreported work by the authors is provided in the context of this review.
1. Introduction
Functional siloxane polymers constitute a large class of reactive materials. Siloxanes with vinyl, silanol, and hydride substitution are the most widely utilized, serving as the basis for the majority of elastomeric silicone products. Functional siloxanes combined with organic monomers form “hybrid” polymers which, despite their demonstrated utility, have comparatively limited commercial applications. Most siloxane polymers are prepared by ring-opening polymerization with high degrees of polydispersion, thereby curtailing their ability to act as precise structural elements. On the other hand, the economics of both the basic building blocks and the polymerization process itself favor equilibrium-derived siloxane polymers. Figure 1 depicts the range of synthetic methods utilized to prepare siloxane polymers.
Due to the intrinsic process as well as the structural control that it provides, Living AROP-derived polymers provide the potential for a broad range of hybrid organic–inorganic materials. Briefly, AROP-derived materials provide a mechanism for translating macromolecular synthetic methods normally associated with organic polymers (and excluded from inorganic polymers) into hybrid polymer structures. Among these AROP-derived siloxane polymers, perhaps the most technologically significant are monofunctional and heterobifunctional silicone macromers. Siloxane macromers are defined as silicon-containing species with a single functional polymerizable group which, although used as monomers, possess sufficiently high molecular weight and enough internal monomer units to be considered polymeric. In another sense, they are siloxane building blocks derived from Living AROP. Siloxane macromers enable the use of technologies other than those associated with siloxane polymerization—e.g., techniques associated with the wider range of synthetic organic polymerization technologies—to incorporate siloxane-associated properties: e.g., techniques associated with the wider range of synthetic organic polymerization technologies. Notably, only controlled “living” AROP provides a path to siloxane polymers with sufficiently controlled structures and functionality to behave as macromers in polymerization with organic monomers. Siloxane macromers thus enable the introduction of selected siloxane properties into higher order structures via macromolecular engineering.
Although the advent of living anionic ring-opening polymerization can be traced back approximately 50 years, commercial applications of this technology have only reached the marketplace within the last 20 and, by our estimate, production has exceeded 100 tons per annum only in the last 5. The availability and range of polymers with tailored molecular weight, polymer backbone structure and basic architecture, and both functional and non-functional alternatives have grown enormously, stimulated by symbiotic application and development efforts. This review relies heavily on patent literature; the global collection of patents, of which there are over 10,000,000 in the US alone, is one of the most comprehensive collections of technical information in the world but is often neglected in scholarly reviews. Nevertheless, the impact of patent technology on how materials are prepared and utilized may equal or exceed that of academic literature.
1.1. Fundamentals/Building Blocks/Architectures
The “living” anionic ring-opening polymerization of cyclosiloxanes is in fact more properly described as kinetically controlled ring-opening polymerization. The features that define the “living” aspects of the polymerization are: a quantitative initiation (as shown in Scheme 1), and the fact that the rate of polymerization propagation is significantly greater than that of the polymer chain randomization processes, particularly the reversion of the degree of polymerization driven by equilibration or “back-biting” processes (as shown in Scheme 2) [1].
Both initiation and back-biting are driven by the catalytic opening of a siloxane bond. The evolution of Living AROP has depended on the recognition of classes of cyclic siloxane monomers that possess ring strain, as well as on “weak” catalysts which are able to rapidly cleave the Si-O-Si bonds of the strained monomers but are relatively slow and ineffective at cleaving Si-O-Si bonds in unstrained systems. The difference in reaction kinetics provides an opportunity to deactivate the catalyst before significant equilibration effects are observed, resulting in the scalable preparation of polymers with polydispersities approaching 1.
The differential polymerization of ring-strained cyclics, as opposed to unstrained cyclic siloxanes, was apparently observed in early industrial development, as is made particularly clear in the example of fluorinated silicones generated from 3,3,3-trifluoropropylmethylcyclic siloxanes [2]. Attempts to polymerize cyclic tetrasiloxanes were ineffective due to the fact that the reversion kinetics apparently matched those of polymerization for the unstrained cyclic tetramer. The polymerization of cyclic trisiloxanes, on the other hand, was effective due to the ability to deactivate the catalyst before significant reversion could occur. The potential for weak catalysts such as lithium phenoxide to produce polymers of ring-strained monomers was first recognized by McVannel [3], while the quantitative, selective formation of a lithium initiator generated from the reaction of organolithium reagents with cyclic trimers was studied by Frye [4], who, surprisingly, observed that, in a 1:1 molar stoichiometry, n-butyl lithium reacted with hexamethylcyclotrisiloxane (D3) to form lithium n-butyldimethylsilanolate, leaving 2/3 of the D3 unreacted. Lee and Frye also noted that polar “promoters” would then cause polymerization to proceed [5]. Finally, Fessler reported the relative effectiveness of promoters in the living polymerization process and provided mechanistic insight into siloxane-silanolate reactions that could result in shifts between triad, Gaussian, and redistribution products, as shown in Scheme 3 [6].
A comparison of a GC (gas chromatograph) of siloxane macromers with equivalent MW and PDIs, showing triad and Gaussian distributions, is provided in Figure 2 (author’s work). In contrast to anionic polymerization with K+ and Na+, in which there is little differentiation between chain scission points, the Li+ redistribution mechanism favors chain termini: i.e., despite chain-end scrambling, narrow polydispersity is maintained.
During the same period when Lee and Frye’s report was published but separate from the discoveries relating to ring-strained siloxane monomers and lithium-based initiators, there was significant interest within the silicone industry in generating block copolymers, with lithium silanolate-based initiators being shown to lead to the sequential polymerization of cyclotrisiloxanes. The combination of the growing interest in forming block copolymers, the development of siloxanes with strained cyclic structures, and quantitative lithium catalyzed polymerization underlies the publication by Saam [7,8] that reviewers point to as establishing the potential of “living” AROP siloxanes—in which he clearly demonstrated initiation, promotion, narrow MW distribution, and the ability to form block copolymers. It was recognized at that time that these polymers had to be terminated before equilibrium processes dominated in order to maintain the target molecular weight, as visualized in Figure 3. Functionalized termination reagents were later used to create siloxane macromers [9,10], although a process for preparing these macromers was not reported until 10 years later [11].
The term “macromer” is a contraction of the word macromonomer and refers to a relatively high molecular weight species with a single functional group which, although used as a monomer, has sufficient internal monomer units to be considered a polymer. The earliest commercial siloxane macromers contained methacrylate functionality and found commercial utility in the formation of organic–inorganic hybrid polymers (Scheme 4). Their termination, or “capping”, functionality was derived from the use of methacryloxypropyldimethylchlorosilane. The general structure for a siloxane is depicted in Scheme 5. Variations of the basic structure are depicted in Scheme 6, Scheme 7 and Scheme 8.
A second class of functionality can be introduced into macromers by using novel initiators, thereby yielding telechelic polymers in which the second functional class—e.g., hydroxyl—is located at the telechelic polymer termini, which are equidistant from the first functional class.
More recently, living AROP has been combined with the concept of functional initiators to generate both monodisperse telechelic and heterobifunctional siloxanes.
This historic overview has only given a condensed description of the chemistry, structure, and function of siloxanes derived via living AROP. For those interested in more details regarding the chemical aspects of siloxanes derived from living AROP, the following references should be consulted [1,12,13]. While the bulk of the literature and commercial applications utilize a lithium anion as a weak base component of the initiator, an intriguing recent series of reports utilize substituted cyclic guanidines in combination with water or silanol that act as initiators for living AROP [14,15].
1.2. Monomeric Building Blocks
The fact that there are only five monomeric building blocks for the living AROP-derived polymers that represent virtually all commercial applications as well as the vast majority of published reports is a consequence both of practical synthetic routes [16,17,18] and the sluggish rates of polymerization reported for cyclotrisiloxanes with greater organic substitution [19]. A preparation has been reported [20] for the simple and highly desirable monomer trimethylcyclotrisiloxane, but its practical isolation has not yet been described. This has led to interest in pentamethylcyclotrisiloxane [21] and hydridotetramethylsiloxanylethyl-substituted cyclotrisiloxanes [22], which can be used directly as monomers for polymerization or reacted with various olefins to form more elaborately substituted trisiloxanes. Similarly, vinylpentamethylcyclotrisiloxane has been prepared, and offers an advantage over trivinyltrimethylcyclotrisiloxane in cases where isolated vinyl substitution on the polymer chain is desired [23]. The primary cyclic monomers used in the production of macromers on both commercial and research levels are listed for convenience in Table 1. Other cyclic monomers have been reported in the literature, and include higher hexaalkylcyclotrisiloxanes [24], chloropropylmethylcyclotrisiloxanes [25], and substituted hexaarylcyclotrisiloxanes [26]. Specialty monomers with limited reference include D4 [27], acrylate [28], cyclic ether [29], trimethylsiloxy-substituted [30] cyclotrisiloxanes, and similarly substituted strained carbosiloxanes [31].
1.3. Termination
Terminating molecules in living AROP can serve several functions. At a minimum, they serve to quench the polymerization before redistribution processes associated with reversion, interchain back-biting, and interchain scrambling; their potential functions can be further divided into the following categories:
- Non-functional termination;
- Non-functional coupling to effect non-functional doubling of MW;
- Non-functional coupling with block insertion;
- Functional termination to form asymmetric macromers;
- Functional coupling to form symmetric macromers;
- Functional coupling to form branched structures.
Terminators in AROP processes are usually chlorosilanes, although other halosilanes and alkoxysilanes have been reported. Apart from the terminators found in the literature associated with living AROP of siloxanes, terminators normally associated with the living anionic polymerization of olefins and other organic polymers can provide similar termination for siloxanes [77,78].
1.4. Macromers Commonly Reported in Literature
The structures and properties listed in Table 3 are an aggregate of the most common siloxane macromers reported in the scholarly and commercial literature. They should not be considered exact values, but rather nominal values for similar materials. Similarly, heterobifunctional macromers are reported in Table 4.
2. Applications
2.1. Compatibility and Reactivity Introduction
The living polymerization of siloxanes provides the basis for synthesizing siloxane macromers capable of acting as precise structural elements, achieving both the requirements of copolymerization with organic monomers as well as the desired properties of copolymers. The incorporation of properties associated with siloxanes—including oxygen and moisture permeability, release, or thermal performance—would thereby improve the performance of a wide range of organic polymers. To date, however, the utilization of silicone macromers in combination with organic monomers has been primarily limited by economic considerations, as well as by structural and compositional challenges.
2.2. Gas/Vapor Permeability
In comparison to organic polymers, siloxanes possess a large free molal volume, a consequence of the length of both silicon–oxygen and silicon–carbon bonds, which allows greater permeation of small molecules. These same factors contribute to the flexibility of silicones which imposes limitations on structural properties. Hybrid polymer systems utilizing both macromer and block polymers generated by living AROP provide the balance of properties required in contact lenses, breathable films, and membranes.
2.2.1. Contact Lens/Hydrogel
Oxygen permeability is a key feature of modern contact lenses, particularly those intended for extended wear. In addition to the bloodstream, the corneal and scleral tissues of the eye take about 1/3 of their respiratory requirement directly from the air. Methacrylate-derived polymers, while possessing good optical and mechanical properties, are occlusive to oxygen transport due to the fact that the permeability of siloxanes is more than 100X greater than that of analogous hydrocarbon structures. Gaylord’s pioneering material work [117,118] under the direction of Seidner led to the commercialization of rigid gas permeable contact lenses by Syntex in 1980 [119]. While these lenses satisfied the physiological requirement for extended wear, broad acceptance of silicone rigid gas permeable (RGP) lenses was not achieved, since user comfort did not match that of hydrogel lenses. Formulation and process challenges in silicone hydrogel contact lens manufacture include combining hydrophobic silicones with hydrophilic monomers while maintaining optical clarity, high water content, and high oxygen transport to the eye in the resulting hydrogels. The first successful silicone hydrogel lens, based on technology disclosed by Vanderlaan, was introduced to the marketplace by Vistakon in 2004 [102]. Monofunctional siloxane macromers polymerized by living anionic polymerization routes are key materials in such silicone hydrogel contact lens formulations: these monodisperse monofunctional materials are used as comonomers with hydrophilic hydrogel monomers such as hydroxyethylmethacrylate (HEMA) and dimethylacrylamide (DMA). The earliest report of soft silicone hydrogel lenses generated from siloxane macromers and HEMA that displayed acceptable performance utilized group transfer polymerization [120]. One material requirement of a silioxane macromer selected for such a formulation is a minimal solubility in the hydrophilic hydrogel monomers. It has been reported that, after curing the silicone hydrogel contact lens reactive monomer mix, an optically clear co-continuous silicone and hydrogel phase membrane suitable for contact lens use is formed [121]. Incorporating low molecular weight (~1000 g mol−1) siloxane macromers into silicone hydrogel formulations enhances oxygen permeability in the resulting contact lenses, achieving superior oxygen transport to the eye compared to hard rigid gas permeable (RGP) and soft HEMA contact lens technologies (Figure 4) [122].
During the contact lens molding process, extraction procedures are employed to remove undesirable impurities. Water insoluble impurities, for example, decrease optical clarity and leach out of the contact lens, causing negative ocular effects such as stinging [92]. Unfortunately, the hydrophobic nature of these impurities makes conventional water extraction procedures insufficient. Silicone hydrogel extraction procedures using alcohols have been reported, but possess drawbacks including increased manufacturing costs, organic solvent waste handling concerns, and potential eye irritation from residual solvent. Living anionic polymerization enables the synthesis of monodisperse monofunctional siloxane macromers that are largely free of impurities. Indeed, newer generation silicone hydrogel formulations employ siloxane macromers of sufficient purity to rely solely on aqueous extraction and hydration steps post-molding. Table 7 gives an overview of the different generations of silicone hydrogel technologies. Generation 1 silicone hydrogel contact lenses did not use siloxane macromers and relied on tris(trimethylsilyl)-silylpropylmethacrylate (TRIS) and difunctional silicone crosslinkers as the oxygen transport enhancers in the formulation. TRIS made control of the lenses’ mechanical properties difficult, resulting in higher modulus lenses that were not comfortable for the wearer [121]. Generation 2 and 3 silicone hydrogel formulations incorporated siloxane macromers as oxygen transport enhancers, which led to greater formulation flexibility and control of the final contact lens material properties (e.g., Dk and modulus) [100,101,123].
Hydrogels comprised of macromers with both polycarbosiloxane units and poly(trifluoropropylmethyl)siloxane units have been reported. The macromers are synthesized by substituting D3 with other ring-strained monomers in a living polymerization process. Awasti describes a monomethacrylate functional polycarbosiloxane synthesized from a 2,2,5,5-tetramethyl-2,5-disila-1-oxacyclopentane monomer as resistant to hydrolysis and therefore suitable for use in silicone hydrogel soft contact lens design [55,128]. Monomethacrylate functional poly(trifluoropropylmethyl)siloxane macromers possess increased polarity, improving their miscibility with hydrophilic monomers and potentially reducing the non-specific binding of proteins on soft contact lens surfaces [59,70].
Low molecular weight symmetric siloxane macromers with hydrophilic termini have increased miscibility with hydrophilic monomers compared to asymmetric siloxane macromers, as the smaller siloxane block size of the symmetric architecture limits phase separated domain formation while still maintaining the oxygen transport-enhancing benefits of asymmetric analogs. The hydrophilic termini reported by Kimble include methoxypropyl and hydroxypropyl groups [59,60]. α-Methacrylate functional, ω-polyalkyleneoxide siloxane macromers have also been described; however, their block copolymer structure results in microphase separation, rendering this macromer structure unsuitable for the production of optically clear hydrogel films [129].
One approach for improving contact lens comfort is to utilize a plasma treatment to improve hydrogel wettability and then apply hydrophilic terpolymers derived from combinations of silicone macromers and hydrophilic monomers such as diethylazetidinium methacrylate ester chloride salt, thereby providing permanent wettability [130].
In a separate but related area, siloxane-based interocular lenses (IOL) provide new opportunities for macromers. Hydride terminated macromers are utilized to control the modulus of cured elastomers used in IOLs [131]. Interestingly, silicone macromers are broadly described as components in laser adjustable IOLs that enable a post-insertion change in dioptric power by altering the refraction of the lens material [132].
Independent of contact lens development, optically clear films were derived from copolymers of styrene, ethylene glycol dimethacrylate (EGDMA), and siloxane-urethane-methacrylate (SiUMA). The SiUMA monomer was synthesized from carbinol-terminated siloxane macromers reacted with isocyanatoethylmethacrylate [133].
2.2.2. Controlled Atmospheric Packaging—Modified Atmosphere Packaging
An appreciation of the role that gases play in maintaining the freshness of meats, fruits, and vegetables during transport has created a role for polymer films with controlled permeability. Optimal oxygen concentration is associated with the appearance of “red” meat and “green” vegetables. In the case of meat, deoxymyoglobin, which is purple, forms when metabolic, diffusion, and other processes deplete oxygen availability. Over time, however, oxymyoglobin is oxidized to amber-brown methemoglobin, which is associated with a lack of freshness [134]. Ethylene is associated with fruit ripening and abscission. Gas permeable films derived from acrylate terpolymers act as overlayers to porous structures, maintaining the oxygen transmission of these structures while providing a barrier to microbial infiltration. Unlike in the case of contact lenses, transparency is usually not required for packaging applications. The use of higher molecular weight monomethacrylate macromers in packaging is therefore acceptable, and has been reported [135]. Breathable films based on this technology have become components in commercial packaging applications offered by BreatheWay, as shown in Figure 5.
2.3. Membranes
The utilization of polydimethylsiloxanes and silicon-rich homopolymers such as poly(trimethylsilylpropyne) [136] and poly(vinyltrimethylsilane) [137] for gas separation membranes has been established. The removal of volatile organic compounds from aqueous mixtures has been more effectively addressed by hybrid polymer systems in which there is formation of siloxane microphases in continuous organic phases. Reports on graft polymer and interpenetrating polymer networks were able to demonstrate selective pervaporation and removal of organics from aqueous streams [138,139]. The control of selectivity and permeation rates has been accomplished with copolymers derived from methacrylate functional macromers copolymerized with a variety of other methacrylate monomers. Uragami demonstrated that macromers with molecular weights of ~4000 Daltons formed copolymers with methyl methacrylate which, depending on comonomer content, allowed selective permeation of water or ethanol from ethanol–water solutions by varying comonomer contents [140]. In a series of reports, Urgami extended these systems to styrene copolymers and the incorporation of ionic liquids, enabling the selective pervaporation of volatile organics, including toluene and chlorinated organics, from aqueous streams [141,142,143].
2.4. Surface Properties/Modification
2.4.1. Dyes, Micelles and Particles for Advanced Printing, Reprographics, and Lithography
Methacrylate functional macromers have been used in both organic dye [89] and pigment-based printing applications [98,99,144]. While these technologies are quite different, the role of the silicone macromer has common features in each. In ink-jet applications, a bulk solvent-based polymerization with dye is accomplished, after which micelles are formed via the addition of water and simultaneous evaporation of solvent. In pigment-based applications, the polymer forms an encapsulant or binder. In both cases, the silicone macromer appears to serve as a hydrophobic outer surface of the micelle or pigment, thereby helping to control particle charging and, secondarily, contributing to the spread and adhesion of the micelles or particles on the substrate. Polymersomes with controlled architecture in both their overall dimensions and membrane thickness have been prepared from diblock polymers of cabinol-termined siloxane macromers and methyloxazolidine [82].
Interestingly, while the ability of styrene-PDMS deblock polymers generated via living AROP to form micelles was recognized during early development efforts [145], no recent commercial applications have been reported.
Colored fluids for electrowetting and electrofluidic applications have also been generated from highly polar dyes by reaction with aminopropyl-terminated siloxane macromers [80]. For example, the yellow dye 2-(4-carboxyphenylazo)acetoacetanalide reacted with a 1000 Mn amino-terminated macromer to form the product 4-(E)-(2,4-dioxopentan-3-yl)diazenyl-N-polydimethylsiloxane-benzamide, which was soluble in non-polar fluids including silicones. Radiation-curable films for adhesive and lithographic applications, in which aminopropyl-terminated siloxane macromers are acrylated, were reported by Leir [146]. Carbinol-terminated siloxane macromers were converted to phosphate-terminated macromers by Tao and then reacted with the surfaces of CdSe quantum dots before incorporation into bulk silicones to form electroluminescent transparent films, as shown in Figure 6 [85].
2.4.2. Coatings Additives—Leveling Agents, Clean Surfaces and Release
Siloxane macromer block polymers are offered commercially as leveling agents to reduce waviness and orange peel in organic coatings. The effect occurs at low concentrations and is based on activity at the liquid–gas interface, in which these polymers are oriented due to limited incompatibility with the actual binder component of the coating system [147,148]. Their versatile chemistry and modular molecular structure make it possible to adjust the properties of these macromers for specific applications. For example, further improvements in levelling have been reported when a fluorinated oxetane is reacted with unsaturated termination in macromers [149].
High silicone content macromers can impart anti-graffiti properties to coated surfaces, though this often requires concentrations higher than those used in levelling applications. Macromers with a lower silicone content can reduce surface tension and improve substrate wetting without impairing recoatability. In the case of automotive coatings, other benefits include retaining the bonding characteristics of films and adhesives, while anti-blocking properties can also be achieved in decorative coatings [150,151].
One of the earliest applications of styryl and methacrylate functional macromers was for controlling release in adhesive tapes [54]. The investigators showed a correlation between molecular weight and release characteristics—e.g., in butyl methacrylate, acrylic acid, and macromer terpolymers—with macromers of low molecular weight proving ineffective but macromers with molecular weights ≥2000 providing control. The same general chemistry has been utilized more recently in dirt resistant coatings [93]. Release coatings, including ice-phobic coatings, have been generated from carbinol-terminated siloxane macromers by reaction with isocyanate and epoxy functional prepolymers to form amphiphilic, self-stratified thin films [152]. A reduction of marine biofouling was observed when aminopropyl-terminated macromers were incorporated into isophorone diiosocyanate-derived urethanes, with low molecular weight macromers (1000 Daltons) providing more favorable results than high molecular weight macromers [81]. Methacrylate-terminated siloxane macromers with embedded hydrophilicity have also been used in this application [108]. Ultraphobic coatings—i.e., coatings exhibiting both superhydrophobic and oleophobic behavior—in which combinations of epoxy functional siloxane telechelics and macromers are reacted with linear and/or branched polyethylenimine (PEI), have been reported by Soane and Ready [94,107]. Hydride-terminated macromers have been used to modify vinyl POSS structures in slippage coatings for ice-phobic applications [91]
2.5. Coatings
2.5.1. Thin Film Silicone Coatings
The inclusion of silicone macromers into the formal polymeric structures of coatings, as distinguished from their use as additives, is a growing area of technology. These applications tend to be associated with release, lubricity, and mechanical properties related to direct physical interactions with humans. Indeed, the sensory appeal of coatings has always been an important driver of consumer applications in which positive tactile interaction is critical to acceptance [87], such as synthetic leather, textile finishes, and hair care.
Older approaches to urethane materials mainly use polydisperse telechelic carbinol-terminated siloxane polymers, in which the two identical functionalities on the termini serve to introduce siloxanes into urethanes as soft-blocks. AROP-derived siloxane macromers (oligomeric materials with functionality on one terminus) represent a newer approach in which the functionality on one terminus of the oligomer allows the formation of a brush polymer with siloxane segments as pendant, allowing the mechanical properties of the urethane backbone to be maintained (Scheme 9) [87]. These structures also demonstrate significantly greater wear resistance as well as lower friction and release properties compared to telechelic controls [153], in which siloxane is incorporated into the urethane backbone as shown in Scheme 10. Table 8 compares tribological and contact angle properties of urethane in which siloxane has been introduced as an unreacted fluid, a soft segment, and a pendant, respectively.
In similar chemistry, self-lubricating cannulae for medical applications have been fabricated from thermoplastic urethanes derived from mono dicarbinol macromers [86].
2.5.2. Thick Film Silicone Coatings
Depending on their formulation, silicone coatings elicit a wide range of tactile responses. Coatings that contain low molecular weight, particularly volatile species are associated with a “silky” feel and exhibit slip. On the other hand, silicone coatings that have been depleted of low molecular weight species are associated with a “tacky” feel. Common to both these experiences is the extreme hydrophobicity of the polymer. When silicone coatings are free of low molecular-weight species, they exhibit high coefficients of friction and, due to their relatively poor mechanical properties, failure by abrasive and adhesive spalling during continuous tactile interaction. Well-defined silicones with a central vinyl functionality and discrete PEG2 (MCS-VX15), PEG3 (MCS-VX16—Scheme 11), or tetrahydrofurfuryl (MCS-VF14—Scheme 12) pendant end-groups can be used as comonomers in addition-cure, platinum-catalyzed 2-part silicone elastomer formulations in order to introduce hydrophilicity [61]. In such formulations, the surface tribological properties are modified by introducing a hydrodynamic lubricating layer of adsorbed water. The modified silicone elastomers retain optical clarity and mechanical performance characteristic of this class of material with up to 15 wt.% comonomer in the 2-part formulation. Contact angle measurements of deionized water on the silicone elastomer surface showed improved wettability with comonomer content: at ~3 wt.% comonomer, the elastomer surface shifts from hydrophobic (contact angle ~120°C) to hydrophilic (contact angle < 90°C). Coefficient of friction measurements for the modified silicone elastomers demonstrate increased surface lubricity with comonomer loadings (Figure 7) [112].
Separately, in the field of dielectric elastomer actuators, monovinyl-terminated PDMS macromers have been used to selectively adjust the network behavior of silicone films between compliant electrodes [111].
2.6. Cosmetics and Hair Care
Hair-care formulations, including shampoos and conditioners with the ability to withstand multiple washings, require good film-forming properties with strong adhesion to the hair cuticle, but must simultaneously offer lubricity in order to provide combability. Copolymers of dimethylaminoethylmethacrylate and low molecular weight methacrylate functional silicone macromers (MW 2000) [89,90] have been used directly or in combination with other polymers [154] to provide increased lubricity and combability of hair. Dispersions of macromer-derived terpolymer particles have been reported in hydrocarbon vehicles such as isododecane, yielding film-forming compositions that are useful in eyeliners and mascaras [97]. Hydride-terminated macromers have been reacted with unsaturated terpenes and cannabidiol (CBD) (Scheme 13 to form emollient compounds with solubility in the siloxane vehicles preferred for skin care [155,156].
2.7. Magneto-Rheological Fluids
Well-defined biocompatible magnetic nanoparticles are of interest as materials for biomedical applications including magnetic field-directed drug delivery, biomolecule separations, and assay devices. Superparamagnetic iron oxide (Fe3O4) nanoparticles (SPION) sterically stabilized with PDMS macromers synthesized by Living AROP produced homogeneous hydrophobic ferrofluids that are stable against precipitation [157,158]. PDMS macromers with a tricarboxylate endgroup capable of binding to the surface of magnetite nanoparticles were synthesized by first making a trivinyl-terminated PDMS via Living AROP, followed by a thiol-ene reaction between the vinyl silane groups and mercaptoacetic acid, as depicted in Figure 8 [159,160]. Molecular effects of the PDMS tail on the stability of the PDMS-magnetite complexes were studied. Magnetic separation methods were developed to narrow the particle size distribution of the magnetite nanoparticles using tricarboxylate PDMS stabilizer while controlling the PDMS surface concentration [161]. In other studies, a monocarboxydecyl-terminated PDMS macromer (MCR-B12) was used to stabilize magnetite nanoparticles; however, the resulting ferrofluids had issues with stability and sedimentation, likely due to the lower number of carboxylate binding groups per macromer chain [88]. The magnetophoretic mobility of the magnetite-PDMS fluids was then studied in different magnetic field conditions (magnetic fields and field gradients), with the results demonstrating that the shape and speed of these droplets in viscous media can be independently manipulated [160,162].
These magnetite-polydimethylsiloxane ferrofluids were proposed by Wilson as a material that could aid in the treatment of retinal detachment disorder [159]. The proposed treatment entailed inserting a pre-aligned magnet into the conjunctiva and then injecting the PDMS-based ferrofluid into the vitreous humor. As shown below in Figure 9, the ferrofluid would close the tear as it moved toward the permanent magnetite, allowing the surgeon to repair the tear.
2.8. Bulk Macroscale Materials
Ultra-High Elongation Elastomer
An ultra-high elongation silicone elastomer has been prepared from a heterobifunctional silicone macromer compounded with reinforcing agent, achieving elongations nearing 5000%, nearly four times greater than conventional silicone elastomers. The cure mechanism of this elastomer is a step-growth polymerization of an α-vinyl-ω-hydride-terminated silicone macromer [62,113,114] via intermolecular hydrosilylation reaction, which yields a linear polymer of exceptionally high molecular weight with no apparent covalent crosslinking (Scheme 14) [73,113].
Atomistic modeling (Figure 10) of the cured silicone macromer shows the probability of knotting within a 50,000 Da segment [115], which correlates to an experimental value of critical molecular weight (Mc) for entanglement of ~42,000 Da [163]. The stress-strain curve is remarkably different from those of crosslinked silicone elastomers, as shown in Figure 11.
Ultra-high elongation elastomers exhibit other behaviors not shared by conventional silicone elastomers [115]: e.g., they undergo extreme multi-axial distortions and return close to their original shape, and display remarkable tear propagation mechanisms, meaning that tear failure occurs at drastically greater elongations; recovery from minor penetration is improved below the failure limits and, finally, pseudo-self-healing behavior is also displayed, as shown in Figure 12 [116].
2.9. Nanoscale Morphology
2.9.1. Photoresist and Contact Printing
Templated self-assembly of a cylinder-forming poly(styrene-b-dimethylsiloxane) (PS−PDMS) diblock copolymer was first described by Saam and Fearon [51]—followed by others [164,165,166,167]—and has been investigated for nanolithography applications [44,145,168,169]. The general structure is depicted in Scheme 15. The general structure (PS−PDMS) diblock copolymer is depicted in structure 11. These copolymers undergo microphase segregation above their Tg, and the large X-polymer-solvent interaction parameter of the blocks is advantageous for achieving long-range ordering as well as for minimizing defect densities. Furthermore, the high Si content in PDMS leads to a robust oxide etch mask after two-step reactive ion etching (RIE) [170], as exemplified in Figure 13.
To address the critical needs of nano-dimensional photoresists, materials that possess dual surface properties are required. When cast or hot-pressed on a high-surface-energy substrate such as silicon, glass, or aluminum, the copolymer film forms both a lower-surface-energy component (PDMS)-enriched air/polymer interface and a higher-surface-energy component (organic block)-dominated polymer/substrate interface [171,172,173].
Interface effects complicate the use of siloxane A-B block copolymers due to the low surface energy and wetting characteristics of the siloxane block.
Silylated styrene block polymers that employ conventional radical polymerization rather than AROP have been proposed [174,175], and azidopropyl functional silicone macromers have been separately “clicked” with alkynyl-terminated ATRP-generated macromers to form PDMS-b-PMMA, producing sub-10 nm structures [174,176]. A second potential solution is star block polymers generated by AROP, which provide a mechanism through which the non-siloxane block can dominate interfacial behavior [52]. The reviewers note that similar morphologies have also been reported for polybutadiene-polydiethylsiloxane block copolymers (PBD-b-PDES) [19]; these copolymers possess less differentiation in surface energy, potentially mitigating issues with the styrenic and methacrylate systems, but have not been evaluated in lithographic systems. Similarly noteworthy in this context, while the synthesis of diblock polymers typically starts with an organic macroanion and then proceeds to a siloxane polymerization, the potential of the reverse process, in which a siloxane macromer starts and proceeds to an acrylate polymerization, was demonstrated using ATRP [177]. Other organic block copolymer polymerization examples initiated by carbinol-terminated macromers include caprolactone and trimethylene carbonate blocks [83,178]. This approach clearly expands the options for generating polymers with varying self-assembly structures.
The use of polystyrene macroanions was further elaborated by Bellas to form triblock and microarm polymers [44], and by Shefelbine to form continuous core–shell gyroid morphologies [179]. This work led to the observation of periodic double gyroid (DG) behavior, as well as a series of publications displaying an appreciation of the potential for extended novel 3D structures (Figure 14) [180,181,182]. As suggested by the DG topological visualizations of Thomas [183], potential structural, dielectric, charge transport, and mass transport material behavior can be controlled by the direct use of DG block polymers structures or by the removal, conversion, and/or infiltration of a DG microphase.
2.9.2. Biomimetic Polymers and Bottle-Brush (BB) Architecture
Recently, grafting-through polymerization and surface-initiated polymerization have led to bottle-brush polymers and particle-brush materials that have shown potential in the fabrication of biomimetic materials. These materials can be broadly considered filamentous structures that exhibit non-linear behavior under deformation, greater relaxation times, and the potential for complex non-covalent interactions leading to the formation of super-molecular structures. Siloxane macromers appear to be of particular interest in creating these structures due to both the palette of functionality and the intrinsic flexibility of the siloxane structures, which allows the length-scale of the filaments to extend beyond the primary polymer backbone or nano-feature without imposing a “hard” structural domain. Siloxane macromers allow entry to classes of materials that possess an unusually low modulus while maintaining mechanical failure properties consistent with the main polymer backbone for both methacrylate [96] and norbornene [84] functional siloxane macromers.
Separately, this recognition revealed such macromers’ potential for generating biomimetic gel structures [95]. The complexity of the potential range and behavioral characteristics associated with bottle-brush polymers in terms of macromer molecular weight, graft density, and final molecular weight is readily apparent. Their tunable physical properties in both crosslinked and uncrosslinked states—based on the Dp of the main chain and grafting density of methacryloxypropyl-terminated polydimethysiloxane (Dp = 10) prepared by Grafting Through Atom Transfer Radical Polymerization—have been investigated in this context [103]. The dynamics of deformation have also been modeled [104,109,110]. A striking extension of this approach can be found in the “chameleon-like” color changes demonstrated in these systems—in which vibrant color, extreme softness, and intense strain stiffening on par with that of skin tissue have been observed as a consequence of placing a heterogeneous polymeric system under varying degrees of strain (Figure 15) [106].
2.9.3. Liquid Crystal Siloxanes
Most polymers that exhibit liquid crystalline behavior have anisotropic side groups. Polydiethylsiloxane, which is distinct from both lost LCPs and its simpler homolog, polydimethylsiloxane, demonstrates mesophasic liquid crystalline behavior at ambient temperatures, as first reported by Beatty [184]. This behavior extends to poly(di-n-alkylsiloxane)s with side chains no longer than seven carbons that are also able to form a columnar mesophase. Such polymers are positionally and orientationally ordered in a two-dimensional hexagonal lattice, but without positional order along the chain. While early work was conducted with polymers of high polydispersity (>2.0), the desire to optimize liquid crystal behavior by controlling the PDI has led to the use of living AROP conditions for the polymerization of polydiethylsiloxane (PDES)—as first reported by Molenberg, who utilized lithium sec-butyldiethylsilanolate as the initiator [41,43]. The phase behavior of diethylsiloxane is shown in Figure 16.
There are few reports of living AROP with higher dialkylsiloxanes, presumably because of either ineffective termination or these systems’ sluggish kinetics.
Molenberg also reported on elastomeric block polymers of butadiene and diethylsiloxane that exhibited mesophase formation under tensile stress [19]. Later work with styrene-diethylsiloxane diblock polymers showed periodic nanoscale lamellar structures with compositions possessing greater than 20% styrene content, as shown in Figure 17 [42].
The vast majority of liquid crystal polysiloxanes based on side chain substitution that have recently been reviewed are polydisperse in nature [185]. Hepenius, however, utilized AROP to form vinylmethylsiloxane copolymers from vinylpentamethylcyclotrisiloxane and then functionalized the positions with mesogenic groups: for example, by reacting 4-cyano-4′-(ω-alkenoxy)biphenyl with an excess of tetramethyldisiloxane and then reacting with the remaining Si-H group (Scheme 16) [46].
3. Conclusions
The accelerating interest in living AROP-derived siloxanes has been spurred by the increasing drive to provide soft matter structures associated with tactile interaction and biocompatibility, particularly in the areas of contact lenses and biomimetic structures, and by these materials’ ability enable control of nano-dimensional physical properties associated with “smart” particles and morphologies associated with self-assembly. Silicone macromers have had great impact in practical applications, primarily due to their ability to cross-over from the paradigm of inorganic siloxane chemistry to the greater paradigm of organic polymerization. Further, the elaboration of simple macromers into heterobifunctional monomers has created opportunities for polymers with new bulk phase properties, including ultra-high elongation and self-healing materials.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
Not applicable.
References
- Chojnowski, J. Kinetically Controlled Siloxane Ring-Opening Polymerization. J. Inorg. Organomet. Polym. 1991, 1, 299–323. [Google Scholar] [CrossRef]
- Johannson, O. Method of Polymerizing Cyclic Diorganosiloxanes. U.S. Patent 3,002,951, 3 October 1961. [Google Scholar]
- McVannel, D. Method of Polymerizing Cylcotrisiloxanes II. U.S. Patent 3,294,740, 27 December 1966. [Google Scholar]
- Frye, C.L.; Salinger, R.M.; Fearon, G.; Klosowski, J.M.; Deyoung, T. Reactions of Organolithium Reagents with Siloxane Substrates. J. Org. Chem. 1970, 35, 1308–1314. [Google Scholar] [CrossRef]
- Lee, C.; Frye, C.; Johansson, O. Selective Polymerization of Reactive Cyclosiloxanes to Give Non-Equilibrium Molecular Weight Distributions. Monodisperse Siloxane Polymers. Polym. Prepr. 1969, 10, 1361. [Google Scholar]
- Fessler, W.A.; Juliano, P.C. Reactivity of Solvated Lithium N-Butyldimethylsilanolate with Organosiloxane Substrates. Ind. Eng. Chem. Prod. Res. Dev. 1972, 11, 407–410. [Google Scholar] [CrossRef]
- Saam, J.C.; Gordon, D.J.; Lindsey, S. Block Copolymers of Polydimethylsiloxane and Polystyrene. Macromolecules 1970, 3, 1–4. [Google Scholar] [CrossRef]
- Saam, J.C.; Ward, A.H. Method for Preparing Improved Polyalphamethylstyrene-Polydiorganosiloxane Copolymers and Products Thereof. U.S. Patent 4,107,227, 15 August 1978. [Google Scholar]
- Kawakami, Y.; Miki, Y.; Tsuda, T.; Murthy, R.; Yamashita, Y. Silicone Macromers for Graft Polymer Synthesis. Polym. J. 1982, 14, 913–917. [Google Scholar] [CrossRef] [Green Version]
- Anderson, R.; Arkles, B.C.; Larson, G.L. Silicon Compounds: Register and Review, 4th ed.; Petrarch Systems: Bristol, PA, USA, 1987. [Google Scholar]
- Nakano, T.; Tsuchiya, K.; Yoshimatsu, S.; Fuchigami, T. Process for Producing Diorganopolysiloxane. U.S. Patent 5,672,671, 30 September 1997. [Google Scholar]
- Chojnowski, J. Polymerization. In Siloxane Polymers; Clarson, S.J., Semlyen, J.A., Eds.; Prentice Hall: Englewood Cliffs, NJ, USA, 1993; pp. 1–62. [Google Scholar]
- Belorgey, G.; Sauvetin, G. Organosilicon Block and Graft Copolymers. In Silicon Containing Polymers; Jones, R.G., Ed.; Springer: Dordrecht, The Netherlands, 2000; pp. 43–78. [Google Scholar]
- Lohmeijer, B.G.G.; Dubois, G.; Leibfart, F.; Pratt, R.C.; Nederberg, F.; Nelson, A.; Waymouth, R.M.; Wade, C.; Hedrick, J.L. Organocatalytic Living Ring-Opening Polymerization of Cyclic Carbosiloxanes. Org. Lett. 2006, 8, 4683–4686. [Google Scholar] [CrossRef]
- Fuchise, K.; Kobayashi, T.; Sato, K.; Igarashi, M. Organocatalytic Ring-Opening Polymerization of Cyclotrisiloxanes Using Silanols as Initiators for the Precise Synthesis of Asymmetric Linear Polysiloxanes. Polym. Chem. 2020, 11, 7625–7636. [Google Scholar] [CrossRef]
- Hunter, M.J.; Hyde, J.F.; Warrick, E.L.; Fletcher, H.J. Organo-Silicon Polymers. The Cyclic Dimethyl Siloxanes. J. Am. Chem. Soc. 1946, 68, 667–672. [Google Scholar] [CrossRef]
- Piccoli, W.A.; Haberland, G.G.; Merker, R.L. Highly Strained Cyclic Paraffin-Siloxanes. J. Am. Chem. Soc. 1960, 82, 1883–1885. [Google Scholar] [CrossRef]
- Pierce, O.R. New Cyclotrisiloxanes. U.S. Patent 2,979,519, 11 April 1961. [Google Scholar]
- Molenberg, A.; Möller, M.; Soden, W.V. Block Copolymers and Networks from Polybutadiene and Polydiethylsiloxane. Acta Polym. 1998, 49, 45–51. [Google Scholar] [CrossRef]
- Crivello, J.V.; Lee, J.L. Synthesis of Cyclic Siloxanes by the Thermal Depolymerization of Linear Poly(Siloxanes). Chem. Mater. 1989, 1, 445–451. [Google Scholar] [CrossRef]
- Paulasaari, J.K.; Weber, W.P. Preparation of Highly Regular Poly(l-Hydrido-l,3,3,5,5-Pentamethyltrisiloxane) and Its Chemical Modification by Hydrosilylation. Macromolecules 1999, 32, 6574–6577. [Google Scholar] [CrossRef]
- Goff, J.; Arkles, B. Hydrodosiloxanyl Substituted Cyclotrisiloxanes, Preparation Method Thereof, and Polymeric Products Derived Therefrom. U.S. Patent 10,875,968, 29 December, 2020. [Google Scholar]
- Rózga-Wijas, K.; Chojnowski, J.; Zundel, T.; Boileau, S. Controlled Synthesis of Siloxane Copolymers Having an Organosulfur Group by Polymerization of Cyclotrisiloxanes with Mixed Units. Macromolecules 1996, 29, 2711–2720. [Google Scholar] [CrossRef]
- Out, G.; Klok, H.-A.; Möller, M. Hexa-n-Alkylcyclotrisiloxanes-Synthesis, Melting Behaviour and Polymerization Gerhardus. Macromol. Chem. Phys. 1995, 196, 195–210. [Google Scholar] [CrossRef] [Green Version]
- Fortuniak, W.; Chojnowski, J.; Sauvet, G. Controlled Synthesis of Siloxane Polymers and Siloxane-Siloxane Block Copolymers with 3-Chloropropyl Groups Pendant to the Siloxane Chain. Macromol. Chem. Phys. 2001, 202, 2306–2313. [Google Scholar] [CrossRef]
- Lee, M.K.; Meier, D.J. Synthesis and Properties of Diarylsiloxane and (Aryl/Methyl)Siloxane Polymers: 3. New Aryl Substituents. Polymer 1994, 35, 4197–4202. [Google Scholar] [CrossRef]
- Gädda, T.M.; Weber, W.P. Ring-Opening and Polymerization of 1-[2′-(Heptaphenylcyclotetrasiloxanyl)Ethyl]-1,3,3,5, 5-Pentamethylcyclotrisiloxane. J. Polym. Sci. Part A Polym. Chem. 2006, 44, 137–146. [Google Scholar] [CrossRef]
- Oba, T.; Naoki, O. Acryloxypropyl-Containing Cyclotrisiloxane, Production of the Polymer Composition and Polymer Composition. Jap. Patent 2856377B2, 8 October 1993. [Google Scholar]
- Minoru, I.; Norio, S.; Masaharu, T. Organopolysiloxane Having Cyclic Ether Group. Jap. Patent 1077346A, 24 March 1998. [Google Scholar]
- Cai, G.P.; Weber, W.P. Synthesis of Polymethyl(Trimethylsiloxy)Siloxane by Anionic Ring-Opening Polymerization of 1,3,5-Trimethyl-1,3,5-Tris(Trimethylsiloxy)Cyclotrisiloxane. Macromolecules 2000, 33, 6310–6314. [Google Scholar] [CrossRef]
- Suryanarayanan, B.; Peace, B.W.; Mayhan, K.G. Anionic Polymerization of a Series of Five-Membered Cyclocarbosiloxanes. J. Polym. Sci. Part A 1 Polym. Chem. 1974, 12, 1109–1123. [Google Scholar] [CrossRef]
- Bostick, E. Ordered Organopolysiloxanes. U.S. Patent 3,337,497, 22 August 1967. [Google Scholar]
- Pike, R. Process for Producing Cyclic Diorganosiloxanes from Diorganodihalosilanes and Alkali and Alkali Earth Metal Oxides and Carbonates. U.S. Patent 3,110,720, 12 November 1963. [Google Scholar]
- Dobay, D. Process for Preparing Hexaethylcyclotrisiloxane. U.S. Patent 2,769,830, 6 November 1956. [Google Scholar]
- Goff, J.; Arkles, B. Alkyl Ether Substituted Cyclotrisiloxanes and Preparation Method Thereof. U.S. Patent 10,669,294, 2 June 2020. [Google Scholar]
- Chojnowski, J.; Cypryk, M.; Fortuniak, W.; Rozga-Wijas, K.; Scibiorek, M. Controlled Synthesis of Vinylmethylsiloxane-Dimethylsiloxane Gradient, Block and Alternate Copolymers by Anionic ROP of Cyclotrisiloxanes. Polymer 2002, 43, 1993–2001. [Google Scholar] [CrossRef]
- Cypryk, M.; Delczyk, B.; Juhari, A.; Koynov, K. Controlled Synthesis of Trifluoropropylmethylsiloxane–Dimethylsiloxane Gradient Copolymers by Anionic ROP of Cyclotrisiloxanes. J. Polym. Sci. Part A Polym. Chem. 2009, 47, 1204–1216. [Google Scholar] [CrossRef]
- Maschke, U.; Wagner, T. Initiation by a Monofunctional Lithium Siloxanolate. Makromol. Chem. 1992, 193, 2453–2466. [Google Scholar] [CrossRef]
- Razzano, J.S.; Simpson, V.G. Fluorosilicone Copolymers and Process for the Preparation Thereof. U.S. Patent 3,974,120, 10 August 1976. [Google Scholar]
- Hawkridge, A.M.; Gardella, J.A. Evaluation of Matrix-Assisted Laser Desorption Ionization Mass Spectrometry for Studying the Sec-Butyllithium and n-Butyllithium Initiated Ring-Opening Polymerization of Hexamethylcyclotrisiloxane (D3). Am. Soc. Mass Spectrom. 2003, 14, 95–101. [Google Scholar] [CrossRef] [Green Version]
- Bellas, V.; Iatrou, H.; Hadjichristidis, N. Controlled Anionic Polymerization of Hexamethylcyclotrisiloxane. Model Linear and Miktoarm Star Co- and Terpolymers of Dimethylsiloxane with Styrene and Isoprene. Macromolecules 2000, 33, 6993–6997. [Google Scholar] [CrossRef]
- Ninago, M.D.; Satti, A.J.; Ressia, J.A.; Ciolino, A.E.; Villar, M.A.; Valles, E.M. Controlled Synthesis of Poly(Dimethylsiloxane) Homopolymers Using High-Vacuum Anionic Polymerization Techniques. J. Polym. Sci. Part A Polym. Chem. 2009, 47, 4774–4783. [Google Scholar] [CrossRef]
- Molenberg, A.; Siffrin, S.; Möller, M.; Boileau, S.; Teyssié, D. Well Defined Columnar Liquid Crystalline Polydiethylsiloxane. Macromol. Symp. 1996, 102, 199–207. [Google Scholar] [CrossRef]
- Molenberg, A.; Möller, M. Polymerization of Cyclotrisiloxanes by Organolithium Compounds and P2-Et Base. Macromol. Chem. Phys. 1997, 198, 717–726. [Google Scholar] [CrossRef]
- Molenberg, A.; Klok, H.A.; Möller, M.; Boileau, S.; Teyssié, D. Controlled Polymerization of Hexa-n-Alkylcyclotrisiloxanes with Long Alkyl Groups. Macromolecules 1997, 30, 792–794. [Google Scholar] [CrossRef]
- Hempenius, M.A.; Lammertink, R.G.H.; Vancso, G.J. Well-Defined Side-Chain Liquid-Crystalline Polysiloxanes. Macromol. Rapid Commun. 1996, 17, 299–303. [Google Scholar] [CrossRef] [Green Version]
- Sieburth, S.M.N.; Mu, W. Silanol Synthesis: Reaction of Hexaphenylcyclotrisiloxane with Organometallic Reagents. J. Org. Chem. 1993, 58, 7584–7586. [Google Scholar] [CrossRef]
- Arkles, B. Silicone Molecular Weight Standards. In Silicon Compounds: Register and Review, 3rd ed.; Arkles, B., Peterson, W., Jr., Anderson, R., Eds.; Petrarch Systems: Bristol, TN, USA, 1984; p. 234. [Google Scholar]
- Boileau, S. Ring Opening Polymerization of Cyclic Organosilicon Compounds: Recent Progress. Makromol. Chem. Macromol. Symp. 1993, 73, 177–181. [Google Scholar] [CrossRef]
- Bauer, J.; Hsing, N.; Kickelbick, G. Preparation of Functionalized Block Copolymers Based on a Polysiloxane Backbone by Anionic Ring-Opening Polymerization. J. Polym. Sci. Part A Polym. Chem. 2002, 40, 1539–1551. [Google Scholar] [CrossRef]
- Saam, J.C.; Gordon Fearon, F.W. Properties of Polystyrene-Polydimethylsiloxane Block Copolymers. Ind. Eng. Chem. Prod. Res. Dev. 1971, 10, 10–14. [Google Scholar]
- Georgopanos, P.; Lo, T.Y.; Ho, R.M.; Avgeropoulos, A. Synthesis, Molecular Characterization and Self-Assembly of (PS-b-PDMS)n Type Linear (n = 1, 2) and Star (n = 3, 4) Block Copolymers. Polym. Chem. 2017, 8, 843–850. [Google Scholar] [CrossRef] [Green Version]
- Noshay, A.; Matzner, M.; Karoly, G.; Stampa, G.B. Poly(α-Methylstyrene)-Poly (Dimethylsiloxane) Block Copolymers. J. Appl. Polym. Sci. 1973, 17, 619–628. [Google Scholar] [CrossRef]
- Clemens, L.; Kantner, S.; Mazurek, M. Polysiloxane-Grafted Copolymer Release Coating Sheets and Adhesive Tapes. U.S. Patent 4,728,571, 1 March 1988. [Google Scholar]
- Awasthi, A.K.; Kunzler, J.F. Ethylenically Unsaturated Polymerizable Group-Containing Polycarbosiloxane Monomers. U.S. Patent 9,039,174, 26 May 2015. [Google Scholar]
- Cypryk, M.; Kaźmierski, K.; Fortuniak, W.; Chojnowski, J. Microstructure of the Copolymer Chain Generated by Anionic Ring-Opening Polymerization of a Model Cyclotrisiloxane with Mixed Siloxane Units. Macromolecules 2000, 33, 1536–1545. [Google Scholar] [CrossRef]
- Leir, C. Radation-Curable Silicone Elastomers and Prssure Sensitive Adhesives. U.S. Patent 5,237,082, 17 August 1993. [Google Scholar]
- Saho, T. Organosiloxane and Process for Preparing the Same. U.S. Patent 4,992,521, 12 February 1991. [Google Scholar]
- Arkles, B.; Kimble, E. Asymmetric Low Molecular Wright Siloxanes with One Functional Group. U.S. Patent 8,455,599, 4 June 2013. [Google Scholar]
- Goff, J.; Kimble, E.; Arkles, B. Living Polymerization Routes to Siloxane Macromers and Higher Order Silicone Structures. In Progress in Silicones and Silicone-Modified Materials: ACS Symposium Series; American Chemical Society: Washington, DC, USA, 2013; Volume 1154, pp. 59–78. [Google Scholar]
- Goff, J.; Arkles, B.; Sulaiman, S. Modification of Silicone Elastomers Using Silicone Comonomers Containing Hydrophilic Surface Active Endgroups. MRS Proc. 2014, 1626, 518055. [Google Scholar] [CrossRef]
- Arkles, B.; Goff, J. Dual Functional Linear Siloxanes, Step-Growth Polymers Derived Therefrom, and Methods of Preparation Thereof. U.S. Patent 8,952,118, 10 February 2015. [Google Scholar]
- Gädda, T.M.; Nelson, A.K.; Weber, W.P. Selectivity in Anionic and Cationic Ring-Opening Polymerizations of Tetramethyl-1-(3′trifluoromethylphenyl)-1-Phenylcyclotrisiloxane and Tetramethyl-1-[3′,5′-Bis(Trifluoromethyl)Phenyl]-1-Phenylcyclotrisiloxane. J. Polym. Sci. Part A Polym. Chem. 2004, 42, 5235–5243. [Google Scholar] [CrossRef]
- Kazama, H.; Tezuka, Y.; Imai, K. A New Bifunctional Initiator for the Living Polymerization of Hexamethylcyclotrisiloxane. Polym. Bull. 1989, 21, 31–37. [Google Scholar] [CrossRef]
- Ogawa, T.; Suzuki, T. Branched Organopolysiloxanes. U.S. Patent 5,272,225, 21 December 1993. [Google Scholar]
- Novozhilov, O.V.; Pavlichenko, I.V.; Demchenko, N.V.; Buzin, A.I.; Vasilenko, N.G.; Muzafarov, A.M. Multiarm Star-like Polydimethylsiloxanes Based on Dendrimers of the Sixth Generation. Russ. Chem. Bull. 2010, 59, 1909–1917. [Google Scholar] [CrossRef]
- Vasilenko, N.G.; Rebrov, E.A.; Muzafarov, A.M.; Eßwein, B.; Striegel, B.; Möller, M. Preparation of Multi-Arm Star Polymers with Polylithiated Carbosilane Dendrimers. Macromol. Chem. Phys. 1998, 199, 889–895. [Google Scholar] [CrossRef]
- Juliano, P.C. Substantially Transparent Polydimethylsiloxane-Polyalkylmethacrylate Compositions and Methods for Making Same. U.S. Patent 3,663,650, 16 May 1972. [Google Scholar]
- Out, G.J.J.; Turetskii, A.A.; Snijder, M.; Möller, M.; Papkov, V.S. Model Polydiethylsiloxane Networks: 1. Synthesis and Phase Behaviour. Polymer 1995, 36, 3213–3221. [Google Scholar] [CrossRef] [Green Version]
- Kimble, E.; Arkles, B. Low Molecular Weight Siloxanes with One Functional Group. U.S. Patent 7,799,888 B2, 21 September 2010. [Google Scholar]
- Elkins, C.L.; Long, T.E. Living Anionic Polymerization of Hexamethylcyclotrisiloxane (D 3) Using Functionalized Initiation. Macromolecules 2004, 37, 6657–6659. [Google Scholar] [CrossRef]
- Babu, J.; Sinai-Zingde, G.; Riffle, J.S. Synthesis of α, ω-Arylamine Functionalized Poly(Dimethylsiloxanes). Polym. Prepr. 1991, 32, 152–153. [Google Scholar] [CrossRef]
- Goff, J.; Sulaiman, S.; Arkles, B.; Lewicki, J.P. Soft Materials with Recoverable Shape Factors from Extreme Distortion States. Adv. Mater. 2016, 28, 2393–2398. [Google Scholar] [CrossRef]
- Okawa, T.; Suzuki, T. Method of Manufacturing Organopolysiloxane Having Terminal Alkenyl Groups. U.S. Patent 4,876,373, 24 October 1989. [Google Scholar]
- Kumar, A.; Eichinger, B.E. Anionic Polymerization of Hexamethylcyclotrisiloxane with Acetylacetone Initiator to Form Telechelic Polymer. Macromolecules 1990, 23, 5358. [Google Scholar] [CrossRef]
- Fuchise, K.; Igarashi, M.; Sato, K.; Shimada, S. Organocatalytic Controlled/Living Ring-Opening Polymerization of Cyclotrisiloxanes Initiated by Water with Strong Organic Base Catalysts. Chem. Sci. 2018, 9, 2879–2891. [Google Scholar] [CrossRef] [Green Version]
- Peters, M.A.; Belu, A.M.; Linton, R.W.; Dupray, L.; Meyer, T.J.; DeSimone, J.M. Termination of Living Anionic Polymerizations Using Chlorosilane Derivatives: A General Synthetic Methodology for the Synthesis of End-Functionalized Polymers. J. Am. Chem. Soc. 1995, 117, 3380–3388. [Google Scholar] [CrossRef]
- Belu, A.M.; DeSimone, J.M.; Linton, R.W.; Lange, G.W.; Friedman, R.M. Evaluation of Matrix-Assisted Laser Desorption Ionization Mass Spectrometry for Polymer Characterization. J. Am. Soc. Mass Spectrom. 1996, 7, 11–24. [Google Scholar] [CrossRef] [Green Version]
- Arkles, B.C.; Kimble, E.; Goff, J. Reactive Silicones: Forging New Polymer Links; Gelest Inc.: Morrisville, PA, USA, 2016. [Google Scholar]
- Vilner, S.; Clapp, L.; Schwartz, R. Colorants for Metalorganic Compounds and Their Application in Electronic Display Technology. U.S. Patent 9,234,101, 12 January 2016. [Google Scholar]
- Sommer, S.; Ekin, A.; Webster, D.C.; Stafslien, S.J.; Daniels, J.; VanderWal, L.J.; Thompson, S.E.M.; Callow, M.E.; Callow, J.A. A Preliminary Study on the Properties and Fouling-Release Performance of Siloxane-Polyurethane Coatings Prepared from Poly(Dimethylsiloxane) (PDMS) Macromers. Biofouling 2010, 26, 961–972. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Spulber, M.; Itel, F.; Chami, M.; Pfohl, T.; Palivan, C.G.; Meier, W. Effect of Molecular Parameters on the Architecture and Membrane Properties of 3D Assemblies of Amphiphilic Copolymers. Macromolecules 2014, 47, 5060–5069. [Google Scholar] [CrossRef]
- Viville, P.; Lazzaroni, R.; Dubois, P.; Kotzev, A.; Geerts, Y.; Borcia, G.; Pireaux, J.J. Impact of Silicone-Based Block Copolymer Surfactants on the Surface and Bulk Microscopic Organization of a Biodegradable Polymer, Poly(ε-Caprolactone). Biomacromolecules 2003, 4, 696–703. [Google Scholar] [CrossRef]
- Reynolds, V.G.; Mukherjee, S.; Xie, R.; Levi, A.E.; Atassi, A.; Uchiyama, T.; Wang, H.; Chabinyc, M.L.; Bates, C.M. Super-Soft Solvent-Free Bottlebrush Elastomers for Touch Sensing. Mater. Horizons 2020, 7, 181–187. [Google Scholar] [CrossRef]
- Tao, P.; Li, Y.; Siegel, R.W.; Schadler, L.S. Transparent Luminescent Silicone Nanocomposites Filled with Bimodal PDMS-Brush-Grafted CdSe Quantum Dots. J. Mater. Chem. C 2013, 1, 86–94. [Google Scholar] [CrossRef]
- Bai, H.; Weimer, M. Self-Lubricating Medical Articles. U.S. Patent Appl. 2020/0093969, 26 March 2020. [Google Scholar]
- Arkles, B.; Goff, J. Positive Tactile Interaction Coatings. Paint Coat. Ind. 2017, 33, 30–36. [Google Scholar]
- Zhou, Q.; Chen, F.; Liu, Y.; Li, J. Preparation and Stability Study of Carboxyl Terminated Silicone Oil Modified Silicone-Oil-Based Ferrofluid. J. Magn. 2019, 24, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Hayama, K.; Narazaki, K.; Kawaguchi, S. Polymer for Hair-Care Products. U.S. Patent 5,166,276, 24 November 1992. [Google Scholar]
- Hayama, K.; Narazaki, K.; Kawaguchi, S. Hair-Care Products Containing Copolymers Formed from Unsaturated Hydrophilic Monomers and Unsaturated Monomers and Unsaturated Monomers Having a Polysiloxane Group. U.S. Patent 5,480,634, 2 January 1996. [Google Scholar]
- Gao, S.; Liu, B.; Peng, J.; Zhu, K.; Zhao, Y.; Li, X.; Yuan, X. Icephobic Durability of Branched PDMS Slippage Coatings Co-Cross-Linked by Functionalized POSS. ACS Appl. Mater. Interfaces 2019, 11, 4654–4666. [Google Scholar] [CrossRef]
- Alli, A.; Zanini, D.; Ford, J.; Mahadevan, S.; Canavan, K.; Turner, D. Process for Making Ophthalmic Lenses. U.S. Patent 8,414,804, 9 April 2013. [Google Scholar]
- Swarup, S.; Xu, S.; Vanier, R.; Endlish, E.; Simpson, D. Acrylic Polymers, Curable Film Forming Compositions Prepared Therefrom, and Method of Mitigating Dirt Build-Up on a Substrate. U.S. Patent 9,120,916, 1 September 2015. [Google Scholar]
- Soane, D.S.; Berg, M.C.; Suddaby, N.A.; Lim, K.J.; Mowers, W.A. Ultraphobic Coating Compositions and Methods of Use. U.S. Patent Appl. 2011/0084421, 14 April 2011. [Google Scholar]
- Daniel, W.F.M.; Burdyńska, J.; Vatankhah-Varnoosfaderani, M.; Matyjaszewski, K.; Paturej, J.; Rubinstein, M.; Dobrynin, A.V.; Sheiko, S.S. Solvent-Free, Supersoft and Superelastic Bottlebrush Melts and Networks. Nat. Mater. 2016, 15, 183–189. [Google Scholar] [CrossRef]
- Nian, S.; Lian, H.; Gong, Z.; Zhernenkov, M.; Qin, J.; Cai, L.-H. Molecular Architecture Directs Linear-Bottlebrush-Linear Triblock Copolymers to Self-Assemble to Soft Reprocessable Elastomers. ACS Macro Lett. 2019, 8, 1528–1534. [Google Scholar] [CrossRef]
- Portal, J.; Schultze, X.; Taupin, S. Dispersion of Polymer Particles in a Non-Aqueous Medium and Cosmetic Use Thereof. U.S. Patent 10,973,753, 13 April 2017. [Google Scholar]
- Akers, C.E., Jr.; Franey, T.E.; Sun, J.; Butler, C. Polymeric Dispersants Used for Aqueous Pigmented Inks for Ink-Jet Printing. U.S. Patent 6,652,634 B1, 25 November 2003. [Google Scholar]
- Tsutsumi, T.; Sawada, M. Aqueous Ink for Inkjet Printing. U.S. Patent 5,852,074, 22 December 1998. [Google Scholar]
- Broad, R.; Gibson, I. Method of Making a Contact Lens. U.S. Patent 9,057,821 B2, 16 June 2006. [Google Scholar]
- Vanderlaan, D.; Turner, D.; Hargiss, M.; Maiden, A.; Love, R.; Ford, J.; Molock, F.; Steffen, R.; Hill, G.; Alli, A.; et al. Soft Contact Lenses. U.S. Patent 6,943,203, 13 September 2005. [Google Scholar]
- Azaam, A.; Vanderlaan, D.; Ford, J.; Joslin, S. Silicone Hydrogels Formed from Zero Diluent Reactive Mixtures. U.S. Patent 10,017,596 B2, 10 July 2018. [Google Scholar]
- Martinez, M.R.; Cong, Y.; Sheiko, S.S.; Matyjaszewski, K. A Thermodynamic Roadmap for the Grafting-through Polymerization of PDMS11MA. ACS Macro Lett. 2020, 9, 1303–1309. [Google Scholar] [CrossRef]
- Cao, Z.; Daniel, W.F.M.; Vatankhah-Varnosfaderani, M.; Sheiko, S.S.; Dobrynin, A.V. Dynamics of Bottlebrush Networks. Macromolecules 2016, 49, 8009–8017. [Google Scholar] [CrossRef]
- Pan, Y.; Maddox, A.; Min, T.; Gonzaga, F.; Goff, J.; Arkles, B. Surface-Triggered Tandem Coupling Reactions of Cyclic Azasilanes. Chem. Asian J. 2017, 12, 1198–1203. [Google Scholar] [CrossRef] [PubMed]
- Vatankhah-Varnosfaderani, M.; Keith, A.N.; Cong, Y.; Liang, H.; Rosenthal, M.; Sztucki, M.; Clair, C.; Magonov, S.; Ivanov, D.A.; Dobrynin, A.V.; et al. Chameleon-like Elastomers with Molecularly Encoded Strain-Adaptive Stiffening and Coloration. Science 2018, 359, 1509–1513. [Google Scholar] [CrossRef] [Green Version]
- Ready, T.E.; Thomas, J.; Choi, S.; Boudjouk, P. New Technologies for the Analysis of Marine Coatings. In New Developments in Coatings Technology ACS Symposium Series; American Chemical Society: Washington, DC, USA, 2007; pp. 69–90. [Google Scholar]
- Rosenberg Read, M.; Dahling, M.; Seim, M.; Oeberg Hed, K. Antifouling Composition. Eur. Patent Appl. 3,489,310 A1, 29 May 2019. [Google Scholar]
- Keith, A.N.; Vatankhah-Varnosfaderani, M.; Clair, C.; Fahimipour, F.; Dashtimoghadam, E.; Lallam, A.; Sztucki, M.; Ivanov, D.A.; Liang, H.; Dobrynin, A.V.; et al. Bottlebrush Bridge between Soft Gels and Firm Tissues. ACS Cent. Sci. 2020, 6, 413–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs, M.; Liang, H.; Dashtimoghadam, E.; Morgan, B.J.; Sheiko, S.S.; Dobrynin, A.V. Nonlinear Elasticity and Swelling of Comb and Bottlebrush Networks. Macromolecules 2019, 52, 5095–5101. [Google Scholar] [CrossRef]
- Biedermann, M.; Blümke, M.; Wegener, M.; Krüger, H. Improved Actuation Strain of PDMS-Based DEA Materials Chemically Modified with Softening Agents. Electroact. Polym. Actuators Devices 2015, 9430, 94301G. [Google Scholar]
- Goff, J.; Arkles, B. Silicone Elastomers with Intrinsic Control of Surface Hydrophilicity. Soc. Biomater. 2013, 24. Available online: http://biomaterials-org.securec7.ezhostingserver.com/abstracts/data/papers/2013/0243-000796.pdf (accessed on 2 April 2021).
- Arkles, B.; Goff, J.D. Method for Producing Silicone Step-Growth Elastomers from Dual Functional Siloxanes. U.S. Patent 9,145,474, 29 September 2015. [Google Scholar]
- Arkles, B.C.; Goff, J. Elastomers with Exceptional Elongation. U.S. Patent Appl. 2016/0319080, 3 November 2016. [Google Scholar]
- Arkles, B.; Goff, J.; Sulaiman, S.; Sikorsky, A. Ultra-High Elongation Silicone Elastomers. Rubber World 2016, 254, 29. [Google Scholar]
- Goff, J.; Sulaiman, S.; Philips, A.; Detwiler, M.; Arkles, B. Soft Tissue Compliant Silicones for Medical Devices. Rubber World 2018, 258, 22–26. [Google Scholar]
- Gaylord, N. Method for Correcting Visual Defects, Compositions and Articles of Manufacture Useful Therein. U.S. Patent 4,120,570, 17 October 1978. [Google Scholar]
- Gaylord, N. Oxygen-Permeable Contact Lens Composition, Methods and Article of Manufacture. U.S. Patent 3,808,178, 30 April 1974. [Google Scholar]
- Schaeffer, J.; Beiting, J. Contact Lens Pioneers. Rev. Optom. 2007, 144, 76. [Google Scholar]
- Spinelli, H.; Anton, W.; Seidner, L.; Coleman, H.; Ali, M.; Weintraub, L.; White, P. Silicone-Containing Polymers, Oxygen Permeable Hydrophilic Contact Lenses and Methods for Making These Lenses and Treating Patients with Visual Impairment. U.S. Patent 5,314,960, 24 May 1994. [Google Scholar]
- Nicolson, P.C.; Vogt, J. Soft Contact Lens Polymers: An Evolution. Biomaterials 2001, 22, 3273–3283. [Google Scholar] [CrossRef]
- Arkles, B.; Redinger, P. Silicones in Biomedical Applications. In Biocompatible Polymers, Metals, and Composites; Technomic: Lancaster, PA, USA, 1983; pp. 749–768. [Google Scholar]
- Vanderlaan, D.; Nunez, I.; Hargiss, M.; Alton, M.; Williams, S. Soft Contact Lenses. U.S. Patent 5,998,498, 7 December 1999. [Google Scholar]
- Nicolson, P.; Baron, R.; Chabreckek, P.; Court, J.; Domschke, A.; Griesser, H.; Ho, A.; Hopken, J.; Laycock, B.; Liu, Q.; et al. Extended Wear Opthalmic Lens. U.S. Patent 5,965,631, 12 October 1999. [Google Scholar]
- Ueyama, H.; Ikawa, S.; Iwata, J. Hydrophilic Polysiloxane Macromonomer, and Product and Use of the Same. U.S. Patent 8,129,442, 6 March 2012. [Google Scholar]
- Iwata, J.; Hoki, T.; Ikawa, S. Long Wearable Soft Contact Lens. U.S. Patent 6,867,245, 15 March 2005. [Google Scholar]
- Awasthi, A.K.; Stanbro, K.; Kunzler, J.F. Linhardt, J. Ethylenically Unsaturated Polymerizable Group-Containing Polycarbosiloxane Monomers. U.S. Patent 8,420,711, 16 April 2013. [Google Scholar]
- Awasthi, A.K.; Meng, F.R.; Künzler, J.F.; Linhardt, J.G.; Papagelis, P.; Oltean, G.; Myers, S.A. Ethylenically Unsaturated Polycarbosiloxanes for Novel Silicone Hydrogels: Synthesis, End-Group Analysis, Contact Lens Formulations, and Structure-Property Correlations. Polym. Adv. Technol. 2013, 24, 557–567. [Google Scholar] [CrossRef]
- Hagiwara, M. One-Terminal Reactive Organopolysiloxane Having a Polyalkyleneoxide Group at the Omega-Terminal and a Method for the Preparation Thereof. U.S. Patent 8,404,882, 26 March 2013. [Google Scholar]
- Qian, X.; Chang, F.; Matsuzawa, Y.; Shankar, V. Soft SIlicone Medical Devices with Durable Lubricious Coatings Thereon. U.S. Patent 10,324,233, 18 June 2019. [Google Scholar]
- Hu, C.; Pham, D.; Lowery, M. Soft Silicone Materials for Ophthalmic Applications. U.S. Patent 8,8530,590, 10 September 2013. [Google Scholar]
- Grubbs, R.; Sandstedt, C. Method for Modifying Power of Light Adjustable Lens. U.S. Patent 9,950,482, 24 April 2018. [Google Scholar]
- Daimatsu, K.; Anno, Y.; Sugimoto, H.; Nakaishi, E.; Inomata, K.; Ikeda, T.; Yokoi, K. Preparation, Morphology, and Physical Properties of Transparent PSt Hybrid Materials Containing Silicone Macromonomer Kazuki. J. Appl. Polym. Sci. 2008, 108, 362–369. [Google Scholar] [CrossRef]
- Kerry, J.P.; O’Grady, M.N.; Hogan, S.A. Past, Current and Potential Utilisation of Active and Intelligent Packaging Systems for Meat and Muscle-Based Products: A Review. Meat Sci. 2006, 74, 113–130. [Google Scholar] [CrossRef]
- Clarke, R. Packaging of Respiring Biological Materials. U.S. Patent 8,092,848 B2, 10 January 2012. [Google Scholar]
- Masuda, T.; Isobe, E.; Higashimura, T.; Takada, K. Poly[1-(Trimethylsilyl)-1-Propyne]: A New High Polymer Synthesized with Transition-Metal Catalysts and Characterized by Extremely High Gas Permeability. J. Am. Chem. Soc. 1983, 105, 7473–7474. [Google Scholar] [CrossRef]
- Volkov, V.V. Free Volume Structure and Transport Properties of Glassy Polymers—Materials for Separating Membranes. Polym. J. 1991, 23, 457–466. [Google Scholar] [CrossRef] [Green Version]
- Liang, L.; Dickson, J.M.; Zhu, Z.; Jiang, J.; Brook, M.A. Removal of 1,2-Dichloroethane from Aqueous Solutions with Novel Composite Polydimethylsiloxane Pervaporation Membranes. J. Appl. Polym. Sci. 2005, 98, 1477–1491. [Google Scholar] [CrossRef]
- Uragami, T.; Sumida, I.; Miyata, T.; Shiraiwa, T.; Tamura, H.; Yajima, T. Pervaporation Characteristics in Removal of Benzene from Water through Polystyrene-Poly (Dimethylsiloxane) IPN Membranes. Mater. Sci. Appl. 2011, 2, 169–179. [Google Scholar] [CrossRef] [Green Version]
- Miyata, T.; Takagi, T.; Kadota, T.; Uragami, T. Characteristics of Permeation and Separation for Aqueous Ethanol Solutions through Methyl Methacrylate-dimethylsiloxane Graft Copolymer Membranes. Macromol. Chem. Phys. 1995, 196, 1211–1220. [Google Scholar] [CrossRef]
- Uragami, T.; Yamada, H.; Miyata, T. Removal of Dilute Volatile Organic Compounds in Water through Graft Copolymer Membranes Consisting of Poly(Alkylmethacrylate) and Poly(Dimethylsiloxane) by Pervaporation and Their Membrane Morphology. J. Memb. Sci. 2001, 187, 255–269. [Google Scholar] [CrossRef]
- Uragami, T.; Matsuoka, Y.; Miyata, T. Permeation and Separation Characteristics in Removal of Dilute Volatile Organic Compounds from Aqueous Solutions through Copolymer Membranes Consisted of Poly(Styrene) and Poly(Dimethylsiloxane) Containing a Hydrophobic Ionic Liquid by Pervaporation. J. Memb. Sci. 2016, 506, 109–118. [Google Scholar] [CrossRef]
- Uragami, T.; Fukuyama, E.; Miyata, T. Selective Removal of Dilute Benzene from Water by Poly(Methyl Methacrylate)-Graft-Poly(Dimethylsiloxane) Membranes Containing Hydrophobic Ionic Liquid by Pervaporation. J. Membr. Sci. 2016, 510, 131–140. [Google Scholar] [CrossRef]
- Sun, J.; Beach, B. Chemically Prepared Toner and Process Therefor. U.S. Patent 6,991,884 B2, 31 January 2006. [Google Scholar]
- Dawkins, J.; Taylor, G. Micelle Formation by AB Block Copolymers of Polystyrene and Poly(Dimethylsiloxane) in N-alkanes. Chem. Macromol. Chem. Phys. 1979, 180, 1737–1741. [Google Scholar] [CrossRef]
- Gobelt, B.; Nagelsdiek, R.; Omeis, J.; Piestert, F. Method for Producing Dispersant Additives. U.S. Patent 9,573,103, 21 February 2017. [Google Scholar]
- Haubennestel, K.; Bubat, A.; Frank, A. Levelling Agents for Surface Coatings. U.S. Patent 6,710,127, 23 March 2004. [Google Scholar]
- Gobelt, B.; Bubat, A.; Frank, A.; Haubennestel, K. Use of Polyacrylate-Modified Polysiloxanes as Levelling Agents in Coating Compositions. U.S. Patent 7,230,051, 12 June 2007. [Google Scholar]
- Van Der Sluis, M.; Hof, W.; Engelbrecht, L.; Onelin, S. Slip and Leveling Agent. U.S. Patent 7,915,343, 29 March 2011. [Google Scholar]
- Nagel, C.; Heekeren, M.; Frank, A. New Silicone Structures. Eur. Coat. J. 2010, 4, 32–34. [Google Scholar]
- BYK-3550 Technical Data Sheet. September 2015. Available online: http://www.uni-trading.com/sub/support/tds.msds/additive/BYK/BYK_etc/TDS/BYK-3550%20TDS(EN).pdf (accessed on 2 April 2021).
- Rahimi, A.R.; Murphy, M.; Upadhyay, V.; Faiyaz, K.; Battocchi, D.; Webster, D.C. Amphiphilically Modified Self-Stratified Siloxane-Glycidyl Carbamate Coatings for Anti-Icing Applications. J. Coat. Technol. Res. 2020, 18, 83–97. [Google Scholar] [CrossRef]
- Kozakiewicz, J. Polysiloxaneurethanes: New Polymers for Potential Coating Applications. Prog. Org. Coat. 1996, 27, 123–131. [Google Scholar] [CrossRef]
- Bolich, R.; Torgerson, P. Hair Conditioning and Styling Compositions. U.S. Patent 5,618,524, 8 April 1997. [Google Scholar]
- Arkles, B.; Cameron, R.; Larson, G.L. Silicones Derived from 2-Propenyl Functional Cyclic Terpenes and Methods of Preparation. U.S. Patent 8,653,294, 18 February 2014. [Google Scholar]
- Arkles, B.C.; Goff, J.D.; Min, T.; Pan, Y.; Abel-Roberman, T. Silicon-Based Cannabadiol Derivatives and Compositions Thereof. U.S. Patent 10,933, 008 March 2021. [Google Scholar]
- Mefford, O.T.; Vadala, M.L.; Goff, J.D.; Carroll, M.R.J.; Mejia-Ariza, R.; Caba, B.L.; Pierre, T.G.S.; Woodward, R.C.; Davis, R.M.; Riffle, J.S. Stability of Polydimethylsiloxane-Magnetite Nanoparticle Dispersions against Flocculation: Interparticle Interactions of Polydisperse Materials. Langmuir 2008, 24, 5060–5069. [Google Scholar] [CrossRef] [PubMed]
- Miles, W.C.; Goff, J.D.; Huffstetler, P.P.; Mefford, O.T.; Riffle, J.S.; Davis, R.M. The Design of Well-Defined PDMS-Magnetite Complexes. Polymer 2010, 51, 482–491. [Google Scholar] [CrossRef]
- Wilson, K.S.; Goff, J.D.; Riffle, J.S.; Harris, L.A.; St Pierre, T.G. Polydimethylsiloxane-Magnetite Nanoparticle Complexes and Dispersions in Polysiloxane Carrier Fluids. Polym. Adv. Technol. 2005, 16, 200–211. [Google Scholar] [CrossRef]
- Goff, J.D. Synthesis and Characterization of Novel Polyethers and Polydimethylsiloxanes for Use in Biomaterials. Ph.D. Thesis, Virginia Tech, Blacksburg, VA, USA, 2009. [Google Scholar]
- Riffle, J.S.; Thompson Mefford, O.; Carroll, M.R.J.; Vadala, M.L.; Goff, J.D.; Mejia-Ariza, R.; Saunders, M.; Woodward, R.C.; St Pierre, T.G.; Davis, R.M. Size Analysis of PDMS-Magnetite Nanoparticle Complexes: Experiment and Theory. Chem. Mater. 2008, 20, 2185–2191. [Google Scholar]
- Mefford, O.T.; Woodward, R.C.; Goff, J.D.; Vadala, T.P.; St. Pierre, T.G.; Dailey, J.P.; Riffle, J.S. Field-Induced Motion of Ferrofluids through Immiscible Viscous Media: Testbed for Restorative Treatment of Retinal Detachment. J. Magn. Magn. Mater. 2007, 311, 347–353. [Google Scholar] [CrossRef]
- Ganachaud, F.; Institut National des Sciences Appliquees de Lyon, France. Private communication, 2017.
- Hadjichistidis, N.; Iatrou, H.; Pispas, S.; Pitsikalis, M. Anionic Polymerization: High Vacuum Techniques. J. Polym. Sci. Part A Polym. Chem. 2000, 38, 3211–3234. [Google Scholar] [CrossRef]
- Varshney, S.K.; Khanna, D.N. Hexamethyl Cyclotrisiloxane–Styrene Block Copolymers and Their Chemical Composition. J. Appl. Polym. Sci. 1980, 25, 2501–2511. [Google Scholar] [CrossRef]
- Dems, A.; Strobin, G. Synthesis and Properties of Well-Defined Multiblock Copolymers: Polystyrene-block-polydimethylsiloxane. Die Makromol. Chem. Macromol. Chem. Phys. 1991, 2537, 2521–2537. [Google Scholar] [CrossRef]
- Chu, H. Morphologies of Strongly Segregated Diblock Copolymers. Polymer. 1995, 36, 1569–1575. [Google Scholar] [CrossRef]
- Borah, D.; Cummins, C.; Rasappa, S.; Senthamaraikannan, R.; Salaun, M.; Zelsmann, M.; Liontos, G.; Ntetsikas, K.; Avgeropoulos, A.; Morris, M.A. Nanopatterning via Self-Assembly of a Lamellar-Forming Polystyrene-Block-Poly(Dimethylsiloxane) Diblock Copolymer on Topographical Substrates Fabricated by Nanoimprint Lithography. Nanomaterials 2018, 8, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.J.; Kim, H.H.; Kim, K.M.; Baek, S.K.; Lee, S.J.; Kim, J.H.; Choi, J.S. Method of Forming a Fine Pattern by Using Block Copolymers. U.S. Patent 9,437,452, 6 September 2016. [Google Scholar]
- Jung, Y.S.; Ross, C.A. Orientation-Controlled Self-Assembled Nanolithography Using a Polystyrene-Polydimethylsiloxane Block Copolymer. Nano Lett. 2007, 7, 2046–2050. [Google Scholar] [CrossRef]
- Jacoby, M. Block Copolymers for Lithography. Chem. Eng. News 2014, 92, 8–12. [Google Scholar] [CrossRef]
- Andersen, T.H.; Tougaard, S.; Larsen, N.B.; Almdal, K.; Johannsen, I. Surface Morphology of PS-PDMS Diblock Copolymer Films. J. Electron. Spectros. Relat. Phenom. 2001, 121, 93–110. [Google Scholar] [CrossRef]
- Choi, P.; Fu, P.F.; Guo, L.J. Siloxane Copolymers for Nanoimprint Lithography. Adv. Funct. Mater. 2007, 17, 65–70. [Google Scholar] [CrossRef] [Green Version]
- Bates, C.M.; Seshimo, T.; Maher, M.J.; Durand, W.J.; Cushen, J.D.; Dean, L.M.; Blachut, G.; Ellison, C.J.; Willson, C.G. Polarity-Switching Top Coats Enable Copolymer Domains. Science 2012, 338, 775–779. [Google Scholar] [CrossRef]
- Maher, M.J.; Bates, C.M.; Blachut, G.; Sirard, S.; Self, J.L.; Carlson, M.C.; Dean, L.M.; Cushen, J.D.; Durand, W.J.; Hayes, C.O.; et al. Interfacial Design for Block Copolymer Thin Films. Chem. Mater. 2014, 26, 1471–1479. [Google Scholar] [CrossRef]
- Luo, Y.; Montarnal, D.; Kim, S.; Shi, W.; Barteau, K.P.; Pester, C.W.; Hustad, P.D.; Christianson, M.D.; Fredrickson, G.H.; Kramer, E.J.; et al. Poly(Dimethylsiloxane-b-Methyl Methacrylate): A Promising Candidate for Sub-10 Nm Patterning. Macromolecules 2015, 48, 3422–3430. [Google Scholar] [CrossRef]
- Miller, P.J.; Matyjaszewski, K. Atom Transfer Radical Polymerization of (Meth)Acrylates from Poly(Dimethylsiloxane) Macroinitiators. Macromolecules 1999, 32, 8760–8767. [Google Scholar] [CrossRef]
- Keul, H.; Müller, A.J.; Höcker, H. Preparation of Polymers with Polycarbonate Sequences and Their Depolymerization; An Example of Thermodynamic Recycling. In Makromolekulare Chemie. Macromolecular Symposia; Hüthig & Wepf Verlag: Basel, Switzerland, 1993; Volume 67, pp. 289–298. [Google Scholar]
- Shefelbine, T.A.; Vigild, M.E.; Matsen, M.W.; Hajduk, D.A.; Hillmyer, M.A.; Cussler, E.L.; Bates, F.S. Core-Shell Gyroid Morphology in a Poly(Isoprene-Block-Styrene-Block- Dimethylsiloxane) Triblock Copolymer. J. Am. Chem. Soc. 1999, 121, 8457–8465. [Google Scholar] [CrossRef]
- Politakos, N.; Ntoukas, E.; Avgeropoulos, A.; Krikorian, V.; Pate, B.D.; Thomas, E.L.; Hill, R.M. Strongly Segregated Cubic Microdomain Morphology Consistent with the Double Gyroid Phase in High Molecular Weight Diblock Copolymers of Polystyrene and Poly(Dimethylsiloxane). J. Polym. Sci. Part B Polym. Phys. 2009, 47, 2419–2427. [Google Scholar] [CrossRef]
- Lo, T.Y.; Chao, C.C.; Ho, R.M.; Georgopanos, P.; Avgeropoulos, A.; Thomas, E.L. Phase Transitions of Polystyrene-b-Poly(Dimethylsiloxane) in Solvents of Varying Selectivity. Macromolecules 2013, 46, 7513–7524. [Google Scholar] [CrossRef]
- Lu, K.Y.; Lo, T.Y.; Georgopanos, P.; Avgeropoulos, A.; Shi, A.C.; Ho, R.M. Orienting Silicon-Containing Block Copolymer Films with Perpendicular Cylinders via Entropy and Surface Plasma Treatment. Macromolecules 2017, 50, 9403–9410. [Google Scholar] [CrossRef]
- Feng, X.; Zhuo, M.; Guo, H.; Thomas, E.L. Visualizing the Double-Gyroid Twin. Proc. Natl. Acad. Sci. USA 2021, 118, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Beatty, C.L.; Pechan, J.M.; Froix, M.F.; Hinman, D.D. Liquid Crystalline Type Order in Polydiethylsiloxane. Macromolecules 1975, 8, 547–551. [Google Scholar] [CrossRef]
- Zhang, L.; Yao, W.; Gao, Y.; Zhang, C.; Yang, H. Polysiloxane-Based Side Chain Liquid Crystal Polymers: From Synthesis to Structure–Phase Transition Behavior Relationships. Polymers 2018, 10, 794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Synthetic methods for preparation of siloxane and siloxane-hybrid polymers.

Scheme 1.
Kinetically Controlled Polymerization.

Scheme 2.
Thermodynamic Equilibrium Driven Processes (i.e., Reversion, Backbiting, Chain-scrambling).
Scheme 2.
Thermodynamic Equilibrium Driven Processes (i.e., Reversion, Backbiting, Chain-scrambling).

Scheme 3.
Ion-pair Complexation by Dimethoxyethane (DME) Promoter Favors Chain-End Over Internal Scrambling [5].
Scheme 3.
Ion-pair Complexation by Dimethoxyethane (DME) Promoter Favors Chain-End Over Internal Scrambling [5].

Figure 2.
Comparison of silicone macromers demonstrating polymer termini scrambling (GC of methacrylate functional macromer (MCR-M11) with a nominal MW of 1000 showing shift from Triad to Gaussian distribution with a change in promoter. (Author’s work)).
Figure 2.
Comparison of silicone macromers demonstrating polymer termini scrambling (GC of methacrylate functional macromer (MCR-M11) with a nominal MW of 1000 showing shift from Triad to Gaussian distribution with a change in promoter. (Author’s work)).

Figure 3.
Time-dependent ROP polymer growth of strained versus unstrained cyclosiloxanes.

Scheme 4.
Polydimethylsiloxane macromers with symmetric architectures, produced via the anionic ring-opening polymerization of a cyclic trisiloxane and subsequent coupling with a functional dihalogen-substituted silane reagent, have recently been described.
Scheme 4.
Polydimethylsiloxane macromers with symmetric architectures, produced via the anionic ring-opening polymerization of a cyclic trisiloxane and subsequent coupling with a functional dihalogen-substituted silane reagent, have recently been described.

Scheme 5.
Siloxane Macromer Structural Segments.

Scheme 6.
Symmetric macromers can contain a pendant methacrylate group bisecting the polydimethylsiloxane backbone.
Scheme 6.
Symmetric macromers can contain a pendant methacrylate group bisecting the polydimethylsiloxane backbone.

Scheme 7.
Symmetric silicone macromer with methacrylate functionality bisection.

Scheme 8.
Carbinol terminated telelechelic with macromeric methacrylate functionality.

Figure 4.
(a) Silicone hydrogel lenses incorporate low MW macromers into otherwise hydrophilic matrices based on hydroxyethylmethacrylate or dimethylacrylamide; (b) comparison of contact lens materials and oxygen transport to the cornea (redrawn with permission from Ref. [122] Copyright 1983 the Technomic Publication). (c) Hydrogel phase structure as a determinant of lens comfort (redrawn with permission from Ref. [121] Copyright 2001 Elsevier Science).
Figure 4.
(a) Silicone hydrogel lenses incorporate low MW macromers into otherwise hydrophilic matrices based on hydroxyethylmethacrylate or dimethylacrylamide; (b) comparison of contact lens materials and oxygen transport to the cornea (redrawn with permission from Ref. [122] Copyright 1983 the Technomic Publication). (c) Hydrogel phase structure as a determinant of lens comfort (redrawn with permission from Ref. [121] Copyright 2001 Elsevier Science).


Figure 5.
Breathable films and packaging (Reprinted courtesy of Breathway Corp).

Figure 6.
Electroluminescent transparent quantum dot composites (Tao) [85]. Left: depiction of macromers (blue) modifying a CdSe QD (red) in a silicone matrix; Right: specimen of composite. (Reprinted with permission from Ref. [85] Copyright 2013 the Royal Society of Chemistry).

Scheme 9.
Urethane Polymer with Pendant Siloxane.

Scheme 10.
Urethane-Siloxane Soft Segment Block Polymer.

Scheme 11.
Vinyl functional Siloxane with PEG Endgroups-Reactive in Two-component Pt-Cure Silicone RTVs.
Scheme 11.
Vinyl functional Siloxane with PEG Endgroups-Reactive in Two-component Pt-Cure Silicone RTVs.

Scheme 12.
Vinyl functional Siloxane with Tetrahydrofurfuryloxy Endgroups-Reactive in Two-component Pt-Cure Silicone RTVs.
Scheme 12.
Vinyl functional Siloxane with Tetrahydrofurfuryloxy Endgroups-Reactive in Two-component Pt-Cure Silicone RTVs.

Figure 7.
Modification of silicone elastomers with symmetric silicone macromers. (a) Effect of comonomer on contact angle and hydrophobicity of silicone elastomer; (b) coefficient of friction of silicone elastomers modified with symmetric silicone macromers (Determined on AR-G2 Rheometer: aqueous, 37 °C, normal force 1N, velocity 1.0 rad/s (1.5 mm/s) and 1.0 rad/s (15 mm/s).)
Figure 7.
Modification of silicone elastomers with symmetric silicone macromers. (a) Effect of comonomer on contact angle and hydrophobicity of silicone elastomer; (b) coefficient of friction of silicone elastomers modified with symmetric silicone macromers (Determined on AR-G2 Rheometer: aqueous, 37 °C, normal force 1N, velocity 1.0 rad/s (1.5 mm/s) and 1.0 rad/s (15 mm/s).)

Scheme 13.
CBD terminated PDMS macromer.

Figure 8.
PDMS macromer-modified SPIONs (Author’s work).

Figure 9.
Retinal repair utilizing macromer-enabled magnetorheological fluids.

Scheme 14.
Step-Growth Polymerization of Heterobifunctional α-Vinyl-ω-Hydride-terminated Silicone Macromer.
Scheme 14.
Step-Growth Polymerization of Heterobifunctional α-Vinyl-ω-Hydride-terminated Silicone Macromer.

Figure 10.
Atomistic model of a 50,000 Da segment of ultra-high elongation elastomer suggesting 1–2 knots. (Author’s work).
Figure 10.
Atomistic model of a 50,000 Da segment of ultra-high elongation elastomer suggesting 1–2 knots. (Author’s work).

Figure 11.
Mechanical properties of ultra-high elongation elastomer (ExSil 100) compared to conventional silicone elastomer.
Figure 11.
Mechanical properties of ultra-high elongation elastomer (ExSil 100) compared to conventional silicone elastomer.

Figure 12.
Pseudo-self-healing demonstration of bisected specimen of ExSil® (Author’s work).

Figure 13.
PS-b-PDMS after a single imprint cycle and annealing provides 60 nm features (Reprinted with permission from Ref. [173] Copyright 2007 Wiley).
Figure 13.
PS-b-PDMS after a single imprint cycle and annealing provides 60 nm features (Reprinted with permission from Ref. [173] Copyright 2007 Wiley).

Scheme 15.
Polystyrene-Polydimethylsiloxane Block Copolymer.

Figure 14.
Reconstruction of a volume containing a Double Gyroid of PS-b-PDMS (Reprinted with permission from Ref. [183] Copyright 2021 National Academy of Sciences).
Figure 14.
Reconstruction of a volume containing a Double Gyroid of PS-b-PDMS (Reprinted with permission from Ref. [183] Copyright 2021 National Academy of Sciences).

Figure 15.
Color alteration from turquoise to dark blue during uniaxial stretching. (Reprinted with permission from Ref. [106] Copyright 2018 AAAS).
Figure 15.
Color alteration from turquoise to dark blue during uniaxial stretching. (Reprinted with permission from Ref. [106] Copyright 2018 AAAS).

Figure 16.
Liquid Crystal-Phase Behavior of Polydiethylsiloxane.

Figure 17.
Bulk structures of PolyStyrene-Polydiethylsiloxane block copolymers. Bulk structures of (a) PS-b-PDES containing 37 wt% PS and (b) PS-b-PDES containing 43 wt% PS (Reprinted with permission from Ref. [19] Copyright 1998 Wiley).
Figure 17.
Bulk structures of PolyStyrene-Polydiethylsiloxane block copolymers. Bulk structures of (a) PS-b-PDES containing 37 wt% PS and (b) PS-b-PDES containing 43 wt% PS (Reprinted with permission from Ref. [19] Copyright 1998 Wiley).

Scheme 16.
Derivatization of AROP Siloxanes with mesogenic pendant groups.


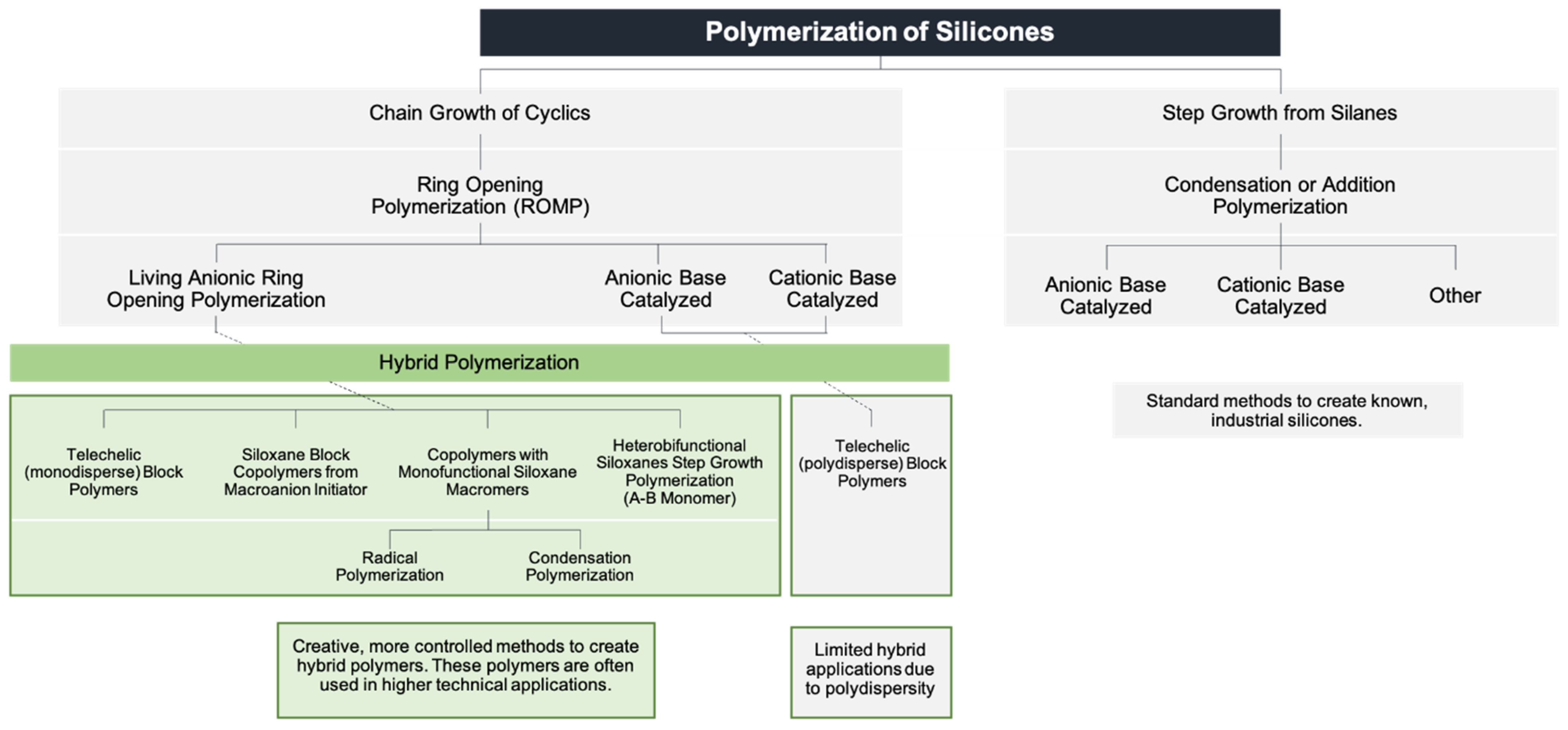{kind=link}
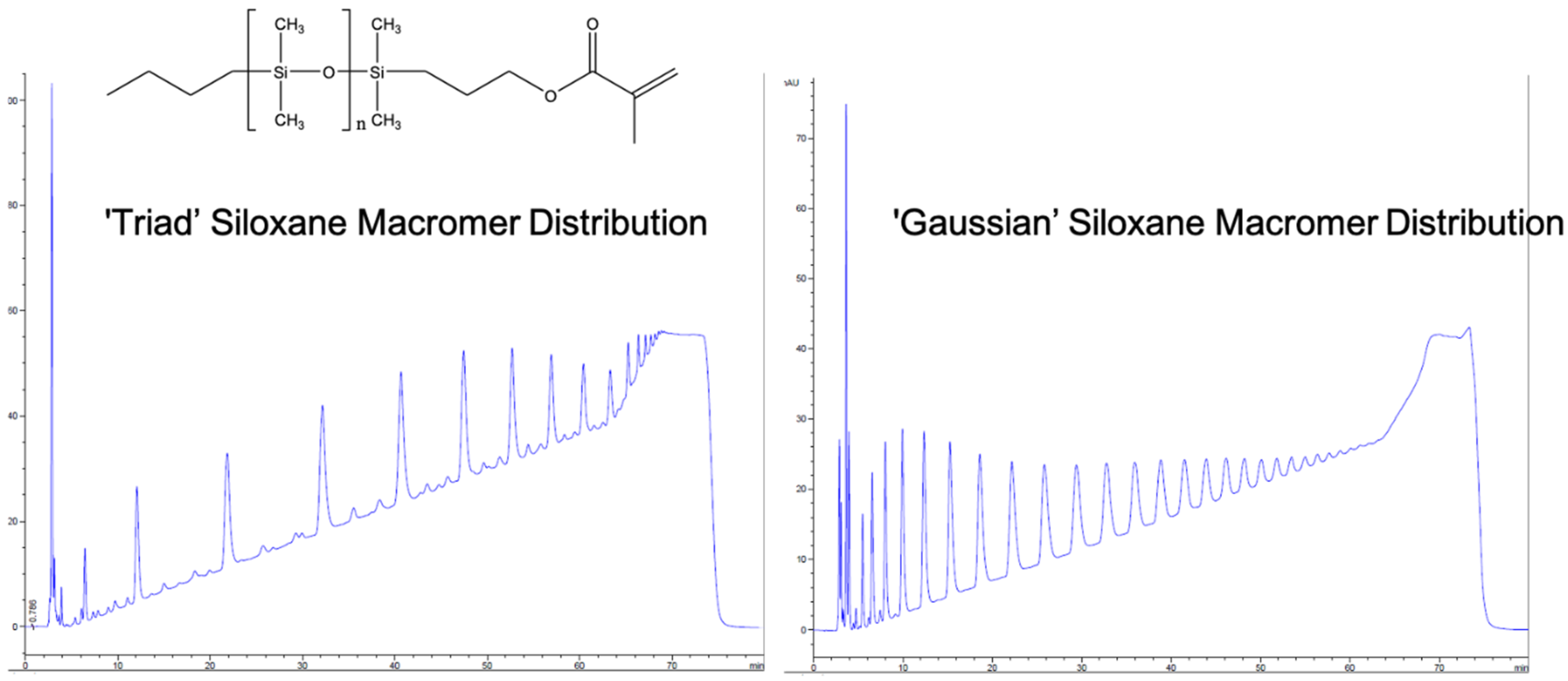{kind=link}
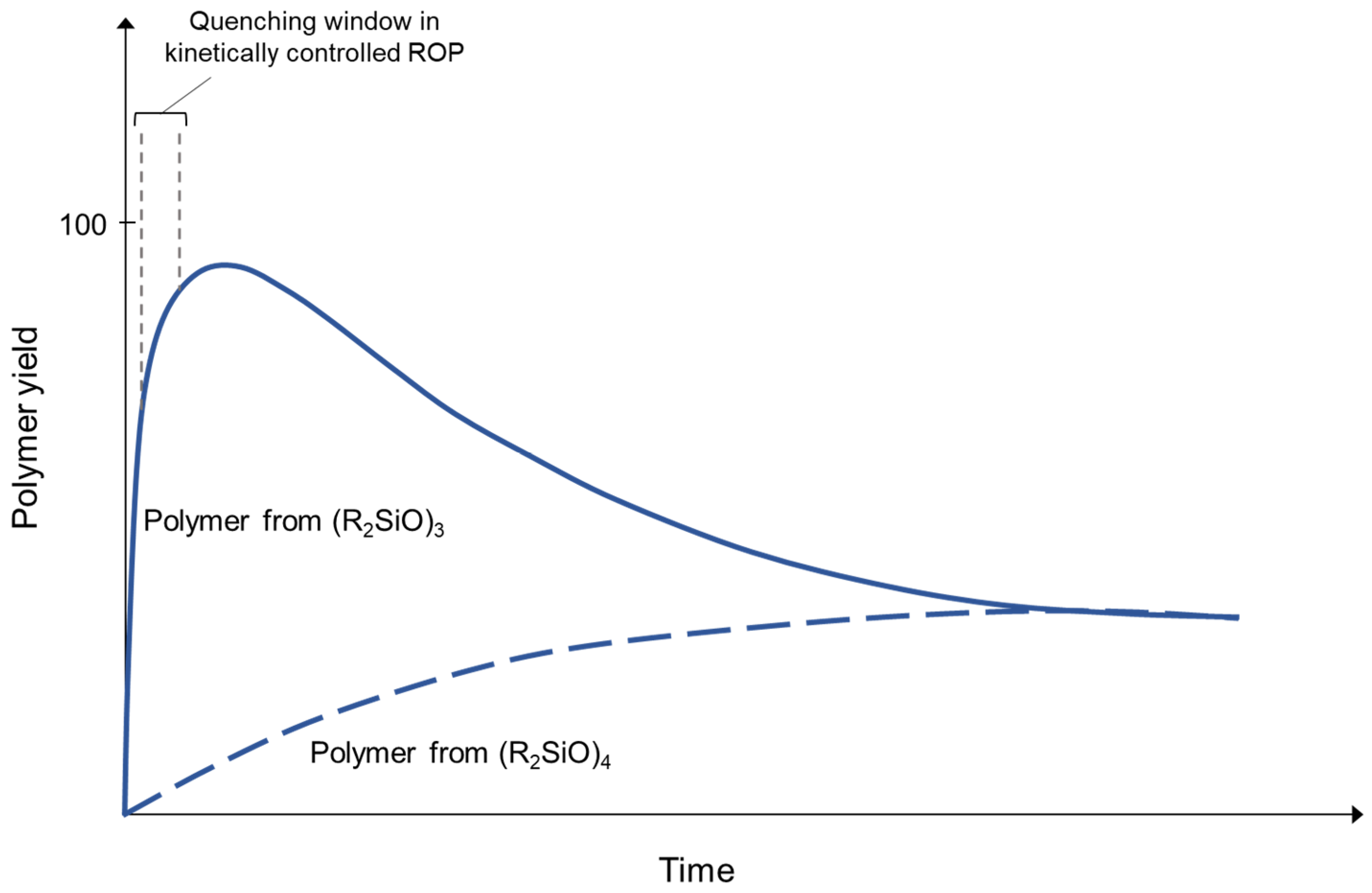{kind=link}
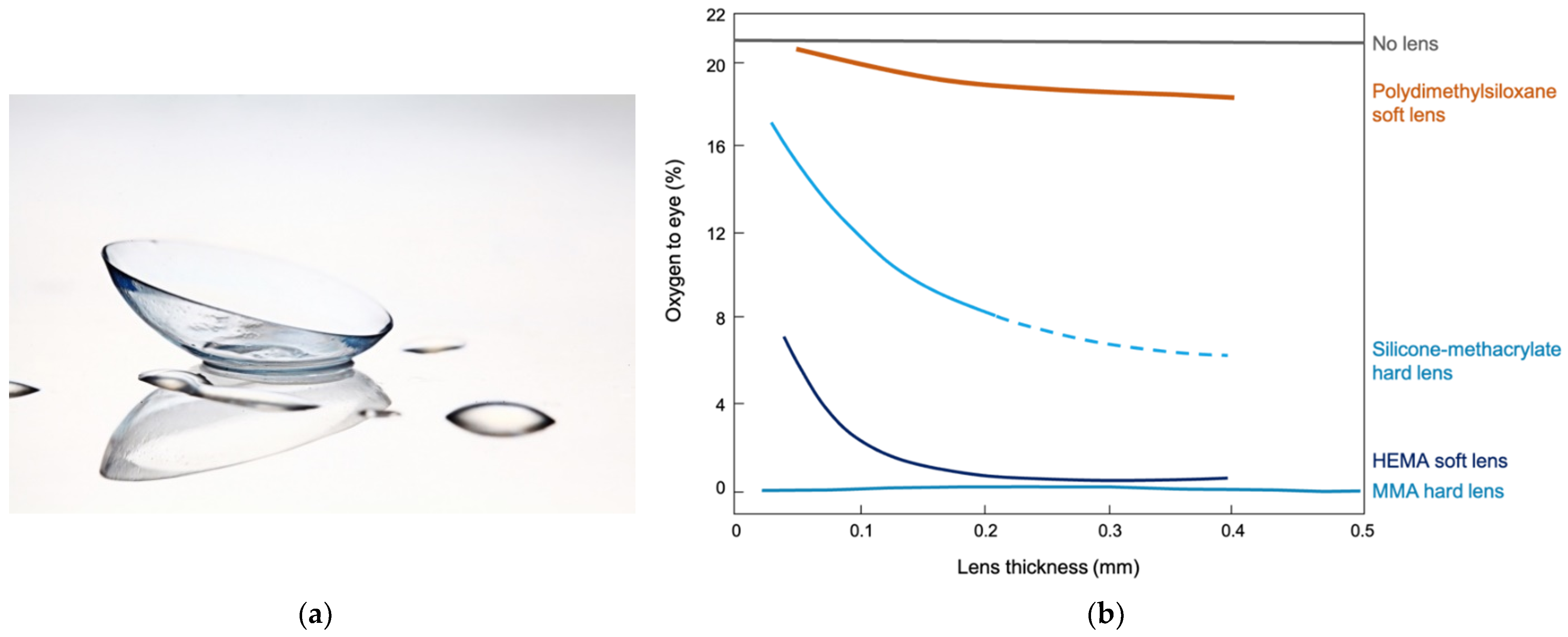{kind=link}
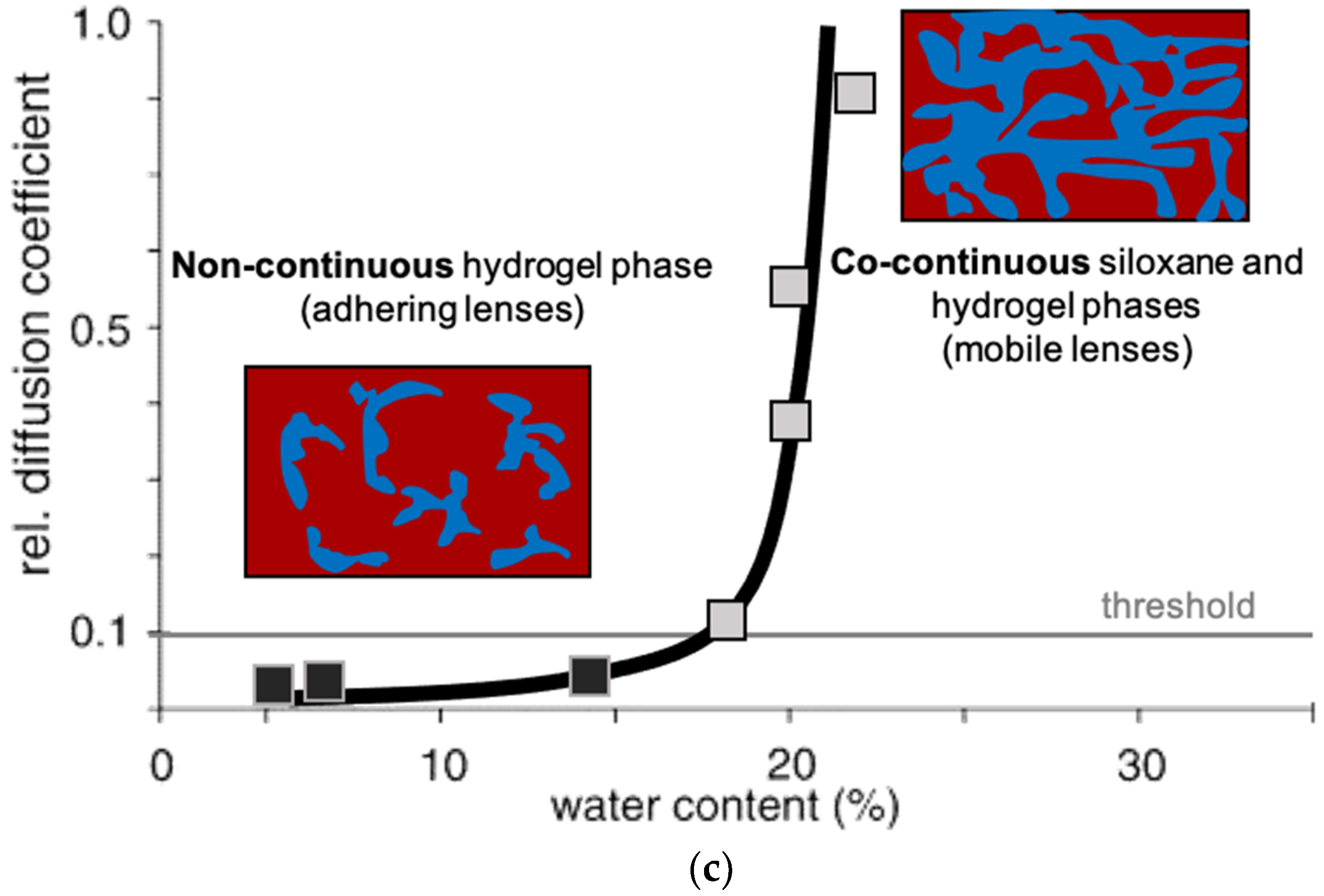{kind=link}
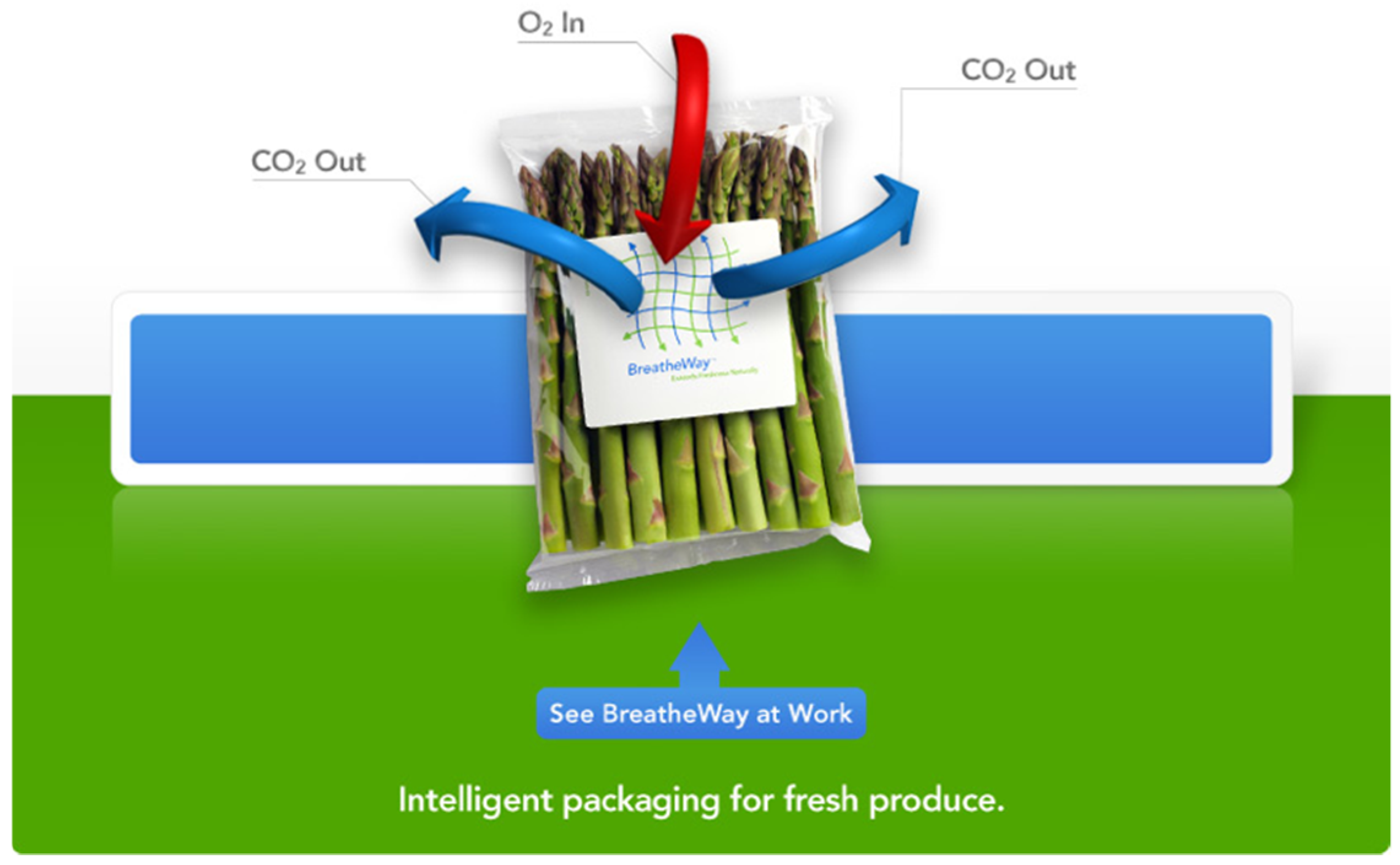{kind=link}
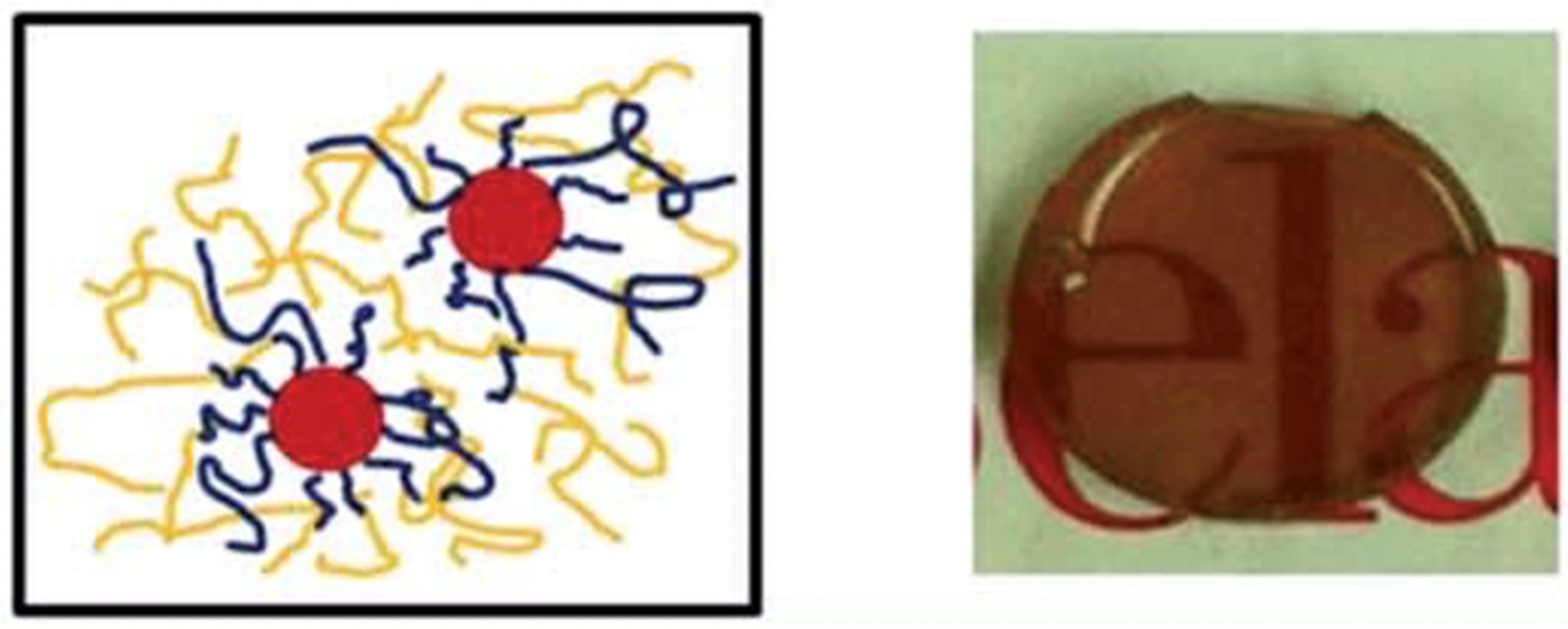{kind=link}
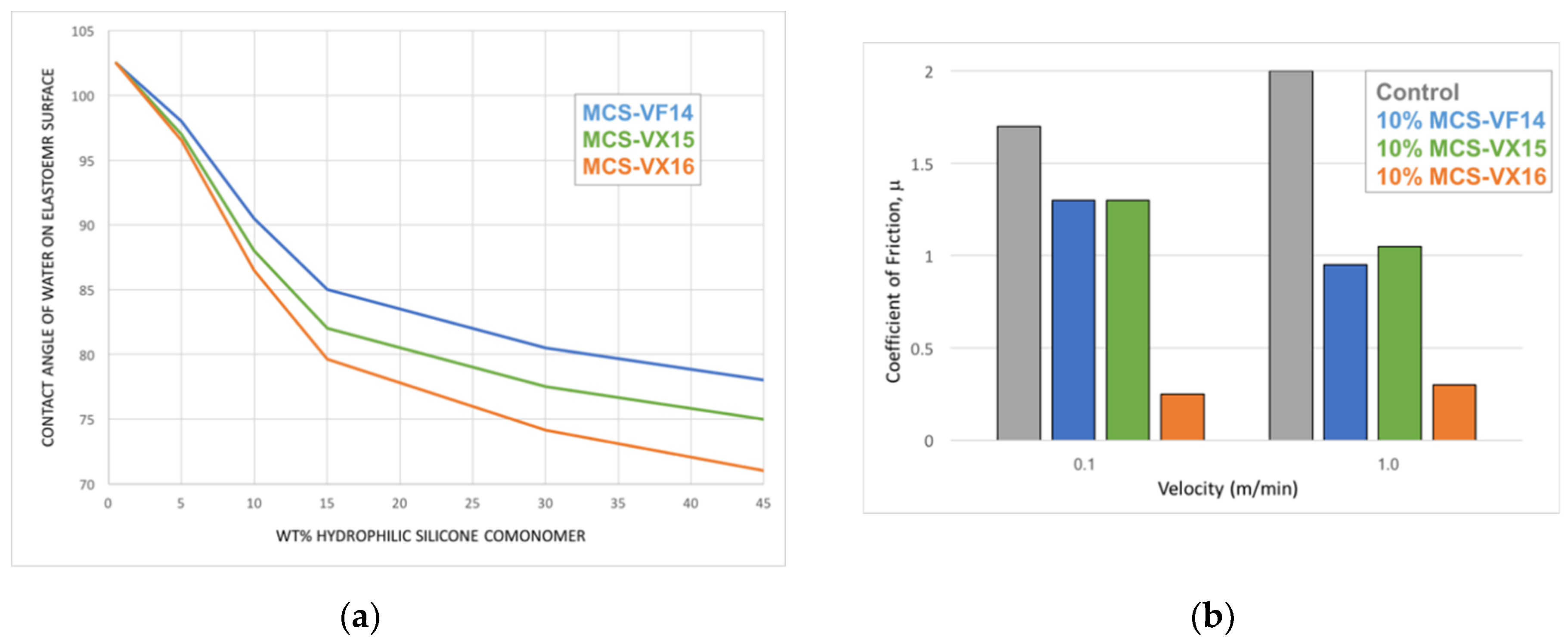{kind=link}
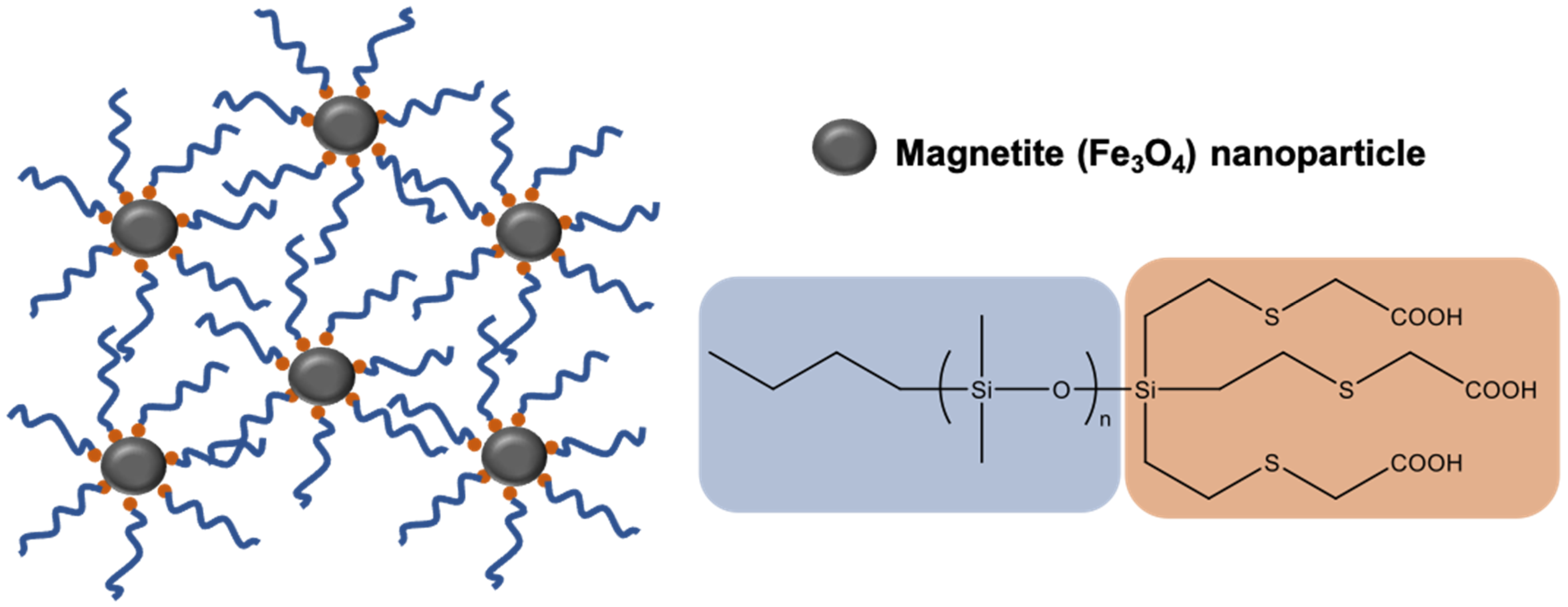{kind=link}
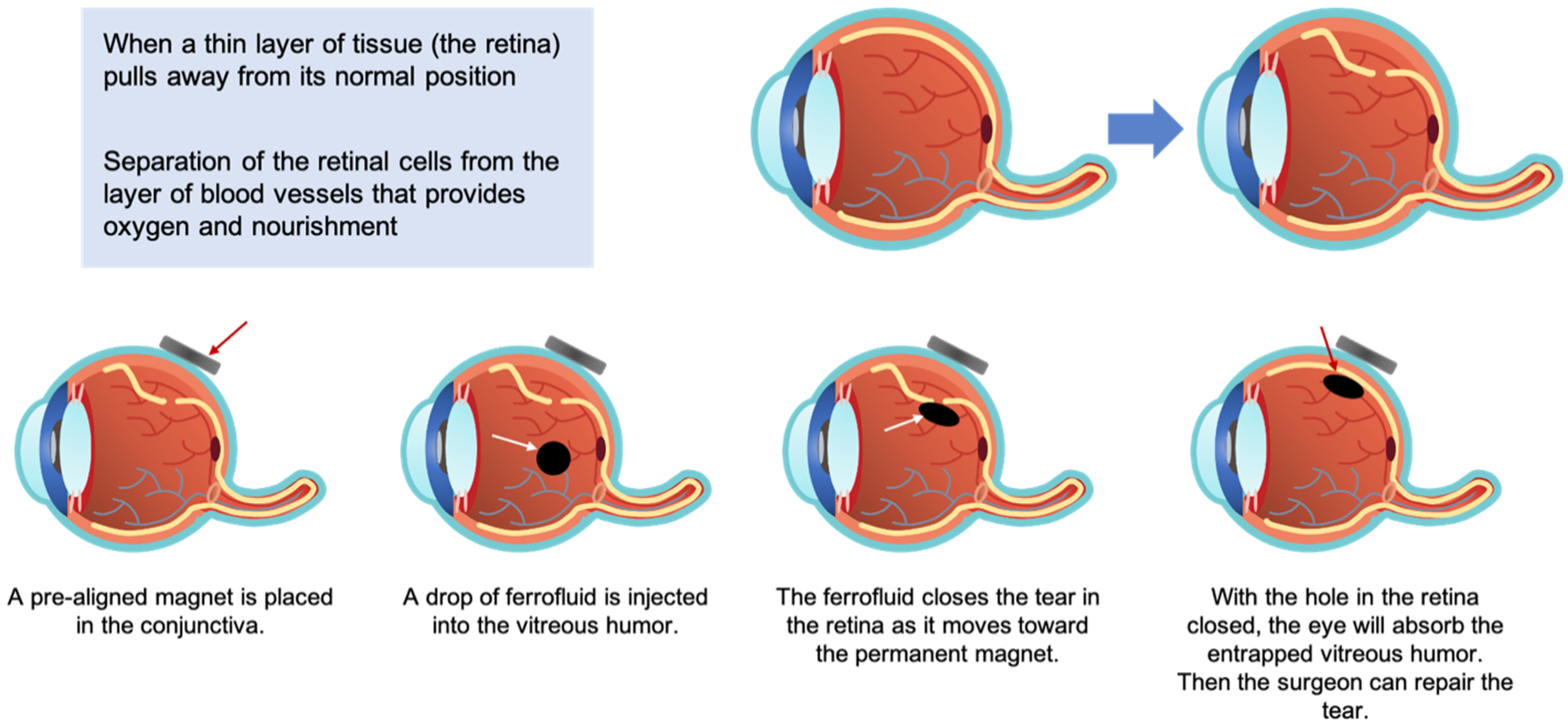{kind=link}
{kind=link}
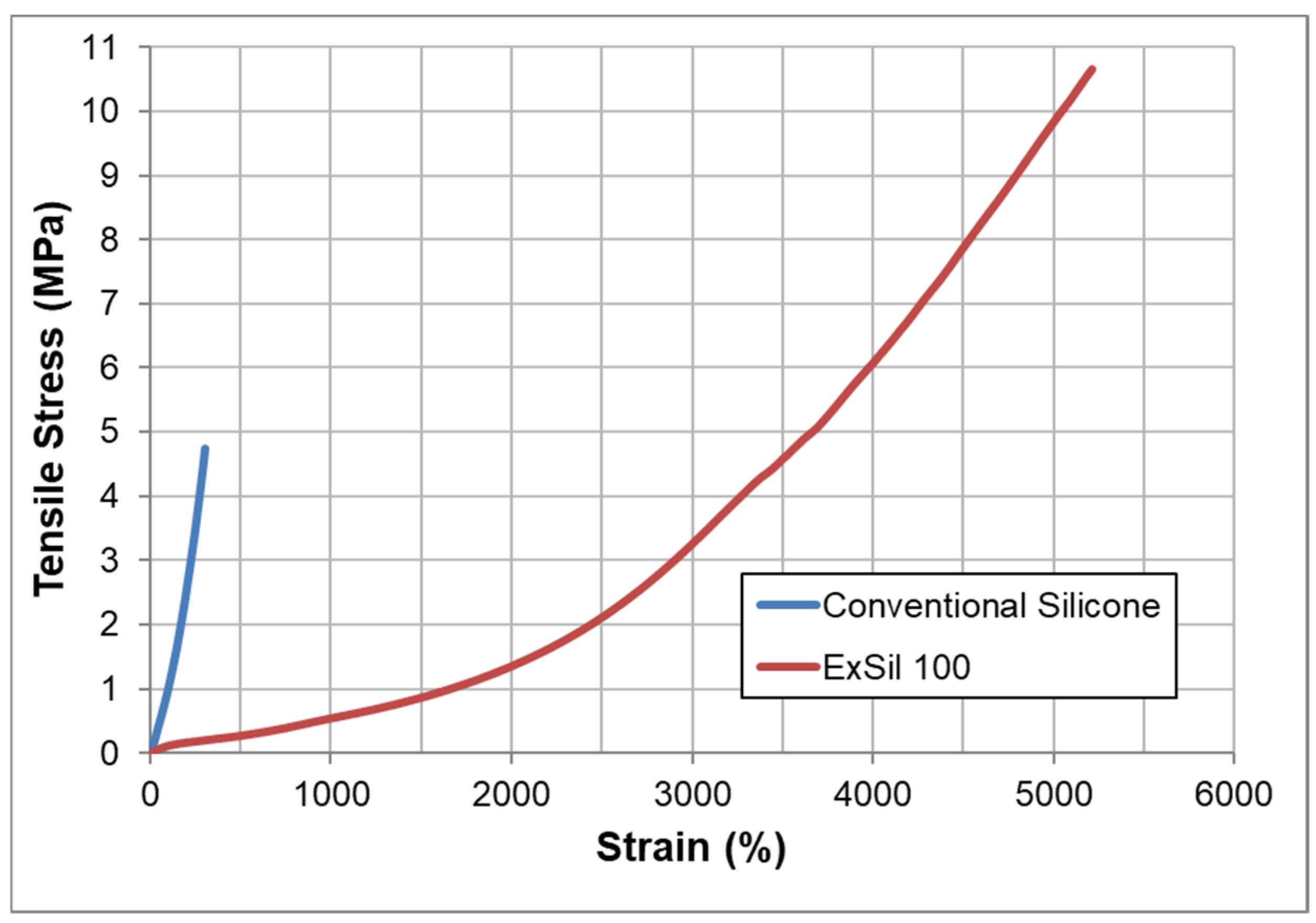{kind=link}
{kind=link}
{kind=link}
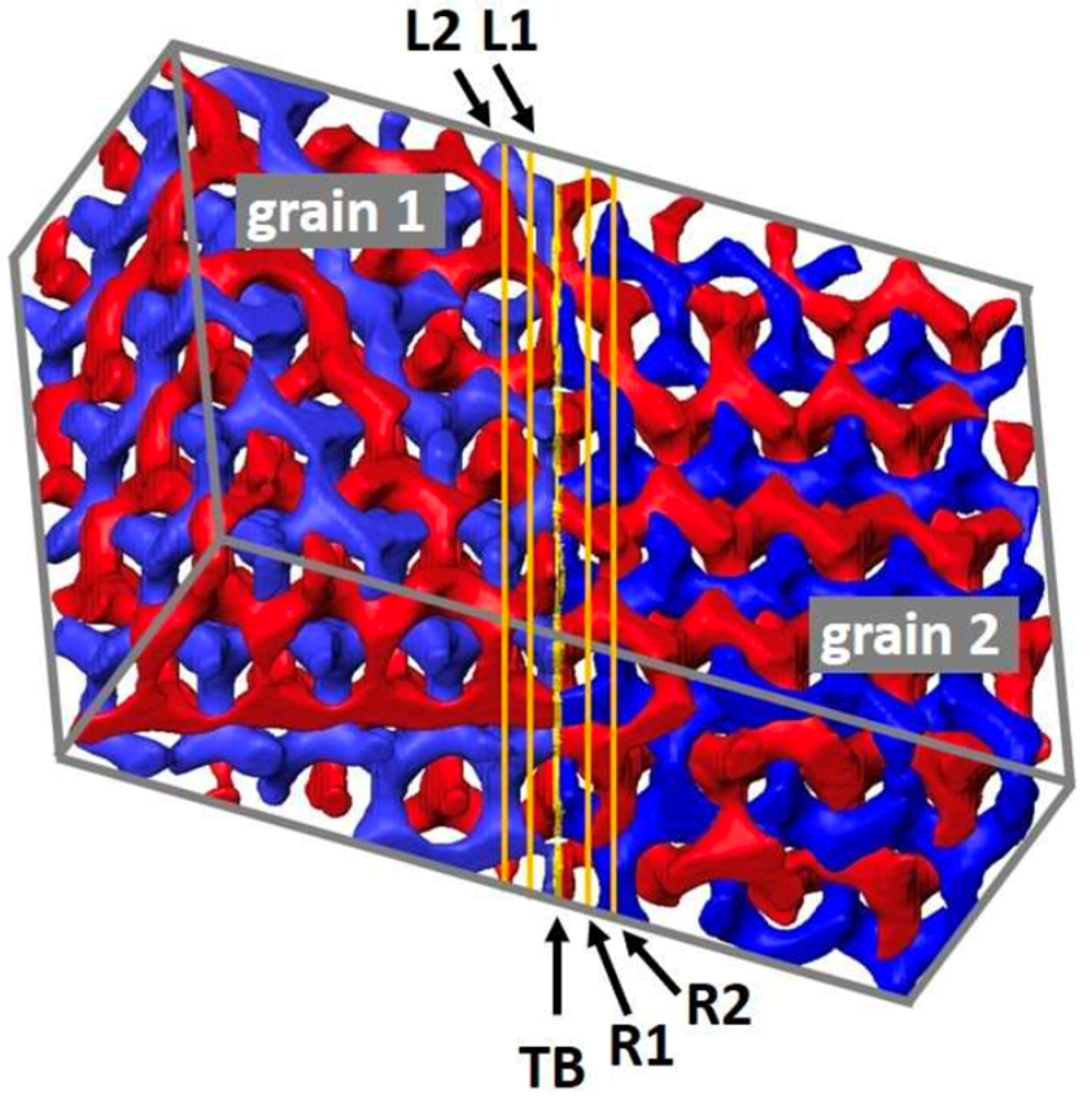{kind=link}
{kind=link}
{kind=link}
{kind=link}
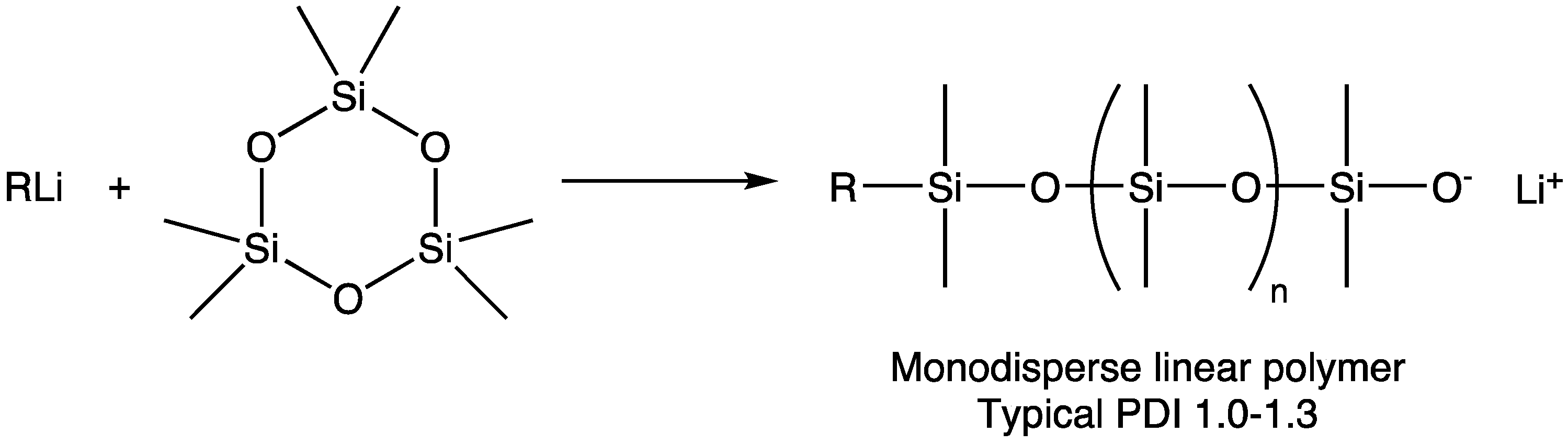{kind=link}
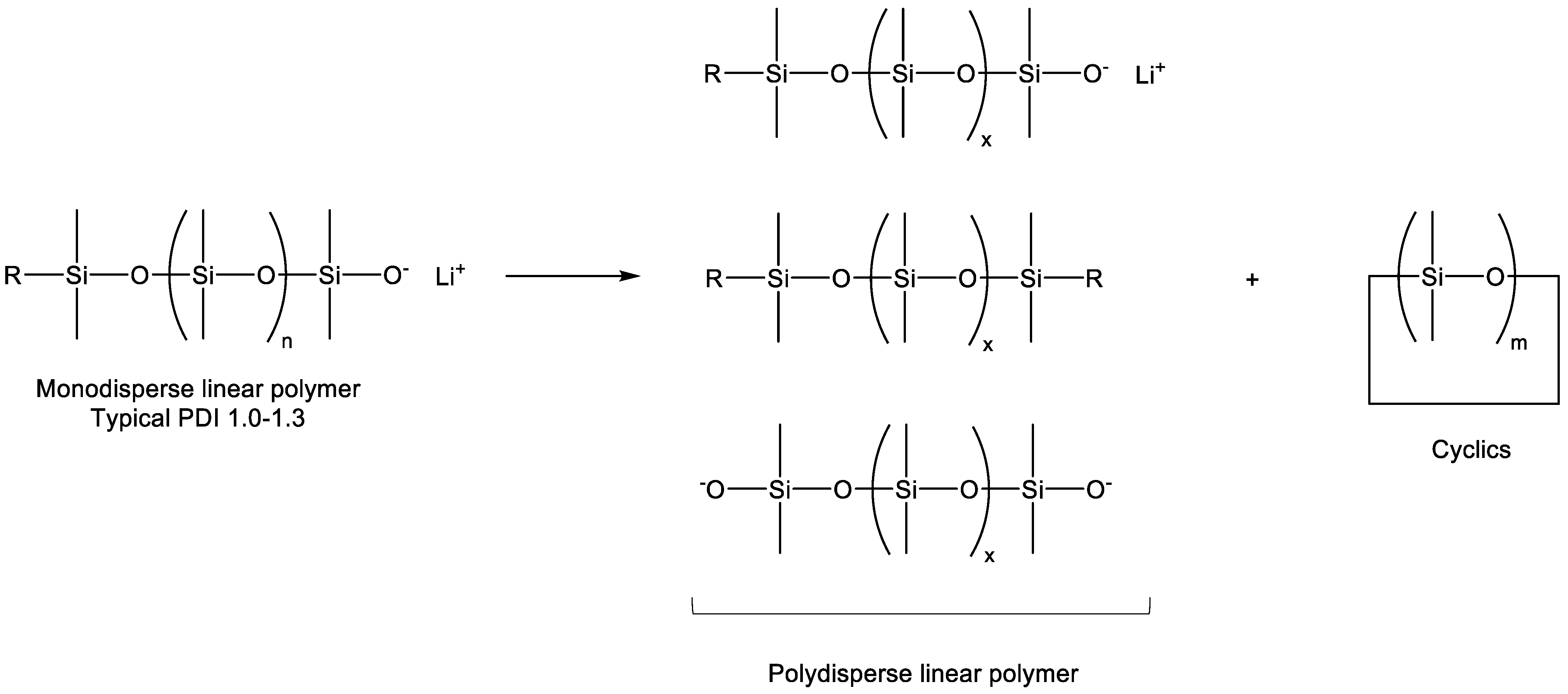{kind=link}
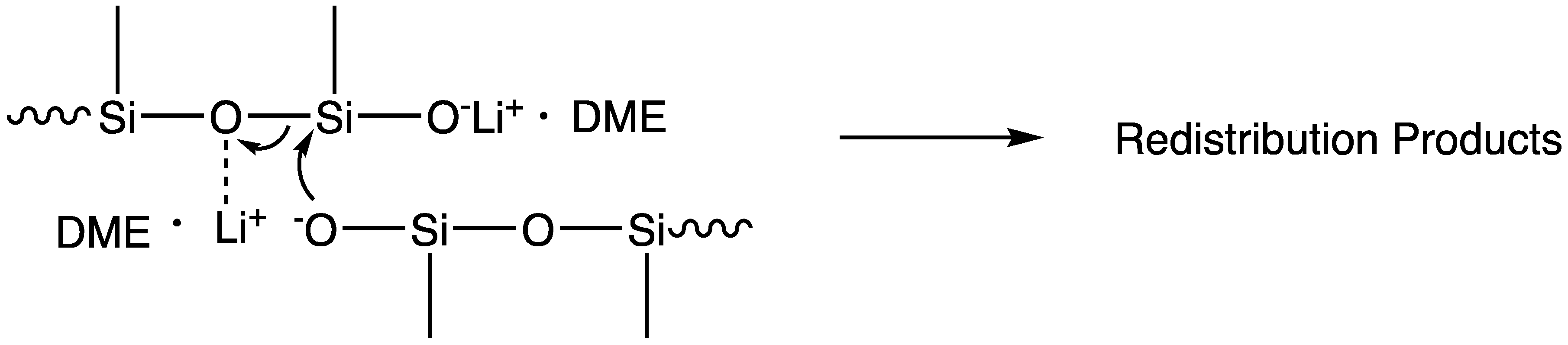{kind=link}
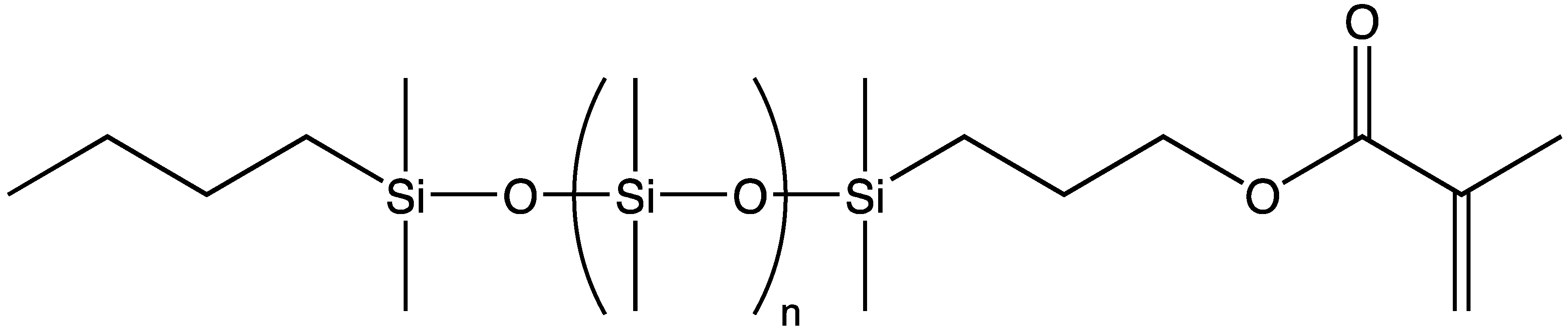{kind=link}
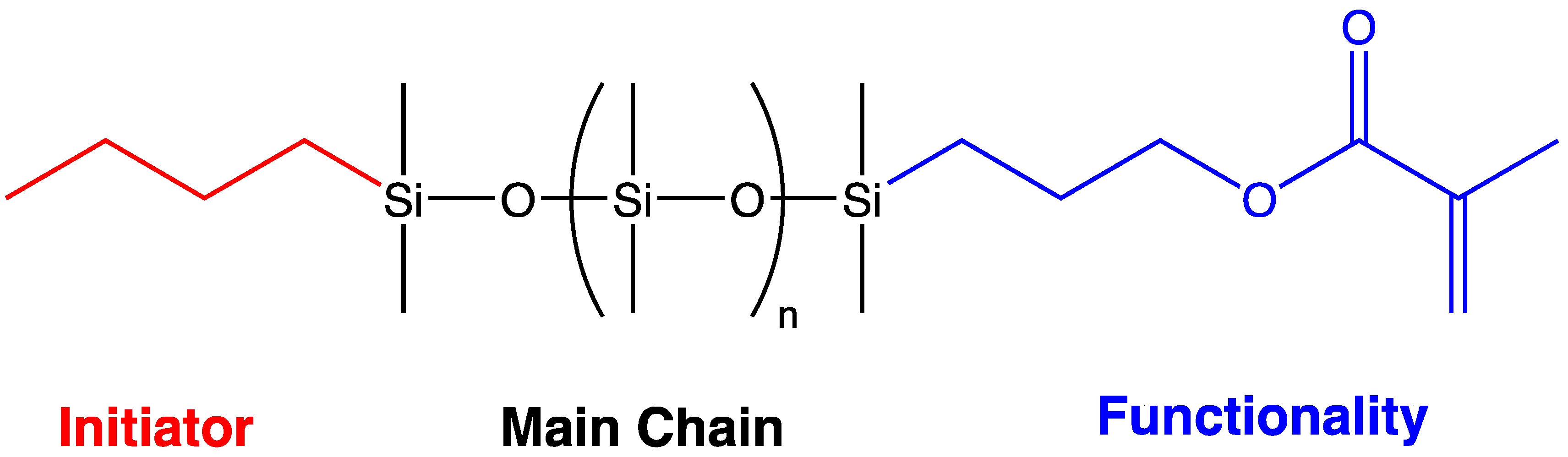{kind=link}
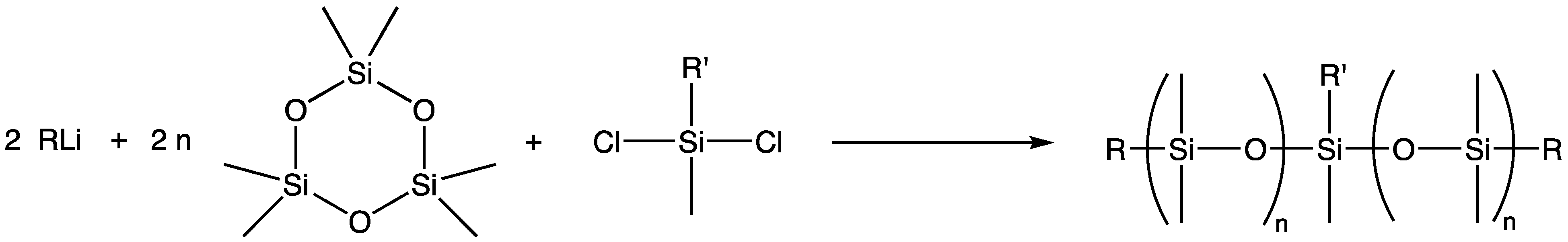{kind=link}
{kind=link}
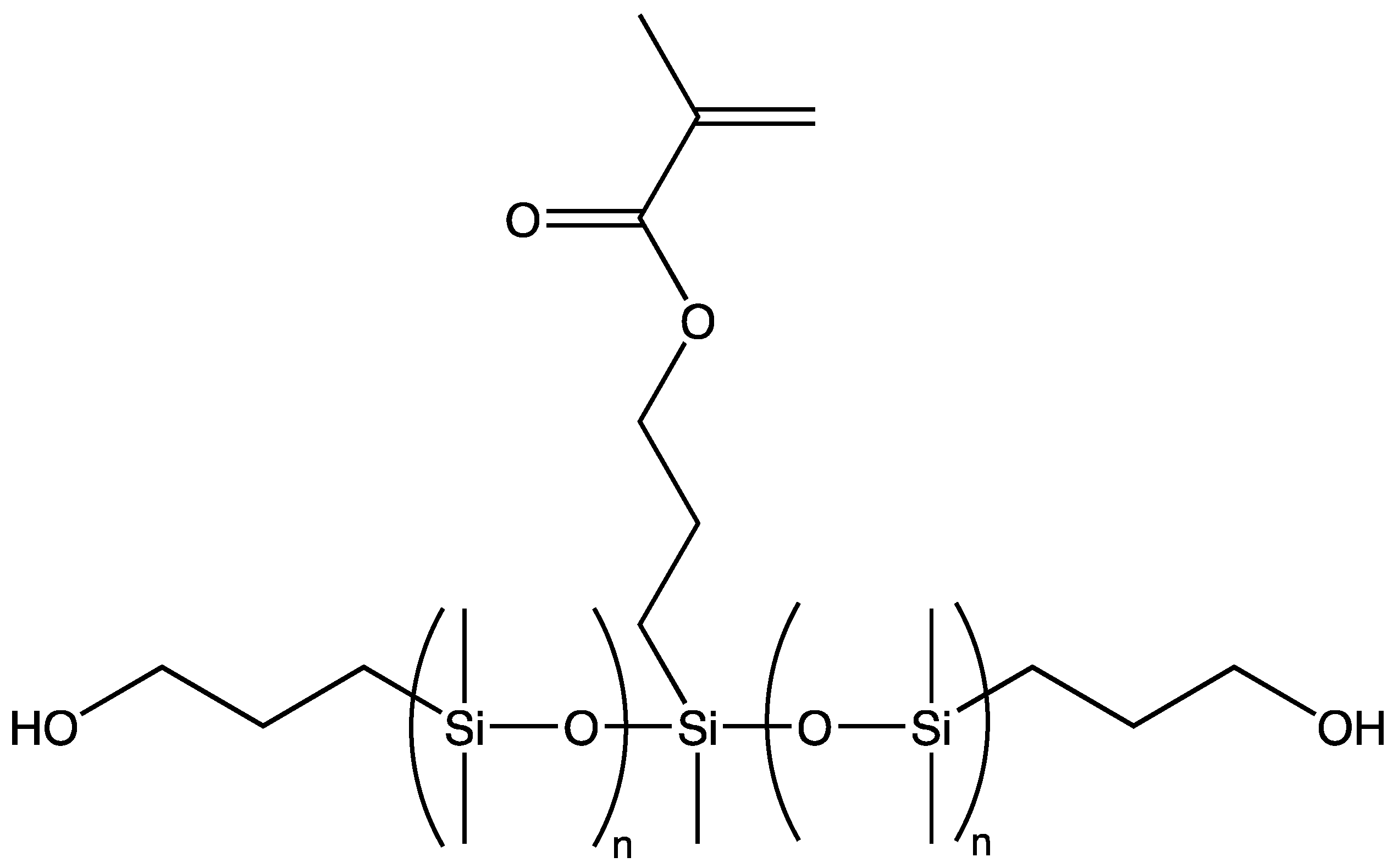{kind=link}
{kind=link}
{kind=link}
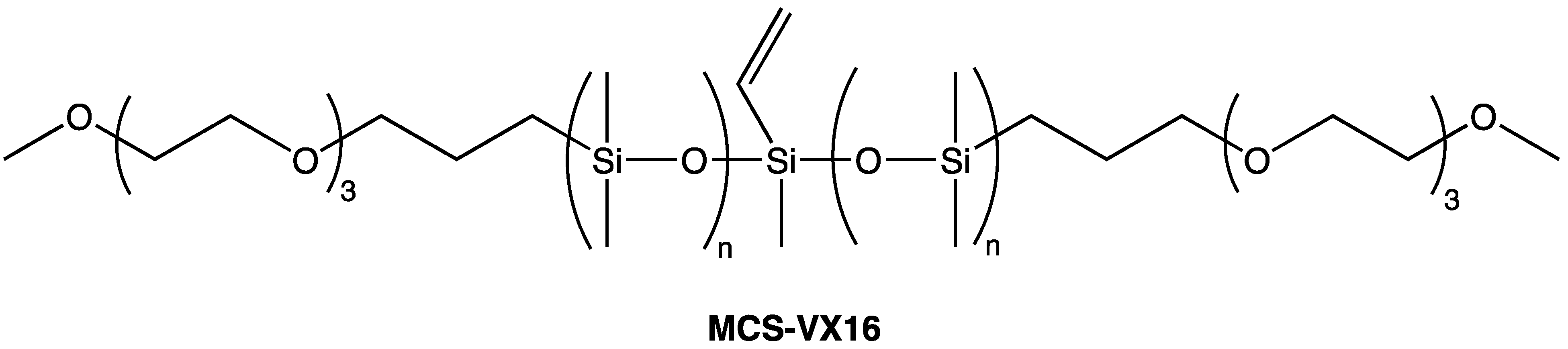{kind=link}
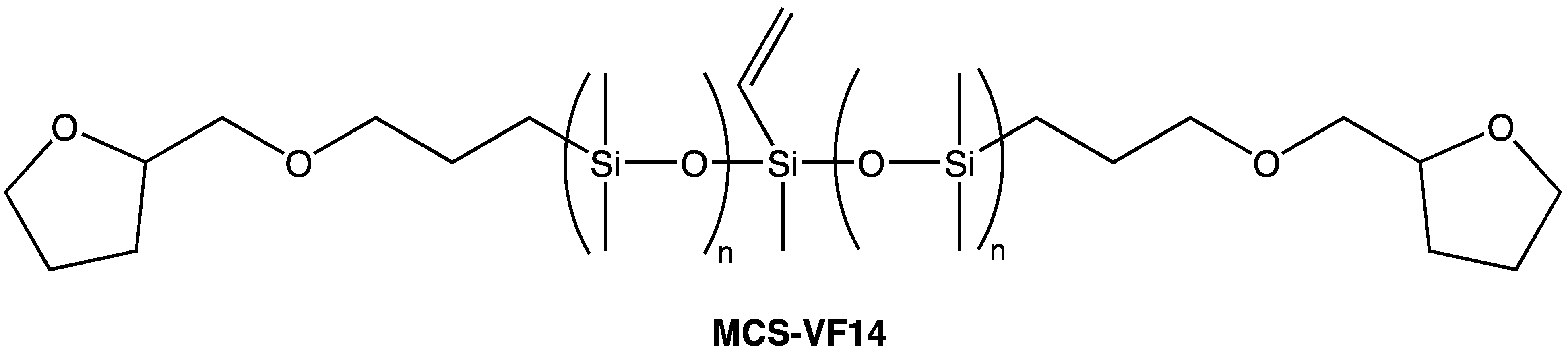{kind=link}
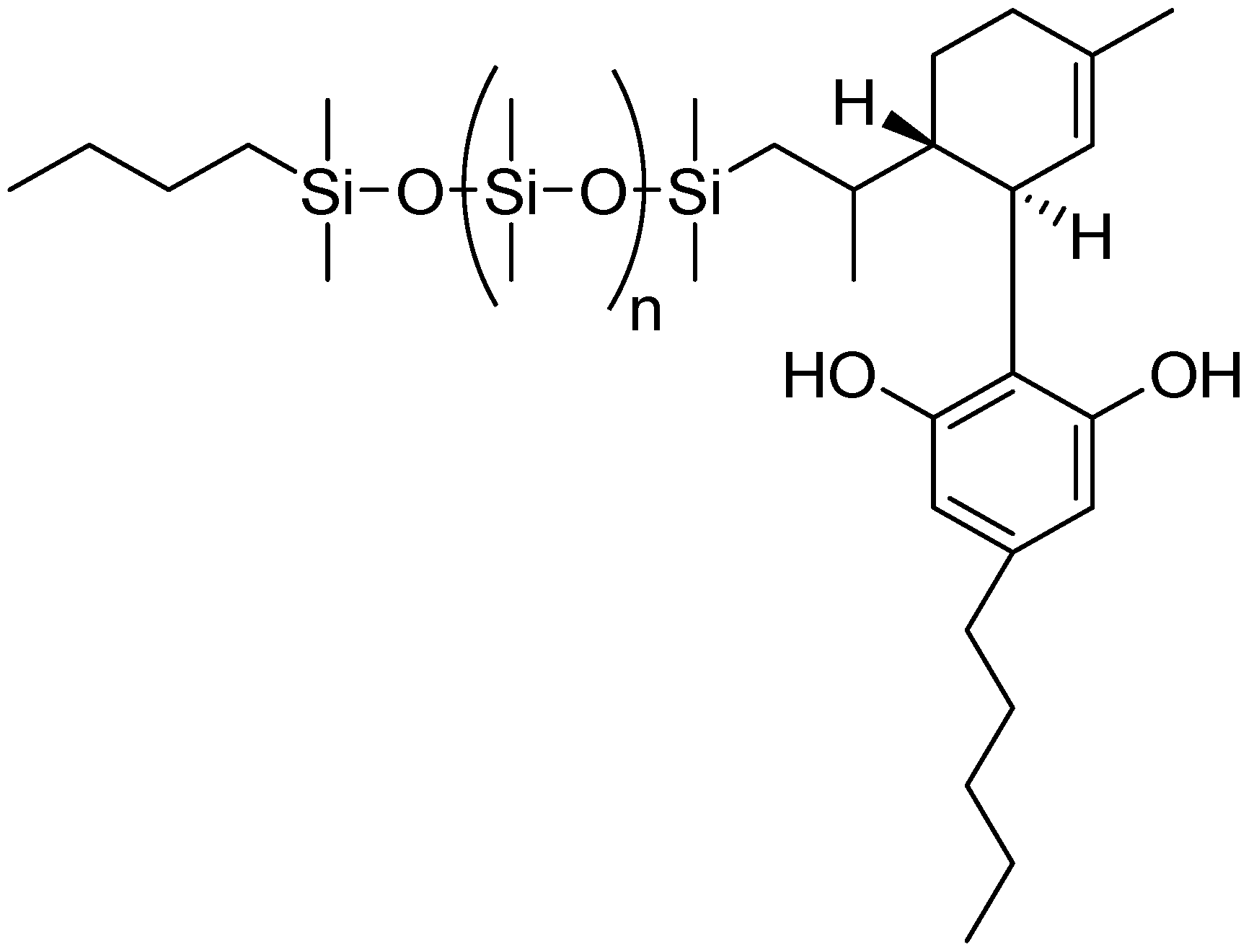{kind=link}
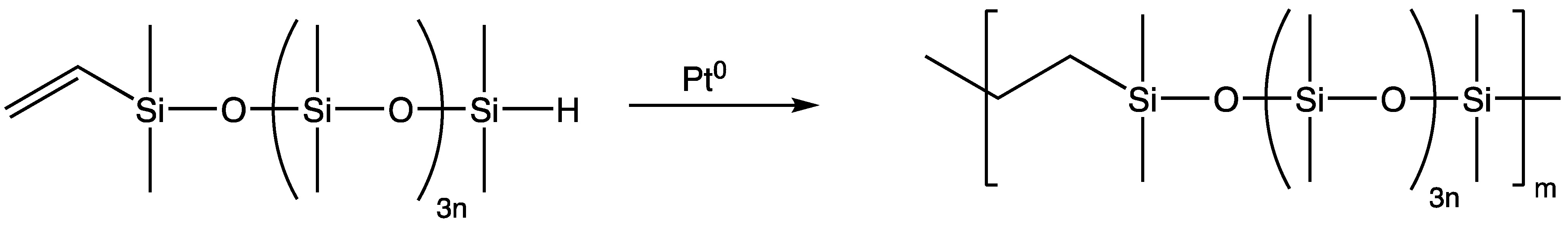{kind=link}
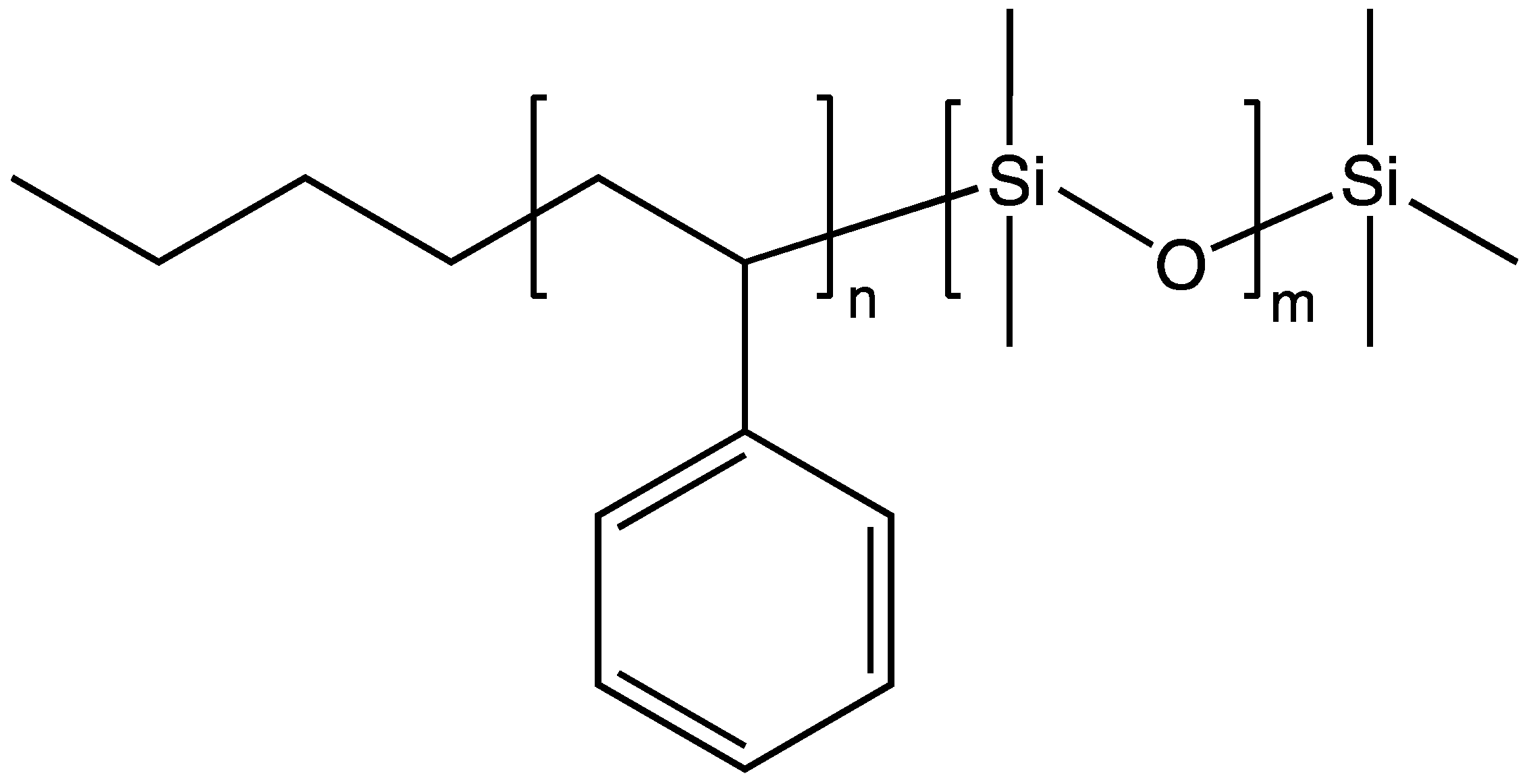{kind=link}
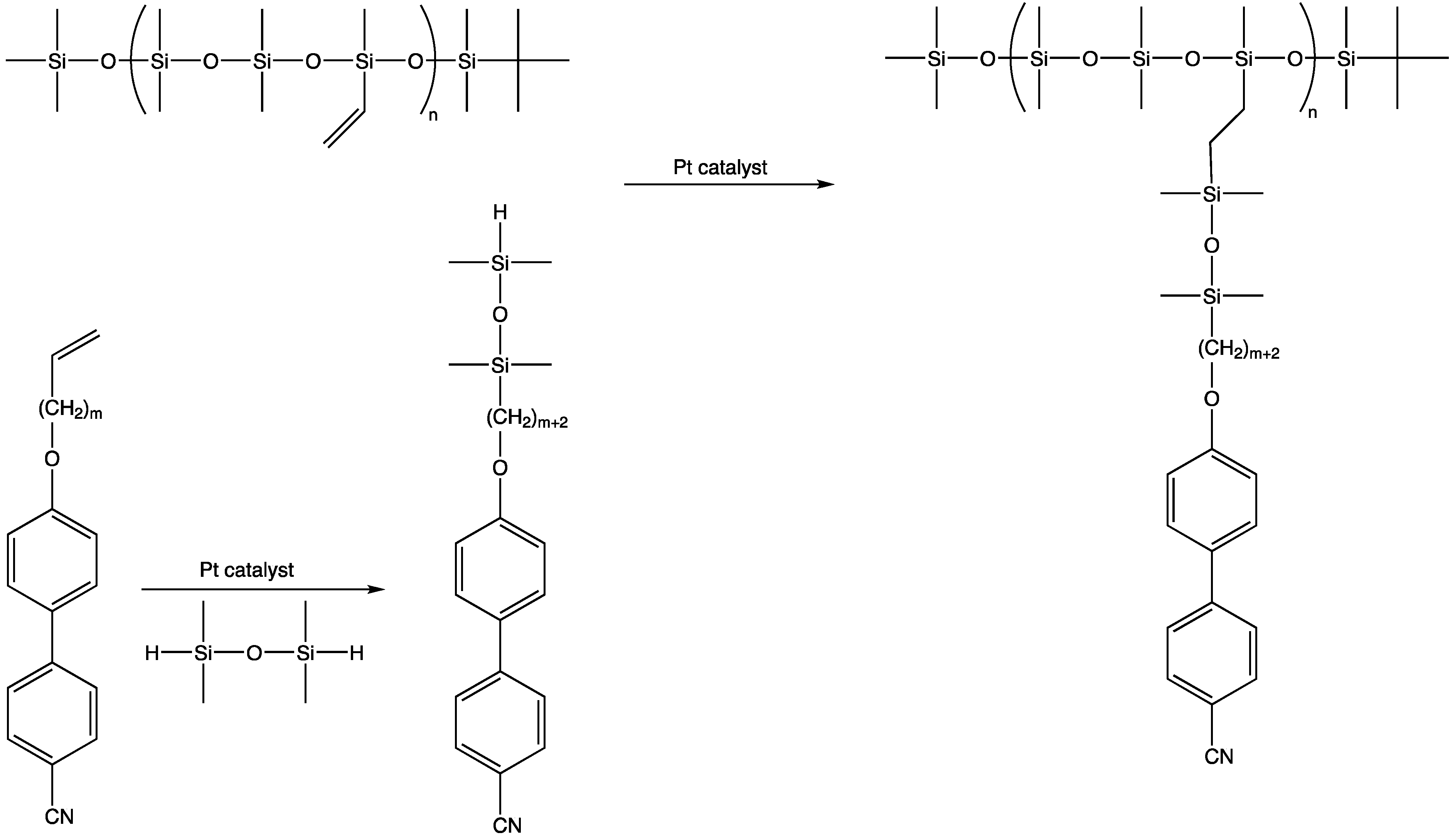{kind=link}
Table 1.
Cyclic Siloxane Macromer “Building Blocks”.
| Cyclic Siloxane Macromer Monomers | |||||
|---|---|---|---|---|---|
| Class | Structure | MW | B.P. | CAS# | References |
| Primary | |||||
| Hexamethylcyclotrisiloxane |  | 222.46 | 134° | 541-05-9 | Hyde [16] |
| Hexaphenylcyclotrisiloxane |  | 594.89 | 300°/1 | 512-63-0 | Bostick [32] |
| Trivinyltrimethylcyclotrisiloxane |  | 258.50 | 80°/20 | 3901-77-7 | Pike [33] |
| Tris(3,3,3-trifluoropropyl)trimethylcyclotrisiloxane |  | 468.55 | 95°/3 | 2374-14-3 | Pierce [18] |
| 2,2,5,5-tetramethyl-2,5-disila-1-oxacyclopentane |  | 160.36 | 124° | 7418-20-4 | Piccolli [17] |
| Minor | |||||
| Hexaethylcyclotrisiloxane |  | 306.82 | 117°/10 | 2031-79-0 | Dobay [34] |
| Vinylpentamethylcyclotrisiloxane |  | 234.47 | 56°/23 | 18395-32-9 | Rózga-Wijas [23] |
| Tris(tetramethylsiloxanylethyl)trimethylcyclotrisiloxane |  | 661.47 | 175°–185°/2 | 2378614-30-1 | Goff [22] |
| 1,3,5-[Tris(methoxyethoxyethoxypropyltetramethyldisiloxanyl)ethyl]-1,3,5-trimethylcyclotrisiloxane |  | 1142.10 | 2378623-20-0 | Goff [35] | |
Table 2.
Initiators.
| Non-Functional Initiators | |||
|---|---|---|---|
| Class | Structure | Comments | References |
| Organolithium | |||
| n-Butyl lithium |  | Most common initiator; stable and economic | Chojnowski [36] Cypryk [37] Maschke [38] Nakano [11] Razzano [39] |
| s-Butyl lithium |  | Less initial back-biting than n-butyl but less stable; used for substituted aryl-cyclic trisiloxane; used for higher alkylcyclo-trisiloxanes | Hawkridge [40] Bellas [41] Ninago [42] Lee [26] Molenberg [43,44,45] |
| t-Butyl lithium |  | Hempenius [46] Sieburth [47] | |
| Phenyl lithium |  | Initiator for PDMS MW standard | Arkles [48] |
| Trimethylsilylmethyl lithium |  | For diMeSiO-ViMeSiO block polymers | Boileau [49] Bauer [50] |
| Macroanions | |||
| Polybutadienyl lithium |  | Molenberg [19] | |
| Polystyryl lithium |  | Formation of block and star polymers—varied block size for nanopatterning | Saam [7,51] Bellas [44] Georgopanos [52] |
| Poly a-methylstyryl lithium |  | Saam [8] Noshay [53] | |
| Lithium Silanolate—Monoanion | |||
| Trimethylsilanol lithium salt |  | Formed from D4 + Methyllithium; initiates tetramethyldisila-oxacyclopentane polymerization | Clemens [54] Kawakami [9] Awasthi [55] |
| n-Butyldimethylsilanol lithium salt |  | Formed from D4 + n-Butyllithium | Fessler [6] Bostick [32] Cypryk [56] Leir [57] |
| Fluoroalkyldimethylsilanol lithium salt |  | Formed in situ from silanol and n-Butyllithium | Saho [58] |
| Methoxypropyldimethylsilanol lithium salt |  | Arkles [59] Goff [60] | |
| Methoxyethoxyethoxypropyldimethylsilanol lithium salt |  | Hydrophilic silicone macromers for contact lenses | Goff [61] Arkles [62] |
| Methoxyethoxyethoxyethoxypropyldimethylsilanol lithium salt |  | Hydrophilic additive for silicone elastomers | Goff [61] Arkles [62] |
| Tetrahydrofurfuryloxpropyldimethylsilanol lithium salt |  | Hydrophilic additive for implantable silicones | Goff [61] Arkles [62] |
| Lithium Silanolate—Multiple Anions | |||
| Diphenylsilanediol, dilithium salt |  | Initiator for pentamethylcyclo trisiloxane; initiator for trifluoropropyltrimethylcyclotrisiloxane | Paulasaari [21] Cypryk [37] |
| Tetramethyldisiloxane-1,3 diol dilithium salt |  | Gadda [63] | |
| 1,3-diphenyl-1,3-dimethylsiloxanediol, dilithium salt |  | Bostick [32] | |
| Bis(p-hydroxydimethylsilyl)phenyl ether, dilithium salt |  | Used to form telechelic vinyl-terminated polymers | Kazama [64] |
| Branched siloxanes, multiple lithium salts | Branched vinyl-terminated siloxanes | Ogawa [65] | |
| Branched carbosilanes, multiple lithium salts | Used to form multi-arm and dendrimer-like structures | Novozhilov [66] Vasilenko [67] | |
| Other | |||
| Dilithiobenzophenone |  | Siloxane methacrylate copolymers | Juliano [68] |
| Dilithiofluorenone |  | Color change when D3 polymerization initiated | Juliano [68] |
| Functional Initiators | |||
| Class | Structure | Comments | References |
| Organo Lithium | |||
| Allyl lithium |  | Functionalized polydiethylsiloxane | Out [69] |
| t-Butyldimethylsiloxypropyl lithium |  | Deprotected to form hydroxyl termination | Arkles [70] Arkles [59] |
| N-Benzyl-N-methylaminopropyl lithium |  | Can be deprotected to provide N-methylaminopropyl termination | Elkins [71] |
| Bis(trimethylsilyl)aminophenyl lithium |  | Deprotected to form aminophenyl termination | Babu [72] |
| Lithium Silanolate | |||
| Vinyldimethylsilanol lithium salt |  | Goff [73] Okawa [73,74] Arkles [62] | |
| Vinylbutylmethylsilanol lithium salt |  | Arkles [62] | |
| Hydridobutylmethylsilanol lithium salt |  | Arkles [62] | |
| 1-Hexenyl, 4-Hydroxyl-octamethyltetrasiloxane lithium salt |  | Okawa [74] | |
| Other | |||
| Lithium anion of Na pentanedionate |  | Forms Na AcAc-terminated polymer, but not stable after neutralization | Kumar [75] |
| 1,5,7-Triazabicyclo [4.4.0] dec-5-ene |  | Used in the polymerization of monodisperse silanol-terminated siloxanes | Lohmeijer [14] |
| 1,3-Trimethylene-n-propylguanidine |  | Used in combination with water and silanols | Fuchise [15,76] |
Table 3.
Commonly Reported Macromers.
| Asymmetric Monofunctional Siloxanes | ||||||
|---|---|---|---|---|---|---|
| Code * | Description | Molecular Weight, Mn | Viscosity, cSt | Density | Refractive Index | Reference |
 | ||||||
| MCR-A11 | monoAMINOPROPYL TERMINATED POLYDIMETHYLSILOXANE | 800–1000 | 8–12 | 0.82 | 1.411 | [79,80,81] |
| MCR-A12 | monoAMINOPROPYL TERMINATED POLYDIMETHYLSILOXANE | 2000 | 18–25 | 0.97 | 1.411 | [79,81] |
 | ||||||
| MCR-W15 | monoACRYLAMIDOPROPYL TERMINATED POLYDIMTHYLSILOXANE | 1000–1500 | 50–75 | 0.96 | 1.418 | [79] |
 | ||||||
| MCR-C12 | monoCARBINOL TERMINATED POLYDIMETHYLSILOXANE | 1000 | 15–20 | 0.96 | 1.409 | [79,82] |
| MCR-C18 | monoCARBINOL TERMINATED POLYDIMETHYLSILOXANE | 5000 | 80–90 | 0.97 | 1.405 | [79,82,83,84] |
| MCR-C22 | monoCARBINOL TERMINATED POLYDIMETHYLSILOXANE | 10,000 | 250 | 0.98 | 1.404 | [79,81,82,85] |
 | ||||||
| MCR-C61 | monoDICARBINOL TERMINATED POLYDIMETHYLSILOXANE | 1000 | 50–60 | 0.97 | 1.417 | [79,86] |
| MCR-C62 | monoDICARBINOL TERMINATED POLYDIMETHYLSILOXANE | 5000 | 100–125 | 0.97 | 1.409 | [79,86,87] |
 | ||||||
| MCR-B12 | monoCARBOXYDECYL TERMINATED POLYDIMETHYLSILOXANE | 1500 | 20 | 0.94 | 1.415 | [79,88] |
 | ||||||
| MCR-E11 | Mono(2,3-EPOXY)PROPYLETHER TERMINATED POLYDIMETHYLSILOXANE | 1000 | 10–15 | 0.96 | 1.410 | [79,89,90] |
| MCR-E21 | Mono(2,3-EPOXY)PROPYLETHER TERMINATED POLYDIMETHYLSILOXANE | 5000 | 120 | 0.97 | 1.408 | [79] |
 | ||||||
| MCR-H07 | MonoHYDRIDE TERMINATED POLYDIMETHYLSILOXANE | 800–900 | 5–8 | 0.96 | 1.404 | [60,79] |
| MCR-H11 | MonoHYDRIDE TERMINATED POLYDIMETHYLSILOXANE | 900–1100 | 8–12 | 0.96 | 1.407 | [60,79,91] |
| MCR-H21 | MonoHYDRIDE TERMINATED POLYDIMETHYLSILOXANE | 4500–5000 | 80–120 | 0.96 | 1.411 | [79] |
| MCR-H22 | MonoHYDRIDE TERMINATED POLYDIMETHYLSILOXANE | 10,000 | 160–220 | 0.98 | 1.411 | [79] |
 | ||||||
| MCR-M07 | MonoMETHACRYLOXYPROPYL TERMINATED POLYDIMETHYLSILOXANE | 600–800 | 6–9 | 0.96 | 1.416 | [60,79,92,93,94,95,96,97] |
| MCR-M11 | MonoMETHACRYLOXYPROPYL TERMINATED POLYDIMETHYLSILOXANE | 800–1000 | 10–11 | 0.96 | 1.411 | [60,70,79,92,93,95,96,97,98,99,100,101,102,103,104,105,106,107,108] |
| MCR-M17 | MonoMETHACRYLOXYPROPYL TERMINATED POLYDIMETHYLSILOXANE | 5000 | 70–80 | 0.97 | 1.406 | [54,70,79,89,90,93,95,96,97,108,109,110] |
| MCR-M22 | MonoMETHACRYLOXYPROPYL TERMINATED POLYDIMETHYLSILOXANE | 10,000 | 150–200 | 0.97 | 1.405 | [54,79,93,94,96,97] |
 | ||||||
| MCR-MXe11 | MonoMETHACRYLOXYETHOXY-PROPYL TERMINATED POLYDIMETHYLSILOXANE | 900–1000 | 9–10 | 0.95 | 1.415 | None? |
 | ||||||
| MCR-MXt11 | MonoMETHACRYLOXYPROPYL, MonoMETHOXYPROPYL TERMINATED POLYDIMETHYLSILOXANE | 800–1000 | 8–12 | 0.96 | 1.430 | [60,70] |
 | ||||||
| MFR-M15 | MonoMETHACRYLOXYPROPYL TERMINATED POLYTRIFLUOROPROPYLMETHYLSILOXANE | 800–1000 | 50–70 | 1.09 | 1.398 | [59,60,70,79,93,100] |
 | ||||||
| MCR-ST11 | MonoSTYRYL TERMINATED POLYDIMETHYLSILOXANE | 800–1000 | 8–15 | 0.95 | 1.429 | [99,102] |
 | ||||||
| MCR-V21 | MonoVINYL TERMINATED POLYDIMETHYLSILOXANE | 5500–6500 | 80–120 | 0.97 | 1.403 | [79,91,111] |
| MCR-V25 | MonoVINYL TERMINATED POLYDIMETHYLSILOXANE | 15,000–20,000 | 400–600 | 0.97 | 1.403 | [79] |
| MCR-V41 | MonoVINYL TERMINATED POLYDIMETHYLSILOXANE | 55,000–65,000 | 8000–12,000 | 0.98 | 1.404 | [79] |
 | ||||||
| MCR-XT11 | MonoTRIETHOXYSILYLETHYL TERMINATED POLYDIMETHYLSILOXANE | 500–1000 | 16–24 | 0.97 | 1.412 | [79] |
| Symmetric Monofunctional Siloxanes | ||||||
 | ||||||
| MCS-C13 | MonoCARBINOL FUNCTIONAL POLYDIMETHYLSILOXANE, symmetric | 550–650 | 35–40 | 1.02 | 1.446 | [79] |
 | ||||||
| MCS-E15 | Mono(2,3-EPOXY)PROPYL ETHER FUNCTIONAL POLYDIMETHYLSILOXANE, symmetric | 800–900 | 45–55 | 1.09 | 1.398 | [59,70,79] |
 | ||||||
| MCS-M11 | MonoMETHACRYLOXYPROPYL FUNCTIONAL POLYDIMETHYLSILOXANE, symmetric | 800–1000 | 7–9 | 0.93 | 1.417 | [59,60,70,79,93] |
 | ||||||
| MCS-MXt11 | MonoMETHACRYLOXYPROPYL FUNCTIONAL POLYDIMETHYLSILOXANE, METHOXYPROPYL TERMINATED, symmetric | 900–1100 | 8–12 | 0.96 | 1.43 | [60,79] |
 | ||||||
| MCS-V212 | MonoVINYL TERMINATED POLYDIMETHYLSILOXANE, symmetric | 1200–1400 | 16–24 | 0.97 | 1.419 | [79] |
 | ||||||
| MCS-VF14 | monoVINYL FUNCTIONAL POLYDIMETHYLSILOXANE, TETRAHYDROFURFURYLOXYPROPYL TERMINATED—symmetric | 1000–1200 | 30–40 | 0.976 | 1.4141 | [61,79,87,112] |
 | ||||||
| MCS-VXt15 | MonoVINYL FUNCTIONAL POLYDIMETHYLSILOXANE, METHOXY(DIETHYLENEOXIDE)PROPYL TERMINATED, symmetric | 1000–1200 | 40–60 | 0.96 | 1.42 | [61,79,87,112] |
* In order to provide a concise description of the macromer systems discussed, the following designation system was adopted: the first two letters, MC, indicate macromer M structure with a C conventional dimethylsiloxane repeat unit; the third letter indicates whether it is asymmetric (R), symmetric (S), or T-structure (T). Macromer functionality and viscosity are indicated after a hyphen, as follows: (1) letters indicate functionality, where M is methacrylate, H is hydride, C is carbinol, V is vinyl, and E is methoxy; (2) numbers are the viscosity ranges of the macromer in powers of 10, followed by the first significant figure. For example, MCR-H25 indicates a macromer, an asymmetric architecture with hydride functionality, and a viscosity of 102 × 5 or 500 cSt. These designations provide a facile method of discussing comparative structures but are not intended to be detailed structural descriptions of the polymers. To remove ambiguity for ethyleneoxy designated X spacer groups, the lower-case letter t, for “tipped”, or e, for “embedded”, is used.
Table 4.
Monodisperse Difunctional Macromers.
| Heterobifunctional Siloxanes | ||||||
|---|---|---|---|---|---|---|
| Code | Description | Molecular Weight, Mn | Viscosity, cSt | Density | Refractive Index | Reference |
 | ||||||
| DMS-HV15 | monoVINYL-monoHYDRIDE terminated POLYDIMETHYLSILOXANE | 2000–3000 | 40–60 | 0.96 | 1.404 | [62,73,79,113,114] |
| DMS-HV22 | monoVINYL-monoHYDRIDE terminated POLYDIMETHYLSILOXANE | 10,000 | 150–250 | 0.97 | 1.403 | [62,73,79,113,114,115,116] |
| DMS-HV31 | monoVINYL-monoHYDRIDE terminated POLYDIMETHYLSILOXANE | 25,000 | 600–1000 | 0.97 | 1.403 | [62,73,79,114] |
 | ||||||
| PMM-HV12 | α-monoVINYL,monoPHENYL-ω-monoHYDRIDE terminated POLYDIMETHYLSILOXANE | 2000 | 20 | 0.97 | 1.414 | [62,79] |
Table 5.
Reactivity Ratios of Methacrylate Functional Siloxane Macromers.
| Siloxane Macromer | Comonomer | Reactivity Ratio (r1:r2) * |
|---|---|---|
| MCR-M11 | Methylmethacrylate | nm **:1.60 |
| MCR-M22 | nm **:2.10 | |
| MCR-M11 | Styrene | 0.26:1.07 |
| MCR-M11 | Acrylonitrile | 5.40:0.89 |
* Rate constants—M1M1/M1M2: M2M2/M2M1; ** no meaningful results.
Table 6.
Solubility of Methacrylate Functional Silicone Macromers *.
| Siloxane Macromer—Methacrylate Functional | Macromer Solubility in Hydrogel Monomers | Water Solubility in Macromer (ppm) | |
|---|---|---|---|
| Dimethylacrylamide (DMA) | Hydroxyethylmethacrylate (HEMA) | ||
| MCR-M11 (asymmetric) | 4% | 1% | 2000 |
| MCS-M11 (symmetric) | 8% | 2% | 3500 |
| MFR-M15 (fluorinated asymmetric) | 100% (miscible) | 2% | 9000 |
| MFS-M15 (fluorinated symmetric) | 100% (miscible) | 3% | 10,000 |
| MCS-MXt11 (methoxy-tipped, symmetric) | 100% (miscible) | 100% (miscible) | 10,000 |
* Author’s work, previously unreported.
Table 7.
Silicone Hydrogel Contact Lens Technology.
| Technology Generation | Proprietary Name | Material | Manufacturer | Launch Date | Water Content (%) | Dk | O2 Transport Enhancer | Hydrophilic Component * | Refs. |
|---|---|---|---|---|---|---|---|---|---|
| 1 | PureVision | Balafilcon A | Bausch & Lomb | 1998 | 36 | 91 | TRIS, Difunctional siloxane | NVP amino acid monomer | |
| Focus Night & Day | Lotrfilcon A | CIBA Vision | 1999 | 24 | 140 | TRIS, Difunctional fluorosiloxane | DMA | Nicolson [124] | |
| AirOptix | Lotrafilcon B | CIBA Vision | 1999 | 33 | 110 | TRIS, Difunctional fluorosiloxane | DMA | Nicolson [124] | |
| 2 | Biofinity | Cofilcon A | Cooper vision | 2007 | 48 | 128 | Monofunctional siloxane macromer | VMA | Ueyama [125] Iwata [126] |
| Acuvue OASYS | Senfilcon A | JJVC | 2004 | 38 | 103 | SiGMA monomer, monofunctional siloxane macromer (MCR-M11) | DMA, HEMA, PVP | Vanderlaan [101,123] | |
| PremuiO | Amofilcon A | Menicon | 2007 | 40 | 129 | Difunctional fluorosiloxane macromer | DMA | ||
| Acuvue Advance | Gayfilcon A | JJVC | 2004 | 47 | 60 | SiGMA monomer, monofunctional siloxane macromer (MCR-M11) | DMA, HEMA, PVP | Vanderlaan [101,123] | |
| 3 | 1-DAY Acuvue TruEYE | Narfilcon A | JJVC | 2008 | 46 | 100 | SiGMA monomer, monofunctional siloxane macromers (OH-mPDMS, MCR-M11) | DMA, HEMA, PVP | Vanderlaan [101,123] |
| Clariti 1-DAY | Somofilcon | Cooper vision | 2014 | 56 | 60 | Monofunctional siloxane macromer, difunctional siloxane | HEMA | Broad [100] | |
| Avaira | Enfilcon | Cooper vision | 2011 | 46 | 100 | Difunctional fluorosiloxane | VMA | ||
| Ultra | Samfilcon A | Bausch & Lomb | 2014 | 46 | 114 | TRIS, Difunctional siloxane, monofunctional carbosiloxane macromer | Unknown | Awasti [55,127] | |
| DAILIES TOTAL1 | Delfilcon A | Alcon | 2013 | 33 | 140 | Unknown | DMPC, DMA | ||
| MyDay | Stenfilcon A | Cooper vision | 2013 | 54 | 80 | Difunctional PEG-PDMS copolymer, monofunctional siloxane macromer | VMA | ||
| Acuvue Vita | Senfilcon C | JJVC | 2016 | 41 | 103 | SiGMA monomer, monofunctional siloxane macromer (MCR-M11) | DMA, HEMA, PVP |
* DMA—Dimethylacrylamide; DMPC—(1,2-dimyristoyl-sn-glycero-3-phosphocholine; HEMA—Hydroxyethylmethacrylate; NVP—N-Vinylpyrrolidone; PVP—Polyvinylpyrrolidone; SiGMA—3-(3-Methacryloxy-2-hydroxypropoxy)propylbis(trimethylsiloxy)methylsilane; TRIS—Methacryloxypropyltris(trimethylsiloxy)silane; VMA—N-Vinylmethylacetamide.
Table 8.
Comparing Contact Angle, Abrasion, and Friction in an IPDI-based Urethane.
| Control | A | B | C | |
|---|---|---|---|---|
| Description | Blend | Block Polymer | Pendant/Side-Chain | |
| Silicone Component | Silicone Fluid | Telechelic Carbinol | Dicarbinol Terminated Asymmetric Macromer | |
| Abrasion Test Failure, cycles | <100 | <100 | <100 | 4200 |
| Coefficient of Friction | 0.61 | 0.12 | 0.74 | 0.16 |
| Contact Angle, water before abrasion | 56° | 83° | 72° | 82° |
| Contact Angle, water after abrasion | 55° | 55° | 72° | 80° |
Control (no siloxane); (A) introduction of 50 cSt dimethylsiloxane fluid as a non-reactive additive; (B) telechelic siloxane MW 5000 (DMS-C21) added to form a soft-block copolymer; (C) macromer MW 5000 (MCR-C62) forming a brush polymer with siloxane pendant.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Goff, J.; Sulaiman, S.; Arkles, B. Applications of Hybrid Polymers Generated from Living Anionic Ring Opening Polymerization. Molecules 2021, 26, 2755. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26092755
AMA Style
Goff J, Sulaiman S, Arkles B. Applications of Hybrid Polymers Generated from Living Anionic Ring Opening Polymerization. Molecules. 2021; 26(9):2755. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26092755
Chicago/Turabian StyleGoff, Jonathan, Santy Sulaiman, and Barry Arkles. 2021. "Applications of Hybrid Polymers Generated from Living Anionic Ring Opening Polymerization" Molecules 26, no. 9: 2755. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26092755